

Since its discovery in 1997 (Li and Sun, 1997; Li et al., 1997; Steck et al., 1997), the phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)P3] phosphatase and tensin homolog (PTEN) has been established as one of the most frequently mutated tumor suppressor genes in human cancer. PTEN is a phosphatase that catalyzes the conversion of the lipid second messenger PtdIns(3,4,5)P3 to phosphatidylinositol (4,5)-bisphosphate [PtdIns(4,5)P2] (Maehama and Dixon, 1998). As such, PTEN functions as a main regulator of the cellular PtdIns(3,4,5)P3 concentration to antagonize the signaling cascades downstream of receptor tyrosine kinases (RTKs) and phosphatidylinositol-3-kinase (PI3K). Interestingly, PTEN might also possess protein phosphatase activity, and several potential protein substrates have been reported (Gu et al., 1999; Mahimainathan and Choudhury, 2004; Raftopoulou et al., 2004; Tamura et al., 1998).
PTEN contains multiple domains, including an N-terminal phosphatase domain, a central C2 domain and a C-terminal tail. The phosphatase and C2 domains form a minimal enzymatic unit that is sufficient for metabolizing PtdIns(3,4,5)P3. The C-terminal tail is a long flexible fragment that is mainly involved in PTEN regulation.
Despite having initially been discovered as a tumor suppressor, PTEN has attracted great interest from various research fields because of its diverse physiological functions. The functional diversity of PTEN demands a collection of delicate regulatory mechanisms, including transcriptional and post-translational regulation in a tissue- and context-dependent manner. This Cell Science at a Glance article summarizes the diversity of PTEN functions, and the mechanisms that underlie the regulation of PTEN expression, its enzymatic activity and subcellular localization.

PTEN has diverse functions in mammalian cells
PTEN as a tumor suppressor
PTEN mutations occur frequently in a variety of human cancers, including endometrial carcinoma, glioblastoma multiforme, skin and prostate cancers (Chalhoub and Baker, 2009). They include a mutational spectrum that is scattered along the entire gene and predominantly represent monoallelic mutations. A majority of PTEN mutations cause truncations of the protein. In addition to cancer-associated somatic mutations, PTEN is often mutated in tumor-prone germline diseases.
Genetic studies in mice have unambiguously demonstrated that PTEN is a potent tumor suppressor in almost all organs. Tissue-specific Pten-knockout mice develop tumors in numerous organs (Knobbe et al., 2008; Suzuki et al., 2008). Intriguingly, subtle changes in PTEN gene expression, mRNA regulation and protein synthesis are sufficient to have a substantial impact on tumorigenesis, a phenomenon that is referred to as quasi-insufficiency (Carracedo et al., 2011). Furthermore, the notion of PTEN haploinsufficiency seems to apply to human cancer because monoallelic mutation of PTEN without loss or mutation of the second allele is prevalent in breast and prostate cancers (Salmena et al., 2008).
PTEN is involved in multiple cellular processes
PTEN is involved in regulating many cellular processes. Many of these functions can be attributed to its lipid phosphatase activity. For example, PTEN regulates cell proliferation and apoptosis. These effects are mediated by suppressing AKT (also known as protein kinase B) activation. Because AKT activation requires PtdIns(3,4,5)P3, dephosphorylation of this lipid second messenger by PTEN results in inhibition of AKT and subsequent alteration of the function of AKT substrates, such as forkhead box protein O (FOXO), the E3 ubiquitin ligase MDM2, and BCL2 antagonist of cell death (BAD) (Tamguney and Stokoe, 2007). PTEN has a crucial role in regulating the self-renewal and differentiation of human embryonic stem cells and hematopoietic stem cells as well as the timing of follicle activation through the regulation of oocyte growth (Reddy et al., 2008). PTEN also regulates the chemotaxis of neutrophils (Heit et al., 2008), and all these functions require its lipid phosphatase activity.
In addition, PTEN can regulate various cellular events independently of its lipid phosphatase activity. For example, it can inhibit cell cycle progression by modulating the activity of the anaphase-promoting complex/cyclosome (APC/C) in the nucleus in a manner that is independent of PTEN enzymatic activity (Song et al., 2011). Furthermore, it can inhibit cell invasion and migration, probably by making use of its protein phosphatase activity (Tamura et al., 1999). PTEN can control the size of DNA-damaged cells by regulating the actin-remodeling process (Kim et al., 2011) through a mechanism that is likely to be independent of its lipid phosphatase activity. Deficiency of any of these functions can contribute to tumorigenesis.
