

Summary
The development of multi-colored fluorescent proteins, nanocrystals and organic fluorophores, along with the resulting engineered biosensors, has revolutionized the study of protein localization and dynamics in living cells. Hyperspectral imaging has proven to be a useful approach for such studies, but this technique is often limited by low signal and insufficient temporal resolution. Here, we present an implementation of a snapshot hyperspectral imaging device, the image mapping spectrometer (IMS), which acquires full spectral information simultaneously from each pixel in the field without scanning. The IMS is capable of real-time signal capture from multiple fluorophores with high collection efficiency (∼65%) and image acquisition rate (up to 7.2 fps). To demonstrate the capabilities of the IMS in cellular applications, we have combined fluorescent protein (FP)-FRET and [Ca2+]i biosensors to measure simultaneously intracellular cAMP and [Ca2+]i signaling in pancreatic β-cells. Additionally, we have compared quantitatively the IMS detection efficiency with a laser-scanning confocal microscope.
Key words: Hyperspectral imaging, Fluorescent biosensors, Image mapping spectrometer, IMS
Introduction
The expanding library of fluorescent probes available for fluorescence microscopy has led to an ever-increasing range of applications accessible to live cell optical microscopy (Pinaud et al., 2006; Lavis and Raines, 2008; Rizzo et al., 2009). Given the limited spectral range of visible and NIR fluorophores, experiments that utilize multiple probes often suffer from significant emission spectral overlap. Thus, optimal utilization of fluorescent probes requires imaging devices that can measure spectral datacubes (x, y, λ) and discriminate probes based on their spectral differences (Zimmermann, 2005). To achieve 3D datacube (x, y, λ) acquisition, current hyperspectral imagers use scanning, either in the spatial domain, e.g. hyperspectral confocal microscope (Sinclair et al., 2006) or in the spectral domain, e.g. liquid-crystal tunable filter or acoustic-optic tunable filter (Morris et al., 1994). The scanning mechanism causes a serious trade-off problem between image acquisition rate and signal throughput – the faster the imager scans, the fewer photons it can collect from each spatial sampling pixel or spectral sampling layer.
To overcome this problem, a snapshot hyperspectral imager, the Image Mapping Spectrometer (IMS), was developed for microscopy (Gao et al., 2009; Gao et al., 2010) and endoscopy (Kester et al., 2011). The IMS is a direct imaging device that can map an object's 3D datacube onto different locations of a 2D detector array and thus allow their measurement in parallel. Since no scanning is employed, the whole datacube is captured in a single snapshot, and optical throughput is maximized. Parallel acquisition along with high optical throughput enables the IMS to capture high dynamic range images at rates up to 7.2 fps.
The operating principle of the IMS is shown in Fig. 1. In this simple example, the object is 4×4 pixels and each pixel has its own color. In the IMS, the object's different rows (y1, y2, y3, y4) are mapped to different parts of the image by a custom fabricated image mapper. Without dispersion, the image of the object would be formed on a 2D detector array (8×8 pixels) directly with blank regions created between adjacent image rows. However, by using a prism as a dispersion unit, the images of the object's rows are dispersed into their adjacent blank regions. In this way, each pixel on the detector array is encoded with the object's unique spatial and spectral information. After applying a simple field remapping algorithm to the captured raw image, the object's 3D datacube (x, y, λ) is acquired.
Fig. 1.
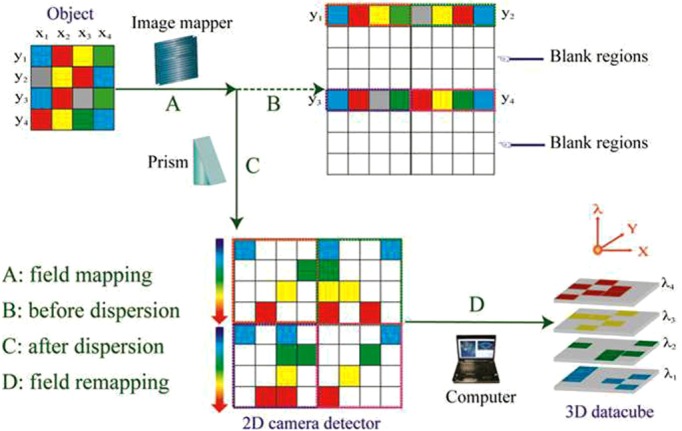
Operation principle and system configuration of the IMS. In the IMS, the object's different image zones are mapped and dispersed onto different locations of a 2D camera detector (the object's rows y1, y2, y3 and y4 are mapped onto the second quadrant, first quadrant, third quadrant, and fourth quadrant of the camera, respectively, in this simple example). The gray pixel in the original object represents a spatial sampling point that has all colors. An x, y, λ datacube is reconstructed by remapping camera pixels encoded with the same color back to the corresponding spectral layer. The full-resolution datacube is acquired by the IMS via a single snapshot.
