

Abstract
All it takes is the Aldol
Nitrogen containing heterocycles have been assembled by means of unprecedented domino processes, designed to take advantage of diversity assembly via strategically decorated Ugi products. The aldol reaction is the second common denominator which enables sequences of up to 5 steps in one pot producing unique molecular architecture in rapid fashion.
Keywords: Aldol reaction, Isonitriles, Molecular diversity, Multicomponent reactions, Nitrogen heterocycles
Nitrogen containing heterocyles represent the vast majority of drugs and biologically relevant molecules. Thus, one of the most investigated areas of present day synthetic organic chemistry is the development of novel and efficient strategies for the assembly of these valuable compounds.1 In this context, isocyanide based multicomponent reactions (IMCRs) have met renewed interest2 due to their unmatched capability to rapidly generate molecular diversity and explore the chemical space.3 In particular, the Ugi reaction (U-4CR) enjoys the lion’s share of this methodology growth because of the extreme versatility it displays.4 As a matter of fact, although this condensation affords a linear peptide backbone, a plethora of strategies are available to rigidify Ugi adducts into more appealing drug-like species.5 To date, Ugi-Diels Alder,6 Ugi Buchwald-Hartwig,7 Ugi-Heck,8 Ugi-nucleophilic additions/substitutions9 and Ugi-ring closing metatheses10 pathways constitute the most well-established routes to gain access into a wide variety of cyclic scaffolds, in line with the post-condensation modification approach.2 Recently, more elaborated processes involving extensive manipulations of Ugi products rendering natural product-like complex frameworks have also been reported.11 Within this blossoming and highly diversified arena of research, it is surprising to notice that Aldol-type reactions have never been exploited.12 Indeed, such studies are reported herein and contain examples of 4 and 5 step one pot post-MCR cascades13 that creatively generate molecular diversity in exquisite non-obvious ways.
The Aldol reaction, first discovered by Wurtz14 more than one century ago, is still one of the most powerful known tools to create carbon-carbon bonds through the use of easily available starting materials,15 namely aldehydes and ketones. Given its outstanding importance in the preparation of both top-selling pharmaceuticals16 and natural products17, we were therefore enticed to investigate the possibility to merge the exploratory power of the U-4CR with the potential to convert Ugi products into novel appealing heterocyclic chemotypes offered by the aldol condensation. In order to endow the multicomponent reaction (MCR) product with functionalities suitable to participate in an aldol condensation, pyruvic aldehyde and phenylglyoxylic acid were employed, along with benzylamine and n-butylisonitrile (Scheme 1). Predictably, this first step proceeded smoothly in methanol under mild conditions overnight to give 1a in good yield (67%). With this acyclic intermediate bearing ketonic carbonyls and an activated methyl in hand, we initiated screening of a panel of solvents and catalysts (Table 1) to achieve the desired Aldol induced cyclization.
Scheme 1.
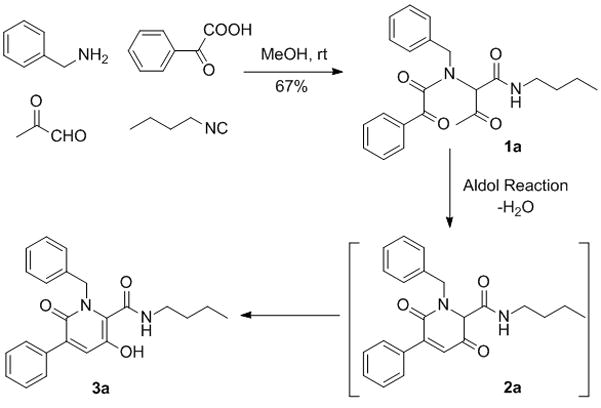
Design of Ugi-Aldol sequence.
Table 1.
Optimization of the aldol reaction.
| Entry | Cat.[a] | Eq. | Solv. | Conditions | Yield % |
|---|---|---|---|---|---|
| 1 | DIPEA | 2.0 | DMSO | 150 °C, 10 min | 5 |
| 2 | DIPA | 2.0 | DMSO | 150 °C, 10 min | 55 |
| 3 | TEA | 2.0 | DMSO | 150 °C, 10 min | 55 |
| 4 | DIPA | 2.0 | DMF | 150 °C, 10 min | 60 |
| 5 | DIPA | 2.0 | DMF | 160 °C, 20 min | 88 |
| 6 | DIPA | 2.0 | THF | 160 °C, 20 min | 55 |
| 7 | DIPA | 2.0 | DCE | 160 °C, 20 min | 9 |
| 8 | 10% TFA/DCE | 150 °C, 10 min | 14 | ||
| 9 | 10% TFA/DCE | 160 °C, 20 min | 30 | ||
DIPEA=N,N-Diisopropylethylamine; DIPA=N,N-Diisopropylamine, TEA=Triethylamine.
