

Abstract
Experimental autoimmune encephalomyelitis (EAE) is generally believed to be an autoimmune disease caused by myelin-specific Th1 and/or Th17 effector cells. The underlying cellular and molecular mechanisms are not fully understood. With mice deficient in IL-9 (IL-9−/−), we showed that IL-9 plays a critical role in EAE. Specifically, IL-9−/− mice were resistant to the induction of EAE both by immunization with PLP180-199 peptide in the presence of Complete Freund’s Adjuvant (CFA) and by adoptive transfer of PLP180-199 peptide-specific effector T cells from WT littermates. EAE-resistant IL-9−/− mice exhibited considerably fewer inflammatory infiltrates in the central nervous system, with lower levels of IL-17 and IFN-λ expression, than did WT littermates. Further studies revealed that null mutation of the IL-9 gene resulted in significantly lower levels of PLP180-199 peptide-specific IL-17 and IFN-λ production. Moreover, IL-9−/− memory/activated T cells decreased chemokine receptors CCR2, CCR5 and CCR6. Interestingly, IL-10 was significantly increased in IL-9−/− mice compared to WT littermates. Importantly, we found that IL-9 mediated Th17 cell differentiation triggers complex STAT signaling pathways.
Keywords: IL-9, Encephalitogenic T cells, STAT signaling pathway
Introduction
Experimental autoimmune encephalomyelitis (EAE), a CD4+ T cell-mediated inflammatory demyelinating disease of the central nervous system (CNS), serves as an experimental model of human multiple sclerosis (MS). EAE can be induced in mice by immunization with myelin antigens or by adoptive transfer of myelin-reactive CD4+ T cells. Recent studies have shown that the pathogenesis of EAE is associated mainly with Th17 cells [1, 2], although a role for IFN-γ producing Th1 cells cannot be absolutely excluded [3, 4]. Indeed, IL-17-deficient mice develop EAE with delayed onset and reduced severity [5, 6]. In humans, Th17 cells have been identified in the CNS of patients with MS [7-10] and a role for IL-17 in the development/pathogenesis of MS has been suggested [7-9, 11]. These findings raise the possibility of suppressing Th17 cells as a potential therapy for MS.
In addition to the compelling evidence that TGF-β and IL-6 in the context of TCR stimulation differentiate Th17 cells from naïve CD4+ T cells [12-16], a recent report indicates that TGF-β and IL-9 may also synergistically drive the differentiation of mouse Th17 cells in vitro [17]. Intriguingly, Th17 cells can produce IL-9, which in turn amplifies and expands differentiated Th17 cells [18]. These data suggest that IL-9 might promote Th17 differentiation, raising the possibility of IL-9 as a potential target for inhibition of Th17 development in vivo. However, data obtained from previous studies in EAE using IL-9R−/− mice are contradictory. One report demonstrated that IL-9R−/− was protective against the development of EAE [18], while another study showed that absence of IL-9 receptor weakens the suppressive activity of nTreg in vivo, leading to an increase in effector cells and worsening of EAE [17]. In addition, IL-9 has an effect on Th17 and Treg through activation of STAT3 and STAT5 signaling. Thus, the role of IL-9 in EAE remains unclear.
STATs play critical roles in controlling help/effector T cells differentiation [19]. Activation of STAT3 is essential for Th17 differentiation [20]; however, the cytokines and factors that influence the phosphorylation of STAT3 are not yet completely elucidated. IL-6 is recognized as a major promoter of STAT3 activation and recently it has been shown that IL-9 treatment also induces tyrosine phosphorylation of STAT3 in T cells, which favors the development of Th17 cells [17]. In human T cells, a point mutation changing YLPQ into YLPA in a region of the cytoplasmic domain of human IL-9Rα was found to greatly reduce IL-9-induced activation of STAT3 and expression of c-myc [21, 22]. These studies suggest an association between IL-9 and Th17 cell differentiation by influencing the phosphorylation of STATs, but which STAT is responsible for the IL-9 effect is unclear. More importantly, it is also not known whether IL-9 signaling is important for T cell activation and differentiation in the development and pathogenesis of EAE.
