

Abstract
Cross-linkers that undergo large changes in length upon photoisomerization may produce large conformational changes, and thereby functional changes, in biomolecules. We have designed and synthesized extended and rigid bis-azobenzene cross-linkers, 4,4’-bis(4-(2-chloroacetamido) phenyl)diazenylbiphenyl (BPDB) and the water-soluble sulfonated analog 4,4’-bis(4-(2-chloroacetamido) phenyl)diazenylbiphenyl-2,2’-disulfonate (BPDBS). These photo-switches can produce a minimum ~ 5 Å and a maximum ~ 23 Å end-to-end distance change upon trans/cis isomerization. They have high absorption coefficients (45-60,000 M-1cm-1) and can produce up to ~80% cis isomers under favorable conditions. The photo-switching behavior of BPDBS cross-linked peptides was found to be highly dependent on the cross-linker attachment site. Upon UV (365 nm) irradiation, a significant decrease in α-helix content was observed for peptides that were cross-linked with BPDBS via Cys residues at i, i+19, and i, i+21 positions. In contrast, a large increase in α-helix content was exhibited by i, i+11 cross-linked peptides. BPDBS thus constitutes a particularly bright and effective photoswitch for biomolecule photo-control.
Keywords: azobenzene, peptide, conformation, photo, control
Intramolecular cross-linking of peptides and proteins can stabilize secondary structures, enhance protease resistance, and improve cell penetration ability. [1, 2] These features of cross-linked (“stapled”) peptides have encouraged their development as peptide-based drugs, for targeting cancer, HIV, and other diseases. [2, 3] When the cross-linker contains an azobenzene moiety, the conformation of the peptide or protein can be altered reversibly using light as an external stimulus. [4, 5] Azobenzene-based photo-switchable cross-linkers have been used to photo-control peptide affinity for protein and DNA targets, to control protein folding, and to control enzyme activity. [6, 7]
The trans and cis isomers of azobenzene differ substantially in ground state stability with the trans isomer being ~10-12 kcal/mol more stable. [8] This large difference in stability can, in principle, drive large changes in the conformations of target biomolecules. For such changes to be realized however, the conformational change of the azobenzene unit must be effectively coupled to peptide or protein folding/unfolding coordinates. Most studies have used the end-to-end distance change that occurs upon azobenzene isomerization as the means to drive conformational transitions. Consequently, azobenzene derivatives that show large end-to-end distance changes are desirable as switches in a variety of systems.
BSBCA (3,3’-bis(sulfonato)-4,4’-bis(chloroacetamide)azobenzene) (Scheme 1), a water soluble diamidoazobenzene derivative, [9] has been used to cross-link peptides through Cys residues spaced at i, i+4; i, i+7 and i, i+11 positions. [5] The first two cross-linking sites lead to an increase in α-helix content upon irradiation, while cross-linking at the i, i+11 position shows decreased α-helix content in the irradiated state. [10-12] For the BSBCA cross-linker, the end-to-end distance change upon photoisomerization is ca. 3 Å.
Scheme 1.
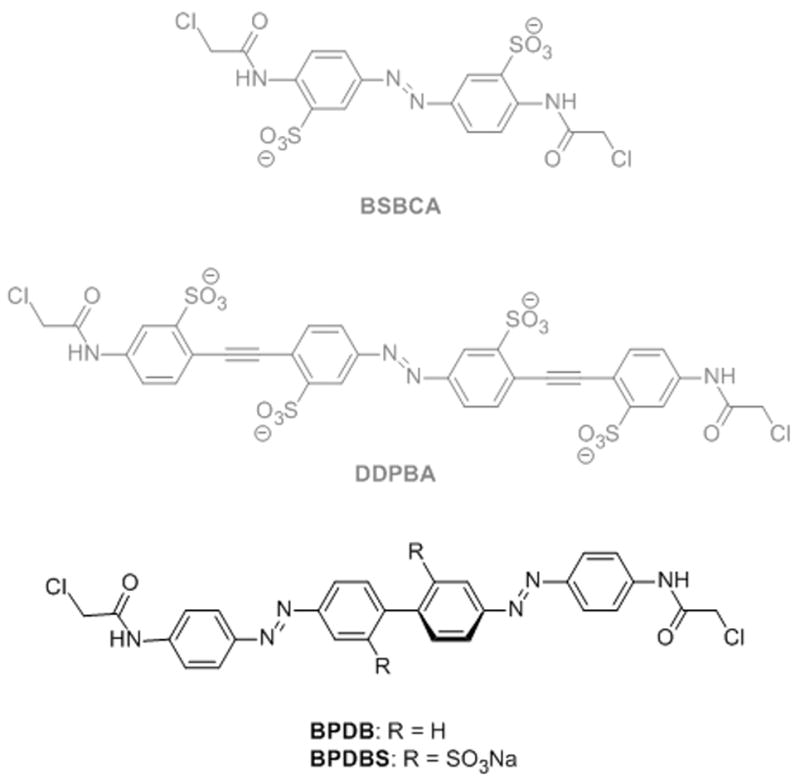
Structures of the new bis-azobenzene structures reported here (BPDB, BPDBS) together with previously studied azobenzene-based cross-linkers BSBCA and DDPBA.
We recently reported a long and rigid para-substituted phenylene ethynylene azobenzene DDPBA (Scheme1), which undergoes a ~13 Å end-to-end distance change upon photoisomerization, however, this derivative gave only ~15% cis isomer in the irradiated state under standard experimental conditions. [13] Hecht and colleagues [14] reported a meta-substituted phenylene ethynylene azobenzene that exhibited better photoswitching properties but meta-substitution (vs. para-substitution) leads to a much larger range of predicted end-to-end distances for the trans isomer, complicating application as a photoswitch for peptides and proteins. Moroder and colleagues designed an extended azobenzene derivative containing alkyne moieties at the para positions[15] and a related derivative was reported by Kuil et al [16]. This switch was successful at photo-controlling conformations of collagen-like peptides and SH2 domain binding peptides despite have a flexible methylene unit as part of the cross-linker. [15, 16] Standaert and Park extended azobenzene by incorporating biphenyl groups. Molecular modeling revealed a difference of 13 Å between the end-to-end distances of trans and cis isomers of a derivative designated mpAbc, [17] however this has yet to be applied as a conformational switch in biomolecules.
In an attempt to couple a high yield of photoisomerization to a relatively large end-to-end distance change, we have now designed two bis-azobenzene derivatives: 4,4’-bis(4-(2-chloroacetamido)phenyl)diazenylbiphenyl (BPDB) and the sulfonated analog 4,4’-bis(4-(2-chloroacetamido)phenyl)diazenylbiphenyl-2,2’-disulfonate (BPDBS) (Scheme 1). Cyclic bis-azo compounds have been studied previously as components of photoswitchable ion chelating systems and cyclic supramolecules. [18] Linear bis-azo compounds similar to the ones reported here with amido substituents at the para positions have been reported in the patent literature as coloring agents (e.g. Vulcan Fast Yellow, Pigment Yellow 5K[19]). In addition, Matĕjeka & Dušak reported photochromic polymers containing amide substituted linear bis-azo compounds.[20] Also, Cisnetti et al reported bis-azo compounds where the central benzene ring was shared between a pair of azobenzene units.[21] Photoisomerization of an azo unit was found to depend on the isomeric state of the other azo unit, particularly in the case of the para substituted derivative. To our knowledge there are no reported applications of bis-azo compounds as conformational switches for biomolecules.
The presence of two azobenzene chromophores means that upon UV irradiation either one or both azo units may isomerize - a feature we expected might complicate the interpretation of the conformational response of stapled peptides or proteins. On the other hand, these bis-azobenzenes were expected to exhibit high molar extinction coefficients, and we hoped, might produce large percentages of cis-isomers upon irradiation. Moreover, their rigidity is expected to lead to substantial end-to-end distance changes even if only one of the two azobenzene chromophores isomerizes (see below). We report here the synthesis and characterization of these bis-azobenzene photo-switches and their effects on helical peptide structures when incorporated via Cys residues spaced at i, i+21; i, i+19; i, i+14 and i, i+11 positions.
Results and Discussion
Cross-linker design
The structures of the designed bis-azobenzenes are shown in Scheme 1. The all-trans forms of both BPDB and BPDBS have a length nearly double that of the well-studied cross-linker BSBCA. The two azobenzene chromophores are linked by a biphenyl unit and the ends are functionalized with an α-chloroacetamido group for covalent attachment of these cross-linkers to peptides via Cys residues. These features are intended to introduce the minimum number of single bonds between the azo bonds that isomerize and the peptide backbone in order to maximize the degree of conformational coupling. The compounds are symmetrical so that only one chemical species is formed upon cross-linking. The incorporation of two sulfonate groups in BPDBS meta to the azo moieties not only increases the water solubility of the cross-linker without directly altering the absorption spectrum, but also inhibits co-planarity of the two aromatic rings of core biphenyl moiety so that each azobenzene chromophore is expected to behave independently. The biphenyl torsion angle in a crystal of the bis-azo dye Congo red varies between zero and 25° [22], whereas Mal et al reported ortho-sulfonated para-amino-substituted biphenyl compounds in which this angle is near 90°. [23]
We used molecular dynamics simulations to estimate the end-to-end distances of trans-trans, trans-cis and cis-cis isomers of the BPDB cross-linker. Simplified models for trans-trans, trans-cis and cis-cis conformers attached to peptides via Cys residues were built in which the β-position of the cysteine side-chain was replaced by a methyl group (Fig. 1). The thermodynamically most stable trans-trans form exhibited an end-to-end (sulfur-to-sulfur) distance of ca. 27-30 Å. In the trans-cis form, this distance varied between 15-25 Å. Rotation around the single bonds linking the azo units to the biphenyl core in the cis-cis isomers can produce two conformational families (cis-cis(l), cis-cis(s)) with very different end-to-end distances so that, overall, the cis-cis form shows a very broad S-S distance distribution in the range of 4.5-25 Å. The most probable end-to-end distances for trans-trans and trans-cis isomers were approximately 28.5 Å and 21 Å, respectively, while approximately 20 Å and 6 Å were observed for cis-cis (l) and cis-cis (s) isomers, respectively.
Figure 1.