Different organic bases were evaluated, as the aldol reaction is most frequently a base-catalyzed transformation proceeding via enolate formation.15 Interestingly, the targeted modification required microwave irradiation at elevated temperatures (≥150°C), but was nevertheless feasible in a straightforward and satisfactorily clean way. Of note, after the nucleophilic attack and dehydration leading to the α,β-unsaturated compound 2a, which was never isolated, further isomerization afforded 5-hydroxypyridine-2-one 3a. Triethylamine (TEA) and N,N-diisopropylamine (DIPA) proved to be equally effective (Entries 2 and 3), whereas use of N,N-diisopropylethylamine (DIPEA) under the same conditions in DMSO afforded only a negligible amount (5%) of the title product (Entry 1). Variation of the solvent showed that DMF (Entries 4 and 5) was beneficial for the desired conversion, especially when irradiating at 160°C for 20 minutes. Conversely, decreasing the polarity of the medium by employing DCE (Entry 7) resulted in a dramatic drop of yield. Since the aldol condensation is also known to be feasible through acid-promoted enol formation, 10% TFA/DCE solution was employed as both the solvent and the catalyst (Entries 8 and 9), but was found to have remarkably less efficacy compared to DIPA in DMF.
After optimizing the second step of the sequence, we exposed different building blocks to our protocol in order to both determine its generality and to prepare a small collection of compounds 3. While the carbonyl input was always represented by pyruvic aldehyde, two different arylglyoxylic acids, three isonitriles and five aromatic and aliphatic amines were evaluated. The Ugi MCR furnished dipeptide-like products 1 in fair to good yields, ranging from 47 to 67% after the well-established method of stirring in a mild chemical environment.18 Acyclic precursors were subsequently purified by column chromatography and treated with DIPA in DMF under microwave-promoted heating to produce 5-hydroxypyridin-2-ones 3 in excellent yields. Gratifyingly, the synthetic route was proved to be robust and tolerated a variety of different substituents. Moreover, the aldol step could be performed in a very clean manner and intermolecular aldol condensations were not observed.
Having developed this novel methodology for the diversification of the Ugi backbone displaying general applicability and providing a 2-pyridone heterocyclic core in two concise straightforward steps, our next goal was to further build the complexity of final products and gain access to more elaborated molecular scaffolds. As a consequence, studies were directed at a further transformation capable of assembling a nitrogen-containing ring (Scheme 2).
Scheme 2.
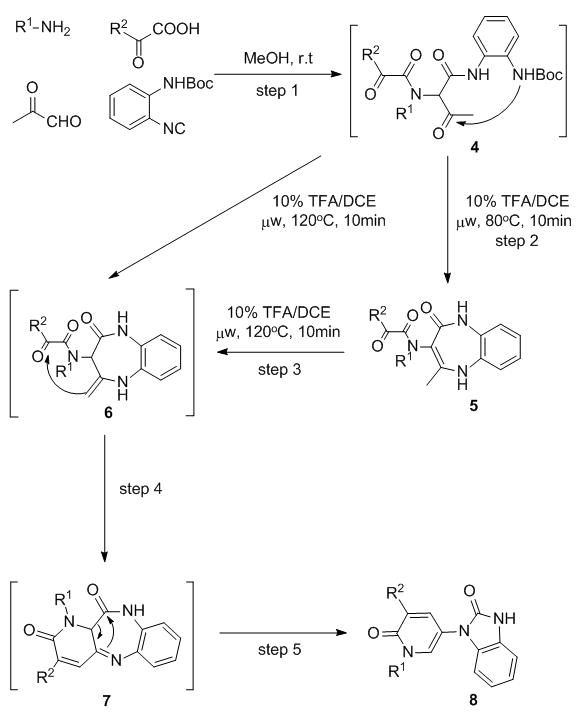
Synthetic route leading to hybrid pyridinone-3-yl-benzimidazol-2-ones 8
Benzodiazepines possess a wide spectrum of biological activities19 that goes well beyond their classic applications as sedatives and anxiolytics. With significant group hindsight in the field of benzodiazepine synthesis,20 it seemed natural to attempt embedding such a valuable scaffold in the Ugi-Aldol products. According to the synthetic plan (Scheme 2) replacement of commercially available isonitriles with ortho-N-Boc-phenylisonitrile, bearing an additional masked amino-group,21 would allow for an anticipated domino process that included two post-MCR modifications in one-pot. Thus, attack of the anilinic amine onto the pyruvaldehyde-derived carbonyl in the presence of 10% TFA/DCE was intended to form intermediate 6, paving the way to a subsequent acid-promoted aldol reaction (imine aldol)22 in this case) rendering the unprecedented tricyclic species 7. To our surprise, microwave irradiation at 120°C generated a different product and X-ray structural analysis23 unambiguously determined the structure to be a pyridinone-3-yl-benzimidazol-2-one chemotype 8. This phenomenon was unanticipated, although over 30 years ago strong thermal conditions were reported to trigger benzodiazepine to benzimidazol-2-one rearrangements.24 On experimenting with lower temperatures (80°C) the expected proposed kinetic product 5h was obtained in an almost quantitative amount (Table 3, Entry 8). Excitingly, the unprecedented bis-heterocycle 8 was of great interest and its preparation was equivalent to a cascade sequence of four distinct transformations occurring after the initial Ugi condensation.
