

Summary
Claudins are critical components of epithelial and endothelial tight junction seals, but their post-transcriptional regulation remains poorly understood. Several studies have implicated phosphorylation in control of claudin localisation and/or function, but these have focused on single sites or pathways with differing results, so that it has been difficult to draw general functional conclusions. In this study, we used mass spectrometry (MS) analysis of purified claudin-2 from MDCK II cells and found that the cytoplasmic tail is multiply phosphorylated on serines, a threonine and tyrosines. Phos-tag SDS PAGE revealed that one site, S208, is heavily constitutively phosphorylated in MDCK II cells and in mouse kidney; this site was targeted for further study. Mutational analysis revealed that the phosphomimetic mutant of claudin-2, S208E, was preferentially localised to the plasma membrane while claudin-2 S208A, which could not be phosphorylated at this site, both immunolocalized and co-fractionated with lysosomal markers. Mutations at sites that were previously reported to interfere with plasma membrane targeting of claudin-2 reduced phosphorylation at S208, suggesting that membrane localisation is required for phosphorylation; however phosphorylation at S208 did not affect binding to ZO-1 or ZO-2 Administration of forskolin or PGE2 resulted in dephosphorylation at S208 and transient small increases in transepithelial electrical resistance (TER). Together these data are consistent with phosphorylation at S208 playing a major role in the retention of claudin-2 at the plasma membrane.
Key words: Tight junction, Claudin, Claudin-2, Phosphorylation
Introduction
Tight junctions are intercellular adhesive cell contacts that form selective barriers between epithelial cells, prohibiting the paracellular movement of most large solutes and variably restricting movement of small solutes and ions. They are composed of a large number of transmembrane and cytoplasmic plaque proteins, with members of the claudin family of integral membrane proteins forming the critical elements creating the ion- and size-selective pores through the barrier (reviewed by Furuse and Tsukita, 2006; Anderson and Van Itallie, 2009; Shen et al., 2011).
The 23 annotated claudins are tetraspan proteins that polymerize to form a continuous paracellular barrier, with two extracellular domains that interact to form the paracellular pores (Elkouby-Naor and Ben-Yosef, 2010). Claudins are small proteins (20–23 kDa) with cytoplasmic carboxyl tails which vary in length from 23 to 56 amino acids. Considerable study has been devoted to the role of specific amino acids in the extracellular domains and how they influence paracellular charge selectivity (Colegio et al., 2002; Hou et al., 2005; Angelow and Yu, 2009) and claudin: claudin interactions (Piontek et al., 2008; Piontek et al., 2011). In addition, the transmembrane helices have been implicated in homo- (Van Itallie et al., 2011) and heterodimerisation (Hou et al., 2009). Most claudins share a highly conserved PDZ-binding motif at the extreme carboxyl terminus which mediates binding to the first PDZ domain of the tight junction scaffolding proteins ZO-1, -2 and -3 (Itoh et al., 1999). Aside from the PDZ binding motif however, the cytoplasmic tails are the least conserved region of claudins. In spite of this lack of sequence identity, the tails share compositional features: first, they contain perimembrane cysteines that have been identified, in claudin-14, as palmitoylation sites (Van Itallie et al., 2005); second, compared to the entire protein they are relatively enriched in serine, threonine, tyrosine, proline and basic residues (supplementary material Table S1).
Along with forming the porous paracellular seal, the tight junction is a nexus for many signalling pathways (González-Mariscal et al., 2008). A large number of serine/threonine and tyrosine kinases have been localised to the area of the junction and phosphorylation of the cytoplasmic scaffolding proteins (Stevenson et al., 1989; Harhaj and Antonetti, 2004; Bal et al., 2012) and of the transmembrane protein occludin (Sakakibara et al., 1997; Jain et al., 2011; Raleigh et al., 2011; Su et al., 2011), which co-polymerizes with claudins, has been reported. In addition, a number of studies have demonstrated regulatable changes in claudin phosphorylation. For example, a wide variety of agents, including phorbol esters (Sjö et al., 2010) and cAMP (Ikari et al., 2006) as well as a number of protein kinases, including protein kinases A (D'Souza et al., 2005; Ikari et al., 2008), C (D'Souza et al., 2007; Aono and Hirai, 2008), EphA2 (Tanaka et al., 2005) and WNK4 (Ohta et al., 2006; Tatum et al., 2007) have all been shown to phosphorylate various claudins; phosphorylation of serine, threonine and tyrosine residues have all been reported. However, there is little commonality among these studies making general conclusions difficult and phosphorylation has been variously reported to increase, decrease or have no effect on junctional localisation and barrier functions.
Claudin-2 is among the best studied of the two dozen claudins. It forms a cation-selective pore (Amasheh et al., 2002; Yu et al., 2009) and its transcription is physiologically regulated by cytokines (Amasheh et al., 2010; Suzuki et al., 2011). Claudin-2 levels are increased in the gut in several models of inflammation (Ridyard et al., 2007; Fries et al., 2008; Weber et al., 2008; Schulzke et al., 2009) and in Crohn's disease (Zeissig et al., 2007) and its expression is associated with increased paracellular leakiness for cations and a decrease in transepithelial electrical resistance (TER). However, most information on the regulation of claudin-2 is at the level of gene expression.
At the level of protein trafficking in MDCK II cells, claudin-2 is reported to be continuously recycled from the tight junction (Dukes et al., 2012) and there are a number of studies which suggest selective retrieval of claudin-2 in response to various stimuli, including EGF treatment (Ikari et al., 2011), exposure to acidosis (Balkovetz et al., 2009) or hydrogen peroxide (Gonzalez et al., 2009). In addition, the interaction of claudin-2 with other tight junction proteins has been reported to be acutely influenced by phosphorylation of occludin (Raleigh et al., 2011). These latter studies suggest that there are likely to be post-translation modifications of claudin-2 allowing regulation of its interactions with other proteins and with the trafficking machinery. Although phosphorylation of claudin-2 has not been reported in the literature, because of its key role in barrier leakiness in health and disease we undertook a systematic analysis of its phosphorylation in order to better understand its regulation. In this study, we used mass spectrometry (MS) analysis of tagged, purified claudin-2 and Phos-tag affinity SDS PAGE (Kinoshita-Kikuta et al., 2007; Kinoshita and Kinoshita-Kikuta, 2011) of endogenous and mutated claudin-2 to identify phosphorylated residues. These techniques allowed us to identify S208 as a major constitutive phosphorylated site that is important in claudin-2 localisation at the plasma membrane.
Results
Claudin-2 is multiply phosphorylated within its carboxyl terminal cytoplasmic domain
Analysis of the cytoplasmic tail of claudin-2 using prediction algorithms such as NetPhos 2.0 (http://www.cbs.dtu.dk/services/NetPhos/ (Blom et al., 1999) suggested that several positions in this 46 residue domain might be targets for phosphorylation; these amino acid are highlighted in Fig. 1A. To determine if any of these, or other sites, were phosphorylated in cultured cells, we expressed tet-regulated mouse claudin-2 Flag-tagged on the N-terminus in Tet-off MDCK II cells, purified the tagged protein by immunoaffinity chromatography followed by SDS-PAGE and performed in-gel proteolysis. MS analysis of tryptic peptides and chymotryptic peptides confirmed the sites identified by NetPhos 2.0, and identified several additional sites; these amino acid residues are identified in Fig. 1A by the use of blue (NetPhos 2.0) or red type (MS).
Fig. 1.
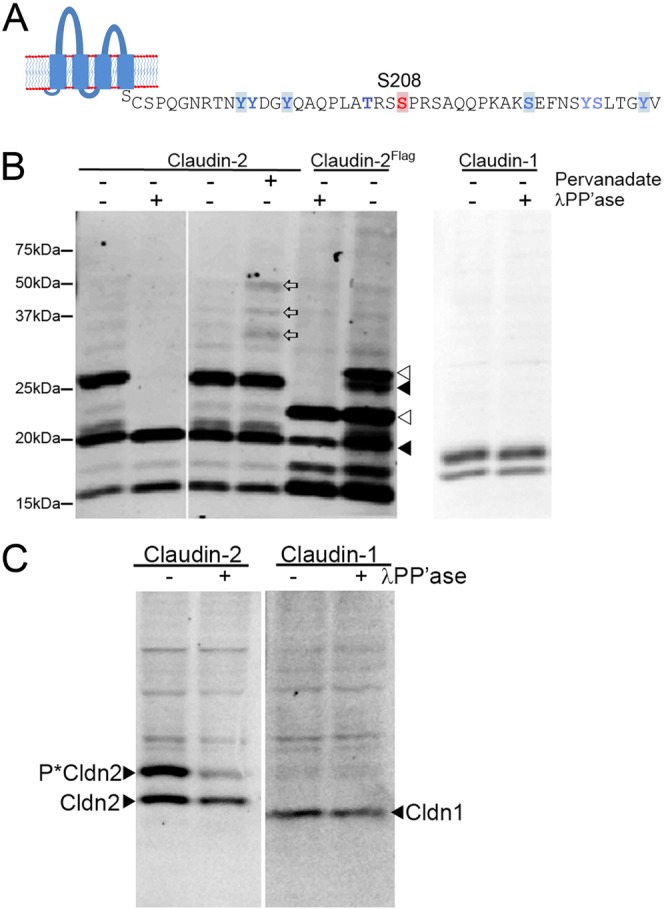
Identification of phosphorylation sites in claudin-2. (A) Use of the Net-Phos algorithm identified the highlighted amino acid residues as potentially modified by phosphorylation; thes and additional sites were confirmed by MS (blue residues). MS additionally identified several other sites, including Y195, T205, Y224 and S225; S208 is indicated in red. (B,C) Phos-tag SDS PAGE reveals that claudin-2 is multiply phosphorylated in MDCK II cells and in mouse kidney. (B, Left panel) Immunoblot of claudin-2 after Phos-tag SDS PAGE shows presence of multiple bands retarded by the Phos-tag-ligand, including a major band that migrates just above the 25 kDa mw marker (1st lane on left). Treatment of lysate with λ phosphatase reduces most of these bands to the size of unphosphorylated claudin-2, ∼20 kDa (2nd lane from left). Treatment of cells for 15 min with 1 mM pervanadate results in the appearance of several new immunoreactive bands (land 4, open arrows) compared with untreated cells (lane 3). N-terminally flag-tagged claudin-2 with (lane 5) and without (lane 6) λ phosphatase treatment shows the same pattern of phosphorylated bands, although they are shifted higher compared (open arrowheads) with the signal from endogenous claudin-2 (closed arrowheads). (B, right panel) Immunoblot of claudin-1 in the same samples reveals no evidence for constitutive phosphorylation; lower band is a proteolysis product. (C, left panel) Claudin-2 immunoblot of Phos-tag SDS PAGE of mouse kidney cell lysate reveals a similar major phosphorylated band as that seen in MDCK II cells; treatment of lysate with λ phosphatase results in decreased signal from this higher MW species; unphosphorylated claudin-2 migrates below the major phosphorylated species. (C, right panel) No constitutive phosphorylation of claudin-1 is seen in the same samples. Upper bands in both blots are non-specific signal from secondary antibody.