For use with biological fluorescence microscopy, we have built an optimized IMS system that is 20 in×10 in×3 in, and can easily be adapted onto a wide-field fluorescence microscope. This device consists of four major optical components: image relay, image mapper, collecting lenses, prisms, and re-imaging lens array (see details in Materials and Methods). The system's optics and opto-mechanics are assembled and fixed in a custom enclosure, making the whole device stable and functional in ambient room light. It acquires a 350×350×46 (x, y, λ, t) 4-D datacube with a spectral window from 450 nm to 650 nm and ∼4 nm sampling interval. The optical design of the presented system is adapted from a previous endoscopic IMS (Kester et al., 2011) with numerical aperture and field of view optimized for high-resolution microscopic imaging. The optical performance of the presented device is described elsewhere (T.S.T., unpublished data). Briefly, it achieves diffraction-limited spatial and spectral resolution in the acquired field of view and spectral range. This device's optical throughput is measured to be ∼65% (from the image input plane to the camera), which is significantly improved compared to the ∼20% measured in the previous IMS system (Gao et al., 2010). We have compared quantitatively the detection efficiency of this new IMS system to that of the Zeiss LSM710 confocal microscope. While the two systems have comparable detection efficiencies, the IMS offers advantages in acquisition speed that increase as larger image sizes are used.
To demonstrate the IMS advantages for imaging dynamic cellular processes, we assayed multiple fluorescent biosensors of pancreatic β-cell signaling pathways. The roles of [Ca2+]i and cAMP in regulating glucose-stimulated insulin secretion from pancreatic β-cells have long been known (Landa et al., 2005; Holz et al., 2008). However, it has been challenging to study temporal relationships between these signaling molecules due to the spectral overlap of most [Ca2+]i and cAMP biosensors (Landa et al., 2005). Here, we use an optimized fluorescent protein (FP)-based FRET biosensor for cAMP called T-Epac-VV, which contains an mTurquoise donor and tandem circularly permuted Venus-Venus (cpVenus-Venus) acceptor (Klarenbeek et al., 2011). To image [Ca2+]i, we use a GCaMP (Wang et al., 2008; Tian et al., 2009) biosensor with a circularly permuted GFP fluorophore, called GCaMP5G (L. L. Looger, personal communication). While the emission spectra of the three FPs (mTurquoise, cpVenus-Venus, and cpGFP) overlap significantly, the IMS can simultaneously measure changes in their intensity and unmix the spectra over time. The resulting 4D dataset (x, y, λ, t) permits detailed examination of the temporal dynamics and relationships between different signaling molecules. However, biosensors like T-Epac-VV based on FP-FRET depend on measurements of small changes in FRET and thus require high signal-to-noise data (Piston and Kremers, 2007; Lam et al., 2010). Using the IMS, we can resolve the effects of glucose and known stimulating drugs on these signaling molecules simultaneously and show that their glucose-induced oscillations are anti-correlated.
Results
Spectral unmixing of triply-labeled HeLa cells
The commonly used fluorescent proteins CFP, GFP, and YFP have considerable overlap in their emission spectra, requiring post-collection unmixing to separate the signals (Kremers et al., 2011). To demonstrate the efficacy of the IMS system for this purpose, we expressed proteins with known localizations fused to ECFP, EGFP, or SYFP (Rizzo et al., 2006) (em 480 nm, 509 nm, and 528 nm respectively) in HeLa cells and fixed them for hyperspectral imaging. To acquire the FPs' reference spectra for spectral unmixing, the singly-labeled control cells were first imaged by the IMS and the resulting base emission spectra (Fig. 2C) were determined independently for all three fluorophores using the IMS. The triply-labeled cells expressing ECFP in the mitochondria, EGFP on the plasma membrane, and SYFP in the nucleus were imaged by the IMS with an integration time of 0.5 seconds (data not shown). The spectral component images (Fig. 2D–F) were acquired by implementing a linear spectral unmixing algorithm (Zimmerman, 2005) on the measured datacubes. To provide a reference image for comparison, we used a color camera to capture an image of the same field of view (Fig. 2A). In this color image, all three components appear as shades of green and cannot be discriminated. In contrast, spectral unmixing reveals the three components clearly and identifies their sub-cellular localization (Fig. 2D–F). Since each channel represents the spectral emission profile for each fluorophore, crosstalk from the other two fluorophores is minimized.
Fig. 2.

Spectral unmixing of ECFP, EGFP and SYFP in triple-labeled HeLa cells. (A) The baseline reference image was captured by a color camera (Infinity 2-1C) directly at a microscope's side image port. (B) A merged image of those shown in D, E and F. (C) The unmixed component spectra. (D–F) The spectral component images were acquired by implementing a linear unmixing algorithm on the IMS measured datacube (captured with 0.5 s integration time); these images are pseudo-color rendered to indicate subcellular localizations of the FPs.