Table 3.
Scope of the Ugi-Aldol based cascade involving ortho-N-Boc-phenylisonitrile.
| Entry | R1 | R2 | Yield % (5) | Yield % (8) |
|---|---|---|---|---|
| 1 | Phenethyl | Ph | - | 8a, 53[a] |
| 2 | Methoxyethyl | Ph | - | 8b, 58[a] |
| 3 | Phenethyl | 2-Thienyl | - | 8c, 52[a] |
| 4 | Ph | i-Bu | - | 8d, 55[a] |
| 5 | 2,6-di-Cl-Bn | Ph | - | 8e, 52[a] |
| 5 | 2-Furyl | 2-Furyl | - | 8f, 47[a] |
| 7 | Ph | 2-Thienyl | - | 8g, 56[a] |
| 8 | 3-Br-Ph | 4-F-Bn | 5h, 95[b] | 8h, 88[c] |
Overall isolated yields.
Isolated yields from Ugi product 4h.
Isolated yields from 5h.
A small collection of compounds with generic structure 8 were thus prepared by means of the 5-step one-pot protocol. After completion of the Ugi reaction, solvent was evaporated in vacuo and the crude material was directly exposed to acidic thermal rearrangement conditions, providing bis-heterocycles 8 (Table 3, Entries 1–7). Very remarkably, excellent yields (47 to 58%) over 5 steps were obtained and only one final step chromatography exercise was required.
Encouraged by the above results that demonstrated high versatility of the new methodology, we envisaged a second strategy exploiting a sequential domino-nucleophilic attack-aldol condensation. In this case, pyruvic aldehyde was replaced by methyl acetoacetate as the source of the activated methylene. Intrigued by the possibility of targeting highly unusual bicyclic five membered rings 12, we postulated a tandem approach entailing nucleophilic substitution onto the ester moiety followed by the aldol step (Scheme 3). Once the MCR step was completed, methanol was removed and crude 9 was subjected to irradiation under basic conditions to directly afford final products 12 (±). Noteworthy, 180°C temperature was essential to drive the process to completion, and when milder conditions were attempted (120°C, 20 mins) the unique pyrrolidine-dione 10a predominated (66% yield). Use of a panel of amines, isonitriles and arylglyoxylic acids allowed for the preparation of a small collection of richly and diversely substituted bicyclic chemotypes (Table 4) 12 in very good yields over four steps. Noteworthy, to the best of our knowledge this represents the first route into this class of tricarbonylic molecules, and the few reported strategies leading to similar scaffolds25 are much less straightforward and diversity-enabling. To our delight, NMR spectra and HPLC-MS traces invariably showed a single set of signals or single peak respectively, suggesting one diastereoisomer was predominating. The stereochemistry was finally resolved by x-ray crystallography,26 and a cis relationship between the hydroxyl and hydrogen and methyl groups at the bicyclic junction was confirmed. This enticing finding was not completely surprising as fused five membered ring bicyclic species are in fact well-known to be preferentially assembled in the cis form, and high diastereoselectivity during formation of such polycyclic systems via similar nucleophilic mechanisms is not unprecedented.27 A crystal structure in the form of two H-bonded enantiomeric units 12a confirmed product structure. Separation of the two optical isomers was performed by chiral analytical HPLC,28 and the ease of the method paves the way to the possibility of recovering optically pure 12 upon running the separation on a preparative scale.