To determine if the endogenous non-tagged claudin-2 were phosphorylated in MDCK cells, we performed phosphate-affinity SDS-PAGE with the acrylamide-pendant Phos-tag reagent; covalent incorporation of this ligand in the acrylamide polymer produces a gel matrix which specifically retards phosphorylated proteins during SDS-PAGE (Kinoshita-Kikuta et al., 2007; Kinoshita and Kinoshita-Kikuta, 2011). Immunoblots of MDCK lysates analysed by Phos-tag SDS-PAGE reveals that a fraction of the endogenous claudin-2 migrates at its characteristic molecular weight (slightly below the 20 kDa prestained molecular weight marker), but that a majority of the claudin-2 migrates more slowly, in one major band (around the 25 kDa molecular weight marker) and several minor bands (Fig. 1B, first lane). Smaller bands (at 15 kDa and below) represent claudin-2 proteolysis products. Treatment of MDCK extract with lambda (λ) phosphatase (Fig. 1B, second lane) resulted in the disappearance of the slower migrating bands and increase in the amount of claudin-2 at 20 kDa, consistent with the upper bands representing phosphorylated forms of claudin-2. For convenience, we refer to this 20 kDa band as nonphosphorylated claudin-2, although we recognise that it may include some phosphorylated claudin-2. Treatment of MDCK cells with the cell-permeant tyrosine phosphatase-inhibitor, pervanadate (Bennett et al., 1993), resulted in the appearance or increase in intensity of several additional higher molecular weight bands (Fig. 1B, compare lanes 3 and 4, open arrows), consistent with the possibility that claudin-2 can be tyrosine phosphorylated as was predicted by NetPhos and demonstrated by MS analyses. Phos-tag SDS-PAGE also demonstrated that Flag-tagged claudin-2 was phosphorylated in a similar pattern to endogenous claudin-2, although the major bands now are shifted up by the additional mass of the Flag epitope (Fig. 1B, lanes 5 and 6 from the left, with and without the addition of lambda phosphatase).
A similarly sized major phosphorylated form of claudin-2 is also present in vivo in mouse kidney (Fig. 1C), although addition of phosphatase was less efficient in dephosphorylation in this tissue sample. In contrast to claudin-2, there was no evidence for constitutive claudin-1 (or claudin-7, not shown) phosphorylation in either MDCK cells or in mouse kidney using the Phos-tag method (Fig. 1B,C, right panels).
Mutational analysis demonstrates that S208 is the major site for claudin-2 phosphorylation
To determine which amino acid residue(s) accounts for the major phosphorylated form of claudin-2, we made mutations in several phosphorylation sites identified by MS analysis. Claudin-2 mutated at different sites was inducibly expressed in MDCK I Tet-off cells, which lack endogenous claudin-2 and consequently develop a high transepithelial resistance Immunoblot analysis of stable cell lines expressing two of the mutant forms (Fig. 2A, top) shows that wild-type claudin-2, claudin-2 with S208 or S219 mutated to a non-phosphorylatable alanine (S208A and S219A) or to a phosphomimetic glutamic acid (S208E and S219E) were all induced and to approximately equal levels. Phos-tag SDS-PAGE (Fig. 2A, bottom panel) reveals that mutation of claudin-2 S208 to either alanine or glutamic acid results in loss of the major claudin-2 phosphorylated band (P*Cldn2), while mutation of S219 produces no discernible change in the phosphorylation pattern compared with wild-type expressing cells; in the example shown in Fig, 2, wild-type claudin-2 and S219E are expressed at a lower level that the other constructs. Because the available antibodies raised against claudin-2 are directed against non-phosphorylated peptides encoding regions of the carboxyl terminus, it was possible that there might be other major phosphorylated forms of claudin-2 that are not recognised in immunoblots. To circumvent the possibility of phosphorylation-dependent inhibition of antibody binding, we expressed N-terminally GFP-tagged wild type and S208A forms of claudin-2 in HEK cells and performed Phos-tag SDS-PAGE on cell lysates. Proteins were directly imaged in the gels, without need for an antibody, using a flat-bed fluorescence scanner. The resulting image (Fig. 2B) reveals two GFP-tagged wild-type protein bands while the GFP-tagged S208A mutant form lacks the upper band. This again supports the idea that the main phosphorylated form of claudin-2 is due to phosphorylation at S208. Other less prominent phosphorylated forms of GFP-tagged claudin-2 are also revealed, suggesting that these minor phospho-forms are easily detectable with our anti-tail antibodies (open arrowheads).
Fig. 2.

Mutational analysis reveals that S208 is the major constitutively phosphorylated site in Claudin-2. (A) MDCK I Tet-off cells were induced (I) or not (U) to express wild-type claudin-2, claudin-2 S208A, claudin-2 S208E, claudin-2 S219A and claudin-2 S219E. Top, immunoblot of SDS-PAGE of cell lysates probed with claudin-2 (cldn2, red) and occludin (ocln, green) antibodies. Bottom, immunoblot of the same samples after Phos-tag SDS PAGE. Unphosphorylated claudin-2 is visible in all (I) lanes as a 20 kDa band. In wild-type and S219 mutants, a major phosphorylated bands is seen above the 25 kDa MW marker position (P*cldn2); this band is absent in the S208 mutants. In the S208 mutants, a diffuse higher molecular weight band was present. Three clones for each construct gave similar results. (B) Lysate from cells expressing GFP alone, GFP-tagged wild-type claudin-2 and GFP-tagged S208A were subjected to Phos-tag electrophoresis and the resulting gels imaged using a Typhoon fluorescent imager. Unphosphorylated GFP-claudin-2 and S208A are detectable in both right hand lanes (lower filled arrowhead), while a strongly phosphorylated band in GFP-claudin-2 but not GFP-claudin-2 S208A (upper filled arrowhead) is consistent with this being the major site of constitutive phosphorylation; upper bands indicated with open arrowheads are likely minor phosphorylated forms of claudin-2. This analysis was repeated three times.
Phosphorylation at S208 increases detergent extractability of claudin-2
To determine if differences in phosphorylation state were associated with differences in biochemical interactions, we measured the Triton X-100 solubility of wild-type claudin-2 (phosphorylated and nonphosphorylated), nonphosphorylatable S208A mutant and phosphomimetic S208E mutant. Comparison of Triton-soluble and insoluble claudin-2 and mutants reveals that wild-type claudin-2 and the phosphomimetic claudin-2 mutant showed a similar level of detergent solubility, while the S208A mutant was less soluble than either form (Fig. 3A,B). In contrast, occludin solubility was similar in all three cells lines (Fig. 3A,B).
Fig. 3.
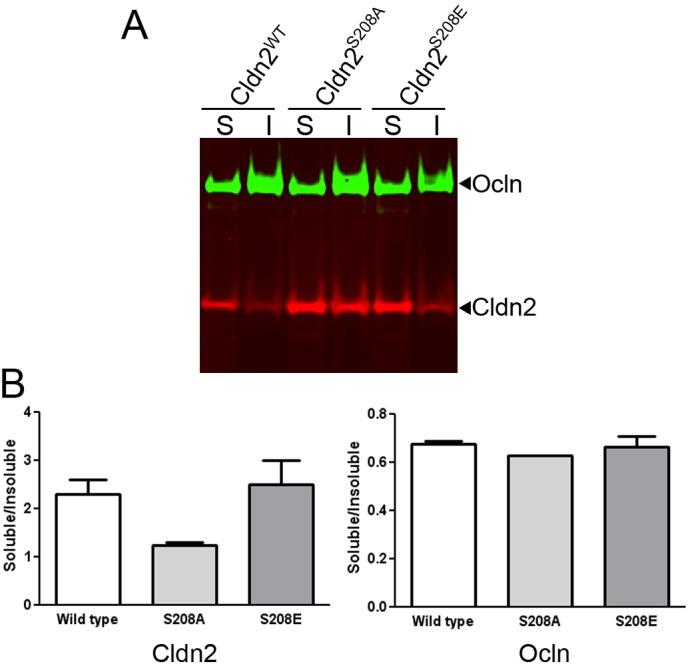
Claudin-2 wild type and S208E are more extracted in detergent than claudin-2 S208A. (A) Immunoblot after SDS PAGE of TX-100-soluble (S) and insoluble (I) claudin-2 extracted from MDCK I Tet-off cells induced to express wild-type claudin-2, claudin-2 S208A and claudin-2 S208E (red) and occludin (ocln, green). (B) Quantification of two replicate experiments reveals that claudin-2 S208A was relatively less soluble in Triton X-100 than either wild-type or claudin-2 S208E; occludin extraction was similar in all samples (mean and range plotted).