Quantitative comparison of IMS and LSM710
To compare quantitatively the detection efficiency (defined as the ratio of photons detected over photons excited, or D = Nd/Ne) of the two systems, we measured their relative excitation intensities and fluorescent photon detection. We imaged the same sample volume on both systems and measured the collected fluorescent photons under matched excitation conditions, which were determined by matching the photobleaching rate in the two systems. The procedure was the following: we matched the total image acquisition time, the image size and the pixel size, then we measured the photobleaching rates for such settings, and their value was used to scale the different excitation intensities. The imaging settings for the experiment on the IMS were the following: integration time 500 ms, pixel size 0.7 µm, image size 348 × 348 pixel, excitation filter 436/20, dichroic T455LP, emission filter HQ460LP. The imaging settings for the experiments on the LSM710 (Zeiss, Oberkochen, Germany) were the following: excitation laser 458 nm, power 5% of 100 mW, scan time 531 ms, dwell time 3.76 µs, pixel size 0.68 µm, pinhole diameter 609 µm corresponding to an optical section of 15.5 µm, image size 348 × 348 pixel, Zeiss Fluar 40×/1.3 oil objective.
We designed two independent sets of experiments to calculate the relative detection efficiency of the two systems, DIMS/D710. In the first set of experiments, we compared the excitation power acting on the sample using microdroplets with a 10 µM aqueous solution of fluorescein (F-1300 from Molecular Probes, Carlsbad, CA) dispersed in hydrophobic medium to create a layer of fluorescent droplets with a thickness less than 3 µm (Kremers and Piston, 2010). The use of microdroplets removes the need to consider lateral diffusion in the sample, and the relatively thin sample thickness is also critical for the comparison. The LSM710 is a confocal system which only collects photons from a section of the sample, while the IMS is coupled to a widefield microscope (Nikon TE 300 inverted microscope, Melville, NY) and therefore receives photons from the whole sample volume. Using a thin sample allows the confocal pinhole size to be set so that all the photons coming from the sample volume are collected. We used the photobleaching rates to estimate the ratio between the excitation intensities delivered onto the sample by the two systems. This in turn is an estimate of the number of incoming photons. For the power levels that are used in fluorescence microscopy, the relationship between excitation intensity and photobleaching rate is generally accepted to be a linear one: k = aI. Under this assumption with matched excitation power P, imaged area A, and imaging time on the two systems, the intensities and photobleaching rates are related by: I710/IIMS = k710/kIMS = npixel.
For the second set of experiments we used a thin film of fluorescein solution to measure the number of photons collected by the two systems. Again, the sample being thinner than the optical section selected on the LSM710 allowed for a direct comparison between the systems. The number of emitted fluorescence photons is now only proportional to the number of excited photons. Moreover in these conditions there is no background to be subtracted from the signal and the only source of noise is the shot noise that follows Poisson's distribution. The number of detected photons (i.e. photo-electrons) Nd equals 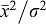 , where
, where  is the mean of the gray level distribution and σ is the standard deviation of the same distribution. For the LSM710 the number of detected photons in a single spectral bin centered at 519 nm, with a spectral width of 9.7 nm, was Nd710 = 78. For the IMS the number of detected photons in an 8 nm spectral bin centered at 519 nm was NdIMS = 1465. Thus, assuming linear photobleaching, the ratio of DIMS/D710 = (NdIMS/Nd710)·(I710/IIMS)·(AIMS/A710) = (NdIMS/Nd710)·(k710/kIMS)·(AIMS/A710) = 8.4.
is the mean of the gray level distribution and σ is the standard deviation of the same distribution. For the LSM710 the number of detected photons in a single spectral bin centered at 519 nm, with a spectral width of 9.7 nm, was Nd710 = 78. For the IMS the number of detected photons in an 8 nm spectral bin centered at 519 nm was NdIMS = 1465. Thus, assuming linear photobleaching, the ratio of DIMS/D710 = (NdIMS/Nd710)·(I710/IIMS)·(AIMS/A710) = (NdIMS/Nd710)·(k710/kIMS)·(AIMS/A710) = 8.4.
This large difference in collection efficiency is surprising considering the improvements introduced to confocal microscopy over the last 25 years. However, these comparisons are based on equal amounts of photobleaching, which is an appropriate comparison for practical imaging but may not accurately reflect actual collection efficiency. It has been shown that the photobleaching rate in laser scanning confocal microscopy is supra-linear (Patterson and Piston, 2000). Assuming this supra-linear dependence k* = aI1.2, we would need to introduce a correction factor in the relation linking intensities and rates: I710/IIMS≈(k*710/kIMS)/(npixel)0.2. For an image of 348×348 pixels, this correction is ∼10, which would bring the overall detection efficiencies of the two systems to nearly the same level. Further optimization of the IMS system, including the use of a high QE camera, will lead to higher collection efficiency in the IMS than is possible with a laser scanning system. It should be noted that if image collection rates are not an issue, any hyperspectral imaging approach could be utilized, including confocal spectral imaging such as we have used with the LSM710. In fact, the main advantages of widefield microscopy over laser scanning confocal imaging are the higher signal collection and resultant increased data acquisition rates. Further, the superior temporal resolution given by the IMS system becomes greater as larger frame sizes are captured.