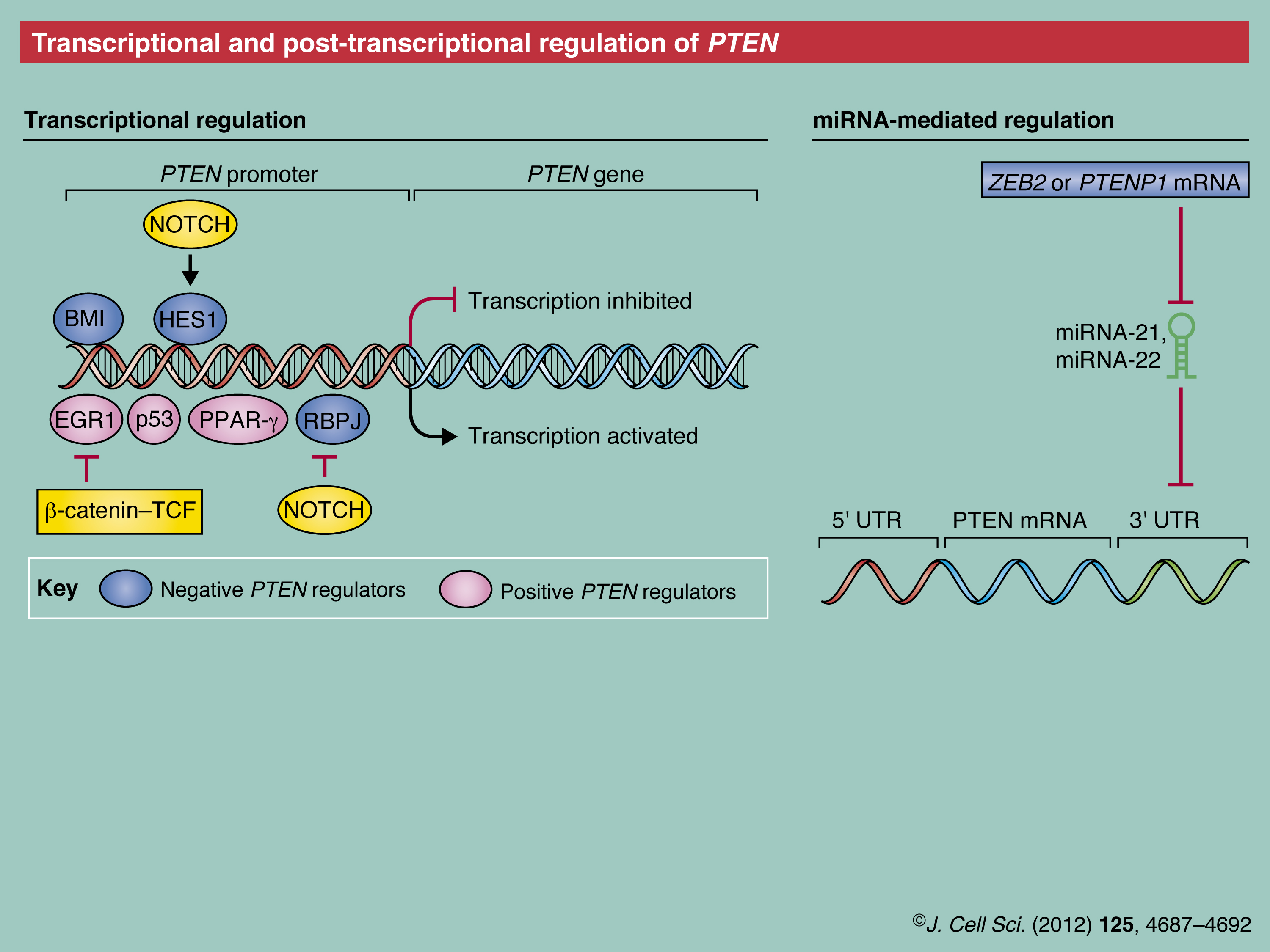
Transcriptional and post-transcriptional regulation of PTEN
As mentioned above, subtle changes in the regulation of gene expression and mRNA of PTEN have a substantial impact on the normal physiology of organisms. Therefore, both are delicately regulated through multiple mechanisms (see Poster).
Transcriptional regulation of PTEN
The PTEN promoter is regulated by many transcription factors that operate at specific times and in different cell types. Transcription factors, such as early growth response protein (EGR1), tumor protein 53 (Tp53, also referred to as p53) and peroxisome proliferator-activated receptor gamma (PPARG) were reported to positively regulate PTEN expression (Martelli et al., 2011). PTEN transcription can also be repressed, for example, by the polycomb complex protein BMI1 during epithelial–mesenchymal transition (EMT) of nasopharyngeal epithelial cells, or through β-catenin/transcription factor (TCF)-mediated downregulation of EGR1 in ovarian cancer cells (Lau et al., 2011; Song et al., 2009). Intriguingly, in a tissue-specific manner, Notch signaling might lead to upregulation of PTEN by inhibiting one of the PTEN downregulators, the recombining binding protein suppressor of hairless (RBPJ, also known as and hereafter referred to as CBF-1) (Chappell et al., 2005; Whelan et al., 2007) or, conversely, to its downregulation by activating another PTEN downregulator, the transcription factor hairy and enhancer of split 1 (HES1) (Palomero et al., 2007). Furthermore, epigenetic silencing by hyper-methylation of the PTEN promoter occurs in some breast and colorectal cancers, and in gastric carcinoma.
Post-transcriptional regulation of PTEN mRNA
PTEN mRNA has been reported to be susceptible to post-transcriptional regulation by a variety of microRNAs (miRNAs), including miRNA-21, miRNA-22 and miRNA-26a (Bar and Dikstein, 2010; Huse et al., 2009; Meng et al., 2007). These miRNAs can, thus, impact both normal biological and pathological functions of PTEN.