Phosphorylation at S208 influences localisation of claudin-2
To determine if this difference in biochemical behaviour might correlate with a difference in subcellular localisation, we used immunofluorescent analysis to compare MDCK I cells induced to express wild-type claudin-2, S208A or S208E. When transgene expression levels were titrated by culturing cells in the presence of a low concentration of doxycycline (0.05 ng/ml), both wild-type claudin-2 and S208E localised preferentially at cell-cell contacts with only scattered intracellular puncta (Fig. 4A, left and right panels). In contrast, although the non-phosphorylatable form of claudin-2, S208A, was also found at cell-cell contacts, there was substantially more intracellular claudin-2 (Fig. 4A, middle panel) than was seen in the wild-type or phosphomimetic S208E mutant. Immunoblot analysis of the doxycycline-titrated cells revealed that the expression levels of the induced claudins differed by less than 20% (supplementary material Fig. S1). In all cases, ZO-1 localisation appeared normal. This difference in localisation between the non-phosphorylatable and phosphomimetic mutants of claudin-2 was also seen when aspartic acid was substituted instead of glutamic acid to mimic negatively-charged phosphate (supplementary material Fig. S2). In contrast, when MDCK I cells were maximally induced to express claudin-2 and the mutant forms, there was no consistent difference in localisation; all cells had considerable intracellular as well as plasma membrane staining for claudin-2 (supplementary material Fig. S3).
Fig. 4.
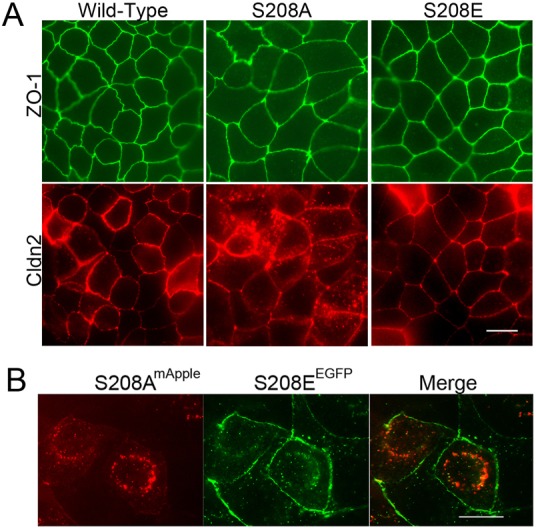
Claudin-2 S208E localises to cell-cell contacts better than non-phosphorylatable claudin-2 mutant S208A. (A) Wild-type claudin-2 (bottom left panel) and claudin-2 S208E (bottom right panel) localised well to cell contacts in MDCK I Tet-off cells induced with a low level of doxycycline (0.05 ng/ml for 7 days), similar to the localisation seen with ZO-1 (top, green). In contrast, some claudin-2 S208A was found at cell:cell contacts, but much was in intracellular vesicles; ZO-1 staining was unaffected by the intracellular claudin-2. Scale bar: 8 µm. This was repeated with several clones with similar results. (B) When N-terminally mApple-labelled claudin-2 S208A and GFP-labelled S208E were co-transfected into MDCK I cells, most of the mApple-labelled claudin-2 S208A was found in intracellular vesicles (left, red), while most of the GFP-labelled claudin-2 S208E was found at the cell membranes, with only a minor amount in intracellular vesicles (middle, green). The merged image (right panel) reveals that there is only minor co-localisation (yellow). Scale bar: 5 µm.
To test if the nonphosphorylatable and phosphomimetic forms of claudin-2 were differentially localised in the same cell, we cotransfected MDCK I cells with mApple-tagged claudin-2 S208A and EGFP-tagged claudin-2 S208E mutants. Although there was considerable overlap between localisation of the two mutants, in many cells claudin-2 S208A was predominately intracellular and S208E was predominately at the plasma membrane (Fig. 4B). Together, the data from both immunolocalization and from fluorescently tagged proteins suggested that claudin-2 S208 phosphorylation is associated with preferential plasma membrane localisation.
In spite of the difference in localisation of the claudin-2 S208A and S208E mutants, we could measure no consistent difference in any physiologic parameters among MDCK I cells expressing wild-type claudin-2 or either of the two mutants. Expression of all three resulted in similar decreases in TER (compared with uninduced cells) over the same time course and did not alter flux of 3 kD dextran, wound healing rates or cell growth (BrdU incorporation) (supplementary material Fig. S4). Additionally, we transfected the S208A and S208E into MDCK II Tet-off cells in which endogenous claudin-2 expression had been depleted using stable shRNA. As expected, the claudin-2 knockdown lines had increased TER (80±4 Ohms×cm2 in KD cells compared with 35±5 Ohms×cm2 in wild-type MDCKs) and decreased dilution potential (6.4±0.4 mV in KD cells compared with 8±1 mV in normal MDCK cells. Expression of wild-type, S208A and S208E claudin-2 forms resulted in equivalent decreases in TER and increases in dilution potential (supplementary material Fig. S5) demonstrating that under these conditions they all had a similar ability to control the barrier.
The S208A mutant of claudin-2 is preferentially associated with LAMP-2, a lysosomal membrane marker
Much of the intracellular punctuate staining of claudin-2 S208A colocalized with LAMP-2 (Fig. 5A, top panels), a lysosomal membrane marker (Nabi et al., 1991), but not with EEA1, an endosomal marker or with caveolin-1 (supplementary material Fig. S6). In contrast, the S208E mutant localised mainly at the plasma membrane and was not found in LAMP-2 positive vesicles (Fig. 5A, bottom panels). Similarly, biochemical enrichment of lysosomes by fractionation with an iodixanol gradient revealed that S208A was concentrated in gradient fractions positive for LAMP-2, while S208E was more evenly distributed throughout the gradient (Fig. 5B,C). Because the S208A appeared preferentially targeted for degradation we compared the half-lives of S208A with S208E after repressing synthesis by restoring doxycyline to the culture media and performing immunoblots over time. In spite of the substantial colocalization of S208A but not S208E with lysosomes, the half-lives of both mutants were similar (supplementary material Fig. S7).
Fig. 5.
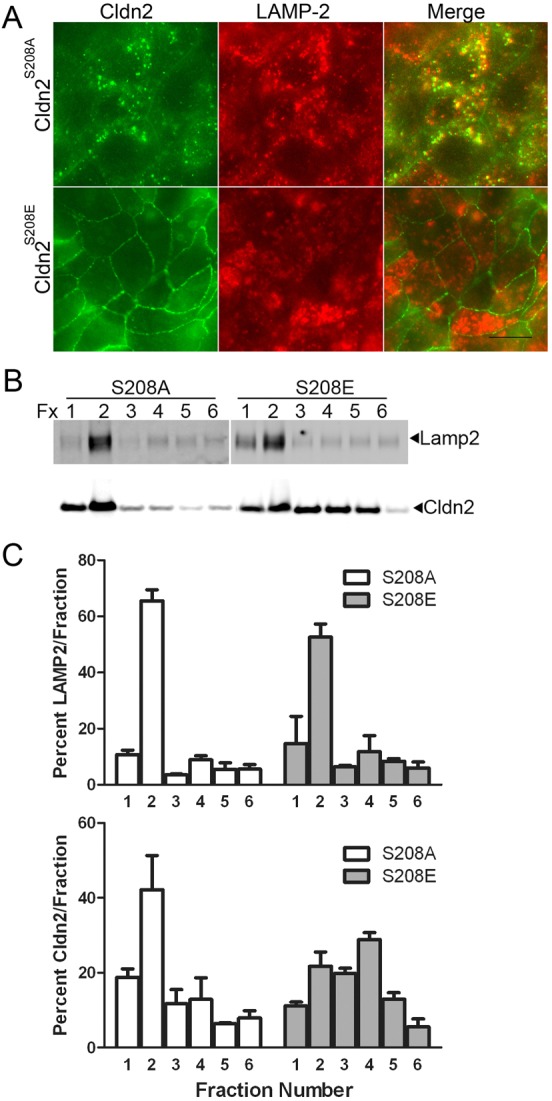
Intracellular S208A mutant of claudin-2 is largely colocalized with LAMP-2, a lysosomal membrane marker. (A) Immunofluorescent co-localisation of claudin-2 mutants expressed in MDCK I cells revealed that claudin-2 S208A (green, top panels) but not S208E (green, bottom panels) colocalized with the lysosomal marker LAMP-2 (red, middle panels). (B) Biochemical enrichment of lysosomes by iodixinal fractionation followed by claudin-2 and LAMP-2 immunoblots demonstrates that claudin-2 S208A concentrates in the same gradient fractions as LAMP-2 (left), while claudin-2 S208E was distributed throughout the gradient. (C) Quantification of the above blot and a separate experiment performed on different claudin-2 S208A and S208E cell clones reveals that most S208A is concentrated in the top two gradient fractions with LAMP-2, while S208E does not show this preferential co-fractionation with the lysosomal marker (mean and range plotted).