Real-time hyperspectral imaging of [Ca2+]i and cAMP oscillations in MIN6 cells
MIN6 tissue culture β-cells exhibit intracellular calcium ([Ca2+]i) pulses with a period of 30–80 s and a duration of 1–4 s that underlie pulsatile insulin secretion from these cells (Holz et al., 2008) . Pulses of [Ca2+]i arise in β-cells due to [Ca2+]i flux across the plasma membrane through voltage-gated [Ca2+]i channels (VGCC) (Fig. 3A). The VGCCs open after depolarization of the membrane caused by the closing of ATP-sensitive K+ channels. Opening of [Ca2+]i-dependent K+ channels then repolarizes the membrane and closes the VGCCs. The observed pulses are a measure of the repeated opening and closing of these ion channels. To demonstrate the ability of the IMS to monitor the temporal dynamics of [Ca2+]i activity in vivo, MIN6 cells were transfected with GCaMP5G (L. L. Looger, personal communication) (ex 495 nm, em 519 nm). GCaMP5G is made of a circularly permuted GFP moiety and a portion of the [Ca2+]i binding protein calmodulin (Baird et al., 1999; Nagai et al., 2001) (Fig. 3B). In the unbound state, the fluorescent signal from the cpGFP is quenched. Upon binding [Ca2+]i, the protein changes conformation, activates the chromophore, and produces fluorescence with peak emission of 519 nm (Fig. 4A). Changes in GCaMP5G fluorescence show an increase in intensity upon stimulation with 20 mM glucose and 20 mM triethanolamine (TEA), and pulses in [Ca2+]i at the expected frequency of 1–2 per min in 20 mM glucose (Fig. 4B). The IMS system provides the sub-second resolution needed to quantify these pulses and study calcium signaling in a time-resolved manner.
Fig. 3.

[Ca2+]i and cAMP signaling in MIN6 cells and biosensors for measuring them. (A) Pulses of [Ca2+]i arise in β-cells due to [Ca2+]i flux across the plasma membrane through voltage-gated [Ca2+]i channels (VGCCs). cAMP is activated either by glucose uptake or activation of GPCRs. AC, adenylate cyclase. The lime green oval represents a mitochondrion. (B) Schematic of the GCaMP5G [Ca2+]i sensor. (C) Schematic of T-Epac-VV cAMP FRET sensor (mTurquoise in blue, cpVenus-Venus in yellow).
Fig. 4.

[Ca2+]i and cAMP signaling in MIN6 cells. (A) The unmixed component spectra of both sensors. (B) Singly-transfected [Ca2+]i intensity collected 5 minutes after stimulation with 20 mM glucose and 20 mM TEA with a 0.5 s integration time every 2 minutes. (C) FRET ratio over time 5 minutes after stimulation with 20 mM glucose and 20 mM TEA collected with a 0.5 s integration time every 2 minutes. (D) Time-resolved components of C with mTurquoise in blue and cpVenus-Venus in yellow.
To test the ability of the IMS to measure an FP-FRET biosensor in live cells, MIN6 cells were transfected with the FRET-based cAMP sensor T-Epac-VV, a fusion protein containing mTurquoise (Goedhart et al., 2010) and cpVenus-Venus connected by a portion of the cAMP binding protein Epac1 (de Rooij et al., 1998). In cells, donor mTurquoise and acceptor cpVenus-Venus are linked and a strong FRET signal is observed in the absence of cAMP (Fig. 3C). Upon activation of cAMP signaling, the sensor binds cAMP and undergoes a conformational change resulting in a loss of FRET (Fig. 3C). As FRET is eliminated with the change in fluorophore conformation, there is an increase in mTurquoise fluorescence and decrease in cpVenus-Venus intensity. Cells were exposed to 20 mM glucose and 20 mM TEA 24 hours after transfection to stimulate cAMP signaling and were monitored for changes in FRET with continuous collection at 2 fps. As expected, glucose and TEA stimulated an increase in cAMP activity, and led to cAMP oscillations (Fig. 4C,D). As with the [Ca2+]i oscillations, these oscillations occur at the expected frequency of 1–2 per minute. The high-throughput of the IMS system allows for extended time-lapse imaging with the low excitation intensities needed to minimize photobleaching and photodamage.
