

Proteins and other biomolecules sequestered in and delivered from polymeric drug delivery systems (DDS) undergo several process and storage-related stresses throughout the life of the product that can result in significant degradation, loss of bioactivity and raise safety concerns.[1–4] Process-related stresses during manufacturing of drug delivery systems can lead to significant protein degradation. These stresses can include elevated temperatures, exposure to liquid and solid hydrophobic interfaces, and vigorous mechanical agitation.[1, 5, 6] A number of approaches have been developed to ameliorate the impact of individual stresses in emulsion-based methods. These include the use of interface stabilizers,[7, 8] protein crystalization[9] and covalent protein modification.[10] Ideally a single approach would protect against all these stresses, yielding excellent encapsulation efficiency and storage stability, while giving burst-free sustained release for any protein and polymer system of interest. Far from meeting this ideal, many approaches for improving one aspect of performance are neutral or deleterious to others. For example, solid-in-oil-in-water (s-o-w) emulsions[3] expose the protein to less solvent-based stress than do water-in-oil-in-water (w-o-w) methods; however, s-o-w methods have thus far led to reduced encapsulation efficiency or significant burst release. S-o-w vehicles incorporate proteins as a solid phase through freeze-drying or related processes, allowing inclusion of buffer salts, sugars, and other stabilizing additives. However, bulk freeze-drying yields relatively large protein particles, leading to poor dispersion in the final drug delivery product and burst release.[2, 9] Smaller particles are necessary to give better dispersion and sustained delivery without burst.[2, 9] Approaches such as spray-drying, spray-freeze drying, or freeze-drying with polyethylene glycol (PEG) have been used to generate smaller stabilizer-bearing protein particles, but these are associated with low protein recovery[11] and can still result in burst release.[12] More recently, small protein particles have been precipitated from organic solvents.[9] However, this technique does not give good protein stability during storage,[9] may yield protein-dependent results, and still needs to be validated for therapeutic proteins.[9, 13]
In addition to the trade-offs in performance with current stabilization methods, many approaches developed are specific to the polymer used, or to particular proteins. This is problematic for drug delivery applications, as new approaches may be needed for each new drug. Furthermore, there is a need in regenerative medicine to develop next generation ‘smart scaffolds’ which could provide microenvironment of biophysical (topography) and biochemical (delivery of single and multiple biomolecules) cues, as a mimic of natural extracellular matrix (ECM) to direct stem cell differentiation.[14, 15] Such tissue engineering applications will likely require a range of polymer / solvent systems.[14, 15] Finding a generic approach to protein stabilization and sustained delivery that will be compatible with a range of polymer and protein systems for DDS is widely expected to be a formidable challenge.[15–17] In addition to being compatible with many polymer / solvent systems, a generic platform must facilitate use of additives to protect the protein from various processing and storage stresses.[1, 3, 4, 13] Such a platform also needs to produce small, preferably nano-sized, surface-functionalized particles to achieve good dispersion and sustained release.[2, 9]
Here we present a novel and generic approach for efficient incorporation and stabilization of proteins in a polymer delivery vehicle. This approach also facilitates excellent dispersion and sustained release. We encapsulate proteins along with sugars and other excipients as needed into nanoparticles of (30 to 160) nm that are functionalized with a surfactant to promote dispersion in organic solvent. A schematic drawing of these protein-bearing nanoparticles is shown in Figure. 1a.
Figure 1.
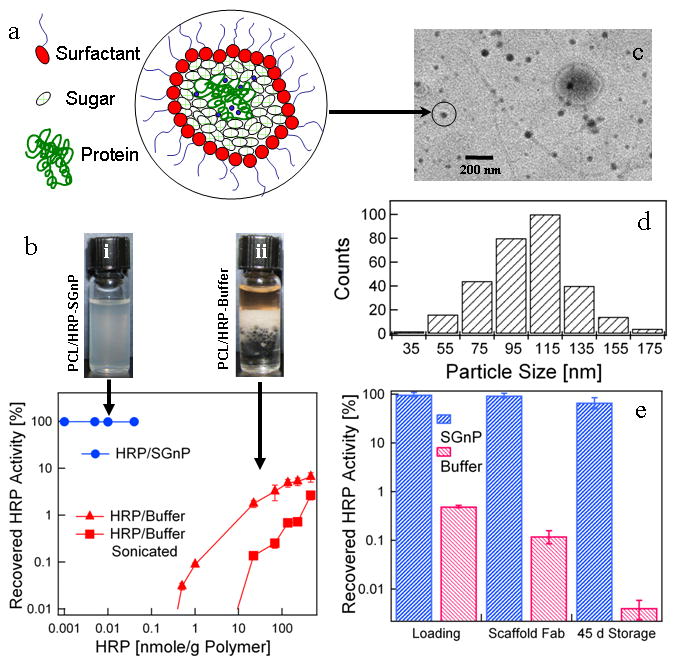
Characterization and efficacy of sugar-glass nanoparticles (SGnPs) in protecting proteins against processing and storage stresses. (a) Schematic presentation of a biomolecule encapsulated in a SGnP. The surfactant coating on the nanoparticle surface provides excellent dispersion properties in organic solvents- polymer solutions. (b) Vials i and ii contain SGnPs suspended in PCL/chloroform, and HRP/buffer after mixing with PCL/chloroform, respectively. The middle phase in vial ii is denatured protein. See text for discussion. The residual activity of the protein (HRP) recovered after incubation with PCL-chloroform solution for 30 min is shown in the graph. Here protein was added into the polymer solution in the form of protein-in-buffer (HRP-buffer) and encapsulated into sugar-glass nanoparticles (HRP-SGnPs). Protein encapsulated into the SGnP shows virtually no activity loss even at very low levels of added protein concentration, whereas HRP in buffer results 100 % of original activity lost at low levels of HRP addition. Large amounts of protein added to the polymer were required to obtain measureable recovered activity of HRP. (c) Representative TEM image of protein-SGnP. (d) Particle size distribution of HRP-SGnP from TEM images. (e) A snapshot of process related activity loss and storage stability at 23 °C for HRP in nanofiber scaffolds electrospun from chloroform- poly(ethylene oxide) (PEO) solutions. Values are average ± one standard deviation of n = 4 samples for each groups.
