

Abstract
The development and application of chiral, non-racemic Re(V)-oxo complexes to the enantioselective reduction of prochiral ketones is described. In addition to the enantioselective reduction of prochiral ketones, we report the application of these complexes to (1) a tandem Meyer-Schuster rearrangement/reduction to access enantioenriched allylic alcohols and (2) the enantioselective reduction of imines.
Keywords: Enantioselective Reduction, Metal-Oxo, Hydrosilylation, Rhenium, Rearrangement
Introduction
Transition metal-oxo complexes are amongst the most important catalysts for oxidative transformations.[i] In contrast, we reported the use of (PPh3)2Re(O)2I as a catalyst in the hydrosilylation of aldehydes and ketones[ii] – an overall reduction of these functional groups. This methodology and those subsequently reported[iii] have highlighted the air-, moisture-, and reagent tolerant nature of these oxidized transition metal complexes. As a result, this class of complexes have become an attractive and practical alternative to many low oxidation state transition metal complexes that are more commonly employed as catalysts in reduction reactions.
A number of rhenium[iv] and molybdenum[v] complexes have been shown to be efficient catalysts for hydrosilylation reactions. Despite these recent reports, enantioselective metal-oxo catalyzed reductions are still rare.[vi] Having previously developed a catalytic system for the hydrosilylation of carbonyls using a rhenium(V)-dioxo complex to yield chiral, racemic silyl ethers, we focused our efforts on expanding this reactivity to enantioselective reductions.
The enantioselective reduction of prochiral ketones and imines by hydrogenation, hydroboration or hydrosilylation has been extensively studied.[vii] A number of different transition metals, including Zn,[viii] Fe,[ix] Ru,[x] Ir,[xi] Ti,[xii] Rh,[xiii] and Cu,[xiv] have successfully been employed in the enantioselective hydrosilylation of prochiral ketones. A similar array of transition metal complexes have been employed as catalysts for the enantioselective hydrosilylation of imines.[xv] Common to the majority of these catalysts is that they are derived from low valent transition metals. In this report, we describe to development and application of chiral, non-racemic Re(V)-oxo complexes to the enantioselective reduction of prochiral ketones and imines. This new class of hydrosilylation catalyst offers opportunities for the creation of novel transformations that take advantage of the unique reactivity of the metal-oxo functionality.[xvi] To this end, we also report the application of metal-oxo complexes to a tandem Meyer-Schuster rearrangement/reduction to access enantioenriched allylic alcohol.
Results and Discussion
Catalyst Design
Evaluation of the mechanistic of the hydrosilylation of carbonyl compounds catalyzed by (PPh3)2(O)2ReI revealed that an open coordination site must be available to allow for the complexation of the carbonyl.[xvii] Consistent with this hypothesis is the failure of Re-oxo complexes with bidentate phosphine ligands to affect the transformation. These neutral, bidentate phosphine ligands, which occupy all available dative (L-type) coordination sites on the metal center, are not conducive to the necessary ligand dissociation. This restriction was considered in the design of a chiral Re(V)-oxo complex for the enantioselective hydrosilylation of ketones. As a result, chiral monoanionic ligands were hypothesized to be suitable ligands for the asymmetric Re(V)-oxo catalyzed hydrosilylation (Scheme 1).
Scheme 1.
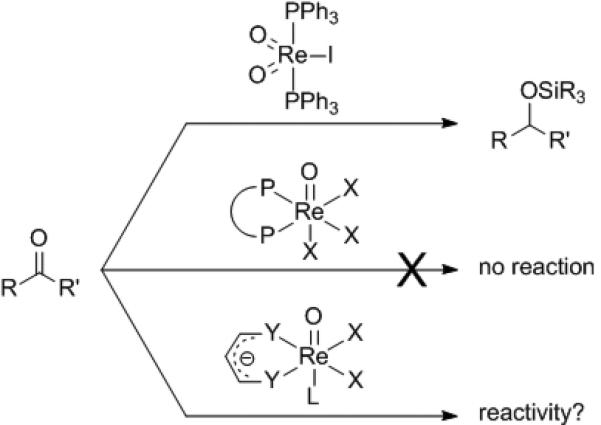
Catalyst Design
C2-symmetric bis(oxazoline) ligands are a privileged class of ligands that have been utilized in numerous transition metal catalyzed enantioselective transformations resulting in high levels of selectivity.[xviii] However, these ligands are most often coordinated to the metal centers as bidentate, neutral ligands.[xix] We envisioned that the addition of an electron withdrawing group on the bridging carbon of the bis(oxazoline) would increase the acidity of the ligand allowing for facile deprotonation and complexation as the corresponding anion ligand. Our investigations began with the commercially available cyanobis(oxazoline) ligand (4S)-(+)-phenyl-α-[(4S)-phenyloxazolidin-2-ylidene]-2-oxazoline-2-acetonitrile (1). Stirring 1 with Re(O)Cl3(OPPh3)(SMe2) (2) in CH2Cl2 at room temperature yielded (CNbox)Re(V)-oxo complex 3 as a emerald green solid (eq 1).
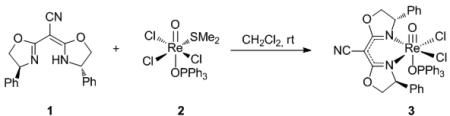 |
(1) |
Initial Optimization
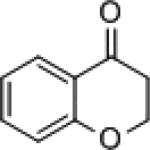
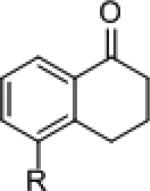
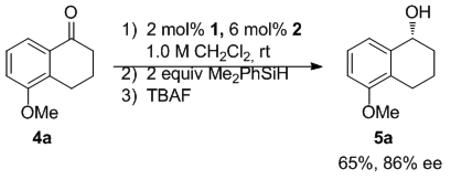
The ability of 3 to catalyze the asymmetric hydrosilylation of ketones was examined. At room temperature, 5-methoxytetralone (4) was reduced with 5 mol% 3 and 1.5 equivalents of Me2PhSiH in 0.5 M CH2Cl2. After TBAF deprotection of the silyl ether, the corresponding alcohol was isolated in 90% yield with 88% enantiomeric excess[xx] (Table 1, entry 1).[xxi] In aromatic solvents, the reaction proved sluggish with only 36% yield of 5a after 48 hours (entry 2). Good reactivity was observed in ethyl acetate; however, the excellent selectivity observed with the reduction of 4a (93% ee entry 3) did not extend to acyclic ketone 4b (40% ee, entry 6). A higher level of enantiomeric enrichment was obtained when acetophenone was reduced in CH2Cl2 (57% yield and 70% ee, entry 5). The neat reduction of 4b yielded the corresponding alcohol in excellent yield, but with low enantioselectivity (22% ee, entry 8). With improved yield compared to dichloromethane, 1,4-dioxane was identified as the optimal solvent for the reduction of both substrates furnishing the resulting alcohols in high enantiomeric excess (entries 4 and 7).