Scheme 3.
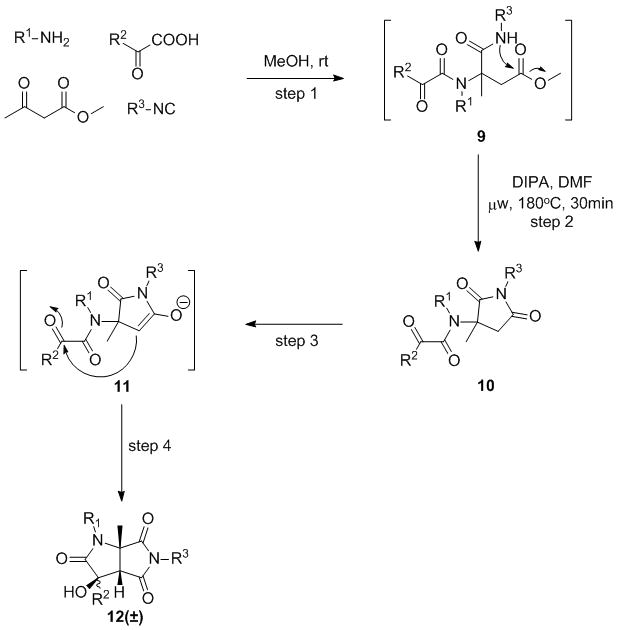
The domino process leading to compounds 12
Table 4.
Scope of the domino process leading to compounds 12.
| Entry | R1 | R2 | R3 | Yield%[a] |
|---|---|---|---|---|
| 1 | Bn | 2-Thienyl | n-Bu | 12a, 56 |
| 2 | Bn | 2-Thienyl | Bn | 12b, 53 |
| 3 | 3,4-di-OMe-Bn | Ph | n-Bu | 12c, 48 |
| 4 | i-Bu | Ph | n-Bu | 12d, 52 |
| 5 | 2-Furyl | 2-Thienyl | Bn | 12e, 52 |
| 6 | Phenethyl | 2-Thienyl | Bn | 12f, 55 |
| 7 | 2-Cl-Bn | 2-Furyl | cy-Hex | 12g, 50 |
| 8 | 2,6-di-Cl-Bn | 2-Furyl | Bn | 12h, 54 |
Overall isolated yields.
In summary, we have developed a novel and first-in-class MCR-based methodology, that exploits post-Ugi Aldol condensations to gain access into unique densely substituted heterocycles. During further elaboration, final products were accessed via a series of unprecedented one pot cascade reactions, assembling scaffolds of high complexity in non-obvious yet operationally friendly ways enabling. Subsequent efforts will be aimed at the application of these strategies toward the total synthesis of natural products.
Supplementary Material
Table 2.
Scope of the Ugi-Aldol route leading to 5-hydroxypyridin-2-ones

| |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yield % (1) | Yield % (3) |
| 1 | Bn | Ph | n-Bu | 1a, 67 | 3a, 88 |
| 2 | Phenethyl | Ph | n-Bu | 1b, 55 | 3b, 91 |
| 3 | i-Bu | 2-Thienyl | Bn | 1c, 53 | 3c, 88 |
| 4 | Ph | 2-Thienyl | Bn | 1d, 48 | 3d, 90 |
| 5 | Ph | 2-Thienyl | n-Bu | 1e, 58 | 3e, 85 |
| 6 | Phenethyl | 2-Thienyl | cy-Pent | 1f, 47 | 3f, 87 |
| 7 | 2-Cl-Bn | 2-Thienyl | cy-Pent | 1g, 51 | 3g, 84 |
| 8 | Bn | 2-Thienyl | cy-Pent | 1h, 53 | 3h, 86 |
Acknowledgments
The authors thank the NIH [5RC2MH090878-02] for financial support, Dr. Gary S. Nichol and Dr. Sue A. Roberts [University of Arizona] for X-ray structural analyses and Kristen Keck for HPLC analyses.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Ruijter E, Scheffelaar R, Orru RVA. Angew Chem. 2011;123:6358–6371. doi: 10.1002/anie.201006515. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed Engl. 2011;50:6234–6246. doi: 10.1002/anie.201006515. [DOI] [PubMed] [Google Scholar]; b) Bonne D, Dekhane M, Zhu J. Angew Chem. 2007;119:2537–2540. doi: 10.1002/anie.200605005. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed Engl. 2007;46:2485–2488. doi: 10.1002/anie.200605005. [DOI] [PubMed] [Google Scholar]; c) Bienaymé H, Bouzid K. Angew Chem. 1998;110:2349–1352. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2234::AID-ANIE2234>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed Engl. 1998;37:2234–2237. [Google Scholar]
- 2.a) Dömling A. Chem Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; b) Zhu J, Bienaymé H, editors. Multicomponent Reactions. WILEY-VCH; Weinheim, Germany: 2005. [Google Scholar]
- 3.Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 4.a) Hulme C, Dietrich J. Mol Divers. 2009;13:195–207. doi: 10.1007/s11030-009-9111-6. [DOI] [PubMed] [Google Scholar]; b) El Kaim L, Grimaud L. Tetrahedron. 2009;65:2153–2171. [Google Scholar]
- 5.For a recent overview see: Ayaz M, De Moliner F, Dietrich J, Hulme C. In: Evolution of Isocyanide Chemistry. Nenajdenko V, editor. WILEY-VCH; Weinheim, Germany: in press.