Additional complexity results from the regulation of PTEN expression through non-coding RNAs, such as the PTEN pseudogene PTENP1 mRNA. PTENP1 genetically resembles PTEN in its protein coding region. Owing to a mutation in its initiator codon PTEN1 mRNA, unlike PTENP mRNA, cannot be translated into a protein. The PTENP1 mRNA is generally subject to same miRNA-mediated regulation and, thus, can function as ‘decoy’ that sequesters miRNA-21. Interestingly, in sporadic colon cancer, PTENP1 undergoes a copy number loss that is concurrent with PTEN downregulation (Poliseno et al., 2010). Similarly, zinc finger E-box-binding homeobox 2 (ZEB2), which encodes another endogenous RNA, has been reported to serve as an miRNA decoy for the PTEN mRNA, and loss of ZEB2 mRNA contributes to melanomagenesis (Karreth et al., 2011). However, it is important to bear in mind that only 25% of cancer patients portray a correlation between the loss of PTEN protein and its mRNA level (Chen et al., 2011), which emphasizes the importance of PTEN regulation at the post-transcriptional and post-translational levels.
Post-translational modification of PTEN
PTEN is subject to various post-translational modifications that regulate its enzymatic activity, interaction with other proteins and subcellular localization (see Poster).
Phosphorylation
The first phosphorylation sites mapped on PTEN were a cluster of serine and threonine residues in its C-terminal tail (Vazquez et al., 2000). Mutation of these residues to alanine leads to elevated membrane affinity, higher enzymatic activity and more-rapid degradation of PTEN. When these residues are phosphorylated, the C-terminal tail can interact with the N-terminal C2 and phosphatase domains (Odriozola et al., 2007; Rahdar et al., 2009), which suggests that phosphorylation of the C-terminal tail functions as an auto-inhibitory mechanism that controls both PTEN membrane recruitment and its lipid phosphatase activity.
Several kinases have been reported to phosphorylate PTEN. Casein kinase 2 (CK2) mainly phosphorylates Ser370 and Ser385 (Torres and Pulido, 2001), whereas glycogen synthase kinase 3 β (GSK3B) targets Ser362 and Thr366 (Al-Khouri et al., 2005). In contrast to the function of C-terminal tail phosphorylation, it seems that Thr366 phosphorylation can promote PTEN degradation (Maccario et al., 2007). Additionally, glioma tumor suppressor candidate region 2 (GLTSCR2, also known as PICT-1) has been shown to interact with PTEN, enhance its phosphorylation at Ser380 and stabilize the phosphatase (Okahara et al., 2006; Yim et al., 2007). Moreover, RhoA-associated kinase (ROCK) has been shown to phosphorylate PTEN at Ser229, Thr232, Thr319 and Thr321, which are all located in the C2 domain and promote membrane targeting of PTEN in chemoattractant-stimulated leukocytes (Li et al., 2005). Recently, a Src family tyrosine kinase, the fyn-related kinase (FRK, also known and hereafter referred to as RAK) has been reported to interact with PTEN and phosphorylate it on Tyr336, thereby protecting PTEN from proteasomal degradation mediated by neural precursor cell expressed, developmentally downregulated-4 (NEDD4) (Yim et al., 2009).
Acetylation
Similar to phosphorylation, acetylation can also regulate PTEN activity. The histone acetyltransferase K(lysine) acetyltransferase 2B (KAT2B, also known and hereafter referred to as PCAF) has been reported to interact with PTEN and promote PTEN acetylation on Lys125 and Lys128 in response to growth factor stimulation (Okumura et al., 2006). As these residues are within the catalytic pocket, PTEN acetylation by PCAF negatively regulates its enzymatic activity. PTEN is also acetylated on Lys402, which is located within the C-terminal PDZ-domain-binding motif Thr-Lys-Val sequence. This, potentially, affects the interaction between PTEN and PDZ-domain-containing proteins (Ikenoue et al., 2008). CREB-binding protein (CREBBP, also known and hereafter referred to as CBP) and the sirtuin SIRT1 have been identified as the main PTEN acetyltransferase and deacetylase, respectively.
Oxidation
Another mechanism that can potentially regulate the catalytic activity of PTEN is direct oxidation by reactive oxygen species (ROS). ROS can oxidize Cys124 in the active site, thereby forming an intramolecular disulfide bond with Cys71 (Lee et al., 2002). Oxidative inactivation of PTEN has been reported in studies describing the use of hydrogen peroxide or endogenous ROS production in macrophages (Kwon et al., 2004; Leslie et al., 2003). PTEN activity can also be indirectly inhibited by oxidation through modulation of PTEN-binding partners. Oxidation of the antioxidant PARK7 (also known as DJ-1) leads to binding to PTEN and the subsequent inhibition of the PTEN lipid phosphatase activity (Kim et al., 2009).
S-nitrosylation
A few studies have demonstrated the importance of another redox mechanism, S-nitrosylation, in the regulation of PTEN. The level of S-nitrosylation on PTEN substantially increases in the early stages of Alzheimer disease, and this correlates with reduced PTEN protein levels and elevated AKT phosphorylation (Kwak et al., 2010a). Nitric oxide (NO) signaling induces PTEN S-nitrosylation, thereby inactivating the lipid phosphatase, downregulating its protein level through NEDD4-mediated degradation and leading to downstream activation of AKT. Another report has shown that PTEN is selectively S-nitrosylated on Cys83 by low concentrations of NO (Numajiri et al., 2011). Moreover, S-nitrosylated PTEN has been detected in the core and penumbra regions of ischemic mouse brains. S-nitrosylation and downregulation of PTEN activity in this region are likely to act as protective mechanisms through AKT activation.