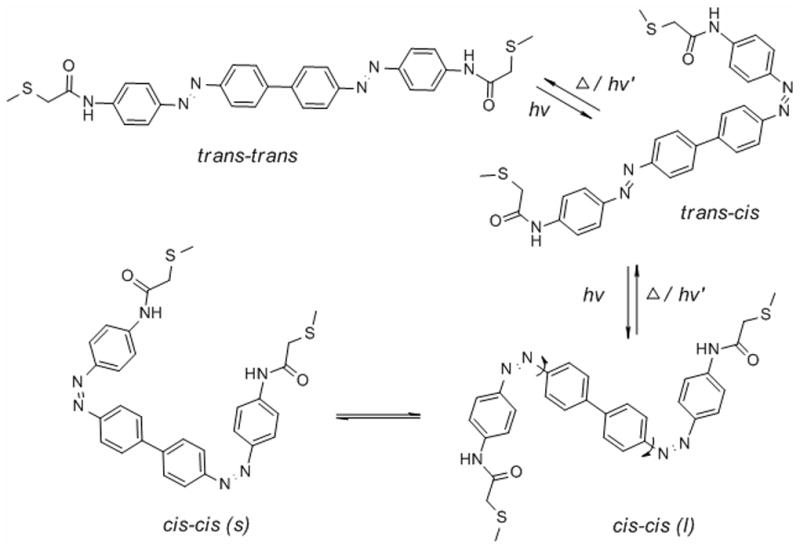

A) Conformational isomers of a BPDB cross-linked peptide where the β-positions of peptide cysteine side-chains are replaced by methyl groups. B) Sulfur to sulfur distances observed during molecular dynamics simulations. Rotation around the bonds indicated in the cis-cis isomer can produce two families of conformers (cis-cis (s) and cis-cis (l)) with very different end-to-end distances.
The conformational properties of these bis-azobenzene cross-linkers are thus significantly more complicated than the single azobenzene cross-linkers used previously, a feature that complicates prediction of conformational effects on target biomolecules. Nevertheless, substantial end-to-end distance changes are possible upon isomerization with these compounds, and even the trans-trans to trans-cis transition can produce a larger end-to-end distance change than the BSBCA cross-linker.
Synthesis of bis-azobenzene cross-linkers
To investigate the photoswitching behavior of both sulfonated and non-sulfonated cross-linkers, we prepared BPDB(r) and BPDBS(r) as reference compounds. The synthetic protocol for the preparation of BPDB and BPDBS as well as BPDB(r) and BPDBS(r) is shown in Scheme 2. Benzidine derivatives, 2a and 2b, were first tetrazotized using NaNO2 and dilute HCl. The tetrazonium solutions were then reacted with the sodium salt of α-anilinophenylmethane sulfonate (3), prepared by the reaction of aniline with formaldehyde in presence of NaHSO3. [24] Hydrolysis of the resulting coupled products using NaOH led to 4,4’-bis((4-aminophenyl) diazenylbiphenyl (1a) and 4,4’-bis(4-aminophenyl)diazenylbiphenyl-2,2’-disulfonate (1b), respectively. The chloroacetamide derivatives BPDB and BPDBS were prepared from 1a and 1b using chloroacetic anhydride in the presence of pyridine-diethyl ether, and chloroacetyl chloride in the presence of aqueous Na2CO3. Similarly, the acetyl derivatives BPDB (r) and BPDBS (r) were generated from their precursor amines 1a and 1b by reaction with acetic anhydride in the presence of pyridine, and aqueous Na2CO3 solution, respectively.
Scheme 2.
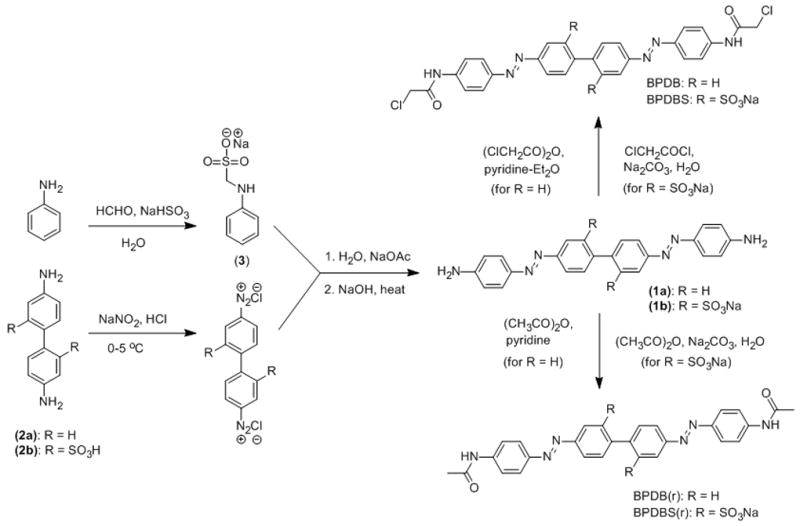
Synthetic route to 4,4’-bis(4-(2-chloroacetamido)phenyl)diazenylbiphenyl (BPDB) and 4,4’-bis(4-(2-chloroacetamido)phenyl)diazenylbiphenyl-2,2’-disulfonate (BPDBS) and the corresponding reference compounds BPDB(r) and BPDBS(r).
UV-Vis spectra and photo-switching of bis-azobenzene cross-linkers
UV-Vis absorption spectroscopy was used to characterize the properties of the reference bis-azobenzene cross-linkers. BPDB(r) showed a strong absorption maximum near 400 nm in DMSO, whereas BPDBS(r) showed a maximum near 380 nm in DMSO and 360 nm in 10 mM sodium phosphate buffer, pH 7.0. (BPDB(r) is not soluble in water). These absorptions are typical of π−π* transitions in amide-substituted trans-azobenzenes. [25] The molar absorption coefficient was determined to be 60,000 M-1cm-1 for BPDB at 400 nm in DMSO, 52,000 M-1cm-1 for BPDBS at 380 nm in DMSO, and 45,000 M-1cm-1 for BPDBS at 360 nm in sodium phosphate buffer, pH 7.0. These values are substantially higher than that for BSBCA (27,000 M-1 cm-1). [26] The 20 nm blue shift in the absorption maximum and the lower molar extinction coefficient of BPDBS(r) compared to BPDB(r), suggest a lack of electronic communication between the two azo chromophores in BPDBS(r) consistent with steric interactions between the two sulfonate groups at 2 and 2’ positions inhibiting co-planarity of the two phenyl rings in the biphenyl core.
When BPDB(r) in DMSO was irradiated with 400 nm light, a photostationary state was obtained in which the absorbance at 400 nm decreased by ~52% (Fig. 2(A)). When BPDBS(r) was irradiated with light of 380 nm (in DMSO) or 365 nm (in 10 mM phosphate buffer, pH 7.0), the absorption maxima decreased by approximately 70% and 83% respectively. New absorption bands with maximum near 450 and 330 nm in DMSO and near 435 and 320 nm in phosphate buffer appeared (Figure 2(B)). We expect that the UV-Vis spectra of trans-cis isomers are similar to those of mixtures of trans-trans and cis-cis isomers so that it is not possible to deconvolute these spectra of irradiated solutions into spectra of individual isomers. Nevertheless, a decrease of more than 50% of the absorption intensity of the trans-trans isomer suggests the appearance of some fraction of cis-cis isomer along with the trans-cis isomer. The thermal decay of irradiated BPDBS(r) in phosphate buffer (pH 7.0) was measured by following the recovery of absorbance at 365 nm as a function of time. Mono-exponential fits of the decay curves gave half-lives of approximately 43 min at 20°C and 11 min at 37°C (Table 1). These half-lives are nearly equal to that exhibited by BSBCA (35 min at 25°C and 12 min at 37°C) [26], and are shorter than that observed for DDPBA (75 min at 20°C and 10 min at 40°C) [13].
Figure 2.
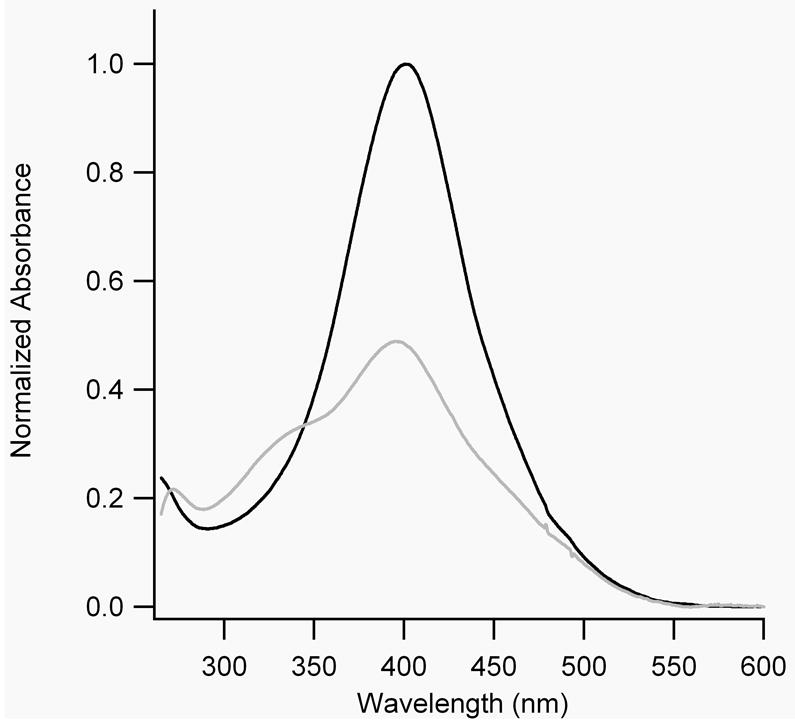
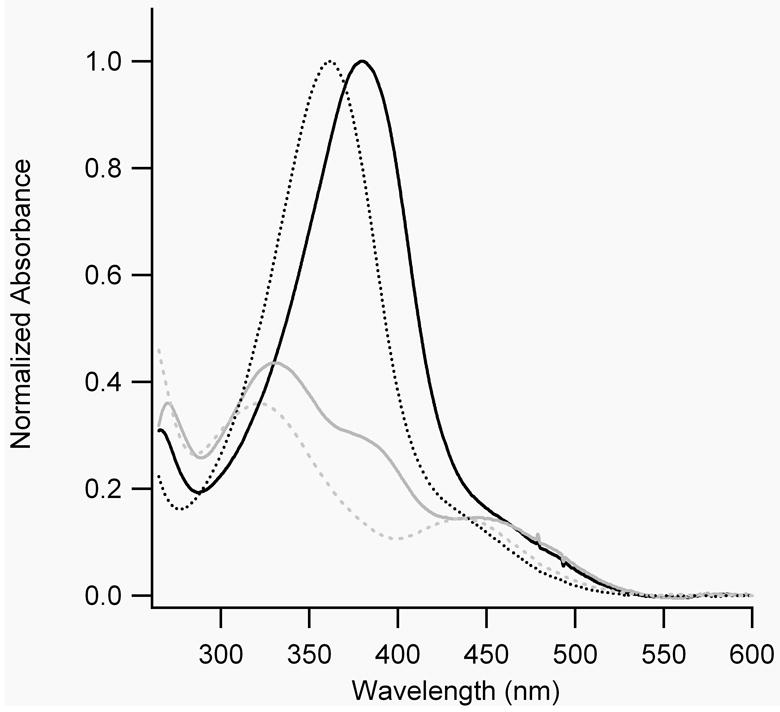
A) UV-Vis spectra of dark-adapted (black lines) and irradiated (grey lines) BPDB(r) in DMSO. B) UV-Vis spectra of dark-adapted (black lines) and irradiated (grey lines) BPDBS(r) in DMSO (—) and in 10 mM sodium phosphate buffer, pH 7.0 (......).