- 6.a) Basso A, Banfi L, Riva R. Eur J Org Chem. 2010:1831–1841. [Google Scholar]; b) Ilyin A, Kysil V, Krasavin M, Kurashvili I, Ivachtchenko AV. J Org Chem. 2006;71:9544–9547. doi: 10.1021/jo061825f. [DOI] [PubMed] [Google Scholar]
- 7.Bonnaterre F, Bois-Choussy M, Zhu J. Org Lett. 2006;8:4351–4354. doi: 10.1021/ol061755z. [DOI] [PubMed] [Google Scholar]
- 8.Salcedo A, Neuville L, Rondot C, Retailleau P, Zhu J. Org Lett. 2008;10:857–860. doi: 10.1021/ol7029799. [DOI] [PubMed] [Google Scholar]
- 9.a) Banfi L, Riva R, Basso A. Synlett. 2010:23–41. [Google Scholar]; b) Rhoden CRB, Rivera DG, Kreye O, Bauer AK, Westermann B, Wessjohann LA. J Comb Chem. 2009;11:1078–1082. doi: 10.1021/cc900106u. [DOI] [PubMed] [Google Scholar]
- 10.Ribelin TP, Judd AS, Akritopoulou-Zanze I, Henry RF, Cross JL, Whittern DN, Djuric SW. Org Lett. 2007;24:5119–5122. doi: 10.1021/ol7023373. [DOI] [PubMed] [Google Scholar]
- 11.a) Pando O, Stark S, Denkert A, Porzel A, Preusentanz R, Wessjohann LA. J Am Chem Soc. 2011;133:7692–7695. doi: 10.1021/ja2022027. [DOI] [PubMed] [Google Scholar]; b) Santra S, Andreana PR. Angew Chem. 2011;123:9590–9594. doi: 10.1002/anie.201103567. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed Engl. 2011;50:9418–9422. doi: 10.1002/anie.201103567. [DOI] [PubMed] [Google Scholar]; (c) Znabet A, Zonneveld J, Janssen E, De Kanter FJJ, Helliwell M, Turner NJ, Ruijter E, Orru RVA. Chem Commun. 2010;46:7706–7708. doi: 10.1039/c0cc02938f. [DOI] [PubMed] [Google Scholar]; (d) Mehta VP, Mohta SG, Ruijter E, Van Hecke K, Van Meerverlt L, Pannecouque C, Balzarini J, Orru RVA, Van der Eycken E. J Org Chem. 2011;76:2828. doi: 10.1021/jo200251q. [DOI] [PubMed] [Google Scholar]
- 12.Only the related Knovenagel reaction has been reported as a post-Ugi modification: Bossio R, Marcaccini S, Pepino R, Torroba T. Heterocycles. 1999;50:463–467.Marcaccini S, Pepino R, Pozo MC, Basurto S, Garcia-Valverde M, Torroba T. Tetrahedron Lett. 2004;45:3999–4001.Zhu J. Eur J Org Chem. 2003:1133–1144.