Generation of these particles combines practices from enzyme chemistry and from biopharmaceutics. In the enzyme chemistry field proteins have been protected from nonpolar solvent exposure by dispersing them in water pools of inverse micelles. This practice provides excellent dispersion of protein while protecting against water-oil interface and, to some extent elevated temperatures.[18–21] Here we incorporate sugars and other stabilizers along with the proteins in inverse micelles. In biopharmaceutics it has long been the practice to freeze-dry proteins in the presence of sugars and other excipients to protect them from chemical and physical degradation during storage.[22] Here we freeze-dry the nanoparticles by flash-freezing the inverse micelle suspension and drying off first the solvent then the water, leaving protein-laden, surfactant-coated sugar-glass nanoparticles (SGnPs).
These freeze-dried nanoparticles can be re-introduced into polymer solutions and organic solvents as a stable suspension, without damage to the protein. Vial i in Figure 1b shows that the nanoparticles suspend homogeneously in a solution of poly(caprolactone) (PCL) in chloroform, and the graph in Figure 1b shows that we recover 98% of protein activity after the horseradish peroxidase (HRP)-containing SGnPs are exposed to chloroform / PCL solution for 30 minutes. In the remainder of this paper we evaluated performance of the SGnPs with respect to process-induced protein damage, protein distribution and release profile, and storage stability.
We compared performance of the SGnPs with results from a parallel study using a conventional water-in-oil emulsion (w-o) process which was recently reported,[23]and is typical of methods that are commonly used for protein encapsulation in tissue engineering and drug delivery.[15, 23] In order to make the comparison, we first demonstrated that the conventional w-o encapsulation process[23] resulted in significant protein loss at initial steps, a fact that is well-known but underreported.[17, 23] We carry out the first steps of the conventional w-o process by adding an aqueous solution of HRP to a 10 % by mass solution of biodegradable poly-caprolactone (PCL) in chloroform. We then emulsified the mixture by vortexing or by sonicating then vortexing. In order to measure residual enzyme activity subsequent to these steps, we allowed the emulsion stand for 30 min at 23 °C, and then extracted the protein out of emulsion by adding more buffer and centrifuging (see Supplementary Information for details method). Although we began with two bulk phases, vial ii in Figure 1b shows that we obtain three bulk phases after processing; the new, middle phase is denatured protein. Such large amounts of denatured protein raise immunogenicity concerns. The residual activity (protein’s enzymatic activity) remaining in the aqueous phase after processing is shown in Figure 1b. No residual activity is observed at low levels of HRP addition. This quantitative loss is likely due to protein denaturation at the oil-water interface.[24] Measurable protein activity was recovered only when the amount of added protein was significantly increased to (0.4 nmole/g PCL) and even then, 99.97 % of activity was lost. Even at the highest protein loading, 100 nmole/g PCL (0.5 % HRP by mass), more than 90 % of protein activity was lost. Note that sonication resulted in even greater protein deactivation at all levels of HRP loading (Figure 1b), probably due to strong shear stress.[17] Thus, significant degradation could occur during conventional processing when the protein was introduced to the solvent from buffer, the usual practice when loading protein into a polymer carrier.[2, 3, 17]
In the approach we have developed, protein was incorporated in the aqueous phase of an inverse micelle suspension stabilized in isooctane using sodium 1,4-bis(2-ethylhexoxy)-1,4-dioxobutane-2-sulfonate (AOT). The suspension was rapidly quenched to 77 K by spraying it into liquid nitrogen, and the resulting glassy mixture was subsequently freeze-dried, leaving only sugar-glass nanoparticles (see Figures SI 1a to 1d for dispersed protein encapsulated particles). The water:surfactant mole ratio (w = [H2O]/[AOT]) determined the equilibrium micelle size, and varied in the range of 10 to 15 depending on the protein used for encapsulation. This choice of ‘w’ resulted in typical particles sizes distributed between 30 nm and 160 nm, as obtained from transmission electron microscopy (TEM) (Figures 1c, d) and, although the particles obtained in this technique had average size of 100 nm, a very few agglomerated particles (> 500 nm) were also obtained (see Figure SI 3) which could be removed by centrifugation (at 100 × g for 3 min). These large agglomerates were associated with sample temperature excursions during freeze-drying, which can be controlled. The mass ratio of protein to trehalose was typically maintained at 1:500 (0.2 mg protein in 100 mg sugar) to provide sufficient coating of sugar-glass around the protein in the nanoparticles (≈ 15 protein molecules in a 100 nm SGnP). The protein:trehalose mass ratio could be increased to 1:200 without adversely effecting performance. Additives such as trehalose and buffer salts are known to stabilize the protein in dry form, and are also thought to keep the protein stable in the water pool within the micelle, and against any adverse interaction from surfactant.[18, 19, 21] We observed only minimal (< 5 %) protein loss associated with the process of encapsulating proteins in SGnPs (see Figure SI 4 for activity of insulin before and after encapsulation into SGnP).