Ubiquitylation
Using a biochemical purification approach, NEDD4 was identified as an E3 ligase that ubiquitylates PTEN (Wang et al., 2007). NEDD4 physically interacts with PTEN and its overexpression leads to both mono- and polyubiquitylation of PTEN. Interestingly, monoubiquitylation of PTEN appears to be crucial for its nuclear import (Trotman et al., 2007). Consistent with the function of the PTEN C-terminal tail in regulating its stability, deletion of this region makes PTEN a stronger binding partner and better substrate for NEDD4 (Wang et al., 2008).
Two other E3 ligases have been reported to target PTEN, X-linked inhibitor of apoptosis protein (XIAP) (Van Themsche et al., 2009) and WWP2, a NEDD4-like protein family member (Maddika et al., 2011). Although the physiological relevance of these E3 ligases with regards to PTEN function has not yet been defined, it is possible that PTEN is regulated by multiple E3 ligases, depending on the context or special physiological circumstances.
However, in most experimental systems, PTEN appears to be a rather stable protein. Thus, what is the biological relevance of PTEN regulation through ubiquitin-mediated proteasomal degradation? It appears that NEDD4 regulates PTEN in a context-dependent manner to achieve specific functions. For example, NEDD4 is required for neuronal axonal branching in retinal ganglion cells (RGCs) and mainly functions through downregulating PTEN (Drinjakovic et al., 2010). Blocking NEDD4 function severely inhibits terminal branching in RGCs, whereas PTEN knockdown rescues the branching deficiency. Also, NEDD4-mediated PTEN ubiquitylation is essential in the regulataion of PI3K-AKT signaling for neuronal survival in response to Zn2+ (Kwak et al., 2010b). Furthermore, in cultured neuronal models, NO signaling not only induces PTEN S-nitrosylation but also results in enhanced PTEN protein degradation through NEDD4-mediated ubiquitylation (Kwak et al., 2010a).
Interestingly, several cellular proteins have been reported to modulate the association between NEDD4 and PTEN, which might provide mechanistic insights into the context-dependent regulation of PTEN by NEDD4. In breast cancer cells, the tyrosine kinase RAK positively regulates PTEN stability by phosphorylating PTEN on Tyr336. This prevents PTEN from binding to NEDD4 and its subsequent degradation (Yim et al., 2009). Recently, the PY (Pro-Pro-x-Tyr)-motif-containing membrane proteins NEDD4-family-interacting proteins 1 and 2 (NDFIP 1 and 2, respectively), which are potent activators of NEDD4 family members, have been shown to promote NEDD4-mediated ubiquitylation and, thus, promote degradation and nuclear import of PTEN (Howitt et al., 2012; Mund and Pelham, 2010).
PTEN regulation by its interacting proteins
The enzymatic activity and the biological effects of PTEN are often regulated by its interaction with other proteins. The lipid phosphatase activity of PTEN can be stimulated by binding to p85, the regulatory subunit of PI3K (Chagpar et al., 2010). By contrast, the binding of shank-interacting protein-like 1 (SIPL1) or PtdIns(3,4,5)P3–RAC-exchanger 2 (PREX2) to PTEN inhibits its lipid phosphatase activity, thereby promoting signaling through the PI3K pathway (Fine et al., 2009; He et al., 2010). Binding of the scaffold protein β-arrestin to PTEN controls the output of distinct PTEN signals during cell proliferation and migration; binding of β-arrestin to PTEN normally activates the lipid phosphatase activity and inhibits cell proliferation. However, following the activation of RhoA as a result of wounding, β-arrestin inhibits the lipid phosphatase-independent anti-migratory function of PTEN. In addition, the binding of several proteins to PTEN can regulate its stability (Lima-Fernandes et al., 2011). These proteins include membrane-associated guanylate kinase inverted 3 (MAGI), GLTSCR2, NDFIP1 and RAK, whose binding affects PTEN phosphorylation status, complex recruitment or NEDD4-mediated ubiquitylation.
Distinct subpopulations of PTEN
The fact that the majority of cellular PTEN is found in the cytosol raises important questions, such as how PTEN is able to execute different cellular functions that often require its membranous lipid phosphatase activity. Accumulating evidence has suggested that a small subpopulation of cellular PTEN is sufficient for each specific biological function. A PTEN subpopulation can be defined by either specific post-translational modification(s) or its interaction with a particular associating factor. It should be emphasized that a functional subpopulation of PTEN might only represent a small portion of total cellular PTEN, thus it might evade evaluation when employing the approaches that are commonly used to monitor total cellular PTEN.
