

Abstract
A Cu-catalyzed enantioselective Claisen rearrangement of easily-accessible enolphosphonates using commercially available (R, R)-PhBOX as the chiral ligand was developed. A wide range of rearrangement products with contiguous tertiary and all carbon quaternary centers were obtained in excellent yields and stereoselectivities. The α-ketophosphonate products could be easily transformed into other functional groups in high efficiency.
Keywords: asymmetric catalysis, catalytic Claisen rearrangement, BOX ligands, copper, quaternary carbon centers
A one-step catalytic method for the enantioselective construction of contiguous tertiary and all-carbon quaternary centers is a challenging task but of great significance.[1,2] Among the limited number of available approaches for the assembly of such sterically congested structures via a single transformation, the Claisen rearrangement, well-known as one of the most effective carbon-carbon bond formation strategies, has shown to be promising. Since its discovery one century ago,[3] the Claisen rearrangement has become a powerful tool widely used by synthetic organic chemists.[4] Despite many efforts made in this field, the development of the catalytic asymmetric Claisen rearrangement is still in an immature stage. Until now, Hiersemann’s copper[5] and Jacobsen’s hydrogen bonding catalysts[6] have realized catalytic versions of asymmetric Claisen rearrangement. Both of their methods are excellent but relied heavily on ketocarboxylic acid derivatives, affording products with limited generality that are in most cases difficult for further functionalization. Moreover, with these reported catalytic systems, there are only few examples of the direct formation of a quaternary carbon center in the presence of a vicinal tertiary center with moderate to good stereoselectivities. In this regard, it remains an extremely significant yet elusive goal for discovering novel catalytic enantioselective Claisen rearrangement as an alternative competent method for rapid creation of vicinal tertiary and quaternary carbon center motifs, which are widely present in complex natural products as well as medicinal and biologically active molecules.[7]
Recently, many groups including our own have reported that the phosphonate group is able to tightly coordinate to metal ions and provide excellent enantiocontrol with additional binding site adjacent to the phosphonate group through chelation with chiral metal catalysts.[8] We thus envisioned that enolphosphonates would be more favored bidentate substrates for metal-catalyzed Claisen rearrangement reactions (Figure 1). More importantly, the corresponding products, α-ketophosphonates, are well-known as one of the most synthetically useful phosphorous motifs and could undergo various transformations with ease and efficiency.[9] Herein, we are glad to report our recent success with the first catalytic enantioselective Claisen rearrangement of enolphosphonate approach which provides a large scope of α-ketophosphonate derivatives with contiguous tertiary and quaternary carbon centers in excellent yields and selectivities.
Figure 1.

Claisen rearrangement of enolphosphonates
Studies were initiated by the development of a convenient protocol for substrates preparation. These compounds could indeed be simply obtained by treating allylic bromides with α-ketophosphonates in the presence of DBU.[10] Next, based on our previous success in catalytic asymmetric reactions of ketophosphonates using metal/TBOx complexes,[11] we investigated the Claisen rearrangement of enolphosphonate S1 using Al/TBOx complexes (Table 1, entry 1).[12] However, no rearrangement product was detected. By switching to the Cu/TBOx complex, the reaction proceeded slowly to give the product with 70% yield but in racemic form (entry 2). Bidentate bisoxazoline ligands instead of tetradentate TBOx ligands were then examined. To our delight, ligand 1 (R, R)-PhBOX gave the product with 88% yield and 93% ee in 2 hours (entry 3).[13,14] Aminoindanol-derived bisoxazoline ligand 2 provided the rearrangement product in 93% ee but lower yield (entry 4). Other metal triflates and copper sources were also tested but none of them gave more satisfying results.[10] A further solvent screening indicated that several solvents were suitable for this reaction and the enantioselectivity could be improved when the reaction was performed in a DCM/hexane (1:5) solvent mixture (entry 5).[10] Lowering catalyst loading to 5 mol% eventually gave the best results (entry 6). Unfortunately, this solvent mixture did not increase the enantioselectivity when ligand 2 was used (entry 7). Interestingly, the catalyst loading could even be lowered to 0.1 mol% without significant detrimental effect on the selectivity and yield, although longer reaction time was required (entry 8). Decreasing the reaction temperature to 0°C failed to give rise to further improved enantiselectivity (entry 9). It’s noteworthy that the size of R group in the phosphonate motif does not have a profound effect on the reaction. Substrates S2 and S3 with either a smaller or a larger R group all gave the desired product with slightly lower selectivities (entries 10 and 11).