In the present study, we investigated the in vivo role of IL-9 in the activation and differentiation of CD4+ T cells in EAE. We show that IL-9−/− mice are resistant to EAE induction by reducing encephalitogenic T cells and inflammatory myeloid cell invasion into the CNS. The suppression of EAE in IL-9−/− mice is attributable to the down regulation of IL-17, IFN-γ, TNF-α, IL-12p70 and inhibition of chemokine receptors CCR2, CCR5 and in particular CCR6 in activated T cells, which are necessary for the migration of pathogenic T cells into the CNS. Importantly, we found that STAT1 and STAT3 are both responsible for IL-9 mediated promotion of Th17 differentiation.
Results
IL-9 deficiency causes resistance to EAE induction
To examine the role of IL-9 in the development of EAE, we induced EAE in mice lacking IL-9, which were generated by targeted disruption of the mouse IL-9 gene in embryonic stem cells [23]. IL-9−/− mice were immunized with PLP180-199 peptide in the presence of CFA and monitored for the development of EAE for 30 days. Surprisingly, IL-9−/− mice were resistant to developing clinical EAE. IL-9−/− mice had not only lower incidence of disease (5/20), but also a much milder clinical course (mean maximal disease grade 0.8 ± 0.5, n = 20) than WT littermates (incidence: 20/20; mean maximal disease grade: 2.5 ± 0.4, p<0.01). In addition, IL-9−/− mice showed a delay in disease onset compared with WT littermates (Fig. 1A).
Figure 1. Figure 1. IL-9-deficient mice are resistant to EAE.

A. EAE was induced in IL-9−/− and WT littermates by immunization with PLP180-199 peptide in CFA. IL-9−/− mice developed attenuated disease characterized by delayed onset, reduced severity and early recovery (n = 5 each group). Data are representative of at least three experiments of five mice per group. B. IL-9−/− and C. WT littermates (n = 5 each group) were immunized with PLP180-199 and daily injected with 10 ng of rIL-9 in 0.3ml PBS, starting at day 0 until day 30 p.i. Administration of rIL-9 restored susceptibility to EAE in IL-9−/− mice to a level similar to that in control mice, while in WT mice rIL-9 exacerbated the disease. Data shown are for five mice per group and representative of at least two experiments. D. Infiltration of leukocytes into the CNS of IL-9−/−, WT, as well as IL-9−/− and WT after rIL-9 treatment. Animals were sacrificed on day 15 p.i. (peak of disease), spinal cords were fixed (10% buffered formalin) and infiltration was assessed by H&E staining and visualized at ×10 magnifications in the arterial region. E. Mean scores of inflammation ± SD in the CNS of IL-9−/−, WT, as well as IL-9−/− and WT littermates after rIL-9 treatment (n =5 each group). *, p %lt; 0.05. F. The mean percentage of infiltrating cells ± SD in all acute lesions analyzed independently of the spinal cord are given in panel (n =5 each group). *, p < 0.05.
To verify that the absence of IL-9 was responsible for the resistance to EAE, we treated IL-9−/− mice daily with rIL-9, starting on day 0 through day 30 post immunization (p.i.) with PLP180-199. This treatment completely restored susceptibility to EAE in IL-9−/− mice (5/5, Fig. 1B). To further establish the role of IL-9 in EAE pathogenesis, we administered IL-9 into WT littermates daily for 30 days p.i. IL-9 treated WT littermates exhibited a tendency toward enhanced disease progression compared to untreated animals (Fig. 1C). Histopathological analysis of the spinal cords revealed that IL-9−/− EAE mice had fewer infiltrating inflammatory cells than EAE WT littermates, a situation that was reversed in knockout mice after treatment with rIL-9 (Fig. 1D). The difference between the pathological scores and numbers of cells per field infiltrating into the spinal cords of IL-9−/− and other groups of mice was significant (Fig. 1E, F).