Table 1.
Solvent Studies of Ketone Reductions

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Solvent | Yield (%) | ee (%) | ||
| 1 | -(CH2)3- | OMe | 4a | CH2Cl2 | 90 | 5a | 88 | |
| 2 | -(CH2)3- | OMe | 4a | benzene | 36 | 5a | 86 | |
| 3 | -(CH2)3- | OMe | 4a | EtOAc | 71 | 5a | 93 | |
| 4 | -(CH2)3- | OMe | 4a | dioxane | 95 | 5a | 89 | |
| 5 | Me | H | H | 4b | CH2Cl2 | 57 | 5b | 70 |
| 6 | Me | H | H | 4b | EtOAc | 79 | 5b | 40 |
| 7 | Me | H | H | 4b | dioxane | 90 | 5b | 77 |
| 8 | Me | H | H | 4b | neat | 97 | 5b | 22 |
A number of mono- and polyhydridic silanes were examined in the enantioselective hydrosilylation of 5-methoxytetralone (Table 1). High enantiomeric excess was also attained with Et3SiH (60% yield and 85% ee, entry 2); however, a significant rate reduction was observed relative to the reaction with Me2PhSiH (95% yield and 89% ee, entry 1). Attenuation of reactivity was more pronounced with Ph2MeSiH (23% yield and 82% ee, entry 3). The use of dihydride Ph2SiH2 led to formation of 5a albeit in lower yield and enantiomeric enrichment (43% yield and 72% ee, entry 4). None of the desired product was observed when bulky silanes (iPr3SiH and t-BuMe3SiH), PMHS or the polyhydridic phenyl silane were incorporated as reducing agents. Me2PhSiH was found to be the optimal stoichiometric reducing agent and, based on these results, was utilized during further reaction optimization.
The effect of temperature was probed in the reduction of 4a with Me2PhSiH catalyzed by 3. At 0 °C, no catalytic activity was observed (Table 2, entry 7). An erosion of enantioselectivity as well as reactivity was observed when the reaction was heated to 60 °C (entry 8). At this elevated temperature, a substantial amount of disilane and disiloxane through decomposition silane was observed.[xxii]
Table 2.
Effect of Silane on Ketone Reductions

| ||||
|---|---|---|---|---|
| Entry | Silane | Temperature | Yield (%) | ee (%) |
| 1 | Me2PhSiH | rt | 95 | 89 |
| 2 | Et3SiH | rt | 60 | 85 |
| 3 | Ph2MeSiH | rt | 23 | 82 |
| 4 | Ph2SiH2 | rt | 43 | 72 |
| 5 | iPr3SiH | rt | nr | n/a |
| 6 | t-BuMe2SiH | rt | nr | n/a |
| 7 | Me2PhSiH | 0 °C | nr | n/a |
| 8 | Me2PhSiH | 60 °C | 71 | 76 |
Ligand Optimization
In an effort to increase the enantioselectivity of the reaction, the effect of changes to steric and electronic composition of the cyanobis(oxazoline) ligand were investigated using the optimized reaction conditions (Me2PhSiH and 3 in dioxane at room temperature). To do so, a number of differentially substituted bis(oxazoline) compounds were synthesized. The synthesis of these compounds was accomplished in two steps from the corresponding chiral, non-racemic amino alcohol (Scheme 2). The amino alcohols were (1) obtained through the reduction of the corresponding amino acid or (2) synthesized by the methods developed by Sharpless[xxiii] or Ellman.[xxiv] The amino alcohols were condensed with diethyl malonimidate dihydrochloride to produce the corresponding bis(oxazoline). Following protocol developed by Corey and Wang, the bis(oxazoline) was deprotonated with nBuLi and treated with p-toluenesulfonyl cyanide to yield the aryl and alkyl substituted cyanonbis(oxazolines) (Scheme 2).[xxv]
Scheme 2.
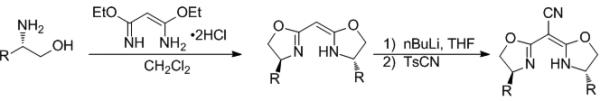
Cyanobis(oxazoline) Synthesis
Complexation of the cyanobis(oxazolines) 6a-g was accomplished through dropwise addition[xxvi] of the ligand as a concentrated solution in CH2Cl2 to a dilute suspension of 2 in CH2Cl2 (eq 2). The reaction underwent a gradual color change to become a clear, emerald green solution. Upon trituration with Et2O or hexanes, the corresponding CN-box Re(V)-oxo complexes 7a-g were isolated as green solids. While formation of complexes 7a-g proceeded readily, complexation of cyanobis(oxazolines) 6h-k to the metal center was unsuccessful. Under the above described conditions, these complexation reactions resulted in the formation of unidentifiable black solids that were isolated and found to be catalytically inactive.[xxvii]
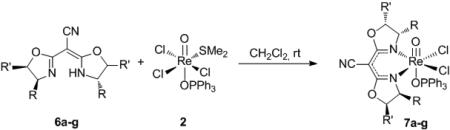 |
(2) |
Cyanobis(oxazoline) ligands of varying electronic and steric composition (6a-g) were readily synthesized from the corresponding amino alcohols and complexed to the metal center, as described above, to yield (CNbox) Re(V)-oxo complexes (7a-g). These complexes were examined in the reduction of 5-methoxytetralone and acetophenone. Moderate enantiomeric excesses were obtained in the hydrosilylation reactions catalyzed by rhenium complexes incorporating cyanobis(oxazoline) ligands with alkyl (53% ee, Table 3, entry 1) and benzyl (53% ee, entry 2), and indenyl (30% ee, entry 3) substitution. Tetraphenyl substituted ligand 6d led to slightly improved levels of enantioenrichment of 5a (67% ee, entry 3) and good selectivity in the reduction of 4b (73%, entry 8). A substantial increase in selectivity was achieved with 2- napthyl cyanobis(oxazoline) Re-oxo complexes 7e (82% and 96% ee, entries 5 and 9). However, markedly improved reactivity was observed with the phenyl and 4-tert-butylphenyl catalysts with consistently high level of enantioselectivity (77-94% ee, entries 6, 7, 10 and 11).