Endogenous claudin-2 in lysosomes is hypophosphorylated
We next tested whether the differential characteristics of claudin-2 S208A and S208E were an artefact of introducing mutated proteins or genuinely revealed that dephosphorylation of wild type endogenous claudin-2 correlated with localisation in lysosomes. Endogenous claudin-2 in MDCK II cells is found both at cell contacts and in intracellular vesicular compartments; a fraction of claudin-2 is normally found in LAMP-2 positive vesicles (Fig. 6A). We used iodixanol gradient fractionation to enrich for lysosomes as above; individual fractions were then analysed by Phos-tag SDS-PAGE to compare phosphorylation at S208 in lysosome-enriched compared with non-enriched fractions (Fig. 6B,C). As above, LAMP-2 was concentrated in the top one or two gradient fractions (Fig. 6B, middle panel, and Fig. 6C, lower graph). Claudin-2 was found in all fractions, but in the LAMP-2 containing fractions it was less phosphorylated than in fractions not enriched for lysosomes (Fig. 6A, top panel; Fig. 6B, top graph). For comparison, distribution of occludin was monitored by SDS-PAGE followed by immunoblotting; like claudin-2, occludin was distributed in all fractions, but most of it (70–75%) was associated with non-lysosomal fractions (Fig. 6A, bottom panel).
Fig. 6.

Biochemical fractionation reveals that endogenous claudin-2 associated with lysosomes is hypophosphorylated relative to nonlysosomal claudin-2. (A) Immunofluorescent analysis of endogenous claudin-2 (left panel, green) in MDCK II cells reveals that this protein is normally distributed both at the sites of cell contact and in intracellular vesicles (arrow); LAMP-2 (middle panel, red) is concentrated in intracellular vesicles. Co-localisation (right panel, yellow) of both proteins reveals that much (example, arrow) intracellular claudin-2 is found in lysosomes. Scale bar: 8 µm. (B) Claudin-2 immunoblot following phos-tag SDS PAGE of iodixinal fractionated MDCK II cells reveals that claudin-2 in LAMP-2-containing fractions (middle panel) is less phosphorylated on S208 than claudin-2 in non-LAMP-2 containing fractions. Occludin (bottom panel) is distributed throughout the gradient fractions. Both LAMP-2 and occludin immunoblots were performed after conventional SDS-PAGE. (C) Quantification of three separate gradient fractionation experiments reveals that claudin-2 fraction 1, which also contains the highest concentration of LAMP-2, is significantly less phosphorylated on S208 than claudin-2 in other fractions (*P<0.05, 1-way ANOVA followed by Dunnett's test.
Phosphorylation of claudin-2 requires efficient integration into the tight junction
The results above suggested the possibility that claudin-2 must be located on the plasma membrane or at the tight junction in order to become phosphorylated on S208. In order to test this possibility we measured the relative phosphorylation at S208 in four different claudin-2 mutant proteins that were previously demonstrated to be compromised in their ability to localise at tight junctions. For endogenous claudin-2, ratio of phosphorylated to nonphosphorylated forms is between 1 and 2∶1 (Fig. 7A,E), depending on time in culture and culture conditions; for example, claudin-2 grown on Transwell filters is more highly phosphorylated on S208 than similarly cultured cells on plastic (supplementary material Fig. S8). Overexpressing wild-type claudin-2 increases the absolute amount of claudin-2 that gets phosphorylated at S208, but decreases the relative fraction of phosphorylated to nonphosphorylated claudin-2 (Fig. 7A,E); we have previously shown that immunofluorescence of cell lines overexpressing wild-type claudin-2 results in relatively larger increases in the cytosolic pool of claudin-2 than at the plasma membrane, although there is also more at the plasma membrane (Colegio et al., 2003).
Fig. 7.

Claudin-2 that is not efficiently incorporated into the tight junction is hypophosphorylated at S208. (A) Phos-tag SDS PAGE of MDCK II cells reveals that endogenous claudin-2 is efficiently phosphorylated on S208 (left lane, U, uninduced), quantified in (E), overexpressed wild-type claudin-2 (right lane, I, induced) is less well phosphorylated; removal of the PDZ-binding motif (-PDZ, last three amino acids) further reduces phosphorylation at S208 (middle lane, quantified in E). (B) Comparison of claudin-2 phosphorylation in transfected MDCK I tet-off cells following Phos-tag SDS PAGE reveals that induced (I) wild-type claudin-2 is more phosphorylated than claudin-2 with mutations in the putative palmitoylation sites (Cldn2 palm mut); quantified in E; this palmitoylation mutant fails to localise to sites of cell:cell contact (C); compare top right (wild-type, green) and bottom right panels (palmitoylation mutant, green); compare with ZO-1 immunofluorescence (red, left, top and bottom). (D) Immunoblots of MDCK II cells uninduced (U) or induced (I) to express vsv-g tagged wild-type claudin-2 or vsv-g tagged claudin-2 with the residues GLW in the first extracellular domain mutated to AAA. Top panel, claudin-2 immunoblot following phos-tag SDS PAGE show efficient phosphorylation of endogenous claudin-2; vsv-g immunoblot shows less phosphorylation of tagged claudin-2 and no apparent phosphorylation of the AAA mutant; quantified in (E). All quantifications represent the mean and range of replicate experiments.
In the first localisation-defective mutant, removal of the PDZ binding motif of claudin-2 (C-terminal 3 residues), which eliminates binding to the scaffolding proteins, ZO-1 and ZO-2, further reduced the phosphorylated fraction of claudin-2 (Fig. 7A,E); the subcellular localisation of this construct is similar to that of overexpressed wild-type claudin-2 (not shown). Second, there is no detectable tight junction localisation of a claudin-2 mutant that cannot be palmitoylated (Fig. 7C); this mutant is very poorly phosphorylated at S208 (Fig. 7B,E). Third, similar to the (-)PDZ binding motif constructs, claudin-2 that is tagged at the carboxyl terminus will likely have decreased ability to interact with ZO-1 or ZO-1; these mutants are phosphorylated slightly less well than overexpressed wild-type claudin-2 mutants (Fig. 7D,E). Fourth, vsv-g tagged claudin-2 containing mutations in the highly conserved GLW in first extracellular domain is neither localised at the tight junction (Van Itallie et al., 2011) nor phosphorylated at S208 (Fig. 7D,E). Thus, the correlation between ability to localise to the tight junction and to become phosphorylated at S208 suggests that the responsible kinase might be located at the plasma membrane or junction.
Phosphorylation at S208 does not alter binding to ZO-1 or ZO-2
Because of the observed correlation of membrane localisation and phosphorylation, we tested the possibility that phosphorylation might enhance the ability of claudin-2 to bind to ZO-1 or ZO-2. Bacterially-expressed MBP/His-tagged fusion proteins encoding the cytoplasmic tail of claudin-2, with and without the S208E phosphomimetic mutation were immobilized on a cobalt affinity resin and incubated with MDCK cell lysate. The bound protein was analysed by immunoblot (supplementary material Fig. S9); both wild-type (non-phosphorylated) and the phosphomimetic S208E bound equal amounts of ZO-1 and ZO-2. This result suggests that PDZ-domain binding of claudin-2 is independent of phosphorylation status on S208.
Endogenous claudin-2 is dephosphorylated on S208 in response to PGE2 or Forskolin
Treatment of MDCK II cells with agents that raise intracellular cAMP levels, including dibutryl cAMP (not shown), PGE2 or forskolin all resulted in decreased phosphorylation at S208. For example, treatment of cells with 1 uM PGE2 resulted in a time depended decrease in the ratio of phosphorylated to non-phosphorylated claudin-2 from nearly 2∶1 to less than 0.4∶1 over a 60 minute incubation period (Fig. 8A,B). This treatment also resulted in a small, but significant increase in monolayer TER and a small but reproducible increase in paracellular flux of 3 kDa fluorescent dextran (Fig. 8C), suggesting the level of claudin-2 in the barrier decreased in parallel with dephosphorylation. Treatment with forskolin or dibutryl cAMP also resulted in similar decreased levels of S208 phosphorylation and small increases in TER (not shown), although we cannot rule out an indirect relationship between these parameters. Analysis of the detergent-extractability of endogenous claudin-2 in PGE2 or forskolin-treated MDCK cells demonstrated that non-phosphorylated claudin-2 was more resistant to detergent extraction than phosphorylated claudin-2 (Fig. 8D,E). This is consistent with the previous demonstration that the S208A mutant was more resistant to detergent extraction than the S208E claudin-2 mutant. Other minor phosphorylated forms of claudin-2 partition more exclusively into detergent-soluble (the minor band just above the non-phosphorylated form of claudin-2, open arrowhead) or insoluble pools (the minor band just below the phosphorylated form of claudin-2, open arrow). Together, these results suggest that increases in cAMP may result in activation of a protein phosphatase that dephosphorylates claudin-2 at S208 and may result in decreased membrane localisation; removal of a fraction of claudin-2 from the tight junction would explain the observed increase in TER and might be responsible for the acute increase in flux. We were unable to detect changes in claudin-2 localisation after PGE-2 treatment by immunofluorescence, but the large pools of both membrane and cytosolic endogenous claudin-2 in MDCK II cells might mask small changes in distribution at the tight junction. However, there was a small but consistent increase in the amount of claudin-2 in lysosomal fractions in an iodixanol gradient following PGE treatment, suggesting that loss of phosphorylation was associated with some movement of claudin-2 from the plasma membrane (supplementary material Fig. S10) into the lysosomal pool.