Regulation of the membrane-associated PTEN subpopulation
A subpopulation of PTEN is responsible for its lipid phosphatase activity at the plasma membrane. A study describing the use of single-molecule microscopy in living cells has shown that the interaction of PTEN with the plasma membrane is a dynamic process (Vazquez et al., 2006). PTEN binds to the membrane for only a few hundred milliseconds, and this time period is sufficient to hydrolyze PtdIns(3,4,5)P3. The interaction of PTEN with the membrane requires the N-terminal lipid-binding motif. Membrane interaction is negatively regulated by C-terminal tail phosphorylation, which appears to constrain PTEN in a closed conformation and to limit its association with the membrane. Another study has demonstrated that phosphorylation of the C-terminal tail regulates an intra-molecular interaction between the phosphatase and C2 domains. This determines the percentage of PTEN that is associated with the membrane as well as the resulting PtdIns(3,4,5)P3 levels (Rahdar et al., 2009). Interestingly, plasma membrane targeting of PTEN greatly enhances PTEN ubiquitylation through NEDD4, and both mono- and polyubiquitylation substantially inhibit PTEN phosphatase activity in vitro, even in the absence of proteasomal degradation (Maccario et al., 2010).
The nuclear PTEN subpopulation
Interestingly, PTEN also localizes into the nucleus, and loss of PTEN nuclear localization has been shown to correlate with cancer progression in certain tissues (Song et al., 2008; Trotman et al., 2007). The nuclear subpopulation of PTEN is controlled by the dynamic regulation of its ubiquitylation, i.e. through NEDD4-mediated monoubiquitylation and deubiquitylation mediated through the network of ubiquitin carboxyl-terminal hydrolase 7 (USP7, also known as HAUSP) and PML (Song et al., 2008; Trotman et al., 2007).
The nuclear subpopulation of PTEN clearly has non-canonical functions (Lindsay et al., 2006). Earlier studies have shown that PTEN regulates p53 protein stability in both phosphatase-dependent and -independent manners (Li et al., 2006). A recent study has shown that genotoxic stress enhances the interaction between PTEN and the p53 family member p73 in the nucleus. This facilitates the binding of p73 to apoptotic promoters and induces the expression of p53-upregulated modulator of apoptosis (PUMA, also known as BBC3) and the apoptosis regulator BAX (Lehman et al., 2011). Nuclear PTEN has also been suggested to maintain chromosomal integrity through the physical interaction with centromere protein C 1 (CENPC1), which is an integral part of the kinetochore (Shen et al., 2007). Whether this function requires the lipid phosphatase activity of PTEN is under debate. More recently, nuclear PTEN has been found to interact with the anaphase-promoting complex/cyclosome (APC/C), an E3 ubiquitin ligase that promotes cell cycle progression by degrading components of the cell cycle machinery. Nuclear PTEN increases APC/C activity by enhancing the association between APC/C and its cofactor, CDH1 (also known as FZR1). This role of nuclear PTEN has been shown to be important for PTEN-mediated tumor suppression (Song et al., 2011).
Conclusions and perspectives
The function of PTEN as a tumor suppressor has been well established over the past decade. Recent studies have revealed more unexpected – particularly lipid-phosphatase-independent – roles of PTEN in both cancer biology and normal physiology. PTEN activity is tightly regulated by various mechanisms on the transcriptional, translational and post-translational levels. Accumulating evidence suggests that PTEN can exert diverse physiological and pathological functions through its context-specific regulation. Furthermore, a highly regulated and dynamic subpopulation of cellular PTEN might be required specifically for a given contextual function of PTEN. In summary, to understand the molecular basis of contextual regulation of PTEN is crucial in order to understand the biological function of PTEN and, subsequently, to develop appropriate approaches to therapeutically target PTEN signaling.
Supplementary Material
Footnotes
Funding
The work in X.J.’s laboratory that is related to this article is supported by a Geoffrey Beene Cancer Research fund, an American Cancer Society scholar award [grant number RSG-07-081-01-MGO] and the National Institutes of Health [grant numbers R01CA113890 and T32CA009072]. Deposited in PMC for release after 6 months.
A high-resolution version of the poster is available for downloading in the online version of this article at jcs.biologists.org. Individual poster panels are available as JPEG files at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.093765/-/DC1.