Table 3.
Ligand Effects on Ketone Reductions

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Ligand | Catalyst | Yield (%) | ee (%) | |
| 1 | -(CH2)3- | OMe | 6a | 7a | 23 | 5a | 53 | |
| 2 | -(CH2)3- | OMe | 6b | 7b | 88 | 5a | 53 | |
| 3 | -(CH2)3- | OMe | 6c | 7c | 71 | 5a | 30 | |
| 4 | -(CH2)3- | OMe | 6d | 7d | 26 | 5a | 67 | |
| 5 | -(CH2)3- | OMe | 6e | 7e | 83 | 5a | 96 | |
| 6 | -(CH2)3- | OMe | 1 | 3 | 95 | 5a | 89 | |
| 7 | -(CH2)3- | OMe | 6f | 7f | 97 | 5a | 94 | |
| 8 | Me | H | H | 6d | 7d | 35 | 5b | 73 |
| 9 | Me | H | H | 6e | 7e | 68 | 5b | 82 |
| 10 | Me | H | H | 1 | 3 | 90 | 5b | 77 |
| 11 | Me | H | H | 6f | 7f | 87 | 5b | 86 |
| 12 | Me | H | H | 6g | 7g | 60 | 5b | 71 |
Scope of Enantioselective Reduction of Ketones
The scope of the hydrosilylation was probed using two equivalents of Me2PhSiH, in dioxane, and 3 mol% of catalyst 3 or 7f. Good to excellent enantioselectivities were obtained in the reduction of aromatic ketones. Acetophenone was reduced with good selectivity (Table 4, entry 1). Good to excellent enantiomeric excess was obtained in the reducation of five-, six-, and seven-membered cyclic ketones (83-94% ee) (entries 2-7). Heteroaromatic ketones were also well tolerated under the reaction conditions; for example, alcohol 9g was obtained in 93% ee (entry 9). In most cases, the 4-tert-butylphenyl catalyst 7f produced greater enantioselectivities than phenyl catalyst 3, although, in some cases, the observed enhancement was not dramatic and the effect can even be reversed (entry 9). The reduction of non-aryl ketones proceeded in good to excellent yield, however, with modest enantioenrichment of the resultant alcohols (6-15% ee, entries 11 and 12).
Table 4.
Asymmetric Reduction of Aryl Ketones

| ||||||
|---|---|---|---|---|---|---|
| Entry | Ketone | Cat | Yield (%) | ee (%) | ||
| 1 |
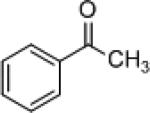
|
4b | (S)-3 (R)-7f |
90 92 |
5b | 78 (R) 86 (S) |
| 2 3 |
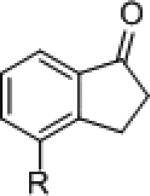
|
8a = H
8b = OTs |
(S)-3 (R)-7f (S)-3 |
59 89 70 |
9a
9b |
88 (R) 86 (S) 83 (R) |
| 4 |
|
8c | (S)-3 (R)-7f |
69 61 |
9c | 87 (R) 86 (S) |
| 5 6 |
|
4a R = OMe
8d = H |
(S)-3 (R)-7f (S)-3 |
71 97 84 |
5a
9d |
93(R) 94(S) 89 (R) |
| 7 |
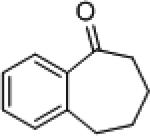
|
8e | (S)-3 (R)-7f |
70 82 |
9e | 75 (R) 88 (S) |
| 8 |
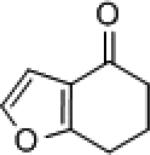
|
8f | (S)-3 (R)-7f |
83 96 |
9f | 78 (R) 81 (S) |
| 9 |
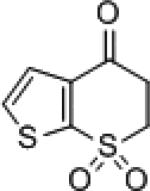
|
8g | (S)-3 (R)-7f |
51 64 |
9g | 93 (R) 82 (S) |
| 10 |
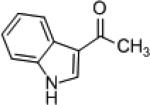
|
8h | (R)-7f | 80 | 9h | 84 (S) |
| 11 |
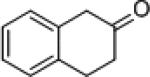
|
8i | (S)-3 | 94 | 9i | 15 (R) |
| 12 |
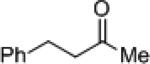
|
8j | (R)-7f | 67 | 9j | 6 (S) |
A convenient, alternate protocol for the hydrosilylation entails the in situ generation of the chiral catalyst. This was achieved by pre-stirring 2 mol% of 2 and 6 mol% of the desired cyanobis(oxazoline) ligand in CH2Cl2 for two hours prior to addition of the ketone and silane. Under these conditions, 4a was hydrosilylated and deprotected yielding the corresponding alcohol 5a with identical enantioselectivity to that observed with the pre-formed catalyst 3 (eq 3).
 |
(3) |
The Re(V)-oxo catalyzed enantioselective hydrosilylation of ketones was extended to the synthesis of chiral allylic alcohols. Modest enantioselectivities, 45-55% ee, were observed in the hydrosilylation of α,β-unsaturated conjugated ketone 10a (Table 5, entry 1). Higher enantiomeric excesses, 55-63% ee, were obtained with α-substituted enone 10b (entry 2). It should be noted that 1,4-reduction of the enones was not observed under these conditions.