Simultaneous imaging of [Ca2+]i and cAMP oscillations in MIN6 cells
A major advantage of hyperspectral imaging is its ability to correlate multiple signals. To image simultaneously multiple live-cell dynamic processes, we transfected MIN6 cells with both GCaMP5G and T-Epac-VV. Using multiple biosensors allows the measurement of both [Ca2+]i and cAMP activation after glucose stimulation in MIN6 cells. We have shown that the two biosensors can be simultaneously measured and spectrally unmixed to indicate the expected dynamics as compared to singly transfected controls (Fig. 5A). Using the unmixed data, two representative cells imaged at 2 fps (Fig. 5B,C) demonstrate that [Ca2+]i and cAMP oscillations occur with a similar frequency after stimulation by glucose and TEA. The oscillations are also observed in the unmixed mTurquoise and cpVenus-Venus spectral components of T-Epac-VV (Fig. 5D,E), verifying that the cAMP oscillations we observe in the ratio are not the result of an unmixing artifact.
Fig. 5.

Simultaneous [Ca2+]i and cAMP signaling in MIN6 cells. (A) The unmixed RGB images of a representative field of view (FOV). (B) [Ca2+]i (green) and cAMP (blue) oscillations after 5 minutes of stimulation with 20 mM glucose and 20 mM TEA stimulation collected at 2 fps. (C) [Ca2+] and cAMP oscillations from a cell in a different FOV stimulated and collected as in B. (D,E) Time-resolved components of T-Epac-VV (mTurquoise in blue, cpVenus-Venus in yellow) from the cells shown in B and C, respectively. Venus traces are corrected to account for the observed photobleaching (see Materials and Methods).
Due to data acquisition restrictions in the IMS computer's memory, collecting data at 2 fps limited the number of oscillation periods in each collection record, and thus prohibited a detailed analysis of the temporal relationship between [Ca2+]i and cAMP oscillations (Fig. 5B,C). To overcome this limitation, we performed experiments with the same 0.5 second integration time, but a frame rate of once every 2 seconds. This protocol allowed us to image for a longer period (400 seconds) and obtain more oscillations (Fig. 6A). The GCaMP5G and T-Epac-VV traces over this time period show anti-correlative oscillations. The component traces from the cAMP sensor demonstrate that these oscillations are not an artifact of unmixing or averaging (Fig. 6B). Finally, we plotted the time variant GCaMP5G against the T-Epac-VV (Fig. 6C) and confirm a negative correlation with a Pearson coefficient of −0.5720 (n = 5 cells). One other cell exhibited similar negative correlation of its smaller-amplitude peaks, but also showed a single large-amplitude peak with a strong positive correlation. For the analysis shown in Fig. 6C, data from this cell was not included since we could not confirm whether the single peak was an artifact or a unique biological event. Additionally, a cross-correlation analysis provides the same negative correlation coefficient of −0.5720 for the GCaMP5G and T-Epac-VV signals. This analysis also demonstrated that the GCaMP5G signal rise precedes the T-Epac-VV decrease. Together, these data show that [Ca2+]i and cAMP signals are anti-correlated in pancreatic β-cells, and [Ca2+]i oscillations lead those of cAMP by 2.5 seconds. Because this difference in rise time is comparable to previously used imaging rates, the relative rise times of these two signals have not been assessed. The observed anti-correlation confirms the predictions of computational models (Holz et al., 2008), and the superior time resolution of the IMS system represents a significant potential for further detailed analysis of the interplay between the [Ca2+]i and cAMP under various treatments.
Fig. 6.

Correlating [Ca2+]i and cAMP oscillations. (A) [Ca2+]i (green) and cAMP (blue) activity average in five cells after stimulation with 20 mM glucose and 20 mM TEA collected with a 0.5 s integration time. (B) Average components of the T-Epac-VV cAMP (mTurquoise in blue, cpVenus-Venus in yellow) sensor from A. (C) Correlation plot of average [Ca2+]i and cAMP traces from A. r = −0.5720.
Discussion
We utilized time-resolved image mapping spectroscopy for real-time hyperspectral imaging of pancreatic β-cell dynamics, and successfully monitored [Ca2+]i and cAMP activity during glucose stimulation. The interplay between these two dynamic signaling pathways has been difficult to determine due to the spatially and spectrally overlapping sensors available to study them in living cells. This is a common hurdle as the overlapping emission spectra of many biosensors prevent the real-time study of signaling dynamics from multiple pathways. A previous study assaying the relationship of [Ca2+]i and cAMP in live cells using spinning disc confocal microscopy suffered from the inability to collect data with a single hardware configuration (Landa et al., 2005). While the fluorophores used in that study (Fura-2 for [Ca2+]i and Epac1-camps) are spectrally separated with excitation and emission, they are both ratiometric, with Fura-2 requiring dual-excitation and Epac1-camps reported as a ratio of emissions. Thus, at least three images were acquired at each time point (two for Fura-2 and one for Epac1) with needed configuration switching between each one, leading to a total acquisition time of a couple of seconds. Our results demonstrate the ability of the IMS approach to address the collection of two biosensors in a snapshot format that is truly simultaneous. This is particularly critical for biological questions that probe temporal relationships and have spectrally overlapping biosensors.