References
- Al–Khouri A. M., Ma Y., Togo S. H., Williams S., Mustelin T. (2005). Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J. Biol. Chem. 280, 35195–35202 10.1074/jbc.M503045200 [DOI] [PubMed] [Google Scholar]
- Bar N., Dikstein R. (2010). miR-22 forms a regulatory loop in PTEN/AKT pathway and modulates signaling kinetics. PLoS ONE 5, e10859 10.1371/journal.pone.0010859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A., Alimonti A., Pandolfi P. P. (2011). PTEN level in tumor suppression: how much is too little? Cancer Res. 71, 629–633 10.1158/0008-5472.CAN-10-2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagpar R. B., Links P. H., Pastor M. C., Furber L. A., Hawrysh A. D., Chamberlain M. D., Anderson D. H. (2010). Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 107, 5471–5476 10.1073/pnas.0908899107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub N., Baker S. J. (2009). PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 4, 127–150 10.1146/annurev.pathol.4.110807.092311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell W. H., Green T. D., Spengeman J. D., McCubrey J. A., Akula S. M., Bertrand F. E.2005). Increased protein expression of the PTEN tumor suppressor in the presence of constitutively active Notch-1. Cell Cycle 41389–1395 10.4161/cc.4.10.2028 [DOI] [PubMed] [Google Scholar]
- Chen M., Pratt C. P., Zeeman M. E., Schultz N., Taylor B. S., O’Neill A., Castillo–Martin M., Nowak D. G., Naguib A., Grace D. M.et al. (2011). Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell 20, 173–186 10.1016/j.ccr.2011.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinjakovic J., Jung H., Campbell D. S., Strochlic L., Dwivedy A., Holt C. E. (2010). E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 65, 341–357 10.1016/j.neuron.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine B., Hodakoski C., Koujak S., Su T., Saal L. H., Maurer M., Hopkins B., Keniry M., Sulis M. L., Mense S.et al. (2009). Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science 325, 1261–1265 10.1126/science.1173569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J., Tamura M., Pankov R., Danen E. H., Takino T., Matsumoto K., Yamada K. M. (1999). Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J. Cell Biol. 146, 389–404 10.1083/jcb.146.2.389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., Ingram A., Rybak A. P., Tang D. (2010). Shank-interacting protein-like 1 promotes tumorigenesis via PTEN inhibition in human tumor cells. J. Clin. Invest. 120, 2094–2108 10.1172/JCI40778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit B., Robbins S. M., Downey C. M., Guan Z., Colarusso P., Miller B. J., Jirik F. R., Kubes P. (2008). PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat. Immunol. 9, 743–752 10.1038/ni.1623 [DOI] [PubMed] [Google Scholar]
- Howitt J., Lackovic J., Low L. H., Naguib A., Macintyre A., Goh C. P., Callaway J. K., Hammond V., Thomas T., Dixon M.et al. (2012). Ndfip1 regulates nuclear Pten import in vivo to promote neuronal survival following cerebral ischemia. J. Cell Biol. 196, 29–36 10.1083/jcb.201105009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse J. T., Brennan C., Hambardzumyan D., Wee B., Pena J., Rouhanifard S. H., Sohn–Lee C., le Sage C., Agami R., Tuschl T.et al. (2009). The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 23, 1327–1337 10.1101/gad.1777409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenoue T., Inoki K., Zhao B., Guan K. L. (2008). PTEN acetylation modulates its interaction with PDZ domain. Cancer Res. 68, 6908–6912 10.1158/0008-5472.CAN-08-1107 [DOI] [PubMed] [Google Scholar]
- Karreth F. A., Tay Y., Perna D., Ala U., Tan S. M., Rust A. G., DeNicola G., Webster K. A., Weiss D., Perez–Mancera P. A.et al. (2011). In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell 147, 382–395 10.1016/j.cell.2011.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. S., Xu X., Li H., Solomon D., Lane W. S., Jin T., Waldman T. (2011). Mechanistic analysis of a DNA damage-induced, PTEN-dependent size checkpoint in human cells. Mol. Cell. Biol. 31, 2756–2771 10.1128/MCB.01323-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. C., Kitaura H., Taira T., Iguchi–Ariga S. M., Ariga H. (2009). Oxidation of DJ-1-dependent cell transformation through direct binding of DJ-1 to PTEN. Int. J. Oncol. 35, 1331–1341 [PubMed] [Google Scholar]
- Knobbe C. B., Lapin V., Suzuki A., Mak T. W. (2008). The roles of PTEN in development, physiology and tumorigenesis in mouse models: a tissue-by-tissue survey. Oncogene 27, 5398–5415 10.1038/onc.2008.238 [DOI] [PubMed] [Google Scholar]
- Kwak Y. D., Ma T., Diao S., Zhang X., Chen Y., Hsu J., Lipton S. A., Masliah E., Xu H., Liao F. F. (2010a). NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol. Neurodegener. 5, 49 10.1186/1750-1326-5-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak Y. D., Wang B., Pan W., Xu H., Jiang X., Liao F. F. (2010b). Functional interaction of phosphatase and tensin homologue (PTEN) with the E3 ligase NEDD4-1 during neuronal response to zinc. J. Biol. Chem. 285, 9847–9857 10.1074/jbc.M109.091637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J., Lee S. R., Yang K. S., Ahn Y., Kim Y. J., Stadtman E. R., Rhee S. G. (2004). Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 101, 16419–16424 10.1073/pnas.0407396101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau M. T., Klausen C., Leung P. C. (2011). E-cadherin inhibits tumor cell growth by suppressing PI3K/Akt signaling via β-catenin-Egr1-mediated PTEN expression. Oncogene 30, 2753–2766 10.1038/onc.2011.6 [DOI] [PubMed] [Google Scholar]