Table 5.
Asymmetric Reduction of Enones

| ||||||
|---|---|---|---|---|---|---|
| Entry | Ketone | Catalyst | Yield (%) | ee(%) | ||
| 1 |
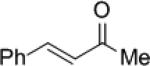
|
10a | (R)-7f | 90 | 11a | 55 (S) |
| 2 |
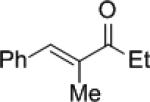
|
10b | (S)-3 (R)-7f |
87 47 |
11b | 63 (R) 55 (S) |
| 3 |
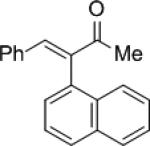
|
10c | (R)-7f | 71 | 11c | 95 (S) |
Tandem 3,3-Rearrangement-Asymmetric Reduction
In the course of examining Re(V)-oxo complexes as catalysts in the etherification of propargyl alcohols,[16e] rearrangement of the alcohol to enone, the Meyer-Schuster rearrangement, was observed to be the prevailing reactivity in non-polar and aromatic solvents.[xxviii] It was envisioned that a Re(V)-oxo catalyzed Meyer-Schuster rearrangement could be coupled with the asymmetric reductions in a one-pot synthesis of chiral allylic alcohols from racemic propargyl alcohols (Scheme 4).
Scheme 4.
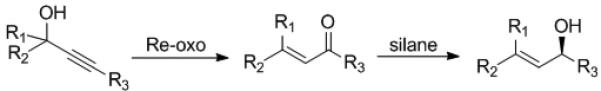
Tandem Meyer-Schuster Rearrangement-Hydrosilylation
Chiral complexes 3 and 7f were tested for there ability to facilitate the Meyer–Schuster rearrangement. Propargyl alcohol 12 was treated with CN-box Re complexes 3 and 7f in toluene at 80 °C.[xxix] The reaction resulted in the formation of cis and trans isomers of the desired enone (13 and 14) as well as a dimerized byproduct (15) (eq 4).
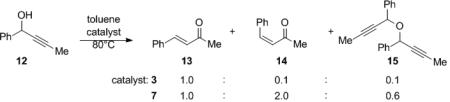 |
(4) |
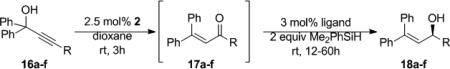
Having observed that the chiral catalyst could be generated in situ (vide supra), we next examined Re-oxo complex 2, the chiral catalyst precursor, as a catalyst for the Meyer-Schuster reaction.[xxx] Gratifyingly, 2 facilitated the rearrangement of the 16a to the corresponding enone (17a). Without isolation of the enone, ligand 1 was added to the reaction solution followed by 2 equivalents of Me2PhSiH. Allyl alcohol 18a was isolated in 43% yield and 49% ee (Table 6, entry 1). After further optimization, it was found that in situ generation of chiral catalyst 7g provided allyl alcohols 18a-f in the highest levels of enantiomeric excess from the one-pot Meyer-Schuster rearrangement–reducion of propargyl alcohols 16b-f.
Table 6.
Tandem Meyer-Schuster Rearrangement-Hydrosilylation
| ||||||
|---|---|---|---|---|---|---|
| Entry | R | Ligand | Yield (%) | ee (%) | ||
| 1 | Ph | 16a | 1 | 43 | 18a | 49 |
| 2 | Ph | 16a | 6g | 38 | 18a | 82 |
| 3 | 2-Nap | 16b | 6g | 42 | 18b | 92 |
| 4 | 2-Cl-Ph | 16c | 6g | 85 | 18c | 64 |
| 5 | 4-Cl-Ph | 16d | 6g | 25 | 18d | 50 |
| 6 | t-Bu | 16e | 6g | 42 | 18e | 63 |
| 7 | n-Bu | 16f | 6g | 73 | 16f | 65 |
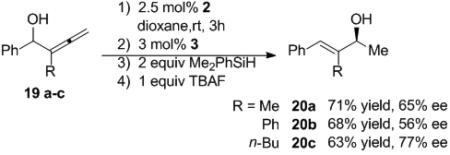
To access α,β-unsaturated enones, the rearrangement of allenyl alcohols was examined. Allenyl alcohols 19a-c were subjected to the tandem reaction conditions (eq 5). Using commercially available ligand 1, the one-pot reactions were performed using Me2PhSiH as the stoichiometric reducing agents. The allylic alcohols 20a-c were obtained in good yields (63-71%) and moderate to good enantiomeric excess (56-77%). As shown above, the ability to generate the chiral catalyst in situ lends provides a highly tunable system for the enantioselective synthesis of allylic alcohols from racemic starting materials.
 |
(5) |
Asymmetric Reduction of Imines
With a system in hand for the asymmetric reduction of ketones, we next turned our attention toward the reduction of imines.vi While efficient systems have been developed, they generally utilize metals in low oxidation states. Representative systems include those catalyzed by low oxidation state late metal complexes[11,15] and titanocene complexes.[15f, xxxi] In order to demonstrate the utility of this air and moisture tolerant catalyst for the synthesis of chiral amines via reduction of the corresponding imine, an appropriate protecting group needed to be selected. Phosphinyl imines have been shown to be stable under ambient conditions and proved suitable for the reaction conditions. The phosphinyl group can easily be cleaved under acidic conditions to yield the chiral amine.[xxxii]
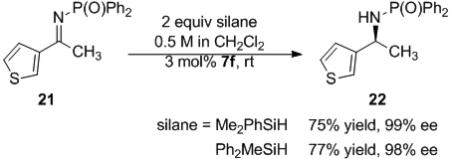
The reduction of methyl thiophene imine 21 in CH2Cl2 with Me2PhSiH proceeded with excellent yield and enantioselectivity at room temperature (eq 6).[xxxiii] The reaction proceeded with the same selectivity in a range of solvents including THF, EtOAc, and toluene. The substitution of Me2PhSiH with Ph2MeSiH did not alter the enantioselectivity; however, Me2PhSiH generally resulted in higher yield of the corresponding phosphinyl amine. No reactivity was observed with Me2t-BuSiH or iPr3SiH. Mild heating of the reaction showed no measurable decrease in enantioselectivity but did result in slightly higher yields (Me2PhSiH at 40° C = 81% yield, 98% ee).