Fig. 8.

Endogenous claudin-2 is dephosphorylated on S208 in response to PGE2 or Forskolin. (A) Immunoblots of claudin-2 after Phos-tag SDS-PAGE following treatment of MDCK II cells with PGE2 reveals a time-dependent decrease in S208 phosphorylation; quantified in (B). (C) PGE2 treatment resulted in slightly but significantly increased TER (top graph, *P<0.05, ANOVA followed by Dunnett's test) and about a 2-fold increase in flux of 3 kDa dextran (*P<0.001, t-test). (D) Immunoblot following Phos-tag SDS PAGE of soluble (left) and insoluble (right) fractions of control, 1 µM PGE2 and 10 µM forskolin-treated MDCK II cells; claudin-2 phosphorylated on S208 is more Triton X-100 (TX) soluble than non-phosphorylated claudin-2. (E) Quantification of relative solubility of S208P to non-phosphorylated claudin-2 after treatments.
Discussion
In this study, we demonstrated that claudin-2 is multiply phosphorylated on serine, threonine and tyrosine residues on the cytoplasmic C-terminal domain, with the major constitutive site of phosphorylation identified as S208. Phosphomimetic mutants of S208 localised to cell-cell contacts better than the non-phosphorylatable S208A mutant; the intracellular pool of S208A was preferentially associated with lysosomes. Similarly, endogenous claudin-2 that co-fractionated with lysosomes was significantly less phosphorylated than non-lysosomal claudin-2. Other types of claudin-2 mutants that were not well localised at the plasma membrane, including mutants of the conserved GLW in the first extracellular domain and mutants than block palmitoylation were poorly phosphorylated at S208, suggesting that plasma membrane localisation was required for phosphorylation at S208. However, in spite of differences in localisation of the S208A and S208E mutants, there were no clear physiologic differences in cell lines expressing the mutants. A small fraction of claudin-2 can likely incorporate into tight junctions independent of phosphorylation status and this fraction is apparently sufficient to be physiologically effective.
Although phosphorylation of the C-terminal tails of other claudins has been reported, both the number of phosphorylation sites and degree of claudin-2 phosphorylation in MDCK II cells was unexpected. We identified phosphorylation of nearly all serines which are conserved among species, the one threonine and all five tyrosines. Recently curated protein phosphorylation sites identified by large scale MS screening from a number of tumours and cancer cell lines collected on PhosphoSitePlus (Hornbeck et al., 2012), confirm all phosphorylated residues identified here except Y229P. Phos-tag SDS PAGE followed by claudin-2 immunoblot also demonstrated the existence of a number of phosphorylated forms; similar constitutive phosphorylation was not observed in other MDCK cell claudins after Phos-tag SDS PAGE and immunoblotting, although the antibodies used might not detect the phosphorylated claudin forms.
The fact that the phosphorylated forms of claudin-2 are differentially retarded in the Phos-tag gels does not imply that more slowly migrating species are more heavily phosphorylated; the degree of retardation using the Phos-tag reagent has been reported to be a function of specific sequences around the phosphorylated sites (Kinoshita-Kikuta et al., 2007). This means that migration rates of individual bands have been shown to be characteristic of specific phosphorylated isoforms (Kinoshita-Kikuta et al., 2007). In addition, not all phosphorylated isoforms are invariably retarded in Phos-tag gels (Kinoshita-Kikuta et al., 2007), since changing the type of gel and chelating agent can alter the number of observed phosphorylated isoforms. Although we saw a similar claudin-2 phosphorylation pattern with both Mn2+ Tris-Gycine SDS PAGE and Zn2+ Neutral SDS PAGE (Kinoshita-Kikuta et al., 2007), it is possible that some phosphorylated claudin-2 forms migrate with the non-phosphorylated claudin-2. We observed a number of minor phosphorylation forms of claudin-2 in unstimulated MDCK cells, some putative tyrosine phosphorylated forms of claudin-2 detectable after treatment of cells with the tyrosine phosphatase inhibitor pervanadate as well as a major phosphorylated claudin-2 form that ran at the size of the 25 kDa molecular weight marker. Mutational analysis demonstrated that this band was due to constitutive phosphorylation of S208.
Using a number of different approaches, we found that phosphorylation of claudin-2 at S208 was associated with plasma membrane localisation. Claudin-2 was likely phosphorylated at the membrane; however, we have not ruled out the possibility that phosphorylation is required for membrane targeting. In any case, S208 phosphorylation did not appear to be an absolute requirement for membrane localisation, since the non-phosphorylatable S208A could still localise, although much less efficiently. The small amount retained on the plasma membrane was still able to lower TER and the selectivity for Na+. Immunofluorescent and biochemical data suggested that S208A was preferentially associated with lysosomes; similarly, biochemical fractionation of endogenous claudin-2 demonstrated a lower level of phosphorylation at S208 in lysosomal fractions than in the heavier non-lysosomal fractions. Since these heavier fractions also contain the majority of the cell occludin, we presume they are relatively enriched in plasma membranes. However, as we had previously observed with mutations made in the palmitoylation sites of claudin-14 (Van Itallie et al., 2005), increased association with lysosomes did not alter protein half-life.
We found that treatment of MDCK II cells with agents that increase cAMP, including dibutryl cAMP, forskolin and PGE2 all resulted in decreased phosphorylation at S208. Treatment with these agents also resulted in slight increases in TER, as has been previously noted at this time point by Flores-Benitez et al. (Flores-Benitez et al., 2009), and which could be explained by transient removal of claudin-2 from the tight junction. However, we were unable to see a change in claudin-2 localisation, which could equally be due to a small effect on localisation in a background of a large pool of endogenous claudin-2 or to a lack of any change. We thus have no direct evidence that the small increases we observe in TER and flux are directly due to dephosphorylation of claudin-2 and its partitioning out of the barrier. Because of the important role of claudin-2 in barrier physiology, we have begun to attempt to identify the kinase(s) responsible for claudin-2 S208 phosphorylation.
Similar to our observations on claudin-2, phosphorylation of transfected claudin-16 S217 was reported to be associated with enhanced tight junction localisation in MDCK II cells. Additionally, both dephosphorylated and a S217A mutant of claudin-16 were localised to lysosomes (Ikari et al., 2006). However, phosphorylation at this site in claudin-16 was increased by cAMP and protein kinase A (Ikari et al., 2008), which is the opposite of what we observed for claudin-2 S208. Additionally, phosphorylation state affected the ability of claudin-16 to interact with ZO-1, which was not the case for S208 phosphorylation of claudin-2. Similar to claudin-16, cAMP treatment results in increased claudin-5 cell contact staining in endothelial cells (Ishizaki et al., 2003) and this change in localisation required T207 (Soma et al., 2004). A requirement for phosphorylation in tight junction localisation has also been reported for claudin-4 S194 in keratinocytes (Aono and Hirai, 2008), but phosphorylation of the same site in ovarian cancer cells correlates with impaired claudin-4 localisation (D'Souza et al., 2007). Phosphorylation of T192 in claudin-3 by protein kinase A disrupts tight junction barriers (D'Souza et al., 2005). Finally, heterologous mutant WNK4 expression in MDCK II cells results in phosphorylation of claudins 1–4 and 7, with no change in localisation of any claudins (Yamauchi et al., 2004), although the phosphorylation site was only identified for claudin-7 (Tatum et al., 2007). The multiplicity of potential phosphorylation sites (Gonzalez-Mariscal et al., 2010) and the lack of sequence conservation among the tails of claudins make it difficult to compare studies. A more comprehensive approach will be required to understand the consequences of claudin phosphorylation.
Most of the reported studies examined the effects of stimulating claudin phosphorylation with a variety of agents, while the present study is mainly focused on constitutive phosphorylation. When we examined other abundant MDCK II cell claudins, including claudin-1, −3, −4 and −7 by Phos-tag SDS PAGE and immunoblot, we saw little evidence for the degree of constitutive phosphorylation seen in claudin-2 (not shown). Why claudin-2 is constitutively phosphorylated is unclear, but as mentioned previously, it is a highly regulated member of the claudin family (Ridyard et al., 2007; Fries et al., 2008; Schulzke et al., 2009; Amasheh et al., 2010; Suzuki et al., 2011). In addition, it is also normally associated with other cell structures aside from tight junctions; depending on cell or tissue type, it is present to a variable extent on the lateral cell membrane (Enck et al., 2001) and in MDCK II cells (this study) (Guillemot et al., 2008; Dukes et al., 2012), a fraction is normally associated with intracellular vesicles. In addition, claudin-2 has been reported in the central cilium (Larre et al., 2011) and in metastatic cells, to be present in integrin-based complexes (Tabariès et al., 2011). Aside from its role in promoting cation permeability at the tight junctions, it has been implicated in regulation of the rate of cell division and metastatic potential (Buchert et al., 2010; Dhawan et al., 2011).
In summary, phosphorylation of claudin-2 at S208 is associated with plasma membrane localisation; one possibility is that phosphorylation at this site may result in release from a dynamic targeting or retrieval mechanism. Claudin-2 dephosphorylated at S208 may thus be targeted for selective removal from the plasma membrane and/or may serve other cellular functions or may be diverted to lysosomes. Our working hypothesis is that an unknown protein-protein interaction is either stabilised or disrupted by phosphorylation of S208; therefore, future studies are aimed at identification of proteins that selectively interact with phosphorylated or dephosphorylated forms of claudin-2. In addition, we plan to explore the relevant signalling pathways and roles of other phosphorylation sites in the regulation of claudin-2 localisation and function.