Table 1.
Thermal relaxation half-lives of all cross-linked peptides and the cross-linker BPDBS(r).
| Peptide | τ½ (min)
|
|
|---|---|---|
| 20°C | 37°C | |
| BPDBS(r) | 42.5 ± 5.0 | 11.2 ± 1.3 |
| FZ21-BPDBS | 16.0 ± 0.5 | 4.9 ± 0.2 |
| SS19-BPDBS | 29.1 ± 1.2 | 7.6 ± 0.2 |
| SS14-BPDBS | 10.6 ± 1.1 | 3.5 ± 0.1 |
| SS11-BPDBS | 12.6 ± 0.3 | 3.5 ± 0.1 |
NMR analysis of bis-azobenzene cross-linkers
To test whether cis-cis isomers of the bis-azobenzene cross-linkers form upon irradiation, we carried out 1H NMR for BPDB(r) in DMSO-d6, and BPDBS(r) in D2O and DMSO-d6 (Figure 3). Irradiated solutions of both cross-linkers showed four acetamido methyl signals, which are denoted by Mett, Metc, Mect and Mecc in Figure 3. The Mecc and Mett signals arise from the two homotopic methyl groups of cis-cis and trans-trans bis-azobenzenes respectively, whereas the trans-cis isomer produces two distinct methyl signals of equal intensity - Metc due to the methyl group nearer the trans azo group and Mect due the methyl group nearer the cis azo group (Fig. 3). Only a small amount of the cis-cis isomer (Mecc) was formed upon irradiation of BPDB(r) (Fig. 3(B)), while irradiation of BPDBS(r) produced a significant amount of the cis-cis isomer, particularly in D2O where a ratio of trans-trans :trans-cis: cis-cis of 1: 3.2 : 3.3 was reached (Fig. 3(D)). This result is consistent with the UV-Vis data presented above (Fig. 2) indicating that the largest fraction of cis isomers is formed with BPDBS in aqueous solution. Irradiation of the concentrated NMR sample leads to less efficient production of cis isomers than in diluted solutions used for recording UV-Vis spectra (Fig. 2) so that irradiation times are longer. We note that although the two azo double bonds are indistinguishable in the reference compounds, they are distinct when part of a cross-linked peptide or protein. Thus a trans/cis cross-linked peptide may differ in stability from a cis/trans cross-linked peptide or protein (see below).
Figure 3.
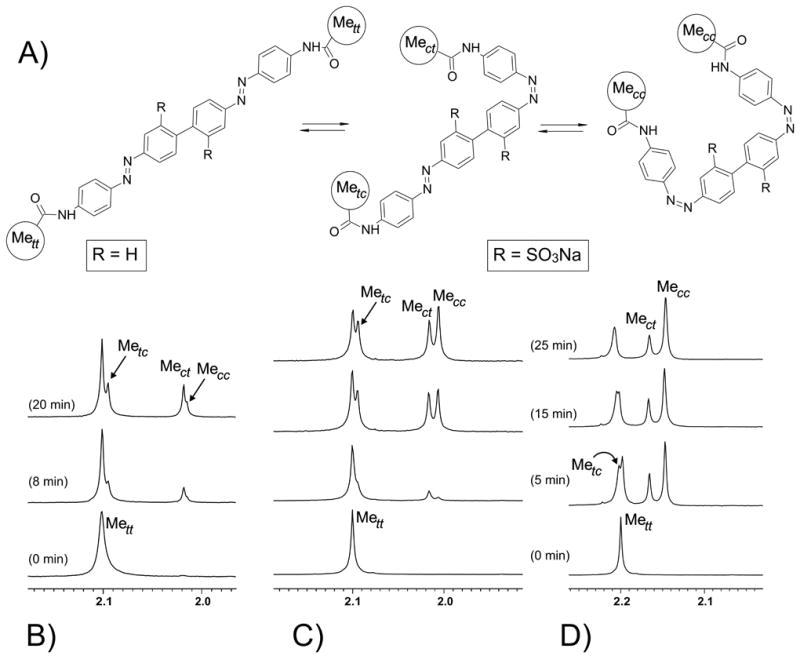
Expanded 1H NMR spectra showing methyl proton signals (highlighted in (A)) of BPDB (r) in DMSO-d6 (B), and BPDBS (r) in DMSO-d6 (C) and in D2O (D) recorded before and after irradiation (at 380 nm or 365 nm respectively) with different irradiation times as indicated.
Owing to the substantially better switching behavior exhibited by the sulfonated derivative, together with its water solubility, we opted to use this compound to test effects of photoisomerization on the conformations of attached peptides.
Design and analysis of bis-azobenzene cross-linked peptides
Using predicted end-to-end distance distributions of the bis-azobenzene cross-linkers (Fig. 1) as preliminary tool, we designed and synthesized four peptide sequences designated FZ21, SS19, SS14, and SS11 where each peptide contained two Cys residues for installing the cross-linker (Figure 4). All of the peptides contain the general sequence Ac-(EAAAR)3-A-NH2 that is known to exist in a monomeric and predominantly helical form in aqueous solution. [10, 27] The spacing between Cys residues when the peptides are in a helical conformation is intended to match the end-to-end distance of the trans-trans, the trans-cis/cis-trans or the cis-cis(l) isomers. FZ21 with Cys residues spaced at i and i+21 positions, which was previously analyzed with the DDPBA cross-linker,[13] has a range of S-S distances from ~29 to 35Å and was expected to accommodate the dark adapted trans-trans cross-linker although the optimal length of trans-trans BPDBS is at the extreme short end of this range. A better fit was predicted between the trans-trans isomer of the cross-linker and the i, i+19 peptide SS19 (S-S range 26.5 to 32.2 Å). In peptide SS14, the distance between S atoms of Cys residues spaced at i and i+14 positions is 19.2-24.5 Å when the peptide is in an ideal α-helical form. This range was expected to be compatible with the end-to-end distances of trans-cis/cis-trans and partly with the cis-cis (l) forms of the cross-linkers. The SS11 peptide was expected to be compatible with all cis-containing forms of the cross-linker (trans-cis/cis-trans and cis-cis (l)). Also included on the histogram (Fig. 4B) are S-S distance ranges for helical peptides with i, i+7 and i, i+4 Cys spacings. Peptides with these Cys spacings cross-linked with the BSBCA linker were studied in detail previously. [10, 11, 13, 28] While the S-S distance ranges of these peptides would be compatible with cis-cis forms of the bis-azo BPDBS cross-linker, the Cys residues are so close in the linear sequence that even if the unfolded peptide were fully extended, the S-S distance would be too short to accommodate an undistorted trans-trans form of the cross-linker. [10, 28] We note that this limitation would not be present in a folded protein where two Cys residues could be close together in three dimensions but far apart in the linear sequence.
Figure 4.
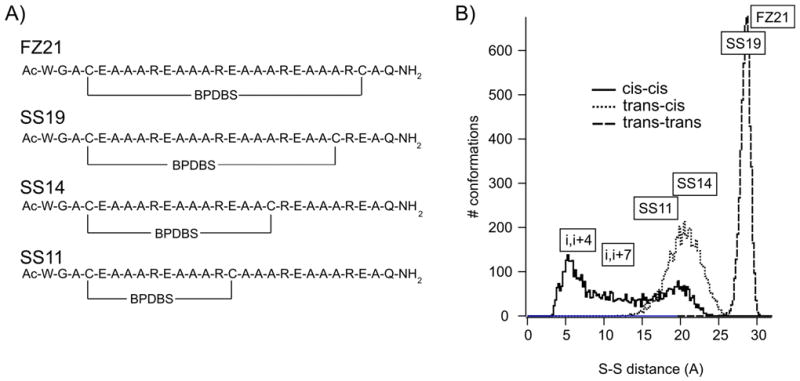
A) Primary sequences of cross-linked peptides: FZ21-BPDBS, SS19-BPDBS, SS14-BPDBS, SS11-BPDBS. B) Histograms of end-to-end distances for trans-trans, trans-cis and cis-cis isomers of BPDB calculated from molecular dynamics simulations with S-S spacing in ideal helical peptides with Cys residues spaced at i, i+21 (FZ21); at i, i+19 (SS19); i, i+14 (SS14); i, i+11 (SS11), i, i+7 and i, i+4 shown.
Each of these peptides was reacted with the cross-linker BPDBS to generate the cross-linked peptides FZ21-BPDBS, SS19-BPDBS, SS14-BPDBS, and SS11-BPDBS. All the cross-linked peptides were HPLC purified and characterized by MALDI/ESI mass spectrometry. The absence of free Cys residues after cross-linking was checked with Ellman’s reagent.[29]
Photoisomerization of bis-azobenzene cross-linked peptides
All BPDBS cross-linked peptides exhibited dark-adapted absorption spectra with maxima near 370 nm, and a shoulder in the range of 420-520 nm. SS19-BPDBS and FZ21-BPDBS had absorption maxima that were slightly red-shifted compared to SS11-BPDBS and SS14-BPDBS. Irradiation of all BPDBS cross-linked peptides with 365 nm light led to a significant decrease in absorbance near 370 nm but the magnitude of the decrease varied from peptide to peptide. Irradiation of FZ21-BPDBS produced a 49% reduction, irradiation of SS19-BPDBS produced a 40% reduction, irradiation of SS14-BPDBS produced a 59% reduction, and irradiation of SS11-BPDBS produced a 66% reduction (Fig. 5A) at 20°C. The magnitude of the absorbance change was temperature dependent with all the BPDBS cross-linked peptides showing larger absorbance changes at higher temperatures indicating a larger fraction of cis isomers was produced. However, the magnitude of this effect varied form peptide to peptide. For instance, irradiation of SS19-BPDBS produced a significantly larger fraction of cis isomers at higher temperatures (Fig 5B) whereas irradiation of SS11-BPDBS produced a large fraction of cis isomers even at 4°C (estimated to be ~80% based on comparison with spectra for pure cis and trans isomers of BSBCA [28]) and this increased only slightly at higher temperatures (Fig. 5C). These results indicate that there are significant effects of peptide structure on the quantum yields for trans/cis photoisomerization of these cross-linkers.
Figure 5.