Table 1.
Screening of reaction conditions for Cu-catalyzed Claisen rearrangement of enolphosphonates

| |||||||
|---|---|---|---|---|---|---|---|
| Entry[a] | Metal | Substrate | Ligand | Solvent | Time | Yield(%)[b] | ee(%)[c] |
| 1 | Me2AICI | S1 | TBOx | DCM | 24 h | N. R. | N. D. |
| 2 | Cu(OTf)2 | S1 | TBOx | DCM | 16 h | 70 | 0 |
| 3 | Cu(OTf)2 | S1 | 1 | DCM | 2 h | 88 | 93 |
| 4 | Cu(OTf)2 | S1 | 2 | DCM | 4 h | 80 | 93 |
| 5 | Cu(OTf)2 | S1 | 1 | -[d] | 4 h | 90 | 96 |
|
|
|||||||
| 6[e] | Cu(OTf)2 | S1 | 1 | -[d] | 4 h | 90 | 97 |
|
|
|||||||
| 7[e] | Cu(OTf)2 | S1 | 2 | -[d] | 24 h | 69 | 83 |
| 8[f] | Cu(OTf)2 | S1 | 1 | -[d] | 72 h | 76 | 93 |
| 9[e],[g] | Cu(OTf)2 | S1 | 1 | -[d] | 72 h | 80 | 97 |
| 10[e] | Cu(OTf)2 | S2 | 1 | -[d] | 2 h | 84 | 95 |
| 11[e] | Cu(OTf)2 | S3 | 1 | -[d] | 2 h | 86 | 95 |
All reactions performed at 0.1 mmol scale.
Isolated yields.
Determined by chiral HPLC.
Dichloromethane(DCM)/hexane=1:5.
5 mol% catalyst loading.
0.1 mol% catalyst loading.
Reaction performed at 0°C.
With the fully optimized reaction conditions in hand, a wide range of substrates were examined and the rearrangement products were obtained with good to excellent yields and stereoselectivities (Table 2). Other than dimethyl substitutions as R1 and R2 groups on the enolphosphonates, substrates bearing various cyclic ring substituents provided 2–4 with similar levels of enantioselectivities. In addition to aryl group, aliphatic R3 groups were also tolerated and products 5, 6 and 7-(S) were obtained with similar results but longer reaction time. The steric bulk of R3 group has no significant effect on the enantioselectivity. Instead of the phenyl group, substrates bearing large naphthyl rings were also examined, generating 8 and 9 with high enantioselectivities respectively. For conjugated substrates, the reaction had to be carried out at 0°C to attenuate the background reaction, providing 10 with 90% ee. It is interesting to note that the existence of substituents R4 did not prevent the rearrangement which has seldom been studied in previous reports. Enolphosphonates bearing either alkyl group or halogen atom as the R4 group all underwent the rearrangement reaction to give α-ketophosphonates 11–13 in high efficiency. Unfortunately, under optimized reaction conditions, substrates bearing either cis-cinnamyl or 3, 3-disubstituted allylic segment failed to proceed rearrangement reactions which indicates that our method still has its own limitations at current stage.[15] However, the utilization of commercially available (S, S)-PhBOX ligand as an alternative strategy enabled to generate the products 1-(R) and 7-(S) with opposite sense of chirality from the same starting materials.
Table 2.