- Lee S. R., Yang K. S., Kwon J., Lee C., Jeong W., Rhee S. G. (2002). Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 277, 20336–20342 10.1074/jbc.M111899200 [DOI] [PubMed] [Google Scholar]
- Lehman J. A., Waning D. L., Batuello C. N., Cipriano R., Kadakia M. P., Mayo L. D. (2011). Induction of apoptotic genes by a p73-phosphatase and tensin homolog (p73-PTEN) protein complex in response to genotoxic stress. J. Biol. Chem. 286, 36631–36640 10.1074/jbc.M110.217620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie N. R., Bennett D., Lindsay Y. E., Stewart H., Gray A., Downes C. P. (2003). Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 22, 5501–5510 10.1093/emboj/cdg513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A. G., Piluso L. G., Cai X., Wei G., Sellers W. R., Liu X. (2006). Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol. Cell 23, 575–587 10.1016/j.molcel.2006.06.028 [DOI] [PubMed] [Google Scholar]
- Li D. M., Sun H. (1997). TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 57, 2124–2129 [PubMed] [Google Scholar]
- Li J., Yen C., Liaw D., Podsypanina K., Bose S., Wang S. I., Puc J., Miliaresis C., Rodgers L., McCombie R.et al. (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 10.1126/science.275.5308.1943 [DOI] [PubMed] [Google Scholar]
- Li Z., Dong X., Wang Z., Liu W., Deng N., Ding Y., Tang L., Hla T., Zeng R., Li L.et al. (2005). Regulation of PTEN by Rho small GTPases. Nat. Cell Biol. 7, 399–404 10.1038/ncb1236 [DOI] [PubMed] [Google Scholar]
- Lima–Fernandes E., Enslen H., Camand E., Kotelevets L., Boularan C., Achour L., Benmerah A., Gibson L. C., Baillie G. S., Pitcher J. A.et al. (2011). Distinct functional outputs of PTEN signalling are controlled by dynamic association with β-arrestins. EMBO J. 30, 2557–2568 10.1038/emboj.2011.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay Y., McCoull D., Davidson L., Leslie N. R., Fairservice A., Gray A., Lucocq J., Downes C. P. (2006). Localization of agonist-sensitive PtdIns(3,4,5)P3 reveals a nuclear pool that is insensitive to PTEN expression. J. Cell Sci. 119, 5160–5168 10.1242/jcs.000133 [DOI] [PubMed] [Google Scholar]
- Maccario H., Perera N. M., Davidson L., Downes C. P., Leslie N. R. (2007). PTEN is destabilized by phosphorylation on Thr366. Biochem. J. 405, 439–444 10.1042/BJ20061837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccario H., Perera N. M., Gray A., Downes C. P., Leslie N. R. (2010). Ubiquitination of PTEN (phosphatase and tensin homolog) inhibits phosphatase activity and is enhanced by membrane targeting and hyperosmotic stress. J. Biol. Chem. 285, 12620–12628 10.1074/jbc.M109.072280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddika S., Kavela S., Rani N., Palicharla V. R., Pokorny J. L., Sarkaria J. N., Chen J. (2011). WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 13, 728–733 10.1038/ncb2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T., Dixon J. E. (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378 10.1074/jbc.273.22.13375 [DOI] [PubMed] [Google Scholar]
- Mahimainathan L., Choudhury G. G. (2004). Inactivation of platelet-derived growth factor receptor by the tumor suppressor PTEN provides a novel mechanism of action of the phosphatase. J. Biol. Chem. 279, 15258–15268 10.1074/jbc.M314328200 [DOI] [PubMed] [Google Scholar]
- Martelli A. M., Evangelisti C., Chappell W., Abrams S. L., Bäsecke J., Stivala F., Donia M., Fagone P., Nicoletti F., Libra M.et al. (2011). Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia 25, 1064–1079 10.1038/leu.2011.46 [DOI] [PubMed] [Google Scholar]
- Meng F., Henson R., Wehbe–Janek H., Ghoshal K., Jacob S. T., Patel T. (2007). MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133, 647–658 10.1053/j.gastro.2007.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mund T., Pelham H. R. (2010). Regulation of PTEN/Akt and MAP kinase signaling pathways by the ubiquitin ligase activators Ndfip1 and Ndfip2. Proc. Natl. Acad. Sci. USA 107, 11429–11434 10.1073/pnas.0911714107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numajiri N., Takasawa K., Nishiya T., Tanaka H., Ohno K., Hayakawa W., Asada M., Matsuda H., Azumi K., Kamata H.et al. (2011). On-off system for PI3-kinase-Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN). Proc. Natl. Acad. Sci. USA 108, 10349–10354 10.1073/pnas.1103503108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odriozola L., Singh G., Hoang T., Chan A. M. (2007). Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J. Biol. Chem. 282, 23306–23315 10.1074/jbc.M611240200 [DOI] [PubMed] [Google Scholar]
- Okahara F., Itoh K., Nakagawara A., Murakami M., Kanaho Y., Maehama T. (2006). Critical role of PICT-1, a tumor suppressor candidate, in phosphatidylinositol 3,4,5-trisphosphate signals and tumorigenic transformation. Mol. Biol. Cell 17, 4888–4895 10.1091/mbc.E06-04-0301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura K., Mendoza M., Bachoo R. M., DePinho R. A., Cavenee W. K., Furnari F. B. (2006). PCAF modulates PTEN activity. J. Biol. Chem. 281, 26562–26568 10.1074/jbc.M605391200 [DOI] [PubMed] [Google Scholar]