Once encapsulated, the sample protein was extremely robust to subsequent solvent exposure and to storage stress. As shown in Figure 1b we obtain a 10,000-fold enhancement of residual protein activity when SGnP-protected protein was exposed to PCL / chloroform as compared to introduction of protein from buffer. The nanoparticles also provide an environment of sugars that stabilizes the proteins against degradation during further processing and storage.[22] Figure 1e shows a snapshot of process-related activity loss and storage stability for HRP in nanofibrous scaffolds electrospun from chloroform-poly(ethylene oxide) (PEO) solutions. In generating these scaffolds, HRP was added to PEO solutions as SGnPs or added directly from buffer. As with the PCL solutions, 95.5% of protein activity was lost when protein is added to the PEO solution from buffer at 8 nMole HRP /g PCL. 75 % of the remaining protein activity was lost during scaffold electrospinning, and almost all of the remaining activity was lost after 45 d storage at 23°C. Conversely, only 5 % of the total activity was lost in all the steps through electrospinning for HRP protected in SGnPs. Additionally, the protein in these scaffolds was much more stable during storage. Including storage-related losses, the protected HRP retained 71 % of its initial activity after 45 d storage at 23 °C, whereas the unprotected HRP retained only 0.003 % of its initial activity
Next we characterized the ability of SGnPs’ to protect proteins against damage from a range of solvents (see Supplementary Information Table SI 1 for solvent details). Non-aqueous solvent exposure is known to induce denaturation and other degradation for many proteins in buffer.[2, 3, 25] Figures 1b and 1e confirm that sequestration in SGnPs largely protected the protein against chloroform exposure. In Figure 2 we present activity retention data for four proteins exposed to nine solvents, either directly from buffer, or while encapsulated in SGnPs (see Figures SI 5 & 6 for experimental procedure). Two of the proteins we tested, HRP and lipase, are model enzymes, commonly used for stability studies. The remaining two, insulin, and bone morphogenetic protein 2 (BMP-2) are clinically important signaling proteins.[26] We challenged these by exposure to organic solvents with a wide range of polarity indices (PI) ranging from 0 to 6.5. As indicated in Table SI 1, we have classified these solvents as water-immiscible and water-miscible.
Figure 2.
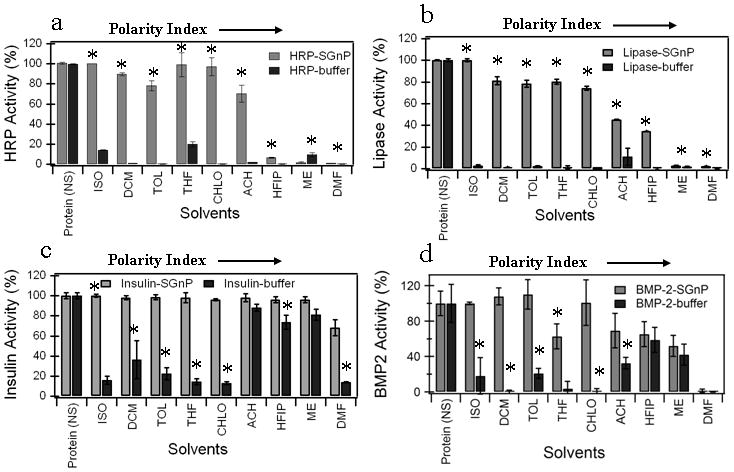
Activity retention data for four proteins; (a) HRP, (b) Lipase, (c) Insulin, and (d) BMP-2 exposed to nine solvents, either directly from buffer, or while encapsulated in SGnPs. HRP and Lipase were used as model proteins to study the enzymatic activity of the proteins whereas insulin and BMP-2 were used to study the biological activity. To compare how the SGnP system protects protein from adverse chemical environments, we incubated HRP-SGnP/lipase-SGnP/insulin-SGnP/BMP-2-SGnP in the different organic solvents. The protein in buffer (HRP-buffer/lipase-buffer/insulin-buffer/BMP-2-buffer) was used to mimic the conventional encapsulation techniques. The residual activity of the protein measured after incubated for 2 hours with different solvents in the form of protein in buffer (protein-buffer) or protein encapsulated into sugar-glass (protein-SGnP). The solvents used for this experiment have wide range of polarity index (0 to 6.4). Protein activity was measured after exposure to solvent and compared to activity values obtained before solvent exposure. Values are average ± one standard deviation of n=4 samples for each groups. Statistically significant differences (P<0.05) in protein activity between protein-buffer and protein-SGnP samples after incubation with a particular solvent are indicated by an asterisk (by t-test). [Protein (NS); protein-SGnP and protein-buffer samples without any solvent treatment, ISO; isooctane, DCM; dichloromethane, TOL; toluene, THF; tetrahydrofuran, CHLO; chloroform, ACH; acetone, HFIP;hexafluoroisopropanol, ME; methanol, DMF;dimethylformamide]