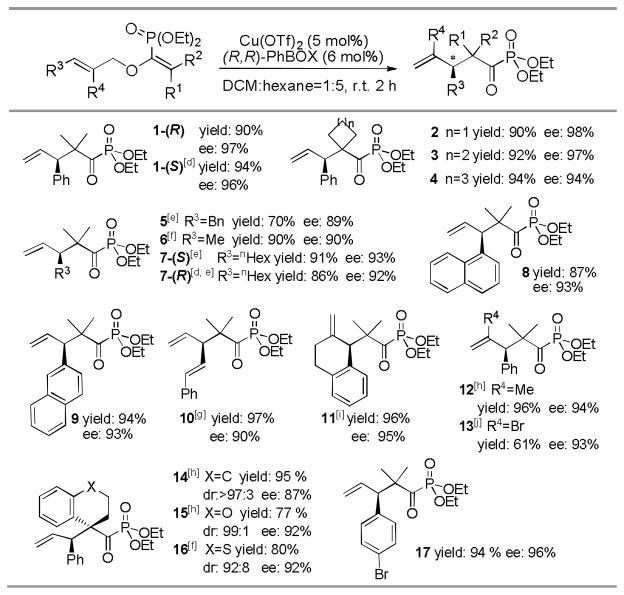
|
All reactions performed at 0.1 mmol scale.
Isolated yields.
Diastereomeric ratio (dr) determined by 1H NMR of the crude mixture. Enantioselectivity determined by chiral HPLC. Absolute configuration was assigned by X-ray crystallographic analysis of the hydrolyzed product of 17 and the rest were assigned by analogy.
(S, S)-PhBOX ligand used instead
Reaction time: 72 h.
Reaction time: 10 h
Reaction performed at 0°C.
Reaction time: 4 h
Reaction time: 24 h
Reaction time: 1 week.
More importantly, the construction of vicinal chiral tertiary and all-carbon quaternary centers was successfully achieved by this rearrangement protocol. With two different R1 and R2 group on the double bond of the enolphosphonates,[16] the substrate bearing tetrahydronaphthyl ring underwent the Claisen rearrangement to give the desired product 14 in 95% yield, >97:3 dr, and 87% ee.[17] Encouraged by this result, we also found that substrates bearing heteroatoms such as oxygen and sulfur were well tolerated under the reaction conditions, furnishing the product 15 and 16 in good yields and stereoselectivities respectively.[17] The absolute stereochemistry of the Claisen rearrangement products was unambiguously assigned on the basis of the X-ray structure of the carboxylic acid obtained from the hydrolysis of rearrangement product 17.[10]
A plausible transition-state model was then proposed as a well-defined tetrahedral substrate-catalyst complex in which bidentate chelation control result in excellent enantioface differentiation (Figure 2).[4k,13b,18] As depicted, the substrate tightly bound to the catalyst was assumed to arrange in a chair-like configuration that the enolphosphonate unit approaches the Si face of allylic ether moiety, which is opposite to the bulky phenyl ring on the BOX ligand.[19] This proposed transition-state model is fully consistent with the observed stereochemical outcome as well as reaction reactivity[20] and was further used for predicting the relative stereochemistry.
Figure 2.