Many questions remain about stimulus-secretion coupling in pancreatic β-cells, especially regarding the role of intracellular second messengers in maintaining proper cell health and function. Significant progress in elucidating these roles in β-cell function will be possible with the simultaneous monitoring provided by the IMS system. Since the 4-D (x, y, λ, t) datacube is captured in a single snapshot, there is no compromise between system throughput and image acquisition rate. Thus, high dynamic range images are captured in real-time imaging experiments, and provide reliable measurements of spectrally overlapped biosensors. While in this study we utilized FRET and cpGFP-based sensors, there are many spatially and spectrally overlapping sensors available for studying dynamic processes. The IMS approach allows simultaneous measurement in real cells of these dynamics, and thus can assist in identifying relationships between multiple cellular processes as well as individual molecular events.
The acquisition rate of our current IMS system is up to 7.2 fps, which is limited by the CCD camera data transfer rate. If acquisition speed was not a limiting issue, these experiments could be done on any spectral imaging device, including the Zeiss LSM710, which provides a spectral resolution to 3.2 nm. However, upon performing the experiments on the LSM710 (data not shown), we found that for spectral resolution comparable to the IMS, the acquisition speeds of >10 sec was insufficient for drawing conclusions about the temporal relationships between the cAMP and [Ca2+]i signaling pathways. Subtle changes in either cAMP or [Ca2+]i with pharmacological modulation require high temporal resolution to identify changes in oscillation frequency, rise time, and the effect these changes have on the relationship between pathways. The IMS system provides a significant improvement in the temporal collection of data from simultaneous measurement of [Ca2+]i and cAMP, and is a powerful tool for multidimensional analysis of signaling pathways. This potential can be even greater considering recent developments in scientific CMOS (sCMOS) detector arrays, which can provide image acquisition rates up to 100 fps, while at the same time, maintain low readout noise (<3 e−1). An IMS imaging system utilizing sCMOS detectors would be an ideal candidate for measuring fast dynamic scenes – e.g. action potentials in neuroscience and cardiology.
Materials and Methods
Cellular sample preparation
MIN6 cells were transiently transfected with plasmid DNA encoding EGFP-EGFR, ECFP-Mito, and SYFP-Nuc. Transfection was accomplished using Lipofectamine2000 (Invitrogen, Carlsbad, CA) transfection reagent according to the manufacturer's instructions. Cells were seeded onto No. 1.5 coverglass bottomed dishes (MatTek, Ashland, MA) and cultured as previously described for microscopy studies (Rizzo et al, 2009). 24 hours after transfection, samples were fixed with 4% paraformaldehyde, washed in DPBS, and mounted on microscope slides with gelvasol.
For live cell experiments, MIN6 cells were transiently transfected with plasmid DNA encoding the GCaMP5G and/or T-Epac-VV biosensors. Reference spectra were collected from cells transiently expressing the individual fluorophores. Transfection was accomplished as described for the fixed cell samples. Cells were seeded onto No. 1.5 coverglass bottomed dishes (MatTek) and cultured as previously described for microscopy studies (Rizzo et al, 2009).
Fluorescence microscopy
The hyperspectral fluorescence imaging experiments were implemented on a Nikon (Melville, NY) inverted microscope TE300 with 40×/1.3 Plan Fluor oil objective. The cells were placed in extracellular-like buffer (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 5.5 mM glucose and 20 mM Hepes, pH 7.4), and were epi-illuminated by a 75 W Xenon lamp. A Chroma (Bellows Falls, VT) filter set (ex: BP 436/20, bs: DCLP455, em: HQ 510/80) was used to separate fluorescence emission from excitation light. The intermediate image is formed at the microscope side image port, which is the image entrance port of the IMS. The hyperspectral images were captured and analyzed by the IMS.
Image mapping spectrometry
Image Mapping Spectrometry is a snapshot spectral imaging method. It is based on the image mapping principle, which maps a sample's 3D datacube (x, y, λ) onto a 2D detector array for parallel measurement. The system setup consists of four major optical components.
(1) Image relay. The image relay has two functions: to transfer the intermediate image from the microscope image port to the image mapper; and to force the light rays to be telecentric at the image side. The telecentricity at the image side is the requirement for correct guidance of light rays reflected from the image mapper. The image relay optics are composed of a Zeiss (Oberkochen, Germany) 2.5×/0.075 Plan-Neofluar objective and Zeiss 130 mm tube lenses (F.L. = 165 mm, Edmund optics, Barrington, NJ, P/N: NT58-452). The distance between the objective back pupil aperture and the tube lens is set to be 165 mm, equal to the focal length of the objective, so that the chief rays are parallel to the optical axis at the image side.
(2) Image mapper. The image mapper is a custom fabricated component, which consists of 350 mirror facets (70 µm wide, 25 mm long) on a 25 mm×25 mm high purity Kobe aluminum substrate. Each mirror facet has a 2D tilt angle (αx, αy) (αx = 0.075, 0.045, 0.015, −0.015, −0.045, −0.075 rad.; αy = 0.045, 0.015, −0.015, −0.045 rad.) to reflect light into different sub-pupils. The image mapper was fabricated by the ruling approach on a 4-axis ultra-precision lathe (Moore Nanotechnology Systems, Swanzey, NH, 250UPL).