Figures 2a and 2b show residual enzyme activity of HRP and lipase respectively, after incubation in solvents. Water-immiscible solvents completely deactivated both proteins when they were introduced from buffer. In contrast, more than 80 % of the initial enzyme activity was retained in all cases for protein in SGnPs. Encapsulation in SGnPs also conferred benefit for exposure to the water-miscible solvents tetrahydrofuran (THF), acetone, and hexafluoroisopropanol (HFIP) (P<0.05). No benefit was observed for exposure to methanol or dimethylformamide (DMF). Figures 2c and 2d show residual protein activity after incubation in solvents for insulin and BMP-2 respectively. Results for these proteins mirrored those of HRP and lipase for water-immiscible solvents. However, unlike the HRP and lipase results, these two proteins showed a moderate level of activity in water-miscible solvent systems even when exposed directly to solvent from buffer.
The primary results of Figure 2 may be interpreted based on the reasonable assumption that trehalose glass absorbs polar solvents to a greater extent than non-polar solvents, which is supported the fact that trehalose glasses swelled considerably in high PI solvents such as methanol.[27] When proteins were presented directly to solvent from buffer, each of the solvents deactivated the proteins to some extent. Thus, the sugar glass was effective at acting as a barrier between the protein and solvent with PI ≤5, but less effective with solvent of PI ≥5. The degree of degradation in a particular solvent varied slightly by protein, likely due to differences in their specific structure and function.[28] However, there was a uniformly high degree of protection from solvents with PI < 5 for all these proteins upon encapsulation in the sugar-glass, suggesting that the SGnP platform may be widely effective for biomolecule protection against non-polar solvents.
In addition to protection from solvent, the nanoencapsulation approach also provides for good dispersion properties in solvents (as is shown in Figure SI 1b to 1e) and homogeneous distribution of the protein throughout the carrier polymer. This property provides a platform to load protein-bearing SGnPs within the DDSs such as nanofiber scaffolds as high as 40 % mass fraction of polymer matrix with essentially 100 % encapsulation efficiency (see Figure IS 7). These loading levels do not impact scaffold preparation outcomes, at least for electrospinning, and gas-foaming. Figure 3a shows a fluorescence micrograph of sugar nanoparticles containing fluorescent dye in electrospun fibers, showing homogeneous distribution of SGnPs within the fiber matrix (see Figure SI 8 for 3D distribution of particles in the matrix). The favorable dispersion seems to be facilitated by the layer of surfactant that persists on the SGnPs. Uniform dispersion is desirable to prevent burst release, and gives uniform sustained release of the biomolecule. Burst release is often observed from carrier systems where separation occurs between the polymer and protein-bearing phases.[23, 26] Many approaches have been described to control drug release kinetics, including tailoring of protein-polymer chemical interactions, use of additional matrix materials, and secondary incorporation of drug into microspheres.[29] [26] As expected from the well dispersion properties of SGnP system the HRP and insulin release profiles from gas-foamed and electrospun nanofibrous scaffolds made of PCL and SGnPs (Figures SI 7 and SI 9 Scanning Electron Microscopy (SEM) micrograph of different scaffolds) were essentially devoid of burst features (Figures 3b and 3c).
Figure 3.