IL-9 deficiency correlates with absence of CD4+ T cell infiltration and IL-17, IFN-γ expression in the CNS
We then examined the effect of IL-9 deficiency on CD4+ T cell infiltration and IL-17 expression in the CNS. Spinal cords were harvested at the peak of disease (15 days p.i.) to analyze CD4+ T cells with immunohistofluorescent staining. We observed many fewer CD4+ T cells in the CNS of IL-9−/− mice (Fig. 2B) compared with WT littermates (Fig. 2A). The absolute number of CD4+ T cells in the CNS of WT littermates was significantly greater than that of IL-9−/− mice. These results indicate that lower disease incidence and less severe disease in IL-9−/− mice are attributable to the absence of pathogenic CD4+ T cells in the CNS. Intracellular staining for the number of IL-17 producing cells was reduced in the spinal cord of IL-9−/− mice than in WT littermates (Fig. 2C). In contrast, an increase of Foxp3+ Treg cells was observed in the spinal cords of IL-9−/− mice (Fig. 2D).
Figure 2. IL-9−/− EAE mice have reduced numbers of CD4+ T cells and lower levels of IL-17 and IFN-γ in the CNS.

EAE was induced in IL-9−/− and WT littermates by immunization with PLP180-199 peptide. Animals were sacrificed at the peak of disease, spinal cords were fixed and tissue sections were immunostained for CD4+ T cells in: A. WT. B. IL-9−/− mice white matter spinal cord tissue sections of mice with EAE immunostained for CD4 (green). C. CD4+ T cells per spinal cord of mice with EAE were determined using flow cytometry. Data shown are representative of ten animals in each group, for IL-17 (C), and CD4+ Foxp3 (D). E. Spinal cord homogenates from IL-9−/− mice and WT control (n=5 per group) from a subsequent EAE were analyzed by ELISA for cytokines. Shown are the mean ± SEM of experiments performed in triplicate. F. IL-10 mRNA levels from spinal cords of WT vs. IL-9−/− mice. G. WT with different clinical scores, and the mean expression of IL-9 relative to β-actin determined by real-time PCR are shown. CD11b mRNA (H) and CD11c mRNA (I) levels from spinal cords of WT vs. IL-9−/− mice. Results are representative of five naive, ten WT EAE littermates and ten IL-9−/− EAE mice obtained from three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We then determined the inflammatory (IFN-γ, TNF-α, IL-12) and regulatory (IL-10, IL-4) cytokines in the CNS in IL-9−/− and WT littermates. IL-9−/− mice showed a significant decrease in production of IFN-γ, TNF-α and IL-12, but IL-10 levels were increased (Fig. 2E, F). Importantly, we observed that IL-9 was indeed expressed in the CNS of WT littermates during the clinical phase of disease, which was positively correlated with the development and severity of the clinical symptoms of EAE (Fig. 2G). IL-9 deficiency also resulted in downregulation of macrophage-DC genes in the spinal cords of IL-9−/− EAE mice compared with WT littermates (Fig. 2H, I).
Deletion of IL-9 results in reduction of antigen-specific T cell proliferation and encephalitogenic T production
To elucidate the mechanism underlying the failure of IL-9−/− mice to develop EAE, we investigated the antigen-specific T cell responses to PLP180-199. At day 10 p.i (prior to the onset of clinical disease), periphery lymph nodes cells were harvested and stimulated with PLP180-199, and the antigen-specific T cell proliferation was determined. IL-9−/− T cells showed significantly lower proliferative response than WT littermates T cells (Fig. 3A). Similar results were obtained when T cell proliferation was examined at day 30 p.i (data not shown). In light of this, the defect of antigen-specific T cell proliferation was restored in IL-9−/− mice and further enhanced in WT littermates, when mice were treated with exogenous rIL-9 (Fig. 3B). To address the question as to whether the decreased proliferative response was antigen specific or due to a generalized defect in the activation or function of T cells, lymph nodes cells were stimulated with the polyclonal activator Con A. Lymph nodes cells from IL-9−/− WT littermates showed no difference in proliferation in response to Con A (Fig. 3A).