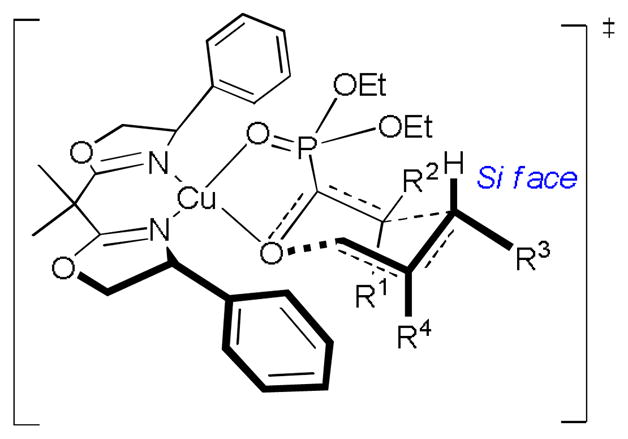
Proposed transition state
To highlight the synthetic utility of this methodology, the Claisen rearrangement product, α-ketophosphonate 1, was examined for further functionalization. Gratifyingly, we found that the phosphonate group could be easily transformed into various functional groups under mild reaction conditions (Scheme 1). First, the α-ketophosphonates 1 acted as a masked acyl chloride and could readily react with alcohols/amines to give the corresponding ester/amide products. Furthermore, these transformations could be carried out in a simple one-pot procedure. Additionally, under basic conditions, α-ketophosphonate 1 hydrolyzed to give the carboxylic acid product without any loss of enantioselectivity. The amino acid derivative can be accessed straightforwardly by treating 1 with L-valine methyl ester in refluxing 1,2-dichloroethane(DCE). Moreover, the carbonyl group of 1 could be functionalized by simple reduction. While DIBAL-H was utilized, the α-ketophosphonate 1 was converted to α-hydroxy phosphonates but with poor diastereoselectivity. When being treated with LiAlH4, 1 underwent complete reduction to give the alcohol product.
Scheme 1.

Synthetic applications of rearrangement product 1 DBU=1,8-Diazabicyclo[5.4.0]undec-7-ene, DIBAL-H= Diisobutylaluminium hydride, LAH=Lithium aluminium hydride
In summary, we have successfully developed the first Cu-catalyzed enantioselective Claisen rearrangement of enolphosphonates. A number of desired α-ketophosphonate products with diverse substitution patterns were obtained in excellent yields and stereoselectivities which easily undergo further functionalization to provide various chiral building blocks effectively. Significantly, this methodology provides a single-step rapid construction of chiral motifs with contiguous tertiary and all-carbon quaternary centers bearing unfunctionalized substituents, which are otherwise difficult to access. Since the starting materials as well as the catalysts are readily available and the highly enantioenriched products can be easily functionalized, this reaction is expected to be widely applicable for asymmetric synthesis. Future efforts will be directed at expanding the substrate scope and synthesis of complex natural products.
Supplementary Material
Acknowledgments
Support of this research was provided by the National Science Foundation (CHE-1049551). We thank Dr. Antoni Jurkiewicz for NMR, Dr. Ian M. Steele for X-ray analysis and Dr. Furong Sun (UIUC) for MS data. Dr David Dickson is acknowledged for his original work for this project.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author. CCDC 877511 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
Contributor Information
Jiajing Tan, Department of Chemistry, The University of Chicago, 5735 South Ellis Avenue, Chicago, IL, 60637 (USA).
Dr. Cheol-Hong Cheon, Department of Chemistry, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 136-701, (Korea)
Prof. Hisashi Yamamoto, Email: yamamoto@uchicago.edu, Department of Chemistry, The University of Chicago, 5735 South Ellis Avenue, Chicago, IL, 60637 (USA)
References
- 1.For reviews, see: Fuji K. Chem Rev. 1993;93:2037.Corey EJ, GuzmanPerez A. Angew Chem Int Ed. 1998;37:388.Christoffers J, Mann A. Angew Chem Int Ed. 2001;40:4591. doi: 10.1002/1521-3773(20011217)40:24<4591::aid-anie4591>3.0.co;2-v.Denissova I, Barriault L. Tetrahedron. 2003;59:10105.Ramon DJ, Yus M. Curr Org Chem. 2004;8:149.Christoffers J, Baro A. Adv Synth Catal. 2005;447:1473.Christoffers J, Bar A, editors. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Wiley-VCH; Weinheim, Germany: 2005. Trost BM, Jiang C. Synthesis. 2006:369.Cozzi PG, Hilgraf R, Zimmermann N. Eur J Org Chem. 2007:5969.Bella M, Gasperi T. Synthesis. 2009:1583.Kumagai N. Chem Pharm Bull. 2011;59:1. doi: 10.1248/cpb.59.1.Das JP, Marek I. Chem Commun. 2011;47:4593. doi: 10.1039/c0cc05222a.