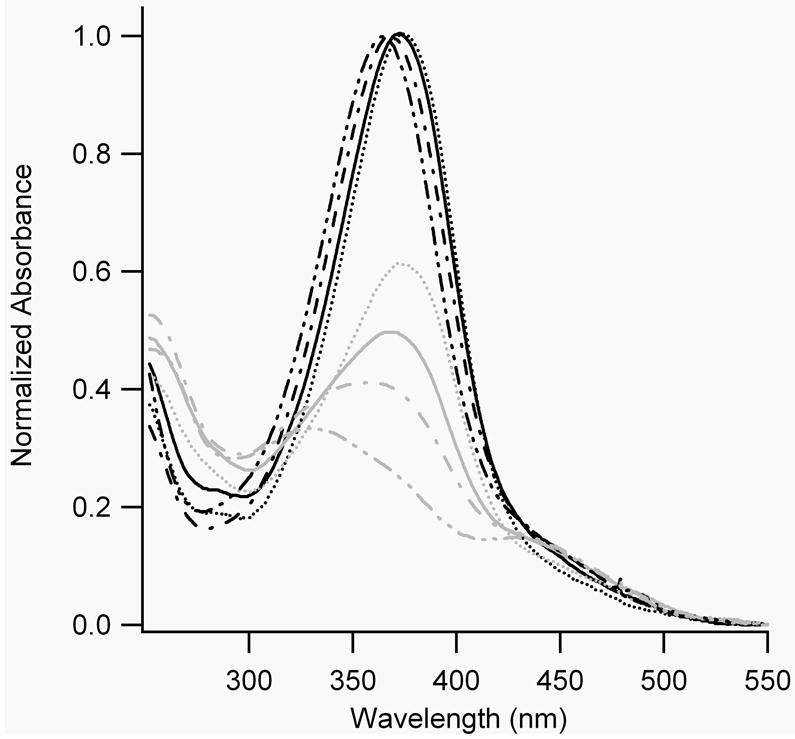
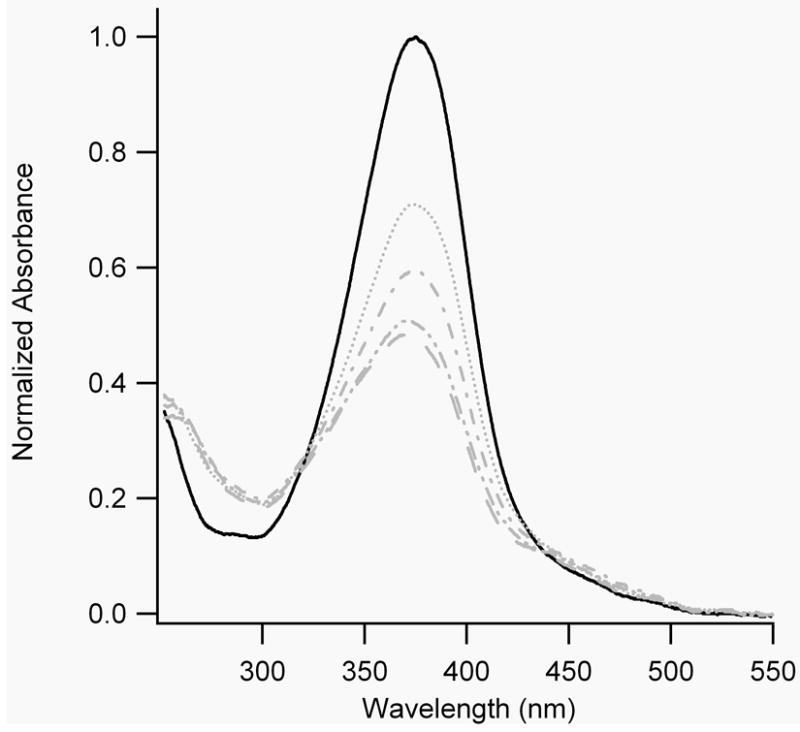


A) UV-Vis absorption spectra of all BPDBS cross-linked peptides without and with continuous irradiation at 365 nm at 20°C. Grey lines represent spectra with continuous irradiation at 365 nm, while black lines represent spectra without 365 nm irradiation. (FZ21-BPDBS (—), SS19-BPDBS (....), SS14-BPDBS (–.–), SS11-BPDBS (–..–)) B) UV-Vis spectra of SS19-BPDBS without (black solid line) and with continuous irradiation (grey lines) at different temperatures (4°C (....), 20°C (–.–), 37°C (–..–), 45°C (– –)). C) UV-Vis spectra of SS11-BPDBS without (black solid line) and with continuous irradiation (grey lines) at different temperatures (4°C (....), 20°C (–.–), 37°C (–..–)). D) Spectra of SS11-BPDBS (20°C) before irradiation (black solid line), after irradiation with 365 nm light (grey solid line), and at different time intervals after irradiation (5, 10, 15, 30, 45, 60, 75, 90, 109 and 120 min)(grey dotted lines). (10-12 μM solutions of cross-linked peptides in 10 mM phosphate buffer at pH 7.0 were used for all experiments)
The thermal reversion of all cross-linked peptides was followed by UV-Vis spectroscopy. Isosbestic points at approximately 249, 305 and 449 nm were observed (Fig. 5D shows the thermal relaxation of SS11-BPDBS as a representative case). As noted above, the trans-cis and cis-trans cross-linked peptides are distinct species. Thus, assuming only cis-to-trans isomerization can occur thermally, the process of thermal reversion may be represented as shown in Scheme 3.
Scheme 3.
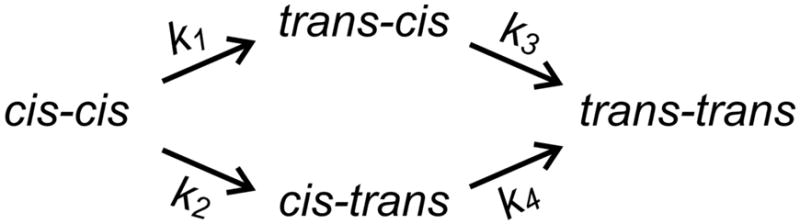
Kinetic processes of thermal reversion in cross-linked peptides
The absorbance at 370 nm is given by:
Where εtrans−trans is assumed to be 50,000 M-1cm-1 for BPDBS cross-linked peptides and εcis−cis can be estimated to be ~4000 M-1cm-1 based on the BSBCA case [28] and εtrans−cis and εcis−trans are each approximately 25,000 M-1cm-1. Numerical simulations using Tenua 2.1 (http://bililite/com/tenua) [30] indicate that such a system can exhibit a variety of behaviors depending on the relative values of the rate constants k1, k2, k3, k4. However, if all rate constants are approximately equal or if k3 ≈ k4 > k1 ≈ k2, an approximately monoexponential increase in absorbance at 370 nm is expected. If instead, k1 ≈ k2 > k3 ≈ k4, or k1 ≠ k2, k3 ≠ k4, a biexponential increase is expected. Both behaviors were observed for different peptides at different temperatures (Fig. 6).
Figure 6.
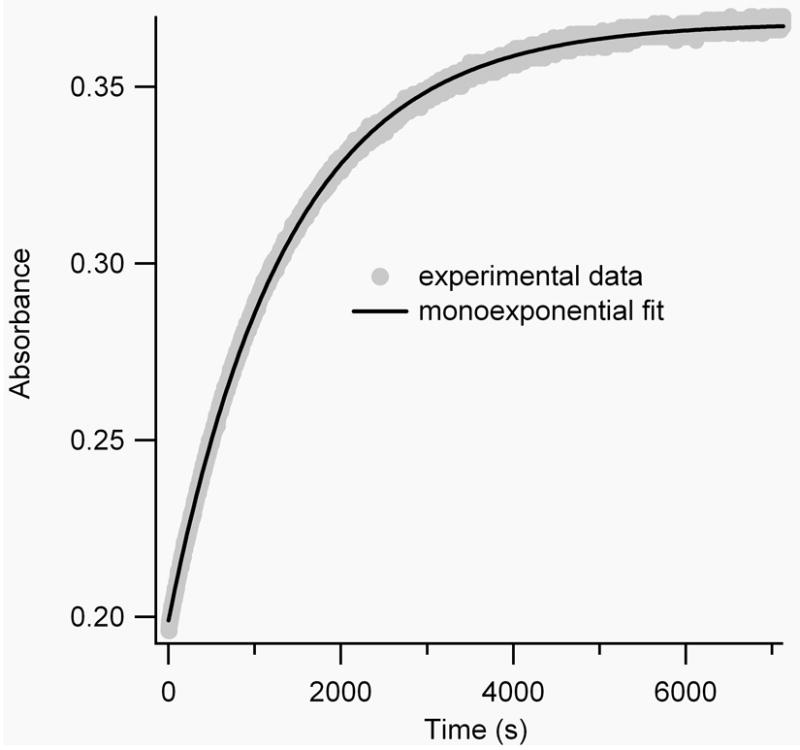
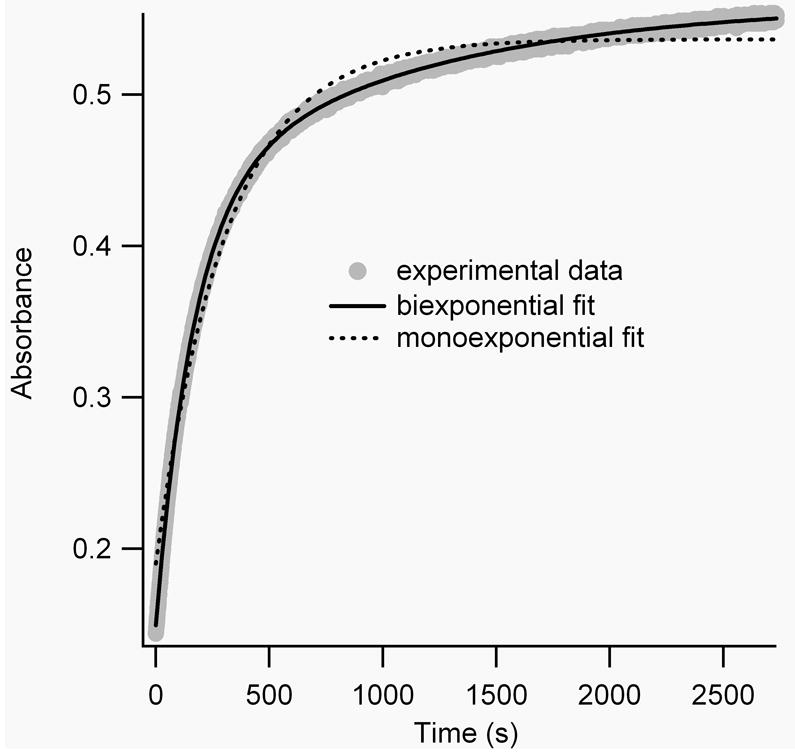
A) Mono-exponential fit (black line) of the measured absorbance decay (grey line) of irradiated FZ21-BPDBS at 20°C. B) Mono-vs. biexponential fits of the measured absorbance decay of irradiated SS11-BPDBS at 37°C.