Figure 3. Decreased PLP180-199-specific Th17 cell response and chemokine receptor expression.

A. Lymph node cells from PLP180-199-immunized IL-9−/− and WT littermates obtained 10 days p.i. were re-stimulated ex vivo with PLP180-199 peptide (10 μg/ml) or with ConA (2 μg/ml). Proliferation was determined by [3H] thymidine incorporation as described in Materials and Methods section. B. Lymph node cells from IL-9−/−, WT littermates, IL-9−/− + rIL-9, or WT + rIL-9 mice (n = 5 each group) were isolated 10 days p.i. in vitro with PLP180-199 peptide (10 μg/ml). Each bar represents the mean stimulation index ± SEM. C-E. Concentrations of IL-17, IFN-γ and IL-10 in supernatants collected at 48 hr (for IFN-γ), 72 hr (for IL-17) and 96 hr (for IL-10) from lymph node cells stimulated with PLP180-199 were assayed by ELISA. Data represent results (mean ± SD) of three independent experiments. *, p < 0.05; ***, p < 0.001. F. Lymph node cells were harvested from mice immunized with PLP180-199 at day 8 p.i. before onset of disease and CD62low CD4+ T cells stained with fluorescently conjugated anti-CC chemokine receptor antibodies. Data are representative of three separate experiments.
Consistent with the T cell proliferative responses, lymph node cells from IL-9−/− mice produced significantly lower levels of antigen-specific inflammatory IL-17 and IFN-γ than those from WT littermates when stimulated with PLP180-199 (Fig. 3C, D). However, the production of Th2 cytokine IL-10 (Fig. 3E) was significantly increased in IL-9−/− mice.
IL-9 deficiency down-regulates chemokine receptor expression of CD62L low CD4+T cells
Polarization of naïve CD4+ T cells into Th17, Th1, or Th2 phenotypes has been shown to modify surface expression of chemokine receptors and adhesion molecules [24-27]. To determine whether these molecules were differentially expressed under IL-9−/− conditions, we investigated surface chemokine receptors CCR2, CCR5, CCR6, as well as P-selectin and E-selectin. Expression of surface CCR2, CCR5 and CCR6 was decreased significantly in IL-9−/− CD62Llow CD4+ T cells compared with WT CD4+ T cells (Fig. 3F). In order to determine whether decreased expression was already present before immunization, we checked expression of CCR2, CCR5 and CCR6 on CD62L+ CD4+ T cells from naïve mice, However, no significant difference was observed in the proportions of CD62L+ CD4+ T cells of naïve mice expressing CCR2, CCR5 and CCR6, P-selectin and E-selectin between IL-9−/− and WT immunized mice (data not shown).
IL-9−/− mice display decreased EAE severity upon adoptive transfer of PLP180-199-specific Th17 cells
We then studied roles of IL-9 in the development of passive EAE. Enriched PLP-specific IL-17-producing T lymphocytes generated from WT mice were transferred into WT or IL-9−/− mice. IL-9−/− recipient mice showed a significantly lower degree of EAE disease severity in comparison to WT littermates (Fig. 4A) (p < 0.01), indicating that IL-9 expressed in the recipients was involved in the encephalitogenicity of transferred Th17 cells during the course of EAE (Fig. 4A).
Figure 4. IL-9 produced by Th17 cells is essential for their encephalitogenicity.