- 13.For the importance of cascade reactions in modern organic synthesis see: Nicolaou KC, Chen JS. Chem Soc Rev. 2009;38:2993–3009. doi: 10.1039/b903290h.Tietze LF, Düfert A, Lotz F, Oum K, Lenzer T, Beck T, Herbst-Imer R. J Am Chem Soc. 2009;131:17879–17884. doi: 10.1021/ja906260x.Rixson JE, Chaloner T, Heath CH, Tietze LF, Stewart SG. Eur J Org Chem. 2012:544–558.Schönhaber J, Frank W, Müller TJJ. Org Lett. 2010;12:4122–4125. doi: 10.1021/ol101709p.
- 14.Wurtz CA. Bull Soc Chim Fr. 1872;17:436–442. [Google Scholar]
- 15.a) Mahrwald R, editor. Modern Aldol Reactions. 1 and 2 WILEY-VCH; Weinheim, Germany: 2004. [Google Scholar]; b) Trost BM, Brindle CS. Chem Soc Rev. 2010;39:1600–1632. doi: 10.1039/b923537j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mlynarski J, Paradowska J. Chem Soc Rev. 2008;37:1502–1511. doi: 10.1039/b710577k. [DOI] [PubMed] [Google Scholar]
- 16.Li JJ, Johnson DS, Sliskovic DR, Roth BD. In: Contemporary Drug Synthesis. Li JJ, Johnson DS, editors. WILEY-INTERSCIENCE; Hoboken, New Jersey: 2004. [Google Scholar]
- 17.a) Schetter B, Mahrwald R. Angew Chem. 2006;118:7668–7687. doi: 10.1002/anie.200602780. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed Engl. 2006;45:7506–7525. doi: 10.1002/anie.200602780. [DOI] [PubMed] [Google Scholar]; b) Brodmann T, Lorenz M, Schäckel R, Simsek S, Kalesse M. Synlett. 2009:174–192. [Google Scholar]
- 18.Marcaccini S, Torroba T. Nature Protoc. 2007;2:632–639. doi: 10.1038/nprot.2007.71. [DOI] [PubMed] [Google Scholar]
- 19.For examples of the different activities displayed by the benzodiazepine core see for instance: Hata M, Marshall GR. J Comp Mol Des. 2006;20:321–331. doi: 10.1007/s10822-006-9059-x.Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, Jose Marugan J, Raboisson P, Schubert C, Koblish HK, Zhao S, Franks CF, Lattanze J, Carver TE, Cummings MD, Maguire D, Grasberger BL, Maroney AC, Lu T. Bioorg Med Chem Lett. 2006;16:3310–3314. doi: 10.1016/j.bmcl.2006.03.055.Koblish HK, Zhao S, Franks CF, Donatelli RR, Tominovich RM, LaFrance LV, Leonard KA, Gushue JM, Parks DJ, Calvo RR, Milkiewicz KL, Marugan JJ, Raboisson P, Cummings MD, Grasberger BL, Johnson DL, Lu T, Molloy CJ, Maroney AC. Mol Cancer Ther. 2006;5:160–169. doi: 10.1158/1535-7163.MCT-05-0199.
- 20.See for instance: Gunawan S, Nichol GS, Chappeta S, Dietrich J, Hulme C. Tetrahedron Lett. 2010;51:4689–4692. doi: 10.1016/j.tetlet.2010.06.131.
- 21.For an example of application of ortho-N-Boc-phenylisonitrile see: Shaw AY, Medda F, Hulme C. Tetrahedron Lett. 2012;53:1313–1315. doi: 10.1016/j.tetlet.2011.12.073.
- 22.Wittig G, Reiff H. Angew Chem. 1968;80:8–15. [Google Scholar]; Angew Chem Int Ed Engl. 1968;7:7–14. [Google Scholar]
- 23.Crystallographic data for 8a are available at CCDC 873779
- 24.a) Israel M, Jones LC, Modest EJ. Tetrahedron Lett. 1968;9:4811–4814. [Google Scholar]; b) Achour R, Essassi E, Zniber R. Tetrahedron Lett. 1988;29:195–198. [Google Scholar]
- 25.a) Kudryavtsev KV. Heterocycles. 2011;83:323–330. [Google Scholar]; b) Trunkfield AE, Gurcha SS, Besra GS, Bugg TDH. Bioorg Med Chem Lett. 2010;18:2651–2663. doi: 10.1016/j.bmc.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crystallographic data for 12a are available at CCDC 873870
- 27.See for instance: Vojkovsky T, Weichsel A, Pátek M. J Org Chem. 1998;63:3162–3163.
- 28.See supporting information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.