Materials and Methods
Antibodies and chemicals
Claudin and occludin antibodies were purchased from Life Technologies (Grand Island, NY); LAMP-2 antibody (MCA2558) was from AbD Serotech (Raleigh, NC); EEA-1 and Caveolin-1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA), BrdU antibody (Bu20a) was from Cell Signaling Technology (Beverly, MA); Flag antibody (M2), M2 resin, 3×Flag peptide, Forskolin, and other miscellaneous chemicals were from Sigma-Aldrich (St. Louis, MO); IR-coupled secondary antibodies were from Rockland (Gilbertsville, PA); fluorescent secondary antibodies were from Jackson Immunoresearch (West Grove, PA).
Halt protease was purchased from Thermo Scientific (Marietta, OH), PhosStop and EDTA-free protease inhibitor tablets were purchased from Roche Diagnostics (Indianapolis, IN), Prostaglandin E2 (PGE2) was from Cayman Chemical (Ann Arbor, MI), Lambda (λ) phosphatase was purchased from New England Biolabs (Ipswich, MA); protein kinase inhibitors were purchased from Tocris Bioscience (Ellisville, MO); Phos-TagTM Acrylamide reagent was purchased from Wako Chemicals (Richmond, VA), precast 4–12% Bis-Tris gels and empty gel cassettes were from Life Technologies.
Cell lines and culture
Tet-off MDCK I cells were generously provided by Dr Alan Yu, University of Kansas, Kansas City, KS; MDCK II Tet-off cells, cells expressing wild-type mouse claudin-2, claudin-2 knockdown cells (Van Itallie et al., 2008), claudin-2 containing the GLW to AAA mutation, N-terminal FLAG-tag (Van Itallie et al., 2011) and (-) PDZ binding motif (Van Itallie et al., 2009b) have been previously described. N-terminally tagged GFP- and mApple claudin-2 were cloned in frame into C-series vectors by PCR; the mApple vector was a gift from Dr Clare Waterman, NIH. Claudin-2 S208A, S208E, S208D, S219A, S219E and T205A mutants were generated using the Agilent Quikchange II site-directed mutagenesis kit with the following sense primers (antisense primers are the reverse complement): 5′-cttgccactaggagcgctccaagatctgctc-3′, 5′-gcctcttgccactaggagcgagccaagatctgctcaacagc-3′, 5′-cctcttgccactaggagcgatccaagatctgctcaaca-3′, 5′-tcaacagcccaaagccaaggctgagttcaactcatacagc-3′, 5′-gctcaacagcccaaagccaaggaagagttcaactcatacagcctg-3′, 5′-gcccagcctcttgccgataggagctctccaag-3′.
Stable cell lines were generated as described previously in both Tet-off MDCK II claudin-2 knockdown cells and in Tet-off MDCK I cells; pTRE claudin-2 vectors were co-transfected with pTKhyg and clones selected by culturing cells in 200 µg/ml hygromycin; at least two clonal cell lines were selected for each construct. For all localisation, barrier assays, phosphorylation studies and pharmacologic treatments, cells were grown on Transwell filters (Corning) for 4–10 days. Measurement of dilution potential, TER and flux of 3 kDa dextran were carried out as previously described (Van Itallie et al., 2010). Detergent solubility was determined by incubation with ice-cold 0.5% Triton X-100, 150 mM NaCl, 20 mM Hepes, pH 7.3 supplemented with phosphatase and protease inhibitors for 30 min. Soluble and insoluble fractions were separated by centrifugation at 12,000×g for 20 min. Stoichiometric amounts of supernatant and sonicated, SDS sample buffer extracted pellet were electrophoresed to compare claudin and occludin levels in each fraction. Mouse kidney was minced and homogenised in 1% Triton-X 100, 150 mM NaCl, 20 mM Hepes, pH 7.3 supplemented with phosphatase and protease inhibitors, and clarified by centrifugation; an aliquot was treated with λ phosphatase.
Phosphorylation sites determination by mass spectrometry
Claudin-2 purification
Ten 150 mm dishes of MDCK II Tet-off cells stably expressing N-terminally Flag-tagged claudin-2 under control of a tet-sensitive promoter were induced for 4 days. Induced cells were washed with PBS and extracted with 1% Triton X-100, 150 mM NaCl, 20 mM Hepes, pH 7.3 supplemented with phosphatase and protease inhibitors on an end-over-end mixer at 4°C for 30 min. Insoluble material was pelleted at 10,000 g for 20 min at 4°C and supernatants transferred to M2-Flag affinity resin. Specifically bound protein was eluted with 3×Flag peptide in four elutions. Eluted material was precipitated with chloroform/methanol and electrophoresed in an SDS-MES gel and stained with Safe-Blue. The claudin-2 band was excised and destained with 50 mM ammonium bicarbonate in 50% methanol before being used for further analysis.
In-gel trypsin digestion
The excised gel band was subjected to previously published trypsin digestion methods with minor modifications (Khositseth et al., 2011). Prior to the reduction and alkylation step, the dried gel band was immersed in 0.25% Anionic Acid Labile Surfactant (AALS), (Protea, Morgantown, WV), in 50 mM ammonium bicarbonate, pH 7.8. After trypsin digestion, the AALS was cleaved by using 10% trifluoroacetic acid (TFA) to a final concentration of 1%. The resulting peptide mixture was concentrated and desalted with C18 Zip-tips (Millipore Corp., Bedford, MA), following the company's protocol.
Phosphopeptide enrichment
The eluted peptide mixture was dried in vacuo before starting the titanium dioxide (TiO2) phosphopeptide enrichment procedure. TiO2 ProteaTip (Catalog SP-125, Protea, Morgantown, WV) was used for enrichment, following company's protocol. The eluted phosphopeptides were dried down and reconstituted in 10 uL of 0.1% Formic Acid in HPLC grade water for mass spectrometry analysis.
LC-MS/MS analysis on LTQ-Orbitrap Velos
Liquid chromatography - tandem mass spectrometry was performed using an Eksigent nanoLC-Ultra 1D plus system (Dublin, CA) coupled to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) using CID fragmentation. Peptides were first loaded onto an Zorbax 300SB-C18 trap column (Agilent, Palo Alto, CA) at a flow rate of 6 uL/min for 6 min, and then separated on a reversed-phase PicoFrit analytical column (New Objective, Woburn, MA) using a 65-min linear gradient of 5–40% acetonitrile in 0.1% formic acid at a flow rate of 250 nL/min. LTQ-Orbitrap Velos settings were as follows: spray voltage 1.5 kV; full MS mass range: m/z 300 to 2000. The LTQ-Orbitrap Velos was operated in a data-dependent mode; i.e. one MS1 high resolution (60,000) scan for precursor ions followed by six data-dependent MS2 scans for precursor ions above a threshold ion count of 500 with collision energy of 35%.
MASCOT database search
The raw file generated from the LTQ Orbitrap Velos was analysed using Proteome Discoverer v1.3 software (Thermo Fisher Scientific, LLC) using Mascot search engine (v2.3). The following Mascot search criteria was set to: database, Swiss-Prot (Swiss Institute of Bioinformatics); taxonomy, Mus musculus (mouse); enzyme, trypsin; miscleavages, 2; variable modifications, Oxidation (M), Deamidation (NQ), Phospho (STY); fixed modifications, carbamidomethyl (C), MS peptide tolerance 10 ppm; MS/MS tolerance as 0.8 Da. The peptide confidence false discovery rate (FDR) was set to 1%.
Phosphopeptide analysis
PhosphoRS (Taus et al., 2011) site localisation tool was used to determine the phophosites of claudin-2, along with manual inspection of spectra.
Phos-tag SDS PAGE, immunoblot and immunofluorescence analyses
Comparison of phosphorylated and dephosphorylated samples: Cells were extracted in 1% Triton X-100, 150 mM NaCl, 20 mM Hepes, pH 7.3 supplemented with protease inhibitors; samples for determination of phosphorylation were supplemented with phosphatase inhibitors as well. Lambda Phosphatase treatment was performed according to the manufacturer's instructions; samples were incubated with enzyme for 30 min at 30°C. Phos-tag SDS PAGE was carried out as described by Kinoshita-Kikuta et al. (Kinoshita-Kikuta et al., 2007; Kinoshita and Kinoshita-Kikuta, 2011) ; both Mn2+ Phos-tag Tris-glycine and Zn2+ Phos-tag Neutral pH gels were used with similar results; all resolving gels were 10% acrylamide (37.5∶1) with 4% stacking gels. Standard SDS-PAGE immunoblotting and immunofluorescence were carried out as described previously (Van Itallie et al., 2009a). Imaging was performed using a Nikon E1000 Upright microscope equipped with a Zeiss Axiocam HRc camera using Zeiss Axiovision software in the NIH/NHLBI light microscope core facility.
Lysosomal fractionation
Lysosomal enrichment was carried out using a commercial kit (Thermo Scientific) with an iodixanol gradient. All samples were extracted using both protease and phosphatase inhibitors. Volumes were modified for centrifugation in a Beckman Optima TL tabletop ultracentrifuge using a TLS-55 rotor. Centrifugation was carried out for 2 h at 50,000 rpm and 0.3 ml fractions were removed, diluted 2-fold with phosphate-buffered saline and recentrifuged at 18,000 g for 30 min at 4°C. The supernatants were discarded and the pellets resuspended in SDS sample buffer, sonicated and subjected to Phos-tag or standard SDS PAGE.