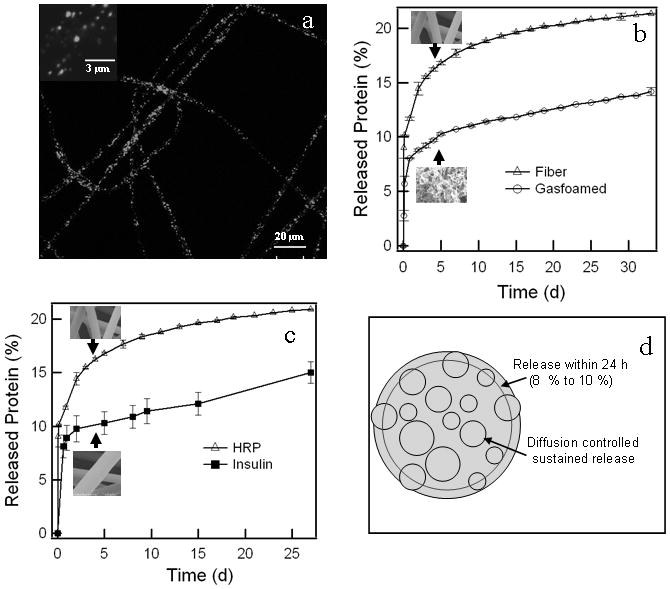
SGnPs distribution within the nanofiber matrix and release profile of the protein from this matrix. (a) Typical confocal image of 2 % FITC-SGnP (mass fraction) loaded PCL nanofiber scaffold. Insert shows higher magnification image of the same fiber showing distinct FITC-SGnP in the fiber matrix. (b) Released profile of HRP from the two types of PCL scaffold, nanofiber and gas-foamed. Insert shows the typical SEM micrograph of the corresponding scaffold. (c) Comparative released profile of insulin and HRP from PCL nanofiber scaffold at 23 °C. Insert shows a typical SEM micrograph of the corresponding scaffold. (d) Schematic presentation of SGnPs distribution across the fiber diameter. Three replicate for each type of scaffold was used for release study. Values are average ± one standard deviation of n=3 samples for each groups.
Figures 3b and 3c show fraction of active protein released from PCL scaffolds as a function of time. In panel b, 11 % and 8 % initial release of HRP was seen from electrospun fibrous and gas-foamed scaffolds respectively. This was followed by a slower released of about 0.85 and 0.33 % / d, up to 9 d from the nanofibrous and gas-foamed scaffold, respectively, and sustained release of HRP (0.1% / d) from the scaffold up to 33 d. Panel c shows that HRP and insulin gave similar release profiles from electrospun nanofiber, i.e., initial release of 8 % to 11 %, followed by slow released up to 9 d and further sustain released up to 33 d. Delivery was expected to be slow as PCL is slow-degrading polymer.[30]
The initial 8 % to 10 % release behavior could be expected from a simple geometric argument as depicted in Figure 3d, given fiber diameters of 1.30 μm ± 0.65 μm, and a homogeneous distribution of nanoparticles with diameters of 100 nm (see Figure SI 7), as approximately 10 % of the particles will have their centers located within one particle radius from the surface of the fiber. In other words, these will have some direct exposure to the medium, not being shielded by a significant amount of PCL. The gas-foamed scaffold has similar dimensions for membrane thickness, so similar argument should hold, although its morphology is membranous rather than fibrous, yielding a smaller surface area. Thus, we attribute the lower initial HRP release in the gas-foamed scaffold to their lower surface to volume ratio.[31]
Finally, we addressed storage stability of the encapsulated protein. Storage stability is crucial for efficacy in long-term release and important for manufacturing. In these studies we used PEO, a water-soluble polymer, as the carrier in order to facilitate extraction of all protein very quickly at specified time points, using only buffer for extraction. The process-related loss of HRP activity in the PEO nanofiber scaffolds fabricated by the conventional technique was similar to that observed in the PCL fiber. The nanofiber scaffolds (see Figure SI 10 SEM images) were stored for 150 d at 23 °C and 35 °C. Residual enzyme activity was measured at 45 d and 150 d. Figure 4 shows that HRP sequestered in SGnPs is approximately 10 X more stable than the unprotected protein. Unprotected and SGnP-protected HRP enzymatic activity decayed to e-1 of its initial value roughly 4 and 34 d, respectively, when stored at 35 °C. At 23 °C storage, activity of unprotected HRP decayed to e-1 of its initial value at 10 d, while SGnP-protected HRP maintained activity above e-1 of its initial value for more than 150 d. Having only three time points we do not attempt to fit the degradation rate to any model; the lines are guides to the eye. The data of Figure 4 show that even if a sufficient amount of active, unprotected protein could be introduce into a tissue scaffold, it would degrade entirely in just a few days, making long-term delivery impossible. Note that protein encapsulated SGnP could be stored at room temperature for at least a month, and several months at −20 °C with <5 % loss of protein activity. Moreover, SGnPs can be transported and stored as isooctane suspension (Figure SI 1b to 1d) which is readily applicable for DDS preparation.
Figure 4.