To simplify comparison with other cross-linkers, thermal reversion curves were fitted as monoexponential processes. All BPDBS cross-linked peptides showed half-lives for thermal reversion between 10-30 min at 20°C and 3-8 min at 37°C (Table 1). These thermal relaxation rates are faster than observed for DDPBA cross-linked peptides (τ½ = 102-159 min at 20°C and 37-54 min at 30°C) [13] and also for BSBCA cross-linked FK11 peptides (τ½ = 35 min at 25°C and 12 min at 37°C). [26] All cross-linked peptides showed faster thermal relaxation rates than the free cross-linker BPDBS(r) indicating that the peptide reduces the activation energy barrier by stabilizing the transition state for thermal reversion and/or destabilizing the cis isomers to varying extents depending on the cross-linker attachment sites.
Effects of photoisomerization on peptide conformations
The secondary structures of all the cross-linked peptides in dark-adapted and irradiated states were investigated using circular dichroism (CD) spectroscopy. Spectra for all cross-linked peptides before and after irradiation at different temperatures are presented in Figure 7. Helical contents were estimated as described in the experimental section.
Figure 7.
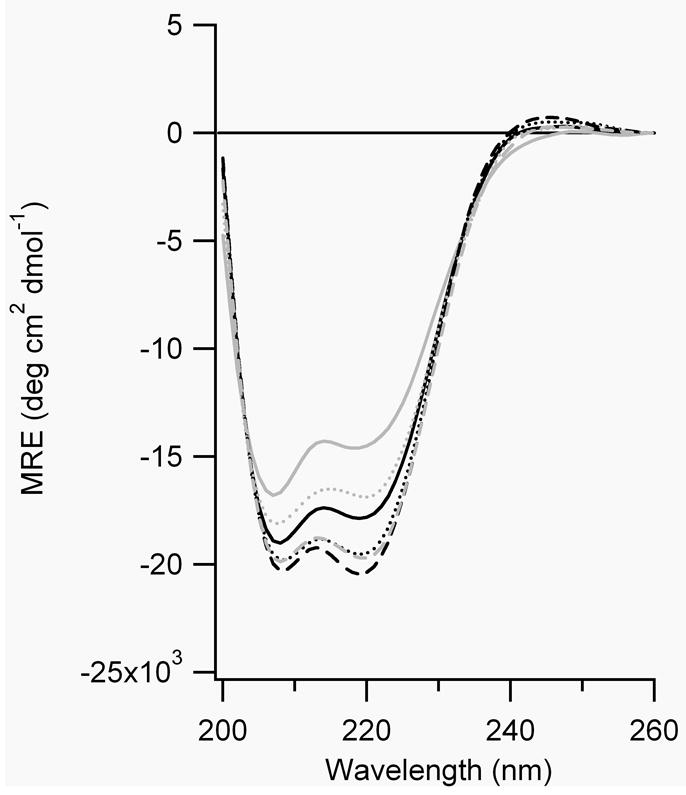
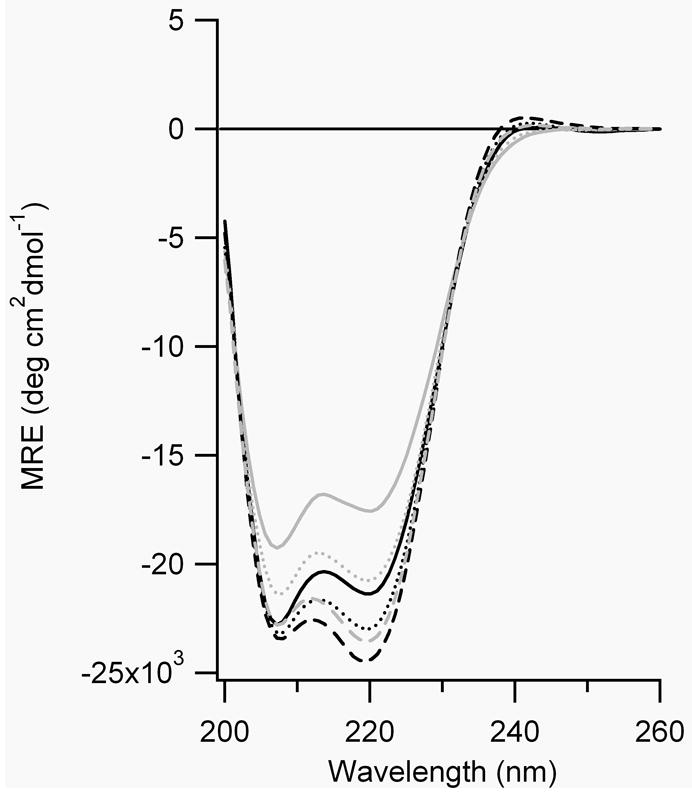

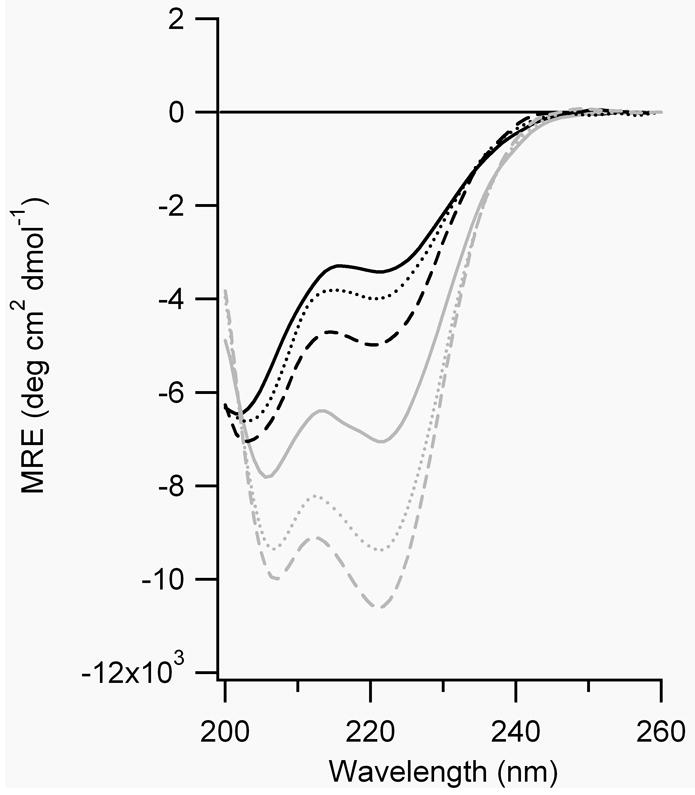
CD spectra of A) FZ21-BPDBS, B) SS19-BPDBS, C) SS14-BPDBS, and D) SS11-BPDBS before (black lines) and after irradiation (grey lines). Each spectrum was scanned at 37°C (—), 20°C (....) and 4°C (– – –). (80-100 μM peptide solutions in 10 mM phosphate buffer at pH 7.0.)
The calculated end-to-end distance (S-S) of the trans-trans BPDBS isomer varies between 27-30 Å (Fig. 1). If FZ21 adopts an ideal α-helical structure, the S-S distances range from ~29 to 35Å whereas the SS19 peptide has an S-S range of 26.5 to 32.2 Å. The dark-adapted BPDBS cross-linked FZ21 and SS19 peptides both show high helical content (Fig. 7, A,B), with that of SS19-BPDBS being somewhat higher consistent with a better match between the trans-trans form of the cross-linker and the helix. The small mismatch in the FZ21-BPDBS case perhaps causes a reduction in helix content since in order to bridge the i, i+21 spacing, the BPDBS cross-linker, like the DDPBA cross-linker, [13] must pack very closely to the FZ21 peptide surface and may thereby distort the helix geometry via steric interactions.
Irradiation of FZ21-BPDBS and SS19-BPDBS led to a decrease in α-helix content consistent with the prediction that irradiated states of the cross-linkers would be even less compatible with an α-helix structure. The decrease in helix content upon irradiation was temperature dependent; higher temperatures produced greater decreases in helix content (Figure 7A,B, Table 2), consistent with the observation that higher fractions of cis content were produced at higher temperatures (Fig. 5). Higher temperatures also decrease the lifetime of cis states (Table 1) so that recording full CD spectra becomes difficult. Instead, Figure 8 shows the CD change at 222 nm as a function of time after irradiation of SS19-BPDBS to produce cis isomers. Helix content is clearly diminished upon irradiation and then restored on a timescale reflecting thermal isomerization to that trans state.
Table 2.
Percentage helicity of cross-linked peptides before and after irradiation.
| peptide |
α-helix content (%)
|
|||||
|---|---|---|---|---|---|---|
| 37°C
|
20°C
|
4°C
|
||||
| dark | irradiated | dark | irradiated | dark | irradiated | |
| FZ21-BPDBS | 51 | 41 | 55 | 49 | 58 | 56 |
| SS19-BPDBS | 63 | 51 | 66 | 60 | 70 | 67 |
| SS14-BPDBS | 37 | 40 | 46 | 49 | 54 | 56 |
| SS11-BPDBS | 10 | 21 | 11 | 28 | 15 | 31 |
Figure 8.
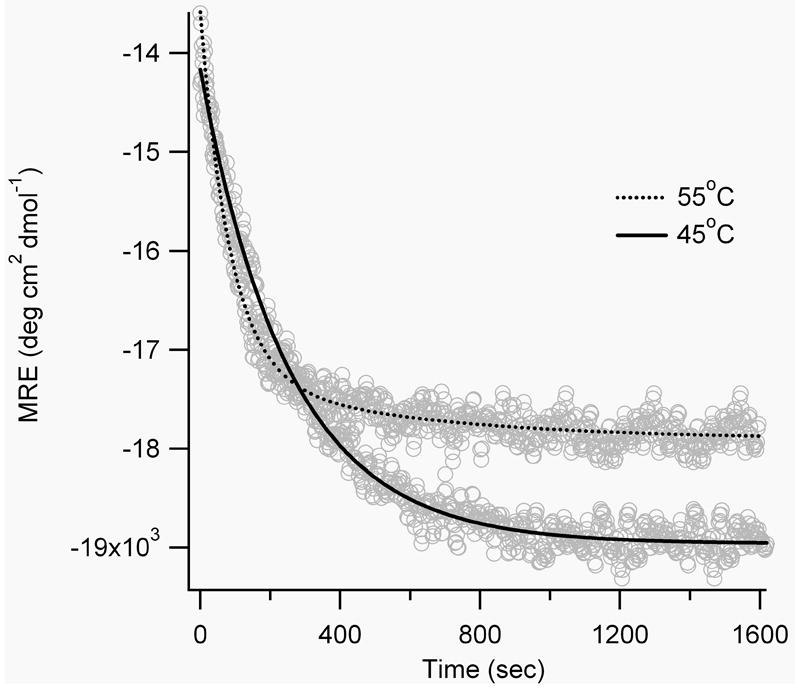
Time-dependent CD signal for SS19-BPDBS at 222 nm after irradiation at 365 nm at higher temperatures.