Supplementary Material
Footnotes
Funding
This research was funded by the Office of the Director through the Intramural Research Program of the National Institutes of Health. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.111237/-/DC1
References
- Amasheh S., Meiri N., Gitter A. H., Schöneberg T., Mankertz J., Schulzke J. D., Fromm M. (2002). Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J. Cell Sci. 115, 4969–4976 10.1242/jcs.00165 [DOI] [PubMed] [Google Scholar]
- Amasheh M., Fromm A., Krug S. M., Amasheh S., Andres S., Zeitz M., Fromm M., Schulzke J. D. (2010). TNFalpha-induced and berberine-antagonized tight junction barrier impairment via tyrosine kinase, Akt and NFkappaB signaling. J. Cell Sci. 123, 4145–4155 10.1242/jcs.070896 [DOI] [PubMed] [Google Scholar]
- Anderson J. M., Van Itallie C. M. (2009). Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 1, a002584 10.1101/cshperspect.a002584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelow S., Yu A. S. (2009). Structure-function studies of claudin extracellular domains by cysteine-scanning mutagenesis. J. Biol. Chem. 284, 29205–29217 10.1074/jbc.M109.043752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aono S., Hirai Y. (2008). Phosphorylation of claudin-4 is required for tight junction formation in a human keratinocyte cell line. Exp. Cell Res. 314, 3326–3339 10.1016/j.yexcr.2008.08.012 [DOI] [PubMed] [Google Scholar]
- Bal M. S., Castro V., Piontek J., Rueckert C., Walter J. K., Shymanets A., Kurig B., Haase H., Nürnberg B., Blasig I. E. (2012). The hinge region of the scaffolding protein of cell contacts, zonula occludens protein 1, regulates interacting with various signaling proteins. J. Cell. Biochem. 113, 934–945 10.1002/jcb.23422 [DOI] [PubMed] [Google Scholar]
- Balkovetz D. F., Chumley P., Amlal H. (2009). Downregulation of claudin-2 expression in renal epithelial cells by metabolic acidosis. Am. J. Physiol. Renal Physiol. 297, F604–F611 10.1152/ajprenal.00043.2009 [DOI] [PubMed] [Google Scholar]
- Bennett P. A., Dixon R. J., Kellie S. (1993). The phosphotyrosine phosphatase inhibitor vanadyl hydroperoxide induces morphological alterations, cytoskeletal rearrangements and increased adhesiveness in rat neutrophil leucocytes. J. Cell Sci. 106, 891–901 [DOI] [PubMed] [Google Scholar]
- Blom N., Gammeltoft S., Brunak S. (1999). Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294, 1351–1362 10.1006/jmbi.1999.3310 [DOI] [PubMed] [Google Scholar]
- Buchert M., Papin M., Bonnans C., Darido C., Raye W. S., Garambois V., Pélegrin A., Bourgaux J. F., Pannequin J., Joubert D.et al. (2010). Symplekin promotes tumorigenicity by up-regulating claudin-2 expression. Proc. Natl. Acad. Sci. USA 107, 2628–2633 10.1073/pnas.0903747107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio O. R., Van Itallie C. M., McCrea H. J., Rahner C., Anderson J. M. (2002). Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am. J. Physiol. Cell Physiol. 283, C142–C147 [DOI] [PubMed] [Google Scholar]
- Colegio O. R., Van Itallie C., Rahner C., Anderson J. M. (2003). Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am. J. Physiol. Cell Physiol. 284, C1346–C1354 [DOI] [PubMed] [Google Scholar]
- D'Souza T., Agarwal R., Morin P. J. (2005). Phosphorylation of claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J. Biol. Chem. 280, 26233–26240 10.1074/jbc.M502003200 [DOI] [PubMed] [Google Scholar]
- D'Souza T., Indig F. E., Morin P. J. (2007). Phosphorylation of claudin-4 by PKCepsilon regulates tight junction barrier function in ovarian cancer cells. Exp. Cell Res. 313, 3364–3375 10.1016/j.yexcr.2007.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan P., Ahmad R., Chaturvedi R., Smith J. J., Midha R., Mittal M. K., Krishnan M., Chen X., Eschrich S., Yeatman T. J.et al. (2011). Claudin-2 expression increases tumorigenicity of colon cancer cells: role of epidermal growth factor receptor activation. Oncogene 30, 3234–3247 10.1038/onc.2011.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukes J. D., Whitley P., Chalmers A. D. (2012). The PIKfyve inhibitor YM201636 blocks the continuous recycling of the tight junction proteins claudin-1 and claudin-2 in MDCK cells. PLoS ONE 7, e28659 10.1371/journal.pone.0028659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkouby–Naor L., Ben–Yosef T. (2010). Functions of claudin tight junction proteins and their complex interactions in various physiological systems. Int. Rev. Cell Mol. Biol. 279, 1–32 10.1016/S1937-6448(10)79001-8 [DOI] [PubMed] [Google Scholar]
- Enck A. H., Berger U. V., Yu A. S. (2001). Claudin-2 is selectively expressed in proximal nephron in mouse kidney. Am. J. Physiol. Renal Physiol. 281, F966–F974 [DOI] [PubMed] [Google Scholar]
- Flores–Benitez D., Rincon–Heredia R., Razgado L. F., Larre I., Cereijido M., Contreras R. G. (2009). Control of tight junctional sealing: roles of epidermal growth factor and prostaglandin E2. Am. J. Physiol. Cell Physiol. 297, C611–C620 10.1152/ajpcell.00622.2008 [DOI] [PubMed] [Google Scholar]
- Fries W., Muja C., Crisafulli C., Cuzzocrea S., Mazzon E. (2008). Dynamics of enterocyte tight junctions: effect of experimental colitis and two different anti-TNF strategies. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G938–G947 10.1152/ajpgi.00469.2007 [DOI] [PubMed] [Google Scholar]
- Furuse M., Tsukita S. (2006). Claudins in occluding junctions of humans and flies. Trends Cell Biol. 16, 181–188 10.1016/j.tcb.2006.02.006 [DOI] [PubMed] [Google Scholar]
- Gonzalez J. E., DiGeronimo R. J., Arthur D. E., King J. M. (2009). Remodeling of the tight junction during recovery from exposure to hydrogen peroxide in kidney epithelial cells. Free Radic. Biol. Med. 47, 1561–1569 10.1016/j.freeradbiomed.2009.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González–Mariscal L., Tapia R., Chamorro D. (2008). Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta 1778, 729–756 10.1016/j.bbamem.2007.08.018 [DOI] [PubMed] [Google Scholar]
- Gonzalez–Mariscal L., Garay E., Quiros M. (2010). Regulation of claudins by post-translational modification and the cell-signaling cascade. Curr. Top. Membra. 65, 113–150 10.1016/S1063-5823(10)65006-5 [DOI] [Google Scholar]
- Guillemot L., Paschoud S., Jond L., Foglia A., Citi S. (2008). Paracingulin regulates the activity of Rac1 and RhoA GTPases by recruiting Tiam1 and GEF-H1 to epithelial junctions. Mol. Biol. Cell 19, 4442–4453 10.1091/mbc.E08-06-0558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harhaj N. S., Antonetti D. A. (2004). Regulation of tight junctions and loss of barrier function in pathophysiology. Int. J. Biochem. Cell Biol. 36, 1206–1237 10.1016/j.biocel.2003.08.007 [DOI] [PubMed] [Google Scholar]
- Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. (2012). PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 10.1093/nar/gkr1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J., Paul D. L., Goodenough D. A. (2005). Paracellin-1 and the modulation of ion selectivity of tight junctions. J. Cell Sci. 118, 5109–5118 10.1242/jcs.02631 [DOI] [PubMed] [Google Scholar]
- Hou J., Renigunta A., Gomes A. S., Hou M., Paul D. L., Waldegger S., Goodenough D. A. (2009). Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc. Natl. Acad. Sci. USA 106, 15350–15355 10.1073/pnas.0907724106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikari A., Matsumoto S., Harada H., Takagi K., Hayashi H., Suzuki Y., Degawa M., Miwa M. (2006). Phosphorylation of paracellin-1 at Ser217 by protein kinase A is essential for localization in tight junctions. J. Cell Sci. 119, 1781–1789 10.1242/jcs.02901 [DOI] [PubMed] [Google Scholar]
- Ikari A., Ito M., Okude C., Sawada H., Harada H., Degawa M., Sakai H., Takahashi T., Sugatani J., Miwa M. (2008). Claudin-16 is directly phosphorylated by protein kinase A independently of a vasodilator-stimulated phosphoprotein-mediated pathway. J. Cell. Physiol. 214, 221–229 10.1002/jcp.21178 [DOI] [PubMed] [Google Scholar]
- Ikari A., Takiguchi A., Atomi K., Sugatani J. (2011). Epidermal growth factor increases clathrin-dependent endocytosis and degradation of claudin-2 protein in MDCK II cells. J. Cell. Physiol. 226, 2448–2456 10.1002/jcp.22590 [DOI] [PubMed] [Google Scholar]
- Ishizaki T., Chiba H., Kojima T., Fujibe M., Soma T., Miyajima H., Nagasawa K., Wada I., Sawada N. (2003). Cyclic AMP induces phosphorylation of claudin-5 immunoprecipitates and expression of claudin-5 gene in blood-brain-barrier endothelial cells via protein kinase A-dependent and -independent pathways. Exp. Cell Res. 290, 275–288 10.1016/S0014-4827(03)00354-9 [DOI] [PubMed] [Google Scholar]