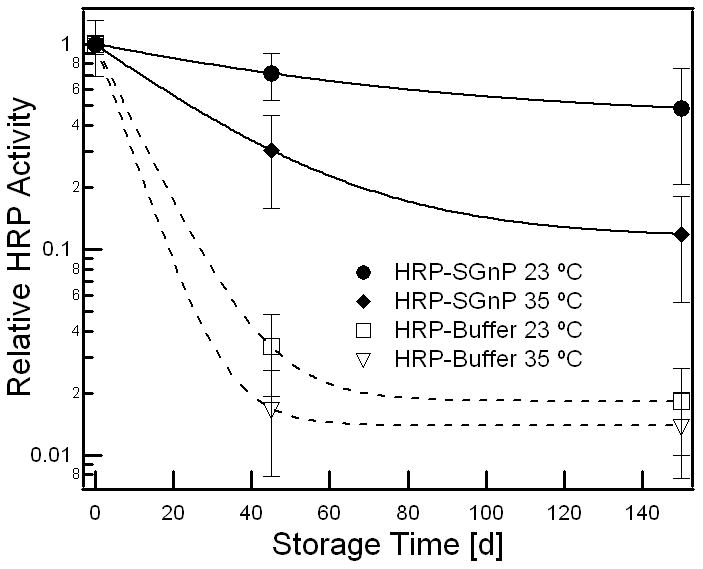
Protein activity loss in protein-polymer construct as a function of storage at two temperatures. Comparative activity loss of HRP in the nanofiber scaffold where protein encapsulated into the scaffold in the form of protein-SGnP or conventional protein-buffer, stored for 150 days at 23 °C and 35 °C. Values are average ± one standard deviation of n=4 samples for each group.
In conclusion, we present an approach to stabilizing proteins that is nearly ideal in that the single approach yields excellent protection from process-related stresses, very good encapsulation efficiency and storage stability, as well as giving burst-free sustained release for essentially any protein and polymer system of interest. The approach borrows from a significant amount of work done in biopharmaceutics towards understanding and controlling protein degradation processes in sugar-based glasses. We have previously demonstrated that proteins are best stabilized only when the environment of the sequestered protein is carefully controlled.[32] The required level of control is not possible with the methods currently used to incorporate biomacromolecules into polymeric carriers, but is readily met by the SGnP nanoencapsulation approach described here. Furthermore, because well-established biopharmaceutical glass technology is used, we expect that this approach will be amenable to other bioactive species such as viruses, DNA or siRNA. Once packaged in SGnPs, distinct bioactive components present identically, so this approach will be applicable to incorporation of multiple bioactive species in a single delivery system (work in progress). This SGnP system provides an ideal platform to prepare next generation ‘smart scaffold’ of any polymer system (soluble in solvents PI ≤5) single/multi-biomolecules encapsulated with required pharmaceutical properties for regenerative medicine.
Experimental Section
Sugar-glass-protein nanoparticle
SGnPs were prepared from inverse micelles of AOT in isooctane. AOT was dissolved in 12 mL of isooctane in a 25 mL centrifuged tube to produce a (0.3 to 0.4) mole/L solution. Aqueous phase of (0.4 to 0.8) mL containing protein and other excipients was then added. The mixture was vortexed for 30 s to 2 min to obtain a clear suspension. The aqueous phase contained protein with other protein-specific excipients (e.g., trehalose, Tween-20, CaCl2, ZnCl2 etc), and its pH was adjusted to provide maximum protein solubility and stability at room temperature. The inverse micelle suspension was flash-frozen by slowly injecting it into a 50 mL vial containing liquid nitrogen (N2). The vial with the frozen nanoparticles and isooctane was then lyophilized to evaporate isooctane and water. After lyophilization, the nanoparticles were washed in isooctane by re-suspending and subsequently centrifuging them at 400 g for 10 min. This washing process was repeated 4 to 5 times and finally the SGnP dispersion was stored in isooctane under desiccation at −20 °C or −80 °C for future use.
Analysis of activity in the presence of different organic solvents
For protein in aqueous phase (i.e., protein-buffer), 0.5 mL of solvent was placed in a 1.5 mL centrifugation tube and 0.050 mL to 0.1 mL (10 % to 20 % volume fraction) of aqueous phase of protein was added into it. For protein in sugar-glass system (i.e., protein-SGnP), 0.010 mL to 0.030 mL of protein-SGnP suspension in isooctane was added to the 0.5 mL of solvent in a 1.5 mL centrifugation tube. As control, no solvent was added to the vial of reference protein samples (protein with no solvent). Vials were placed on a vortexer or shaker for 2 h to achieve homogeneous mixing of the aqueous phase and the organic solvent phase. The schematic presentation of the general process is described in Figures SI 4 and SI 5. All solvents used in this experiment were dehydrated by pretreatment with molecular sieves 4A (Sigma, USA). Once shaken or vortexed, the samples were directly reconstituted with suitable buffer or lyophilized to remove the solvent from the system in the presence/absence of trehalose (0.012 mL of 23 % trehalose in water) followed by reconstitution with buffer. Reconstituted proteins samples (protein-buffer and protein-SGnP samples with or without solvent treated) were used for their specific activity assay (see Supplementary Information for details).[33, 34] [35] Catalytic activity was measured for reconstituted HRP/lipase samples. In vitro bioactivity assay of reconstituted insulin and BMP-2 samples was performed using rat myoblast L6 cell line and mouse stromal cell line W-20-17 cells respectively. The protein activity recovered after solvent treatment was calculated with respect to control protein samples without any solvent exposure.