(3) Collecting lenses. We use an Olympus (Center Valley, PA) 1×objective (P/N: MVPLAPO, F.L. = 90 mm, N.A. = 0.189) to collect light reflected from the image mapper. A 6×4 sub-pupil array is formed at the back focal plane of the collecting lenses.
(4) Prism array and re-imaging lens array. The role of the prisms and re-imaging lenses is to disperse the sub-pupils and re-image them onto the CCD detector. The prisms are constructed as a 6×1 array of double-Amici prisms fabricated by Tower Optical, Inc. (Boynton Beach, FL). Each double-Amici prism consists of two identical Amici prisms separated by a pupil mask (dia. = 3 mm). The designed spectral range is from 450 nm to 650 nm. The re-imaging lens array is composed of 6×4 achromatic doublets (Edmund optics, Barrington, NJ, P/N: 45-408, dia. = 5 mm, F.L. = 20 mm). The field of view of each re-imaging lens on the CCD detector is of size 5.55 mm×5.55 mm. When the PSF on the image mapper matches the mirror facet width, the PSF on the CCD detector is about 16.8 µm in diameter, and is sampled by ∼2 camera pixels (pixel size: 7.4 µm×7.4 µm).
Time-resolved measurements of cellular dynamics
We used MetaMorph (Molecular Devices, Sunnyvale, CA) as the controller software to synchronize the acquisition of the IMS with the illumination shutter. The IMS camera and illumination shutter were working under “rising edge triggering mode”, in which the frame rate is controlled by trigger pulse frequency while the frame exposure time is controlled by trigger pulse duration.
The [Ca2+]i and cAMP dynamics were measured in two imaging modalities. The first, continuous, was utilized for all 2 fps imaging and involved the IMS software driving the camera shutter. For the second modality, interval, MetaMorph was used to control the acquisition at 0.5 s integration time and 2 second interval sampling. A total of 200 images were captured during 400 seconds of acquisition time. For [Ca2+]i oscillations, the same region of interest (ROI) was chosen in acquired datacubes at the spectral page whose nominal wavelength (519 nm) is close to the GCaMP5G's peak emission. The GCaMP5G intensity at each temporal sampling point was calculated by averaging all the pixels in the selected ROI at corresponding frame, and then drawn versus time to show [Ca2+]i oscillations (Fig. 4B).
For combined GCaMP5G and T-Epac-VV imaging, the acquired datacubes were processed by a linear spectral unmixing algorithm (Zimmermann, 2005), which unmixed mTurquoise, cpVenus-Venus, and GCaMP5G based on their spectral differences and generated time-lapsed intensity changes for all three biosensors. Then, the abundance of each unmixed biosensor inside a cell is calculated by averaging all pixels within the cellular boundary. The processed FRET data was plotted as the ratio of donor (mTurquoise) to corrected acceptor (cpVenus-Venus) and both FRET and GCaMP5G traces were normalized to the average of the first 5 seconds of data collected before glucose stimulation. cpVenus-Venus curves were corrected using a linear detrending algorithm to account for photobleaching. Correlations were defined by Pearson correlation coefficient from plotting the normalized GCaMP5G signal against the normalized FRET ratio. Cross-correlation analysis was performed by the crosscorr function in MatLab with undefined bounds and 200 lags for T-Epac-VV and GCaMP5G signals.
Acknowledgments
We thank K. Jalink, and L. Looger and L. Tian for the gifts of the T-Epac-VV and GCaMP5G plasmids, respectively. We also thank Rebellion Photonics Inc. for supplying software used for setting up an IMS.
Footnotes
Funding
This work was supported by the National Institutes of Health [grant numbers R21EB009186, R01DK53434, R01DK85064, R01CA124319]. Deposited in PMC for release after 12 months.