- Itoh M., Furuse M., Morita K., Kubota K., Saitou M., Tsukita S. (1999). Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J. Cell Biol. 147, 1351–1363 10.1083/jcb.147.6.1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S., Suzuki T., Seth A., Samak G., Rao R. (2011). Protein kinase Cζ phosphorylates occludin and promotes assembly of epithelial tight junctions. Biochem. J. 437, 289–299 10.1042/BJ20110587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khositseth S., Pisitkun T., Slentz D. H., Wang G., Hoffert J. D., Knepper M. A., Yu M. J. (2011). Quantitative protein and mRNA profiling shows selective post-transcriptional control of protein expression by vasopressin in kidney cells. Mol. Cell Proteomics 10, M110 004036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E., Kinoshita–Kikuta E. (2011). Improved Phos-tag SDS-PAGE under neutral pH conditions for advanced protein phosphorylation profiling. Proteomics 11, 319–323 10.1002/pmic.201000472 [DOI] [PubMed] [Google Scholar]
- Kinoshita–Kikuta E., Aoki Y., Kinoshita E., Koike T. (2007). Label-free kinase profiling using phosphate affinity polyacrylamide gel electrophoresis. Mol. Cell. Proteomics 6, 356–366 10.1074/mcp.T600044-MCP200 [DOI] [PubMed] [Google Scholar]
- Larre I., Castillo A., Flores–Maldonado C., Contreras R. G., Galvan I., Muñoz–Estrada J., Cereijido M. (2011). Ouabain modulates ciliogenesis in epithelial cells. Proc. Natl. Acad. Sci. USA 108, 20591–20596 10.1073/pnas.1102617108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabi I. R., Le Bivic A., Fambrough D., Rodriguez–Boulan E. (1991). An endogenous MDCK lysosomal membrane glycoprotein is targeted basolaterally before delivery to lysosomes. J. Cell Biol. 115, 1573–1584 10.1083/jcb.115.6.1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta A., Yang S. S., Rai T., Chiga M., Sasaki S., Uchida S. (2006). Overexpression of human WNK1 increases paracellular chloride permeability and phosphorylation of claudin-4 in MDCKII cells. Biochem. Biophys. Res. Commun. 349, 804–808 10.1016/j.bbrc.2006.08.101 [DOI] [PubMed] [Google Scholar]
- Piontek J., Winkler L., Wolburg H., Müller S. L., Zuleger N., Piehl C., Wiesner B., Krause G., Blasig I. E. (2008). Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J. 22, 146–158 10.1096/fj.07-8319com [DOI] [PubMed] [Google Scholar]
- Piontek J., Fritzsche S., Cording J., Richter S., Hartwig J., Walter M., Yu D., Turner J. R., Gehring C., Rahn H. P.et al. (2011). Elucidating the principles of the molecular organization of heteropolymeric tight junction strands. Cell. Mol. Life Sci. 68, 3903–3918 10.1007/s00018-011-0680-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raleigh D. R., Boe D. M., Yu D., Weber C. R., Marchiando A. M., Bradford E. M., Wang Y., Wu L., Schneeberger E. E., Shen L.et al. (2011). Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J. Cell Biol. 193, 565–582 10.1083/jcb.201010065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridyard A. E., Brown J. K., Rhind S. M., Else R. W., Simpson J. W., Miller H. R. (2007). Apical junction complex protein expression in the canine colon: differential expression of claudin-2 in the colonic mucosa in dogs with idiopathic colitis. J. Histochem. Cytochem. 55, 1049–1058 10.1369/jhc.7A7211.2007 [DOI] [PubMed] [Google Scholar]
- Sakakibara A., Furuse M., Saitou M., Ando–Akatsuka Y., Tsukita S. (1997). Possible involvement of phosphorylation of occludin in tight junction formation. J. Cell Biol. 137, 1393–1401 10.1083/jcb.137.6.1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulzke J. D., Ploeger S., Amasheh M., Fromm A., Zeissig S., Troeger H., Richter J., Bojarski C., Schumann M., Fromm M. (2009). Epithelial tight junctions in intestinal inflammation. Ann. N. Y. Acad. Sci. 1165, 294–300 10.1111/j.1749-6632.2009.04062.x [DOI] [PubMed] [Google Scholar]
- Shen L., Weber C. R., Raleigh D. R., Yu D., Turner J. R. (2011). Tight junction pore and leak pathways: a dynamic duo. Annu. Rev. Physiol. 73, 283–309 10.1146/annurev-physiol-012110-142150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjö A., Magnusson K. E., Peterson K. H. (2010). Protein kinase C activation has distinct effects on the localization, phosphorylation and detergent solubility of the claudin protein family in tight and leaky epithelial cells. J. Membr. Biol. 236, 181–189 10.1007/s00232-010-9289-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma T., Chiba H., Kato–Mori Y., Wada T., Yamashita T., Kojima T., Sawada N. (2004). Thr(207) of claudin-5 is involved in size-selective loosening of the endothelial barrier by cyclic AMP. Exp. Cell Res. 300, 202–212 10.1016/j.yexcr.2004.07.012 [DOI] [PubMed] [Google Scholar]
- Stevenson B. R., Anderson J. M., Braun I. D., Mooseker M. S. (1989). Phosphorylation of the tight-junction protein ZO-1 in two strains of Madin-Darby canine kidney cells which differ in transepithelial resistance. Biochem. J. 263, 597–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L., Mruk D. D., Lui W. Y., Lee W. M., Cheng C. Y. (2011). P-glycoprotein regulates blood-testis barrier dynamics via its effects on the occludin/zonula occludens 1 (ZO-1) protein complex mediated by focal adhesion kinase (FAK). Proc. Natl. Acad. Sci. USA 108, 19623–19628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Yoshinaga N., Tanabe S. (2011). Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J. Biol. Chem. 286, 31263–31271 10.1074/jbc.M111.238147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabariès S., Dong Z., Annis M. G., Omeroglu A., Pepin F., Ouellet V., Russo C., Hassanain M., Metrakos P., Diaz Z.et al. (2011). Claudin-2 is selectively enriched in and promotes the formation of breast cancer liver metastases through engagement of integrin complexes. Oncogene 30, 1318–1328 10.1038/onc.2010.518 [DOI] [PubMed] [Google Scholar]
- Tanaka M., Kamata R., Sakai R. (2005). EphA2 phosphorylates the cytoplasmic tail of Claudin-4 and mediates paracellular permeability. J. Biol. Chem. 280, 42375–42382 10.1074/jbc.M503786200 [DOI] [PubMed] [Google Scholar]
- Tatum R., Zhang Y., Lu Q., Kim K., Jeansonne B. G., Chen Y. H. (2007). WNK4 phosphorylates ser(206) of claudin-7 and promotes paracellular Cl(-) permeability. FEBS Lett. 581, 3887–3891 10.1016/j.febslet.2007.07.014 [DOI] [PubMed] [Google Scholar]
- Taus T., Köcher T., Pichler P., Paschke C., Schmidt A., Henrich C., Mechtler K. (2011). Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 10.1021/pr200611n [DOI] [PubMed] [Google Scholar]
- Van Itallie C. M., Gambling T. M., Carson J. L., Anderson J. M. (2005). Palmitoylation of claudins is required for efficient tight-junction localization. J. Cell Sci. 118, 1427–1436 10.1242/jcs.01735 [DOI] [PubMed] [Google Scholar]
- Van Itallie C. M., Holmes J., Bridges A., Gookin J. L., Coccaro M. R., Proctor W., Colegio O. R., Anderson J. M. (2008). The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J. Cell Sci. 121, 298–305 10.1242/jcs.021485 [DOI] [PubMed] [Google Scholar]
- Van Itallie C. M., Fanning A. S., Bridges A., Anderson J. M. (2009a). ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol. Biol. Cell 20, 3930–3940 10.1091/mbc.E09-04-0320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie C. M., Holmes J., Bridges A., Anderson J. M. (2009b). Claudin-2-dependent changes in noncharged solute flux are mediated by the extracellular domains and require attachment to the PDZ-scaffold. Ann. N. Y. Acad. Sci. 1165, 82–87 10.1111/j.1749-6632.2009.04052.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie C. M., Fanning A. S., Holmes J., Anderson J. M. (2010). Occludin is required for cytokine-induced regulation of tight junction barriers. J. Cell Sci. 123, 2844–2852 10.1242/jcs.065581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie C. M., Mitic L. L., Anderson J. M. (2011). Claudin-2 forms homodimers and is a component of a high molecular weight protein complex. J. Biol. Chem. 286, 3442–3450 10.1074/jbc.M110.195578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C. R., Nalle S. C., Tretiakova M., Rubin D. T., Turner J. R. (2008). Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Invest. 88, 1110–1120 10.1038/labinvest.2008.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi K., Rai T., Kobayashi K., Sohara E., Suzuki T., Itoh T., Suda S., Hayama A., Sasaki S., Uchida S. (2004). Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc. Natl. Acad. Sci. USA 101, 4690–4694 10.1073/pnas.0306924101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A. S., Cheng M. H., Angelow S., Günzel D., Kanzawa S. A., Schneeberger E. E., Fromm M., Coalson R. D. (2009). Molecular basis for cation selectivity in claudin-2-based paracellular pores: identification of an electrostatic interaction site. J. Gen. Physiol. 133, 111–127 10.1085/jgp.200810154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeissig S., Bürgel N., Günzel D., Richter J., Mankertz J., Wahnschaffe U., Kroesen A. J., Zeitz M., Fromm M., Schulzke J. D. (2007). Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut 56, 61–72 10.1136/gut.2006.094375 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.