 |
(6) |
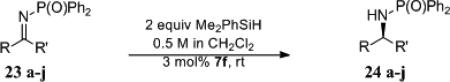
The reaction successfully led to the reduction of aryl phosphinyl imines. Acyclic ketimines (Table 7, entries 1-7) reduced good yield with excellent enantioselectivity. Similar selectivity was observed with cyclic ketimines (entries 8-10). Notably, imine 23j exists primarily as the enamine and the reduction proceeded in 71% yield with 96% ee without any additional reaction time or heating. Additionally, heteraromatic compounds performed well under the reaction conditions and were reduced in very good yield with excellent selectivity (entries 6 and 7). Interestingly, N-methylpyrrole methyl ketimine was unreactive when subject to the reduction conditions. Aliphatic ketimines did not display the same selectivity as the aryl imines. Cyclohexyl methyl imine 23k and isopropyl ethyl imine 23l were reduced with 32% and 17% ee respectively (table 7, entries 11 and 12). The bulky tert-butyl methyl imine was unreactive (entry 13).
Table 7.
Reduction of Aryl and Alkyl Phosphinyl Imines
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Imine | Yield (%) | ee (%) | ||||
| 1 2 3 4 5 |
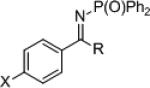
|
23a
23b 23c 23d 23e |
X = H OMe CF3 I H |
R = Me Me Me Me Bu |
51 61 78 71 68 |
24a
24b 24c 24d 24e |
>99 98 98 99 >99 |
| 6 |
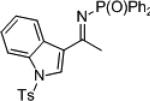
|
23f | 81 | 24f | > 99 | ||
| 7 |
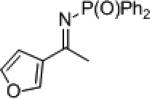
|
23g | 76 | 24g | 99 | ||
| 8 9 10 |
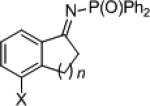
|
23h
23i 23j |
n = 1 2 3 |
X = H OMe H |
89 69 71 |
24h
24i 24j |
95 95 96 |
| 11 12 13 |
|
23k
23l 23m |
R = Me Et We |
R’ = Cy i-Pr t-Pr |
69 73 nr |
24k
24l 24m |
32 17 n/a |
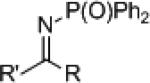
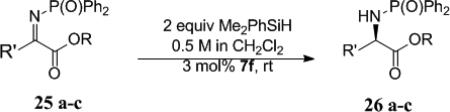
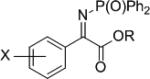
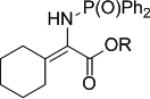
The synthesis of phenyl glycine derivatives was achieved through the reduction of α-imino esters by 7f. The reduction of these imines proceeded in moderate to good yield with excellent enantioselectivity (Table 8, entries 1-3). Non-aromatic α-imino esters bearing α-protons existed as the α-amino acrylate and were unreactive (entries 4 and 5). Similarly, β-imino ester 25f did not yield the desired amine 26f (entry 6).
Table 8.
Reduction of α-Imino Esters
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Imine | Yield (%) | ee (%) | ||||
| 1 2 3 |
|
25a
25b 25c |
X = Ph 4-OMePh 2-MePh |
R = Et Me Me |
83 47 69 |
26a
26b 26c |
>99 95 99 |
| 4 |
|
25d | R = Me | nr | 26d | n/a | |
| 5 |
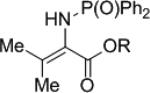
|
25e | R = Me | nr | 26e | n/a | |
| 6 |
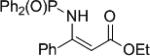
|
25f | nr | 26f | n/a | ||
Synthesis of chiral allylic amines was achieved through chemo- and enantioselective reduction of the corresponding imine. Conjugated aromatic imines were reduced in good yield with excellent selectivity (Table 9, entries 1-2). Unconjugated vinyl imines were reduced with good enantioselectivity in moderate yield. For this substrate, extension of the reaction time led to reduction of the olefin to yield the alkyl amine.
Table 9.
Synthesis of Allylic Amines
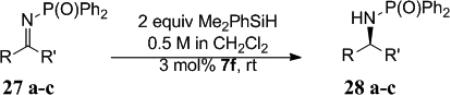
| |||||
|---|---|---|---|---|---|
| Entry | Imine | Yield (%) | ee (%) | ||
| 1 |
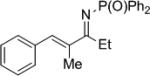
|
27a | 71 | 28a | >99 |
| 2 |
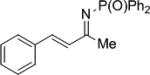
|
27b | 62 | 28b | 75 |
| 3 |
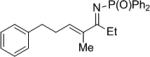
|
27c | 48 | 28c | 83 |
Conclusion
In summary, a series of chiral, non-racemic (CN-box)Re(V)-oxo complexes have been prepared and employed as versatile and efficient catalysts for the hydrosilylation of ketones and imines. These reductions proceed under an ambient air atmosphere without the need for exclusion of advantageous water. The mild reaction is highly functional group tolerant and has been extended to include the formation phenyl glycine derivative, allylic alcohol and allylic amines. The (CNbox)Re(V)-oxo complexes can be pre-formed and isolated as benchtop stable solids or generated in situ from a single precursor. The utility of in situ generation of the chiral catalyst and the unique reactivity of metal-oxo complexes was highlighted in tandem Meyer-Schuster–hydrosilylation reactions providing access to enantioenriched allylic alcohols from racemic propargyl and allenyl alcohols.