- 2.For selected communications, see: Taylor MS, Jacobsen EN. J Am Chem Soc. 2003;125:11204. doi: 10.1021/ja037177o.Mase N, Thayumanavan R, Tanaka F, Barbas CF., III Org Lett. 2004;6:2527. doi: 10.1021/ol049196o.Austin JF, Kim SG, Sinz CJ, Xiao WJ, MacMillan DWC. Proc Natl Acad Sci USA. 2004;101:5482. doi: 10.1073/pnas.0308177101.Okino T, Hoashi Y, Furukawa T, Xu X, Takemoto Y. J Am Chem Soc. 2005;127:119. doi: 10.1021/ja044370p.Li H, Wang Y, Tang L, Wu F, Liu X, Guo C, Foxman BM, Deng L. Angew Chem Int Ed. 2005;44:105. doi: 10.1002/anie.200461923.Taylor MS, Zalatan DN, Lerchner AM, Jacobsen EN. J Am Chem Soc. 2005;127:1313. doi: 10.1021/ja044999s.Poulsen TB, Alemparte C, Saaby S, Bella M, Jørgensen KA. Angew Chem Int Ed. 2005;44:2896. doi: 10.1002/anie.200500144.Bartoli G, Bosco M, Carlone A, Cavalli A, Locatelli M, Mazzanti A, Ricci P, Sambri L, Melchiorre P. Angew Chem Int Ed. 2006;45:4966. doi: 10.1002/anie.200600370.Wu F, Li H, Hong R, Deng L. Angew Chem Int Ed. 2006;45:947. doi: 10.1002/anie.200502658.Lalonde MP, Chen Y, Jacobsen EN. Angew Chem, Int Ed. 2006;45:6366. doi: 10.1002/anie.200602221.Qin Y, Stivala CE, Zakarian A. Angew Chem Int Ed. 2007;46:7466. doi: 10.1002/anie.200702142.Wang Y, Li HM, Wang YQ, Liu Y, Foxman BM, Deng L. J Am Chem Soc. 2007;129:6364. doi: 10.1021/ja070859h.Chen ZH, Morimoto H, Matsunga S, Shibasaki M. J Am Chem Soc. 2008;129:2170. doi: 10.1021/ja710398q.Bencivenni G, Wu LY, Mazzanti A, Giannichi B, Pesciaioli F, Song MP, Bartoli G, Melchiorre P. Angew Chem Int Ed. 2009;48:7200. doi: 10.1002/anie.200903192.Streuff J, White DE, Virgil SC, Stoltz BM. Nat Chem. 2010;2:192. doi: 10.1038/nchem.518.Singh RP, Foxman BM, Deng L. J Am Chem Soc. 2010;132:9558. doi: 10.1021/ja103331t.Quintard A, Lefranc A, Alexakis A. Org Lett. 2011;13:1540. doi: 10.1021/ol200235j.Trost BM, Miller JR, Hoffman CM., Jr J Am Chem Soc. 2011;133:8165. doi: 10.1021/ja2029602.Fujiwara Y, Fu GC. J Am Chem Soc. 2011;133:12293. doi: 10.1021/ja2049012.Terada M, Moriya K, Kanomata K, Sorimachi K. Angew Chem Int Ed. 2011;50:12586. doi: 10.1002/anie.201105562.