References
- Baird G. S., Zacharias D. A., Tsien R. Y. (1999). Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 96, 11241–11246 10.1073/pnas.96.20.11241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J., Zwartkruis F. J., Verheijen M. H., Cool R. H., Nijman S. M., Wittinghofer A., Bos J. L. (1998). Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477 10.1038/24884 [DOI] [PubMed] [Google Scholar]
- Gao L., Kester R. T., Tkaczyk T. S. (2009). Compact Image Slicing Spectrometer (ISS) for hyperspectral fluorescence microscopy. Opt. Express 17, 12293–12308 10.1364/OE.17.012293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L. A., Kester R. T., Hagen N., Tkaczyk T. S. (2010). Snapshot Image Mapping Spectrometer (IMS) with high sampling density for hyperspectral microscopy. Opt. Express 18, 14330–14344 10.1364/OE.18.014330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J., van Weeren L., Hink M. A., Vischer N. O., Jalink K., Gadella T. W., Jr (2010). Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat. Methods 7, 137–139 10.1038/nmeth.1415 [DOI] [PubMed] [Google Scholar]
- Holz G. G., Heart E., Leech C. A. (2008). Synchronizing Ca2+ and cAMP oscillations in pancreatic beta-cells: a role for glucose metabolism and GLP-1 receptors? Focus on “regulation of cAMP dynamics by Ca2+ and G protein-coupled receptors in the pancreatic beta-cell: a computational approach”. Am. J. Physiol. Cell Physiol. 294, C4–C6 10.1152/ajpcell.00522.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kester R. T., Bedard N., Gao L., Tkaczyk T. S. (2011). Real-time snapshot hyperspectral imaging endoscope. J. Biomed. Opt. 16, 056005 10.1117/1.3574756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarenbeek J. B., Goedhart J., Hink M. A., Gadella T. W., Jalink K. (2011). A mTurquoise-based cAMP sensor for both FLIM and ratiometric read-out has improved dynamic range. PLoS ONE 6, e19170 10.1371/journal.pone.0019170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers G. J., Piston D. (2010). Photoconversion of purified fluorescent proteins and dual-probe optical highlighting in live cells. J. Vis. Exp. (40). 10.3791/1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers G. J., Gilbert S. G., Cranfill P. J., Davidson M. W., Piston D. W. (2011). Fluorescent proteins at a glance. J. Cell Sci. 124, 157–160 10.1242/jcs.072744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam A. J., St–Pierre F., Gong Y., Marshall J D., Cranfill P J., Baird M A., McKeown M R., Wiedenmann J., Davidson M W., Schnitzer M J., Tsien R Y., Lin M. Z.2010). Improving FRET dynamic range with bright green and red fluorescent proteins. Nat Methods. 9 1005–1012 10.1038/nmeth.2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landa L. R., Jr, Harbeck M., Kaihara K., Chepurny O., Kitiphongspattana K., Graf O., Nikolaev V. O., Lohse M. J., Holz G. G., Roe M. W. (2005). Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 beta-cell line. J. Biol. Chem. 280, 31294–31302 10.1074/jbc.M505657200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavis L. D., Raines R. T. (2008). Bright ideas for chemical biology. ACS Chem. Biol. 3, 142–155 10.1021/cb700248m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris H. R., Hoyt C. C., Treado P. J. (1994). Imaging Spectrometers for Fluorescence and Raman Microscopy: Acoustooptic and Liquid-Crystal Tunable Filters. Appl. Spectrosc. 48, 857–866 10.1366/0003702944029820 [DOI] [Google Scholar]
- Nagai T., Sawano A., Park E. S., Miyawaki A. (2001). Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 98, 3197–3202 10.1073/pnas.051636098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson G. H., Piston D. W. (2000). Photobleaching in two-photon excitation microscopy. Biophys. J. 78, 2159–2162 10.1016/S0006-3495(00)76762-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinaud F., Michalet X., Bentolila L. A., Tsay J. M., Doose S., Li J. J., Iyer G., Weiss S. (2006). Advances in fluorescence imaging with quantum dot bio-probes. Biomaterials 27, 1679–1687 10.1016/j.biomaterials.2005.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piston D. W., Kremers G. J. (2007). Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem. Sci. 32, 407–414 10.1016/j.tibs.2007.08.003 [DOI] [PubMed] [Google Scholar]
- Rizzo M. A., Springer G., Segawa K., Zipfel W. R., Piston D. W. (2006). Optimization of pairings and detection conditions for measurement of FRET between cyan and yellow fluorescent proteins. Microsc. Microanal. 12, 238–254 10.1017/S1431927606060235 [DOI] [PubMed] [Google Scholar]
- Rizzo M. A., Davidson M. W., Piston D. W. (2009). Fluorescent Protein Tracking and Detection: Fluorescent Protein Structure and Color Variants. Cold Spring Harbor Protocols 2009, pdb.top63 10.1101/pdb.top63 [DOI] [PubMed] [Google Scholar]
- Sinclair M. B., Haaland D. M., Timlin J. A., Jones H. D. T. (2006). Hyperspectral confocal microscope. Appl. Opt. 45, 6283–6291 10.1364/AO.45.006283 [DOI] [PubMed] [Google Scholar]
- Tian L., Hires S. A., Mao T., Huber D., Chiappe M. E., Chalasani S. H., Petreanu L., Akerboom J., McKinney S. A., Schreiter E. R.et al. (2009). Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881 10.1038/nmeth.1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Shui B., Kotlikoff M. I., Sondermann H.2008). Structural basis for calcium sensing by GCaMP2. Structure 16, 1817–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann T. (2005). Spectral imaging and linear unmixing in light microscopy. Adv. Biochem. Eng. Biotechnol. 95, 245–265 [DOI] [PubMed] [Google Scholar]