Analysis of loss of HRP in the presence of polymer solution
PCL solution in chloroform of 10 % mass fraction was placed (300 μL) in a 1.5 mL microcentrifuge tube and 30 μL of HRP-Tris HCl solution (0.001 nmole to 1000 nmole HRP/g polymer) or HRP-SGnP (0.001 to 0.1 nmole HRP/g polymer) suspension with various protein concentrations was added, and subsequently vortexed (1 min to 5 min) and optionally sonicated (15 s to 30 s) followed by 30 min incubation at room temperature. After incubation, 470 μL buffer was added and vortexed (30 s to 1 min), followed by centrifugation at 9000 g for 5 min to separate the protein-buffer phase from polymer-solvent (see Figures SI 5 and SI 6). Active protein was quantified by activity assay.
Scaffold Fabrication
Scaffolds with two distinct structures made of PCL or PEO were prepared using electrospinning[36] or gas-foaming[37] (see Supplementary Information for further details of scaffold fabrication.)
Analysis of activity loss in protein-polymer construct as a function of storage
Disks of 5 mm to 7 mm diameter were cut randomly from protein encapsulated nanofiber scaffold sheets prepared by either conventional encapsulation method (i.e., protein-buffer in polymer solution) or using protein-SGnP and stored desiccated for up to 150 d at different temperature (23 °C and 35 °C). The fiber samples were dissolved in buffer to extract encapsulated protein into the buffer followed by their activity measurement using suitable assay (see Supplementary Information). Protein activity in the scaffold was measured immediately after scaffold preparation to establish the baseline activity. The protein activity change during storage was presented as % of residual activity compared to the activity just after casting of scaffold, considering as 100 % activity.
In vitro release study
HRP and insulin were used as model proteins to study the release profile from PCL-based scaffolds prepared by two different techniques. Approximately 20 mg to 30 mg of nanofiber (5 mm to 10 mm circular disk) or gas-foamed scaffold were used for these release studies (see Supplementary Information for further details).
Supplementary Material
Acknowledgments
We acknowledge funding from NIH/NIBIB under grant R01 EB006398-01A1. JG acknowledges postdoctoral fellowship support from the National Research Council of the National Academy of Sciences in the Joint NIH/NIST Postdoctoral Programs. The authors gratefully acknowledge assistance from Steve Hudson (NIST) for TEM imaging. This article, a contribution of NIST, is not subject to US copyright.
Footnotes
Supporting Information is available online from Wiley InterScience or from the author.
Contributor Information
Dr. Jyotsnendu Giri, Email: jgiri@nist.gov, Polymers Division, National Institute of Standards and Technology, Gaithersburg MD 20899, USA, National Institute of Arthritis, and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD 20892
Prof. Wan-Ju Li, Email: li@ortho.wisc.edu, Department of Orthopedics and Rehabilitation, and Department of Biomedical Engineering, University of Wisconsin-Madison, Madison, WI 53705 USA
Prof. Rocky S Tuan, Email: rst13@pitt.edu, Center for Cellular and Molecular Engineering, Department of Orthopedic Surgery, University of Pittsburgh School of Medicine, Pittsburgh, PA 15219
Dr. Marcus T Cicerone, Email: cicerone@nist.gov, Polymers Division, National Institute of Standards and Technology, Gaithersburg MD 20899, USA, Phone: 301.975.8104
References
- 1.Fu K, Klibanov AM, Langer R. Nat Biotechnol. 2000;18:24. doi: 10.1038/71875. [DOI] [PubMed] [Google Scholar]
- 2.Perez C, Castellanos IJ, Costantino HR, Al-Azzam W, Griebenow K. Journal of Pharmacy and Pharmacology. 2002;54:301. doi: 10.1211/0022357021778448. [DOI] [PubMed] [Google Scholar]
- 3.Putney SD, Burke PA. Nat Biotechnol. 1998;16:153. doi: 10.1038/nbt0298-153. [DOI] [PubMed] [Google Scholar]
- 4.Zhu GZ, Mallery SR, Schwendeman SP. Nat Biotechnol. 2000;18:52. doi: 10.1038/71916. [DOI] [PubMed] [Google Scholar]
- 5.Bartus RT, Tracy MA, Emerich DF, Zale SE. Science. 1998;281:1161. doi: 10.1126/science.281.5380.1161. [DOI] [PubMed] [Google Scholar]
- 6.Cleland JL, Langer R. Formulation and Delivery of Proteins and Peptides. 1994;567:1. [Google Scholar]