Experimental Section
General Procedure for Synthesis of Re(V)-Oxo Complexes 3 and 7a-g
Re(V)dimethylsulfide complex 2 (1 equiv) was added to a round-bottom flask charged with a magnetic stir bar. CH2Cl2 was added to a reaction flask to a concentration of 5 mM. In a scintillation vial, 1.2 equiv of cyanobis(oxazoline) ligand 6f was diluted in CH2Cl2 (5 mM). Ligand solution was added slowly to Re suspension. Reaction solution quickly changes from light green to very dark green. Reaction stirred at rt for ~5 h at which timethe solution is a bright emerald green. (Note: This reaction time can be reduced to 30 minutes by adding 1 drop of DMSO; however, isolation of the product becomes more difficult.) Solvent was evaporated and the dark green film was diluted with a small amount of CH2Cl2 then triturated with Et2O. Filtration with a Buchner funnel yields Re complex 7f, a bright green solid. The trituration was repeated 4 times or until no significant material was isolated. The resultant solid was driedin vacuo.
General Procedure for Asymmetric Reduction of Ketones with Isolated Asymmetric Catalyst
To a 7.4 mL Fischer vial charged with a solution of ketone (50 mg, 1 equiv) in dioxane (1 M), was added silane (2 equiv) followed by catalyst3 or 7f (3 mol%). The reaction was monitored by TLC. Upon completion or 72 hours, the reaction was quenched with 1 equiv of tetrabutylammonium fluoride (1.0 M in THF). The reaction mixture was loaded directly on to a silica gel column and chromatographed with the 10-20% ethyl acetate in hexanes (9g: 50% ethyl acetate in hexanes) to give the alcohol. NMR and HPLC analyses of the compounds listed below were consistent with previously reported values.[xxxiv] Absolute configuration of 5b, 9a, and 9d were assigned by comparison of the optical rotation and HPLC retention times to literature values.[xxxv]
General Procedure for Tandem Meyer-Schuster-Asymmetric Reduction Reaction
Re-DMS complex 2 (2.5 mol%) was added to scintillation vial charged with 1 equivalent of propargyl alcohol in 0.5 M dioxane. The reaction solution is stirred at room temperature for 3 hours. Upon complextion, 3 mol% of cyanobis(oxazoline) ligand was added and the reaction solution was stirred at room temperature for 5 hours resulting in a clear solution then 2 equivalents of Me2PhSiH were added. The reaction solution was stirred at room temperature and monitored by TLC. After 60 hours or reaction completion, the silyl ether was deprotected with 1 equiv of tetrabutylammonium fluoride (1.0 M in THF). Direct purification reaction mixture by column chromatography on a silica gel column with the 10-20% ethyl acetate in hexanes gave the allylic alcohol. The enantiomeric excess was determined by chiral HPLC.
General Procedure for Asymmetric Reduction of Phosphinyl Imines with Asymmetric Catalyst
To a 7.4 mL Fischer vial charged with a solution of imine (50 mg, 1 equiv) in CH2Cl2 (1 M), was added Me2PhSiH (2 equiv) followed by catalyst 7f (3 mol%). The reaction was monitored by TLC. Upon completion or 72 hours, the reaction was chromatographed. The reaction mixture was loaded directly on to a silica gel column and chromatographed with the 10-50% acetone in CH2Cl2 to give the amine. Absolute configurations were assigned by comparison of the HPLC retention times to literature values.[33]
Scheme 3.
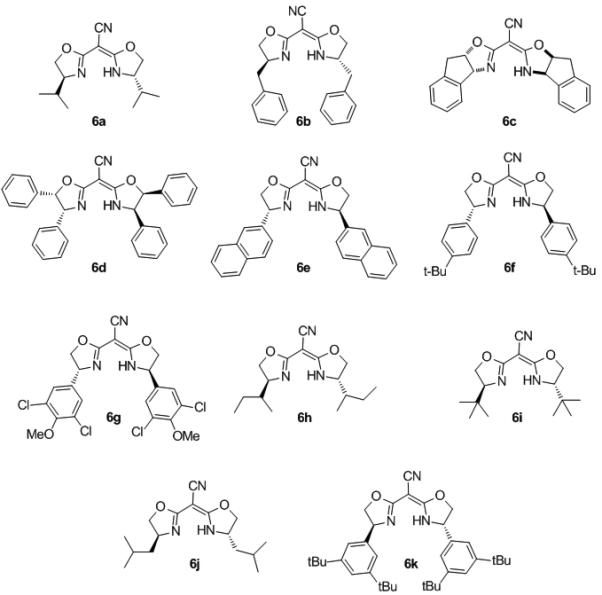
Cyanobis(oxazoline) Ligands
Acknowledgements
We gratefully acknowledge NIHGMS (GM074774), the Camille and Henry Dreyfus Foundation and Research Corporation (Research Innovation Award) for financial support. K.A.N. thanks Novartis for a graduate fellowship and Y.K. is grateful for a research fellowship from JSPS for young scientists.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- i.Hua R, Jiang J-L. Curr. Org. Synth. 2007;4:151–174. [Google Scholar]
- ii.Kennedy-Smith JJ, Nolin KA, Gunterman HP, Toste FD. J. Am. Chem. Soc. 2003;125:4056–4057. doi: 10.1021/ja029498q. [DOI] [PubMed] [Google Scholar]
- iii.Roy AK. Adv. Organomet. Chem. 2008;55:1–59. For a review see: Du G, Abu-Omar MM. Curr. Org. Chem. 2008;12:1185–1198.Sanz R, Pedrosa MR. Curr. Org. Syn. 2009;6:239–263.
- iv.For examples of reduction of various functional groups, with silanes, catalyzed by rhenium-oxo complexes. Aldehyde/Ketone: Reis PM, Royo B. Catal. Commun. 2007;8:1057–1059.Du GD, Abu-Omar MM. Organometallics. 2006;25:4920–4923.Ison EA, Cessarich JE, Du GD, Fanwich PE, Abu-Omar MM. Inorg. Chem. 2006;45:2385–2387. doi: 10.1021/ic0520768. Sulfoxide: Sousa SCA, Fernandes, A. C. AC. Tetrahedron Lett. 2009;50:6872–6876. Nitro: de Noronha RG, Romao CC, Fernandes AC. J. Org. Chem. 2009;74:6960–6964. doi: 10.1021/jo9008657.