- 3.Claisen L. Ber Dtsch Chem Ges. 1912;45:3157. [Google Scholar]
- 4.For reviews, see: Ito H, Taguchi T. Chem Soc Rev. 1999;28:43.Hiersemann M, Abraham L. Eur J Org Chem. 2002;9:1461.Chai Y, Hong SP, Lindsay HA, McFarland C, McIntosh MC. Tetrahedron. 2002;58:2905.Martin Castro AM. Chem Rev. 2004;104:2939. doi: 10.1021/cr020703u.Hiersemann M, Nubbemeyer U, editors. The Claisen Rearrangment. Wiley-VCH; Weinheim, Germany: 2007. Ilardi EA, Stivala CE, Zakarian A. Chem Soc Rev. 2009;38:3133. doi: 10.1039/b901177n.For selected communication, see: Yoon TP, MacMillan DWC. J Am Chem Soc. 2001;123:2911. doi: 10.1021/ja015612d.Helmboldt H, Hiersemann M. Tetrahedron. 2003;59:4031.Anderson CE, Overman LE. J Am Chem Soc. 2003;125:12412. doi: 10.1021/ja037086r.Afarinkia K, Twist AJ, Yu HW. J Org Chem. 2004;69:6500. doi: 10.1021/jo049133j.Abraham L, Koerner M, Schwab P, Hiersemann M. Adv Synth Catal. 2004;346:1281.Afarinkia K, Twist AJ, Yu HW. J Organomet Chem. 2005;690:2688.Linton EC, Kozlowski MC. J Am Chem Soc. 2008;130:16162. doi: 10.1021/ja807026z.Geherty ME, Dura RD, Nelson SG. J Am Chem Soc. 2010;132:11875. doi: 10.1021/ja1039314.Kaeobamrung J, Mahatthananchai J, Zheng P, Bode JW. J Am Chem Soc. 2010;132:8810. doi: 10.1021/ja103631u.
- 5.a) Abraham L, Czerwonka R, Hiersemann M. Angew Chem Int Ed. 2001;40:4700. doi: 10.1002/1521-3773(20011217)40:24<4700::aid-anie4700>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]; b) Abraham L, Koerner M, Hiersemann M. Tetrahedron Lett. 2004;45:3647. [Google Scholar]
- 6.a) Uyeda C, Jacobsen EN. J Am Chem Soc. 2008;130:9228. doi: 10.1021/ja803370x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Uyeda C, Rotheli AR, Jacobsen EN. Angew Chem Int Ed. 2010;49:9753. doi: 10.1002/anie.201005183. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Uyeda C, Jacobsen EN. J Am Chem Soc. 2011;133:5062. doi: 10.1021/ja110842s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Douglas CJ, Overman LE. Proc Natl Acad Sci USA. 2004;101:5363. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For representative references, see: Evans DA, Johnson JS. J Am Chem Soc. 1998;120:4895.Evans DA, Johnson JS, Burgey CS, Campos KR. Tetrahedron Lett. 1999;40:2879.Evans DA, Scheidt KA, Fandrick KR, Lam HW, Wu J. J Am Chem Soc. 2003;125:10780. doi: 10.1021/ja036985c.
- 9.a) Breuer E. The Chemistry of Organophosphorus Compounds. Vol. 4. John Wiley & Sons; New York: 1996. [Google Scholar]; b) Iorga B, Eymery F, Mouries V, Savignac P. Tetrahedron. 1998;4:14637. [Google Scholar]; c) McKenna CE, Kashemirov BA. Top Curr Chem. 2002;220:201. [Google Scholar]
- 10.For details, see Supporting Information.
- 11.a) Takenaka N, Xia G, Yamamoto H. J Am Chem Soc. 2004;126:13198. doi: 10.1021/ja045430u. [DOI] [PubMed] [Google Scholar]; b) Xia G, Yamamoto H. J Am Chem Soc. 2006;128:2554. doi: 10.1021/ja058454p. [DOI] [PubMed] [Google Scholar]; c) Xia G, Yamamoto H. J Am Chem Soc. 2007;129:496. doi: 10.1021/ja0679578. [DOI] [PubMed] [Google Scholar]; d) Takenaka N, Abell JP, Yamamoto H. J Am Chem Soc. 2007;129:742. doi: 10.1021/ja0668320. [DOI] [PubMed] [Google Scholar]; e) Abell JP, Yamamoto H. J Am Chem Soc. 2008;130:10521. doi: 10.1021/ja803859p. [DOI] [PubMed] [Google Scholar]; f) Abell JP, Yamamoto H. J Am Chem Soc. 2009;131:15118. doi: 10.1021/ja907268g. [DOI] [PubMed] [Google Scholar]; g) Usanov DL, Yamamoto H. Angew Chem Int Ed. 2010;49:8169. doi: 10.1002/anie.201002751. [DOI] [PubMed] [Google Scholar]; h) Usanov DL, Yamamoto H. J Am Chem Soc. 2011;133:1286. doi: 10.1021/ja1102822. [DOI] [PubMed] [Google Scholar]
- 12.For representative references, see: Yamamoto H, Maruoka K. Pure Appl Chem. 1990;62:2063.Nonoshita K, Banno H, Maruoka K, Yamamoto H. J Am Chem Soc. 1990;112:316.