- Palomero T., Sulis M. L., Cortina M., Real P. J., Barnes K., Ciofani M., Caparros E., Buteau J., Brown K., Perkins S. L.et al. 2007). Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 131203–1210 10.1038/nm1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliseno L., Salmena L., Zhang J., Carver B., Haveman W. J., Pandolfi P. P. (2010). A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465, 1033–1038 10.1038/nature09144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M., Etienne–Manneville S., Self A., Nicholls S., Hall A. (2004). Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science 303, 1179–1181 10.1126/science.1092089 [DOI] [PubMed] [Google Scholar]
- Rahdar M., Inoue T., Meyer T., Zhang J., Vazquez F., Devreotes P. N. (2009). A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc. Natl. Acad. Sci. USA 106, 480–485 10.1073/pnas.0811212106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P., Liu L., Adhikari D., Jagarlamudi K., Rajareddy S., Shen Y., Du C., Tang W., Hämäläinen T., Peng S. L.et al. (2008). Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science 319, 611–613 10.1126/science.1152257 [DOI] [PubMed] [Google Scholar]
- Salmena L., Carracedo A., Pandolfi P. P. (2008). Tenets of PTEN tumor suppression. Cell 133, 403–414 10.1016/j.cell.2008.04.013 [DOI] [PubMed] [Google Scholar]
- Shen W. H., Balajee A. S., Wang J., Wu H., Eng C., Pandolfi P. P., Yin Y. (2007). Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128, 157–170 10.1016/j.cell.2006.11.042 [DOI] [PubMed] [Google Scholar]
- Song L. B., Li J., Liao W. T., Feng Y., Yu C. P., Hu L. J., Kong Q. L., Xu L. H., Zhang X., Liu W. L.et al. (2009). The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Invest. 119, 3626–3636 10.1172/JCI39374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M. S., Salmena L., Carracedo A., Egia A., Lo–Coco F., Teruya–Feldstein J., Pandolfi P. P. (2008). The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455, 813–817 10.1038/nature07290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M. S., Carracedo A., Salmena L., Song S. J., Egia A., Malumbres M., Pandolfi P. P. (2011). Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144, 187–199 10.1016/j.cell.2010.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck P. A., Pershouse M. A., Jasser S. A., Yung W. K., Lin H., Ligon A. H., Langford L. A., Baumgard M. L., Hattier T., Davis T.et al. (1997). Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362 10.1038/ng0497-356 [DOI] [PubMed] [Google Scholar]
- Suzuki A., Nakano T., Mak T. W., Sasaki T. (2008). Portrait of PTEN: messages from mutant mice. Cancer Sci. 99, 209–213 10.1111/j.1349-7006.2007.00670.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamguney T., Stokoe D. (2007). New insights into PTEN. J. Cell Sci. 120, 4071–4079 10.1242/jcs.015230 [DOI] [PubMed] [Google Scholar]
- Tamura M., Gu J., Matsumoto K., Aota S., Parsons R., Yamada K. M. (1998). Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280, 1614–1617 10.1126/science.280.5369.1614 [DOI] [PubMed] [Google Scholar]
- Tamura M., Gu J., Takino T., Yamada K. M. (1999). Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 59, 442–449 [PubMed] [Google Scholar]
- Torres J., Pulido R. (2001). The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J. Biol. Chem. 276, 993–998 10.1074/jbc.M009134200 [DOI] [PubMed] [Google Scholar]
- Trotman L. C., Wang X., Alimonti A., Chen Z., Teruya–Feldstein J., Yang H., Pavletich N. P., Carver B. S., Cordon–Cardo C., Erdjument–Bromage H.et al. (2007). Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156 10.1016/j.cell.2006.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Themsche C., Leblanc V., Parent S., Asselin E. (2009). X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J. Biol. Chem. 284, 20462–20466 10.1074/jbc.C109.009522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F., Ramaswamy S., Nakamura N., Sellers W. R. (2000). Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell. Biol. 20, 5010–5018 10.1128/MCB.20.14.5010-5018.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F., Matsuoka S., Sellers W. R., Yanagida T., Ueda M., Devreotes P. N. (2006). Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc. Natl. Acad. Sci. USA 103, 3633–3638 10.1073/pnas.0510570103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Trotman L. C., Koppie T., Alimonti A., Chen Z., Gao Z., Wang J., Erdjument–Bromage H., Tempst P., Cordon–Cardo C.et al. (2007). NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128, 129–139 10.1016/j.cell.2006.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Shi Y., Wang J., Huang G., Jiang X. (2008). Crucial role of the C-terminus of PTEN in antagonizing NEDD4-1-mediated PTEN ubiquitination and degradation. Biochem. J. 414, 221–229 10.1042/BJ20080674 [DOI] [PubMed] [Google Scholar]
- Whelan J. T., Forbes S. L., Bertrand F. E.2007). CBF-1 (RBP-J kappa) binds to the PTEN promoter and regulates PTEN gene expression. Cell Cycle 680–84 10.4161/cc.6.1.3648 [DOI] [PubMed] [Google Scholar]
- Yim E. K., Peng G., Dai H., Hu R., Li K., Lu Y., Mills G. B., Meric–Bernstam F., Hennessy B. T., Craven R. J.et al. (2009). Rak functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell 15, 304–314 10.1016/j.ccr.2009.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim J. H., Kim Y. J., Ko J. H., Cho Y. E., Kim S. M., Kim J. Y., Lee S., Park J. H. (2007). The putative tumor suppressor gene GLTSCR2 induces PTEN-modulated cell death. Cell Death Differ. 14, 1872–1879 10.1038/sj.cdd.4402204 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}