Based on predicted end-to-end distance distributions (Fig. 4) we expected that the SS14-BPDBS peptide would show relatively low helix content in the dark (trans-trans form) but increased helicity upon irradiation to produce trans-cis/cis-trans isomers. The helix content of the dark-adapted SS14-BPDBS peptide was higher than expected and the changes in CD spectra upon irradiation were very small (Fig. 7C, Table 2). The small change may be due to the formation of cis-cis isomers which may be too short to be compatible with ideal helical structure.
The SS11 peptide contains Cys residues at i and i+11 positions and was expected to accommodate the trans-cis/cis-trans and cis-cis (l) cross-linker isomers. The observed helix content of the dark-adapted state of SS11-BPDBS, was <10 %, consistent with a poor match to the dark-adapted trans-trans isomer. Upon irradiation with 365 nm light, a large increase in the helix content of SS11-BPDBS was observed (>50%)(Fig. 7D, Table 2).
Moreover, unlike the other cross-linked peptides, irradiated SS11-BPDBS exhibited a larger change in helix content at lower temperatures, although a somewhat lower percentage of cis isomers is formed at lower temperatures (Figure 5C). This may be due to stabilization of α-helix structure as the temperature is lowered.
Stability of the cross-linked peptides in a reducing cellular environment
The cellular environment is reducing in nature due to the presence of small tripeptide glutathione in its reduced form in 1-10 mM concentration. [31] We therefore tested the photo-switching behavior of BPDBS cross-linked peptides in phosphate buffer pH 7.0 at 37°C in the presence of 10 mM reduced glutathione. To mimic the conditions for using such peptides in cells in culture, [7] cross-linked peptide was incubated with glutathione in the dark for 24 hours at 37°C. Photo-switching remained unaffected (data not shown), which suggests that BPDBS is inert to glutathione, and hence BPDBS-linked peptides should be functional in typical intracellular environments.
Summary
We have designed and synthesized biphenyl based diamidobisazobenzene cross-linkers BPDB and BPDBS, and examined their photoswitching behaviors both as free cross-linkers and when attached to a series of peptides. Compared to previously studied mono-azo cross-linkers BSBCA and DDPBA, these bis-azo species offer certain advantages for photo-control. First, the end-to-end distance change upon trans-to-cis isomerization is substantially larger for the bis-azo species than the mono-azo BSBCA compound. Second, the fraction of cis isomers produced upon irradiation for the bis-azo species, particularly the sulfonated BPDBS version, is substantially greater than for DDPBA, reaching ~80 % in favourable cases. Third, the molar extinction coefficients are high (>45,000 M-1cm-1) making these bright photoswitches.
As with previously studied azobenzene based cross-linkers, the new bis-azo species can be used to photocontrol peptide conformation and the effects on helical content of designed helical peptides can be approximately predicted by comparing the end-to-end distance distributions of the cross-linkers with Cys spacings in the target peptides. Because these bis-azo species are substantially longer than BSBCA, the extent of conformational change upon isomerization is necessarily larger. Compared to the mono-azo BSBCA cross-linker however, the interplay between cross-linker isomerization and peptide conformation is more complicated. First, we observed a strong dependence of the yield of cis isomers in the photostationary state on the nature of the peptide attachment. Whereas the SS11-BPDBS cross-linked peptide gave ~ 80 % cis, the SS19-BPDBS gave only ~50%.
A further complication of the bis-azo cross-linkers is that one cannot tell from the UV spectra what fraction of cis containing species are cis-cis versus cis-trans/trans-cis. Thus, for a case like the SS19-BPDBS peptide, it is unclear whether the decrease in helix content in the irradiated state reflects a mixture of very helical trans-trans species and very disordered cis-cis species, or a large fraction of cis-trans/trans-cis species of intermediate helicity. For a peptide or protein with a particular biological activity, these two scenarios would be expected to have very different functional consequences. The conformational response of an attached peptide or protein is thus likely to be case specific. In favorable cases however, such as SS11-BPDBS, substantial conformational change is possible. Peptides with Cys residues spaced i, i+11 in the sequence have been cross-linked with monoazo BSBCA cross-linker and studied in detail. [11, 32] Such peptides are predominantly helical in the dark and undergo a substantial loss of helical structure upon UV irradiation. The SS11-BPDBS peptide shows precisely the opposite behavior as diagrammed in Figure 9. In the dark, the long bis-azo BPDBS cross-linker strongly inhibits helical formation. UV irradiation to produce trans-cis, cis-trans, and cis-cis peptides leads to increased helical content.
Figure 9.

Models showing the increase in helical content of the SS11-BPDBS peptide upon UV irradiation.
Experimental Section
Synthesis of the cross-linkers
Compounds 2a (benzidine), 2b (4,4’-diaminobiphenyl-2,2’-disulfonic acid) and aniline were purchased from Sigma-Aldrich, and were used. All 1H and 13C NMR spectra were recorded using a Varian Unity 400 or Bruker 400 spectrometer. High resolution mass spectra were obtained by electron spray ionization (ESI), and mass spectra of peptides were obtained by MALDI TOF mass spectrometry.
1) Synthesis of sodium α-anilinophenylmethanesulfonate (3)
Following a literature procedure [24] formaldehyde (0.65 g, 21.5 mmol) was added to a solution of sodium bisulfite (2.2 g, 21.5 mmol) in water (12 mL). After stirring the reaction for 1h at room temperature, aniline (2.0 g, 21.5 mmol) was introduced into the reaction and it was stirred for 6 h at 75°C. The reaction was first cooled to room temperature and then in an ice bath. The precipitate obtained was filtered, washed with cold water and dried in vacuo. The product, α-anilinophenylmethanesulfonate (2.4 g, 53%) was pure enough to use for further reaction. 1H NMR (400 MHz, D2O): δ = 4.45 (s, 2H), 6.87 (t, J = 7.4 Hz, 1H), 6.93 (d, J = 8.7 Hz, 2H), 7.30 (t, J = 8.7 Hz, 1H).
2) Synthesis of 4,4’-bis(4-aminophenyl)diazenylbiphenyl (1a)
To a round-bottomed flask containing benzidine, 2a (0.8 g, 3.1 mmol) was added a mixture of ice (10 g) and concentrated hydrochloric acid (1.3 mL). A solution of sodium nitrite (0.46 g, 6.6 mmol) in water (2 mL) was added slowly with stirring at 0-5 °C. After 1 h of stirring at this temperature, the excess nitrous acid was consumed by adding urea. In parallel, a coupler solution was prepared by dissolving α-anilinophenylmethanesulfonate (1.6 g, 7.8 mmol) in 20 mL of water and stirred at 0-5°C. The diazo solution was kept at 0-5 °C and was added dropwise with stirring. The pH of the reaction mixture was maintained in the range of 4 to 7 with an aqueous NaOAc solution. The resulting reaction mixture was then allowed to stir at 0-5°C for 2 h and at room temperature for a further 12 h. After the reaction, the precipitate was removed by filtration and washed with water. The cake obtained was boiled together with a 20% aqueous NaOH solution for 2 h. The resulting precipitate was filtered, washed thoroughly with water until the washing water become neutral and dried in vacuo to yield 1a (0.23 g, 20%) as an orange solid. 1H NMR (400 MHz, DMSO-d6): δ = 6.14 (br s, NH2), 6.68 (d, J = 9.0 Hz, 4H), 7.69 (d, J = 8.7 Hz, 4H), 7.84 (d, J = 9.0 Hz, 4H), 7.90 (d, J = 8.7 Hz, 4H); 13C NMR (100 MHz, DMSO-d6): δ = 114.1, 123.1, 126.0, 128.1, 140.6, 143.7, 152.5, 153.7; ESI-HRMS: m/z calc’d for C24H21N6 : 393.1822 [M+H]+, found: 393.1807.
3) Synthesis of 4,4’-bis(4-aminophenyl)diazenylbiphenyl-2,2’-disulfonate (1b)
4,4’-Diaminobiphenyl-2,2’-disulfonic acid, 2b (1.6 g, 4.6 mmol) was dissolved in water (20 mL) with sodium carbonate (0.49 g, 4.6 mmol), and a solution of sodium nitrite (0.76 g, 11.0 mmol) in water (1 mL) was added. This solution was added dropwise to a mixture of ice (30.0 g) and concentrated hydrochloric acid (2.0 mL), and the reaction was stirred at 0-5°C for 1 h. Excess nitrous acid in the reaction mixture was decomposed with urea. To this tetrazonium solution was added dropwise an ice cold solution of α-anilinophenylmethanesulfonate (2.4 g, 11.5 mmol) in water (10 mL). After stirring the coupling reaction mixture for 2 h at 0-5°C and at room temperature for 12 h, a 20% aqueous NaOH solution was added. The reaction was then heated at 80°C for 2 h. After cooling to room temperature, the aqueous solution was saturated with NaCl, which led to the precipitation of the required product. This was filtered, thoroughly washed with aqueous saturated NaCl solution, and dried in vacuo to yield 1b (1.2 g, 43% yield) as a red solid. 1H NMR (400 MHz, DMSO-d6): δ = 6.09 (br s, NH2), 6.68 (d, J = 8.7 Hz, 4H), 7.42 (d, J = 8.4 Hz, 2H), 7.61 (dd, J1 = 8.4 Hz, J2 = 2.0 Hz, 2H), 7.69 (d, J = 8.7 Hz, 4H), 8.24 (d, J = 2.0 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ = 114.1, 120.5, 121.8, 125.8, 133.8, 139.7, 143.7, 146.3, 151.2, 153.5; ESI-HRMS: m/z calc’d for C24H21N6O6S2: 553.0958 [M+H]+, found: 553.0942.
4) Synthesis of 4,4’-bis(4-(2-chloroacetamido)phenyl)diazenylbiphenyl, BPDB
To an ice cold solution of 1a (80 mg, 0.20 mmol) in an ether/pyridine mixture (1:1, 5 mL) was added dropwise a solution of chloroacetic anhydride (105 mg, 0.61 mmol) in ether (2 mL). The resulting reaction was warmed to room temperature and allowed to stir for 1 h. The resulting precipitate was filtered and then washed with ether. The crude product was recrystallized from dimethylformamide/water to yield BPDB (78.8 mg, 70% yield) as orange solid. 1H NMR (400 MHz, DMSO-d6): δ = 4.32 (s, 4H), 7.84 (d, J = 9.0 Hz, 4H), 7.95 (d, J = 8.7 Hz, 4H), 7.99 (d, J = 9.0 Hz, 4H), 8.02 (d, J = 8.7 Hz, 4H), 10.67 (br s, NH); 13C NMR (100 MHz, DMSO-d6 at 60 °C): δ = 44.2, 120.5, 123.8, 124.4, 128.5, 142.1, 142.2, 148.9, 152.4, 165.7; ESI-HRMS: m/z calc’d for C28H23N6O2Cl2 : 545.1254 [M+H]+, found: 545.1269.