- 7.He JT, Su HB, Li GP, Tao XM, Mo W, Song HY. International Journal of Pharmaceutics. 2006;309:101. doi: 10.1016/j.ijpharm.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 8.Kokai LE, Tan HP, Jhunjhunwala S, Little SR, Frank JW, Marra KG. Journal of Controlled Release. 2010;141:168. doi: 10.1016/j.jconrel.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Giteau A, Venier-Julienne MC, Marchal S, Courthaudon JL, Sergent M, Montero-Menei C, Verdier JM, Benoit JP. European Journal of Pharmaceutics and Biopharmaceutics. 2008;70:127. doi: 10.1016/j.ejpb.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Castellanos IJ, Flores G, Griebenow K. Journal of Pharmacy and Pharmacology. 2005;57:1261. doi: 10.1211/jpp.57.10.0004. [DOI] [PubMed] [Google Scholar]
- 11.Wang JJ, Chua KM, Wang CH. Journal of Colloid and Interface Science. 2004;271:92. doi: 10.1016/j.jcis.2003.08.072. [DOI] [PubMed] [Google Scholar]
- 12.Morita T, Sakamura Y, Horikiri Y, Suzuki T, Yoshino H. Journal of Controlled Release. 2000;69:435. doi: 10.1016/s0168-3659(00)00326-6. [DOI] [PubMed] [Google Scholar]
- 13.Paillard-Giteau A, Tran VT, Thomas O, Garric X, Coudane J, Marchal S, Chourpa I, Benoit JP, Montero-Menei CN, Venier-Julienne MC. European Journal of Pharmaceutics and Biopharmaceutics. 2010;75:128. doi: 10.1016/j.ejpb.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Lee KY, Mooney DJ. Advances in Controlled Drug Delivery: Science, Technology, and Products. 2003;846:73. [Google Scholar]
- 15.Tayalia P, Mooney DJ. Advanced Materials. 2009;21:3269. doi: 10.1002/adma.200900241. [DOI] [PubMed] [Google Scholar]
- 16.Tessmar JK, Gopferich AM. Adv Drug Deliv Rev. 2007;59:274. doi: 10.1016/j.addr.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 17.Yang SX, Yuan WE, Jin T. Expert Opin Drug Deliv. 2009;6:1123. doi: 10.1517/17425240903156374. [DOI] [PubMed] [Google Scholar]
- 18.Anarbaev RO, Rogozina AL, Lavrik OI. J Mol Catal B-Enzym. 2009;59:64. [Google Scholar]
- 19.Mathew DS, Juang RS. Biochem Eng J. 2005;25:219. [Google Scholar]
- 20.Naoe K, Noda K, Kawagoe M, Imai M. Colloid Surf B-Biointerfaces. 2004;38:179. doi: 10.1016/j.colsurfb.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 21.Savelli G, Spreti N, Di Profio P. Current Opinion in Colloid & Interface Science. 2000;5:111. [Google Scholar]
- 22.Wang BQ, Tchessalov S, Warne NW, Pikal MJ. Journal of Pharmaceutical Sciences. 2009;98:3131. doi: 10.1002/jps.21621. [DOI] [PubMed] [Google Scholar]
- 23.Sy JC, Klemm AS, Shastri VP. Advanced Materials. 2009;21:1814. [Google Scholar]
- 24.Phillips MC. Chemistry & Industry. 1977:170. [Google Scholar]
- 25.Schwendeman SP. Critical Reviews in Therapeutic Drug Carrier Systems. 2002;19:73. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 26.Chen FM, Zhang M, Wu ZF. Biomaterials. 31:6279. doi: 10.1016/j.biomaterials.2010.04.053. [DOI] [PubMed] [Google Scholar]
- 27.Engasser JM, Chamouleau F, Chebil L, Ghoul M. Biochem Eng J. 2008;42:159. [Google Scholar]
- 28.Doukyu N, Ogino H. Biochem Eng J. 48:270. [Google Scholar]
- 29.Chew SY, Wen Y, Dzenis Y, Leong KW. Current Pharmaceutical Design. 2006;12:4751. doi: 10.2174/138161206779026326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valmikinathan CM, Defroda S, Yu XJ. Biomacromolecules. 2009;10:1084. doi: 10.1021/bm8012499. [DOI] [PubMed] [Google Scholar]
- 31.Acharya G, Shin CS, Vedantham K, McDermott M, Rish T, Hansen K, Fu YR, Park K. Journal of Controlled Release. 2010;146:201. doi: 10.1016/j.jconrel.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 32.Cicerone MT, Soles CL. Biophysical Journal. 2004;86:3836. doi: 10.1529/biophysj.103.035519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bovaird JH, Ngo TT, Lenhoff HM. Clinical Chemistry. 1982;28:2423. [PubMed] [Google Scholar]
- 34.Choi SJ, Hwang JM, Kim SI. Journal of Biochemistry and Molecular Biology. 2003;36:417. doi: 10.5483/bmbrep.2003.36.4.417. [DOI] [PubMed] [Google Scholar]
- 35.Patel N, Craddock BL, Staniforth JN, Tobyn MJ, Welham MJ. Journal of Pharmacy and Pharmacology. 2001;53:1415. doi: 10.1211/0022357011777774. [DOI] [PubMed] [Google Scholar]
- 36.Li WJ, Cooper JA, Mauck RL, Tuan RS. Acta Biomaterialia. 2006;2:377. doi: 10.1016/j.actbio.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 37.Harris LD, Kim BS, Mooney DJ. J Biomed Mater Res. 1998;42:396. doi: 10.1002/(sici)1097-4636(19981205)42:3<396::aid-jbm7>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.