- v.For examples of reduction with molybdenum-oxo complexes, see: Aldehyde/Ketone: Ziegler JE, Du G, Fanwick PE, Abu-Omar MM. Inorg. Chem. 2009;48:11290–11296. doi: 10.1021/ic901794h.Reis PM, Romao CC, Royo B. Dalton Trans. 2006:1842–1846. doi: 10.1039/b514930d.Fernandes AC, Fernandes R, Romao CC, Royo B. Chem. Commun. 2005:213–214. doi: 10.1039/b414145h. Imines: Fernandes AC, Romao CC. Tetrahedron Lett. 2005;46:8881–8883. Amides: Fernandes AC, Romao CC. J. Mol. Catal. A. 2007;272:60–63. Esters: Fernandes AC, Romao CC. J. Mol. Catal. A. 2006;253:96–98.
- vi.a Nolin KA, Ahn RW, Toste FD. J. Am. Chem Soc. 2005;127:12462–12463. doi: 10.1021/ja050831a. [DOI] [PubMed] [Google Scholar]; b Du G, Abu-Omar MM. Inorg. Chim. Acta. 2008;361:3184–3192. [Google Scholar]
- vii.a Nishiyama H, Itoh K. In: Catalytic Asymmetric Synthesis. Ojima I, editor. John Wiley & Sons; New York: 2000. [Google Scholar]; b Ohkuma T, Kitamura M, Noyori, R R. In: Catalytic Asymmetric Synthesis 2nd Edition. Ojima I, editor. Wiley-VCH; New York: 2000. [Google Scholar]; c Ohkuma T, Noyori R. In: Comprehensive Asymmetric Catalysis. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 1. Springer; 1999. p. 199. [Google Scholar]; d Riant O, Mostefai N, Courmarcel, J J. Synthesis. 2004:2943–2958. [Google Scholar]; e Diez-Gonzalez S, Nolan SP. Org. Prep. Proced. Int. 2007;39:523. [Google Scholar]; f Arena CG. Mini-Rev. Org. Chem. 2009;6:159. [Google Scholar]
- viii.a Mastranzo VM, Quintero L, Anaya de Parrodi C, Juaristi E, Walsh PJ. Tetrahedron. 2004;60:1781–1789. [Google Scholar]; b Bette V, Mortreux A, Savoia D, Carpentier J-F. Adv. Synth. Catal. 2005;347:289–302. [Google Scholar]; c Mimoun H, de Saint Laumer JY, Giannini L, Scopelliti R, Floriani C. J. Am. Chem. Soc. 1999;121:6158–6166. [Google Scholar]
- ix.a Nishiyama H, Furuta A; Chem. Commun. 2007:760–762. doi: 10.1039/b617388h. [DOI] [PubMed] [Google Scholar]; b Shaikh NR, Enthaler S, Junge K, Beller M. Angew. Chem. Int. Ed. 2008;47:2497–2501. doi: 10.1002/anie.200705624. [DOI] [PubMed] [Google Scholar]; c Inagaki T, Phong LT, Furuta A, Ito J, Nishiyama, H H. Chem–Eur. J. 2010;16:3090–3096. doi: 10.1002/chem.200903118. [DOI] [PubMed] [Google Scholar]
- x.Nishibayashi Y, Takei L, Uemura S, Hidai M. Organometallics. 1998;17:3420–3422. [Google Scholar]
- xi.Takei I, Nishibayashi Y, Arikawa Y, Uemura S, Hidai M. Organometallics. 1999;18:2271–2274. [Google Scholar]
- xii.Carter MB, Schlott B, Gutierrez A, Buchwald SL. J. Am. Chem. Soc. 1994;116:11667–11670. [Google Scholar]
- xiii.a Carpentier J-F, Bette V. Curr. Org. Chem. 2002;6:913–936. [Google Scholar]; b Corriu RJ, Poulin JC, Dang TP, Kagan HB; J. Chem. Soc., Chem. Commun. 1973:38. [Google Scholar]; c Dumont W, Poulin JC, Dang TP, Kagan HB. J. Am. Chem. Soc. 1973;95:8295. [Google Scholar]; d Ojima I, Nihonyanagi M, Nagai Y. J. Chem. Soc., Chem. Commun. 1972:938. [Google Scholar]; e Ojima I, Kogure T, Nihonyanagi M, Nagai Y. Bull. Chem. Soc. Jpn. 1972;45:3506. [Google Scholar]; f Ojima I, Kogure T, Nihonyanagi M, Nagai Y. Bull. Chem. Soc. Jpn. 1972;45:3722. [Google Scholar]; g Tao B, Fu GC. Angew. Chem. Int. Ed. 2002;41:3892–3894. doi: 10.1002/1521-3773(20021018)41:20<3892::AID-ANIE3892>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]; h Nishibayahi Y, Segawa K, Ohe K, Uemura S. Organometallics. 1995;14:5486–5487. [Google Scholar]
- xiv.Lipshutz BH, Noson K, Chrisman W, Lower A. J. Am. Chem. Soc. 2003;125:8779–8789. doi: 10.1021/ja021391f. [DOI] [PubMed] [Google Scholar]
- xv.Riant O. In: Modern Reduction Methods. Andersson PG, Munslow IJ, editors. Wiley-VCH: Weinheim; Germany: 2008. pp. 321–337. Rh: Becker R, Brunner H, Mahboobi S, Wiegrebe W. Angew. Chem. Int. Ed. 1985;97:969.Langlois N, Dang TP, Kagan HB. Tetrahedron Lett. 1973:4865. Ir: Xiao D, Zhang X. Angew. Chem. Int. Ed. 2001;40:3425. doi: 10.1002/1521-3773(20010917)40:18<3425::aid-anie3425>3.0.co;2-o. Ru: Nishibayshi Y, Takei I, Uemura S, Hidai M. Organometallics. 1998;17:3420. Ti Verdaguer X, Lange UEW, Reding MT, Buchwald SL. J. Am. Chem. Soc. 1996;118:6784–6785. Cu Lipshutz BH, Shimizu, H H. Angew. Chem. Int. Ed. 2004;43:2228–2230. doi: 10.1002/anie.200353294. Zn Park B-M, Mun S, Yun J. Adv. Syn. Catal. 2006;348:1029.