5) Synthesis of 4,4’-bis(4-acetamidophenyl)diazenylbiphenyl, BPDB(r)
To a solution of 1a (33.0 mg, 0.08 mmol) in pyridine (2 mL) was added acetic anhydride (26.0 mg, 0.25 mmol) dropwise in ice cold conditions. The reaction mixture was allowed to warm to room temperature and then allowed to stir for 1 h. The reaction was quenched with water. The resulting precipitate was filtered, washed with water and finally recrystallized from dimethylformamide/water to yield BPDB(r) (27.3 mg, 68% yield) as yellow solid. 1H NMR (400 MHz, DMSO-d6): δ = 2.10 (s, 6H), 7.81 (d, J = 8.6 Hz, 4H), 7.91 (d, J = 9.0 Hz, 4H), 7.97 (d, J = 8.6 Hz, 4H), 8.00 (d, J = 8.6 Hz, 4H), 10.31(br s, NH); 13C NMR (100 MHz, DMSO-d6 at 60 °C): δ = 24.5, 119.7, 123.4, 124.1, 128.2, 141.7, 143.0, 148.2, 152.2, 169.2; ESI-HRMS: m/z calc’d for C28H24N6O2: 476.1947 [M+H]+, found: 476.1961.
6) Synthesis of 4,4’-bis(4-(2-chloroacetamido)phenyl)diazenylbiphenyl-2,2’-disulfonate, BPDBS
1b (135 mg, 0.23 mmol) was dissolved in water (4 mL) with sodium carbonate (25.0 mg, 0.23 mmol), and chloroacetyl chloride (126 mg, 1.1 mmol) was added dropwise in ice cold conditions. After stirring the reaction for 15 min, another aliquot of chloroacetyl chloride (126 mg, 1.1 mmol) was introduced dropwise without further cooling and the resulting reaction was allowed to stir at room temperature for 30 min. The aqueous reaction mixture was saturated with NaCl, which led to precipitation of the desired product. The precipitate was filtered, washed with saturated aqueous NaCl solution and ether, and dried in vacuo to yield BPDBS (141 mg, 80% yield, <5 % trans-cis isomer) as an orange red solid. 1H NMR (400 MHz, DMSO-d6): δ = 4.33 (s, 4H), 7.54 (d, J = 8.0 Hz, 2H), 7.76 (dd, J1 = 8.0 Hz, J2 = 2.0 Hz, 2H), 7.83 (d, J = 9.0 Hz, 4H), 7.94 (d, J = 9.0 Hz, 4H), 8.35 (d, 2H, J = 2.0 Hz, 2H), 10.66 (br s, NH); 13C NMR (100 MHz, DMSO-d6): δ = 44.3, 120.3, 121.2, 122.4, 124.4, 133.9, 141.2, 142.1, 146.6, 148.8, 150.9, 165.8; ESI-HRMS: m/z calc’d for C28H23N6O8S2Cl2: 705.0390 [M+H]+, found: 705.0370.
7) Synthesis of 4,4’-bis(4-acetamidophenyl)diazenylbiphenyl-2,2’-disulfonate, BPDBS(r)
To an ice cold aqueous solution of 1b (36.0 mg, 0.06 mmol) containing sodium carbonate (7.2 mg, 0.06 mmol) was added acetic anhydride (60.0 mg, 0.6 mmol) dropwise with stirring. The resulting reaction was stirred at room temperature for 2 h. The same work up procedure as above led to a solid orange product BPDBS(r) (29.0 mg, 76% yield). The product was further purified by a reverse phase column chromatography using Varian Bond Elut C18. The product was eluted with a 20-30% methanol-water mixture. 1H NMR (400 MHz, D2O): δ = 2.20 (s, 6H), 7.34 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 9.0 Hz, 4H), 7.67 (dd, J1 = 8.2 Hz, J2 = 2.0 Hz, 2H), 7.78 (d, J = 9.0 Hz, 4H), 8.41 (d, J = 2.0 Hz, 2H); 13C NMR (100 MHz, D2O): δ = 23.4, 120.8, 121.9, 122.9, 124.0, 133.2, 139.5, 140.9, 142.0, 148.4, 151.0, 172.7; ESI-HRMS: m/z calc’d for C28H23N6O8S2: 635.1024 [M+H]+, found: 635.1051.
Peptide synthesis
The peptides FZ21 (Ac-WGACEAAAREAAAREAAAREAAARCAQ-NH2), SS19 (Ac-WGACEAAAREAAAREAAAREAACREAQ-NH2), SS14 (Ac-WGACEAAAREAAAREAACREAAAREAQ-NH2, FK11 (Ac-WGEACAREAAAREAACRQ-NH2) and SS7 (Ac-WGEAAARECAAREAACRQ-NH2) were prepared by using standard protocols for Fmoc-based solid-phase peptide synthesis. All peptides were HPLC-purified on a Zorbax SB-18 column with linear gradient of 5-70% acetonitrile/water containing (+0.1% trifluoroacetic acid) over a course of 35 min. The molecular compositions of peptides were confirmed by MALDI-MS [M+]: FZ21 (C112 H182 N42 O37 S2) calc’d: 2773.1 Da, found: 2773.6 Da; SS19 (C114 H185 N42 O39 S2) calc’d: 2832.1 Da, found: 2832.2 Da; SS14 (C114 H185 N42 O39 S2) calc’d: 2832.1 Da, found: 2832.2 Da; SS11 (C112 H182 N42 O37 S2) calc’d: 2773.1 Da, found: 2773.5 Da.
Peptide cross-linking
Intramolecular cross-linking of Cys residues in FZ21 peptide with BPDBS cross-linker was performed as follows: to a final concentration of 0.5 mM peptide in sodium phosphate buffer (50 mM, pH 8.5) was added tris(carboxyethyl)phosphine (10 mM), and the solution was stirred under nitrogen for 45 min to ensure that cysteine residues were in their reduced state. A solution of the cross-linker BPDB in DMSO was added to the reaction to a final concentration of 2 mM, and the solvent system for the reaction was adjusted to 15-20 % DMSO/water. The reaction mixture was allowed to stir at 45°C protected from light for 24 h. A similar protocol was used for intramolecular cross-linking of Cys residues in SS19, SS14 and SS11with BPDBS. The cross-linked peptides were HPLC-purified using the same conditions as described above and were characterized by ESI-MS: m/z calc’d for FZ21-BPDBS (C140H202N48O45S4): 3406.70 [M+H]+, found: 3405.69; SS19-BPDBS (C142H204N48O47S4): 3464.74 [M+H]+, found: 3463.40; m/z calc’d for SS14-BPDBS (C142H204N48O47S4): 3464.74 [M+H]+, found: 3463.60; m/z calc’d for SS11-BPDBS (C140H202N48O45S4): 3406.70 [M+H]+, found: 3405.47.
UV-Vis spectra and photoisomerization
UV-Vis absorption spectra were obtained using either a Perkin-Elmer Lambda 35 spectrophotometer or using a diode array UV-Vis spectrophotometer (Ocean Optics Inc., USB4000) coupled to a temperature controlled cuvette holder (Quantum Northwest, Inc. Spokane, WA, USA). The latter arrangement was used to measure thermal relaxation rates, and steady-state spectra under UV-Vis illumination. For steady state spectra, typically, a 8-10 μM solution of cross-linked peptide in 10 mM sodium phosphate buffer (pH 7.0), contained in a 1cm path length quartz cuvette in the temperature controlled cuvette holder, was irradiated (at 90° to the measuring beam) with LEDs emitting at 365 nm or 385 nm at 4°C, 20°C and 37°C for 5 min until no further decrease in absorbance was observed, and under this steady-state irradiation condition the spectra were recorded.
Rates of thermal reversion of the irradiated samples were measured for a series of temperatures by monitoring absorbance at 365 nm or 385 nm with time. All curves were fitted to mono-exponential and/or biexponential decay kinetics. The light used for the absorbance measurement was of sufficiently low intensity to cause negligible isomerization.
The concentrations of all cross-linked peptides were measured by using the molar extinction coefficient of cross-linkers, which were determined as follows: 1H NMR spectra of solutions of the cross-linkers BPDB and BPDBS in DMSO-d6 containing a known concentration of 1,2-dichloroethane (DCE) were recorded. By comparing the intensity of methylene signal of DCE with the cross-linker’s α-methylene signal, the concentration of the cross-linker in the NMR sample was determined. These NMR solutions were later diluted in DMSO for BPDB and in phosphate buffer (10 mM, pH 7.0) for BPDBS, and the UV-Vis spectra were recorded to calculate the molar extinction coefficient.
Circular dichroism measurements
Circular dichroism (CD) measurements were performed on an Olis RSM 1000 circular dichroism spectrophotometer with a Quantum Northwest Peltier accessory. 80-100 μM solutions of cross-linked peptides in 10 mM phosphate buffer (pH 7.0) contained in 1 mm path length quartz cuvette were used to record the CD spectra at 4°C, 20°C and 37°C. Tris(carboxyethyl)phosphine (1 mM) was used in the uncross-linked peptide solutions to ensure that cysteine residues were in their reduced form. Each spectrum was scanned from 260 to 190 nm with an integration time of 2 s at each wavelength. Spectra for non-irradiated samples were the average of two individual experiments of two scans each, with appropriate background spectrum subtracted. At different temperatures, the cross-linked peptides were irradiated for 5 min, and the CD spectra immediately recorded. In this case, the spectra were the average of two individual experiments of one scan each, with the appropriate background spectrum subtracted. Mean residual ellipticities were calculated using this standard equation: [θ]r = θ / (10 ncl); where θ is the measured ellipticity in mdeg, n is the number of backbone amide bonds, c is the concentration in M, and l is the pathlength in cm. The percentage of helix content was calculated by using this standard equation: % helicity = −100 n [θ]r, 222 /40 000 (n–4); where ‘n’ is the number of amide bonds in the peptide. [33] [θ]r, 222 is the measured ellipticity in mdeg at 222 nm. Peptide concentrations were determined using a value of ε = 50,000 M-1cm-1 at the absorption maximum of the cross-linked dark-adapted peptides. This value was used since the absorption spectrum of the cross-linker attached to a peptide was more similar to the free cross-linker in DMSO (λmax=380 nm, ε = 52,000 M-1cm-1) than that of the free cross-linker in phosphate buffer (λmax=360 nm, ε = 45,000 M-1cm-1).