- 13.For reviews, see: Evans DA, Chapman KT, Bisaha J. J Am Chem Soc. 1988;110:1238.Johnson JS, Evans DA. Acc Chem Res. 2000;33:325. doi: 10.1021/ar960062n.Desimoni G, Faita G, Quadrelli P. Chem Rev. 2003;103:3119. doi: 10.1021/cr020004h.Desimoni G, Faita G, Jørgensen KA. Chem Rev. 2006;106:3561. doi: 10.1021/cr0505324.Rasappan R, Laventine D, Reiser O. Coord Chem Rev. 2008;252:702.Hargaden GC, Guiry PJ. Chem Rev. 2009;109:2505. doi: 10.1021/cr800400z.
- 14.For selected communications, see: Evans DA, Miller SJ, Lectka T. J Am Chem Soc. 1993;115:6460.Evans DA, Johnson JS, Olhava EJ. J Am Chem Soc. 2000;122:1635.Evans DA, Rovis T, Kozlowski MC, Downey CW, Tedrow JS. J Am Chem Soc. 2000;122:9134.Evans DA, Scheidt KA, Johnston JN, Willis MC. J Am Chem Soc. 2001;123:4480. doi: 10.1021/ja010302g.Evans DA, Seidel D, Rueping M, Lam HW, Shaw JT, Downey CW. J Am Chem Soc. 2003;125:12692. doi: 10.1021/ja0373871.Palomo C, Oiarbide M, Kardak BG, García JM, Linden A. J Am Chem Soc. 2005;127:4154. doi: 10.1021/ja0423217.Magdziak D, Lalic G, Lee HM, Fortner KC, Aloise AD, Shair MD. J Am Chem Soc. 2005;127:7284. doi: 10.1021/ja051759j.Sibi MP, Stanley LM, Nie X, Venkatraman L, Liu M, Jasperse CP. J Am Chem Soc. 2007;129:395. doi: 10.1021/ja066425o.Sun Z, Yu S, Ding Z, Ma D. J Am Chem Soc. 2007;129:9300. doi: 10.1021/ja0734849.Sakakura A, Kondo R, Akakura M, Ishihara K. J Am Chem Soc. 2009;131:17762. doi: 10.1021/ja906098b.Xu X, Hu WH, Doyle MP. Angew Chem Int Ed. 2011;50:6392. doi: 10.1002/anie.201102405.
- 15.A detailed explanation of substrate scope as well as its limitation can be found in Supporting Information.
- 16.Due to the relative steric bulk of the phosphonate group, we were able to synthesize and isolate E-isomers of enolphosphonates.
- 17.The relative stereochemistry was determined by the proposed transition state as shown in Figure 2.
- 18.a) Johannsen M, Jørgensen KA. J Org Chem. 1995;60:5757. [Google Scholar]; b) Johannsen M, Yao S, Graven A, Jørgensen KA. Pure Appl Chem. 1998;70:1117. [Google Scholar]; c) Evans DA, Johnson JS, Burgey CS, Campos KR. Tetrahedron Lett. 1999;40:2879. [Google Scholar]
- 19.A possible boat-like transition state cannot be excluded.
- 20.Substrates bearing either cis-Cinnamyl or 3,3′-disubstituted allylic ether segment might have a severe 1,3-diaxial repulsion in the chair-like transition state, which possibly leads no reaction activity for Claisen rearrangement.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.