- xvi.For examples non-oxidative transformation catalyzed by rhenium-oxo complexes, developed in our group see, Ohri RV, Radosevich AT, Hrovat KJ, Musich C, Huang D, Holman TR, Toste FD. Org. Lett. 2005;7:2501. doi: 10.1021/ol050897a.Kennedy-Smith JJ, Young LA, Toste FD. Org. Lett. 2004;6:1325. doi: 10.1021/ol049649p.Sherry BD, Loy RN, Toste FD. J. Am. Chem. Soc. 2004;126:4510. doi: 10.1021/ja031895t.Luzung MR, Toste FD. J. Am. Chem. Soc. 2003;125:15760. doi: 10.1021/ja039124c.Sherry BD, Radosevich AT, Toste FD. J. Am. Chem. Soc. 2003;125:6076. doi: 10.1021/ja0343050.
- xvii.Nolin KA, Krumper JR, Pluth MD, Bergman RG, Toste FD. J. Am. Chem. Soc. 2007;129:14684–14696. doi: 10.1021/ja074477n. [DOI] [PubMed] [Google Scholar]
- xviii.Desimoni G, Faita G, Jorgensen KA. Chem. Rev. 2006;106:3561. doi: 10.1021/cr0505324. [DOI] [PubMed] [Google Scholar]
- xix.Dagorne S, Bellemin-Laponnaz S, Maisse-Francois A. Eur. J. Inorg. Chem. 2007:913–925. doi: 10.1002/chem.200601112. [DOI] [PubMed] [Google Scholar]
- xx.Enantiomeric excess of ketone reductions determined by chiral HPLC.
- xxi.Absolute configuration of 5b and 9b assigned by comparison of HPLC retention times to those reported by: Hannedouche J, Clarkson G, Wills M. J. Am. Chem. Soc. 2004;126:986. doi: 10.1021/ja0392768. The absolute stereochemistry of remaining products assigned by analogy.
- xxii.Ison EA, Corbin RA, Obu-Omar MM. J. Am. Chem. Soc. 2005;127:11938–11939. doi: 10.1021/ja053860u. [DOI] [PubMed] [Google Scholar]
- xxiii.Chang H-T, Sharpless K-B; Tetrahedron Lett. 1996;37:3219–3222. [Google Scholar]
- xxiv.Tang TP, Volkman SK, Ellman JA. J. Org. Chem. 2001;66:8772–8778. doi: 10.1021/jo0156868. [DOI] [PubMed] [Google Scholar]
- xxv.Corey EJ, Wang Z. Tetrahedron Lett. 1993;34:4001–4004. [Google Scholar]
- xxvi.Faster addition results in a blackening of solution and the isolation of an unreactive complex.
- xxvii.Modifications to the standard complexation conditions where unsuccessful. (CNbox) Re-oxo complex synthesis was also attempted in acetonitrile and THF albeit with similar results.
- xxviii.Lorber CY, Osborn JA, Tetrahedron Lett JA. 1996;37:853–856. and references therein.
- xxix.Previous studies within the group found that toluene was a suitable solvent for Re(V)-oxo catalyzed Meyer-Schuster reaction.
- xxx.During the course of this work, a related rhenium-oxo catalyzed rearrangement of propargyl alcohols was reported: Stefanoni M, Luparia M, Porta A, Zanoni G, Vidari G. Chem. – Eur. J. 2009;15:3940–3944. doi: 10.1002/chem.200802622.
- xxxi.a Hansen MC, Buchwald SL. Org. Lett. 2000;2:713–715. doi: 10.1021/ol005583w. [DOI] [PubMed] [Google Scholar]; b Verdaguer X, Lange UEW, Buchwald SL. Angew. Chem. Int. Ed. 1998;37:1103–1107. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1103::AID-ANIE1103>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- xxxii.a Krzyzanowska B, W. J. Synthesis. 1978;1978:521. [Google Scholar]; b Masumoto S, Usuda H;, Suzuki M, Kanai M, Shibaski, M M. J. Am. Chem. Soc. 2003;125:5634. doi: 10.1021/ja034980+. [DOI] [PubMed] [Google Scholar]; c Spindler F, Blaser H-U. Adv. Synth. Catal. 2001;343:68–70. [Google Scholar]; d Sugi KD, Nagata T, Yamada T, Mukaiyama, T T. Chem Lett. 1997:493–494. [Google Scholar]; e Weix DJ, Shi Y, Ellman JA. J. Am. Chem. Soc. 2005;127:1092–1093. doi: 10.1021/ja044003d. [DOI] [PubMed] [Google Scholar]
- xxxiii.Absolute configuration was assigned by comparison of HPLC retention times to those reported by: Yamada T, Nagata T, Sugi K, Yorozu K, Ikeno T, Ohtsuka Y, Miyazaki D, Mukaiyama, T T. Chem. – Eur. J. 2003;9:4485–4509. doi: 10.1002/chem.200304794.
- xxxiv.a Brown HC, Chandrasekharan J, Ramachandran P. J. Am. Chem. Soc. 1988;110:1539. [Google Scholar]; b Corey EJ, Bakshi R, Shibata S, Chen C, Singh V. J. Am. Chem. Soc. 1987;109:7925. [Google Scholar]; c Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R. J. Am. Chem. Soc. 1996;118:2521. [Google Scholar]
- xxxv.Hannedouche J, Clarkson G, Wills M. J. Am. Chem. Soc. 2004;126:986. doi: 10.1021/ja0392768. [DOI] [PubMed] [Google Scholar]