Molecular modeling
Models of trans-trans, trans-cis and cis-cis cross-linkers were built using HyperChem (v.8, Hypercube Inc.) with the linker terminated with methyl groups representing the β carbon of Cys in the cross-linked peptide, and minimized using the Amber99 forcefield. Restraints were added to the azo bond for the trans-cis and cis-cis conformations (force constant 16). Molecular dynamics runs were performed in vacuo essentially as described previously [34] with a distant dependent dielectric, and 1–4 scale factors of 0.833 for electrostatic and 0.5 for van der Waals interactions, a step size of 1 fs and 300 K as the simulation temperature. Trajectories were analyzed to verify that numerous torsion angle changes occurred for all single bonds during the course of the simulation to ensure that conformational space was adequately sampled. All points histograms were then produced for the S–S distance during the full set of simulations for each isomer.
Acknowledgments
We wish to thank the Natural Sciences and Engineering Research Council of Canada, the Canadian Institutes of Health Research and the National Institutes of Health (U.S.A)(R01 MH086379) for financial support.
Contributor Information
Dr. Subhas Samanta, Department of Chemistry, University of Toronto, 80 St. George St., Toronto, ON, M5S 3H6, Canada
Prof. G. Andrew Woolley, Department of Chemistry, University of Toronto, 80 St. George St., Toronto, ON, M5S 3H6, Canada, awoolley@chem.utoronto.ca, telephone: (416) 978-0675, fax: (416) 978-8775
References
- 1.Garner J, Harding MM. Org Biomol Chem. 2007;5:3577–3585. doi: 10.1039/b710425a. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharya S, Zhang H, Debnath AK, Cowburn D. J Biol Chem. 2008;283:16274–16278. doi: 10.1074/jbc.C800048200. [DOI] [PMC free article] [PubMed] [Google Scholar]; Henchey LK, Jochim AL, Arora PS. Curr Opin Chem Biol. 2008;12:692–697. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, Kung AL, Gilliland DG, Verdine GL, Bradner JE. Nature. 2009;462:182–188. doi: 10.1038/nature08543. [DOI] [PMC free article] [PubMed] [Google Scholar]; Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. Mol Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 3.Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJ, Datta SR, Pitter KL, Bird GH, Wikstrom JD, Deeney JT, Robertson K, Morash J, Kulkarni A, Neschen S, Kim S, Greenberg ME, Corkey BE, Shirihai OS, Shulman GI, Lowell BB, Korsmeyer SJ. Nat Med. 2008;14:144–153. doi: 10.1038/nm1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renner C, Moroder L. ChemBioChem. 2006;7:868–878. doi: 10.1002/cbic.200500531. [DOI] [PubMed] [Google Scholar]
- 5.Woolley GA. Acc Chem Res. 2005;38:486–493. doi: 10.1021/ar040091v. [DOI] [PubMed] [Google Scholar]
- 6.Woolley GA, Jaikaran AS, Berezovski M, Calarco JP, Krylov SN, Smart OS, Kumita JR. Biochemistry. 2006;45:6075–6084. doi: 10.1021/bi060142r. [DOI] [PubMed] [Google Scholar]; Guerrero L, Smart OS, Weston CJ, Burns DC, Woolley GA, Allemann RK. Angew Chem Int Ed Engl. 2005;44:7778–7782. doi: 10.1002/anie.200502666. [DOI] [PubMed] [Google Scholar]; Guerrero L, Smart OS, Woolley GA, Allemann RK. J Am Chem Soc. 2005;127:15624–15629. doi: 10.1021/ja0550428. [DOI] [PubMed] [Google Scholar]; Kneissl S, Loveridge EJ, Williams C, Crump MP, Allemann RK. ChemBioChem. 2008;9:3046–3054. doi: 10.1002/cbic.200800502. [DOI] [PubMed] [Google Scholar]; Zhang F, Zarrine-Afsar A, Al-Abdul-Wahid MS, Prosser RS, Davidson AR, Woolley GA. J Am Chem Soc. 2009;131:2283–2289. doi: 10.1021/ja807938v. [DOI] [PubMed] [Google Scholar]; Schierling B, Noel AJ, Wende W, Hien le T, Volkov E, Kubareva E, Oretskaya T, Kokkinidis M, Rompp A, Spengler B, Pingoud A. Proc Natl Acad Sci U S A. 2010;107:1361–1366. doi: 10.1073/pnas.0909444107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang F, Timm KA, Arndt KM, Woolley GA. Angew Chem Int Ed Engl. 2010;49:3943–3946. doi: 10.1002/anie.201000909. [DOI] [PubMed] [Google Scholar]
- 8.Dias AR, Minas da Piedade ME, Martinho Simoes JA, Simoni JA, Teixeira C, Diogo HP, Meng-Yan Y, Pilcher G. J Chem Thermodynamics. 1992;24:439–447. [Google Scholar]; Dokic J, Gothe M, Wirth J, Peters MV, Schwarz J, Hecht S, Saalfrank P. J Phys Chem A. 2009;113:6763–6773. doi: 10.1021/jp9021344. [DOI] [PubMed] [Google Scholar]; Rau H. Angew Chem Int Ed Engl. 1973;12:224–235. [Google Scholar]
- 9.Burns DC, Zhang F, Woolley GA. Nat Protoc. 2007;2:251–258. doi: 10.1038/nprot.2007.21. [DOI] [PubMed] [Google Scholar]
- 10.Burns DC, Flint DG, Kumita JR, Feldman HJ, Serrano L, Zhang Z, Smart OS, Woolley GA. Biochemistry. 2004;43:15329–15338. doi: 10.1021/bi048152k. [DOI] [PubMed] [Google Scholar]
- 11.Flint DG, Kumita JR, Smart OS, Woolley GA. Chem Biol. 2002;9:391–397. doi: 10.1016/s1074-5521(02)00109-6. [DOI] [PubMed] [Google Scholar]
- 12.Kumita JR, Smart OS, Woolley GA. Proc Natl Acad Sci USA. 2000;97:3803–3808. doi: 10.1073/pnas.97.8.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang F, Sadovski O, Woolley GA. ChemBioChem. 2008;9:2147–2154. doi: 10.1002/cbic.200800196. [DOI] [PubMed] [Google Scholar]
- 14.Khan A, Hecht S. Chemistry. 2006;12:4764–4774. doi: 10.1002/chem.200501564. [DOI] [PubMed] [Google Scholar]; Yu Z, Hecht S. Angew Chem Int Ed Engl. 2011 [Google Scholar]
- 15.Kusebauch U, Cadamuro SA, Musiol HJ, Lenz MO, Wachtveitl J, Moroder L, Renner C. Angew Chem Int Ed Engl. 2006;45:7015–7018. doi: 10.1002/anie.200601432. [DOI] [PubMed] [Google Scholar]
- 16.Kuil J, van Wandelen LT, de Mol NJ, Liskamp RM. J Pept Sci. 2009;15:685–691. doi: 10.1002/psc.1173. [DOI] [PubMed] [Google Scholar]
- 17.Standaert RF, Park SB. J Org Chem. 2006;71:7952–7966. doi: 10.1021/jo060763q. [DOI] [PubMed] [Google Scholar]
- 18.Shinkai S, Manabe O. Top Curr Chem. 1984;121:67–104. [Google Scholar]; Tamaoki N, Ogata K, Koseki K, Yamaoka T. Tetrahedron. 1990;46:5931–5942. [Google Scholar]; Norikane Y, Katoh R, Tamaoki N. Chem Comm. 2008:1898–1900. doi: 10.1039/b718813g. [DOI] [PubMed] [Google Scholar]; Vogtle F, Muller WM, Muller U, Bauer M, Rissanen K. Angew Chem Int Ed Engl. 1993;32:1295–1297. [Google Scholar]; Rau H, Rottger D. Mol Cryst Liq Cryst Sci Tech A: Mol Cryst Liq Cryst. 1994;246:143–146. [Google Scholar]; Bencini A, Bernardo MA, Bianchi A, Ciampolini M, Fusi V, Nardi N, Parola AJ, Pina F, Valtancoli B. J Chem Soc Perkin Trans II. 1998:413–418. [Google Scholar]
- 19.May EM, Fono A. U. S. P. Office. CAplus. Otto B. May Inc.; U.S.A.: 1966. p. 4. [Google Scholar]; Claussen U. In: CAplus. Offen G, editor. Bayer, A.G.; Germany: 1982. p. 26. [Google Scholar]
- 20.Matejka L, Dusek K. Makromol Chem Macromol Chem Phys. 1981;182:3223–3236. [Google Scholar]
- 21.Cisnetti F, Ballardini R, Credi A, Gandolfi MT, Masiero S, Negri F, Pieraccini S, Spada GP. Chem Eur J. 2004;10:2011–2021. doi: 10.1002/chem.200305590. [DOI] [PubMed] [Google Scholar]
- 22.Ojala WH, Ojala CR, Gleason WB. Antiviral Chem Chemother. 1995;6:25–33. [Google Scholar]
- 23.Mal P, Breiner B, Rissanen K, Nitschke JR. Science. 2009;324:1697–1699. doi: 10.1126/science.1175313. [DOI] [PubMed] [Google Scholar]
- 24.Neelakantan L, Hartung WH. J Org Chem. 1959;24:1943–1948. [Google Scholar]
- 25.Knoll H. In: CRC Handbook of organic photochemistry and photobiology. 2. Horspool W, Lenci F, editors. CRC Press; Boca Raton: 2004. pp. 1–16. [Google Scholar]
- 26.Zhang Z, Burns DC, Kumita JR, Smart OS, Woolley GA. Bioconjug Chem. 2003;14:824–829. doi: 10.1021/bc0340161. [DOI] [PubMed] [Google Scholar]
- 27.Merutka G, Shalongo W, Stellwagen E. Biochemistry. 1991;30:4245–4248. doi: 10.1021/bi00231a020. [DOI] [PubMed] [Google Scholar]
- 28.Borisenko V, Woolley GA. J Photochem Photobiol A Chem. 2005;173:21–28. [Google Scholar]
- 29.Riener CK, Kada G, Gruber HJ. Anal Bioanal Chem. 2002;373:266–276. doi: 10.1007/s00216-002-1347-2. [DOI] [PubMed] [Google Scholar]
- 30.Barshop BA, Wrenn RF, Frieden C. Anal Biochem. 1983;130:134–145. doi: 10.1016/0003-2697(83)90660-7. [DOI] [PubMed] [Google Scholar]
- 31.Lopez-Mirabal HR, Winther JR. Biochim Biophys Acta. 2008;1783:629–640. doi: 10.1016/j.bbamcr.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 32.Ihalainen JA, Paoli B, Muff S, Backus EH, Bredenbeck J, Woolley GA, Caflisch A, Hamm P. Proc Natl Acad Sci U S A. 2008;105:9588–9593. doi: 10.1073/pnas.0712099105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson D, King D, Chmielewski J, Singh S, Schultz P. J Am Chem Soc. 1991;113:9391–9392. [Google Scholar]
- 34.Chi L, Sadovski O, Woolley GA. Bioconjug Chem. 2006;17:670–676. doi: 10.1021/bc050363u. [DOI] [PubMed] [Google Scholar]