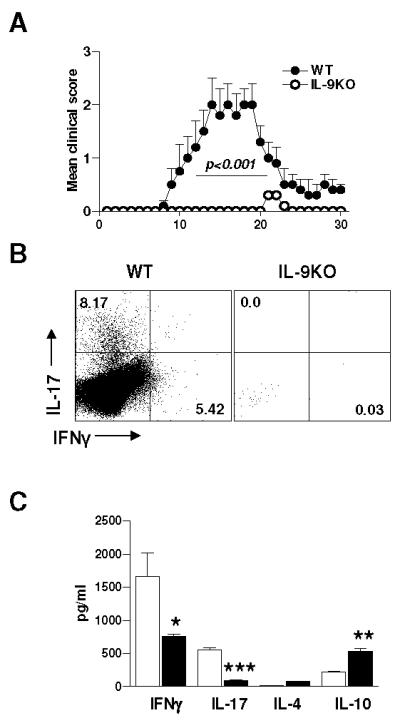
A. Passive EAE was induced in IL-9−/− and WT littermates (n=10) by adoptive transfer of IL-17 enriched PLP180-199-sensitized T cells. IL-17 enriched PLP180-199-sensitized T cells were i.v. transferred into IL-9−/− and WT littermates (5 × 106 cells /mouse; n= 5 per group). Mean clinical EAE scores were determined on a daily basis. B. Inflammatory cells were isolated by Percoll gradient from pooled spinal cords of IL-9−/− versus WT littermates (n=5) at EAE peak disease and were stained for CD4+CD8-IL-17+IFN-γ+. C. ELISA analysis of cytokines from lymph node cells stimulated with PLP180-199 was assayed. Results shown are mean ± SEM of triplicate experiments. PLP180-199 peptide add at 30ng/ml. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
At the peak of disease, analysis of spinal cord-infiltrating inflammatory cells was performed by flow cytometry to evaluate IL-17+IFN-γ+-producing T cells as well as surface markers of T-cell activation. The results showed a significant decrease of the IL-17+IFN-γ+-Tcell population in IL-9−/− mice as compared with WT littermates (Fig. 4B). We found consistently lower levels of IL-17, IFN-γ, and TNF-α and a higher level of IL-10 in the supernatants of antigen-specific cultures from IL-9−/− mice (Fig. 4C).
STAT1 and STAT3 regulate IL-9 mediated Th17 differentiation
Given that the critical role of IL-9 was originally described as a T cell growth factor[28-30], we next examined whether IL-9 deficiency had any effect on Th17 cell differentiation. Indeed, naïve IL-9−/− T cells showed significantly lower levels (three-fold decrease) of IL-17 production compared with T cells from WT littermates when cultured with IL-6 plus TGF-β (Fig. 5A).
Figure 5. STATs responsible for IL-9 mediated Th17 differentiation.

A. Naïve CD4+ T cells from naïve IL-9−/− and WT littermates were stimulated with anti-CD3 and anti-CD28 antibodies for 5 days in the presence of TGF-β (3 ng /ml), IL-6 (30 ng/ml) and anti-IL-4 (10 μg/ml). IL-17 production was determined by ELISA. B-D. Lymph node naïve CD4+ T cells from STAT1-, STAT3- and STAT5-deficient mice and naive WT littermates were stimulated with anti-CD3, anti-CD28 and TGF-β for 4 days in the presence or absence of IL-9 (10 ng/ml), cells were supplemented with recombinant IL-2(50U/ml) at day 2; IL-17 levels in the culture supernatant were measured by ELISA. Data are mean cytokine production ± SD of three mice and are representative of three independent experiments. *, p < 0.05; **, p < 0.01.
Because IL-9 has been reported to induce activation of a STAT complex containing STAT3 (Th17), STAT1 (Th1), and STAT5 (Treg) [31], we analyzed the function of STAT1, STAT3, and STAT5 in IL-17 production in response to IL-9. CD4+ T cells from naive STAT1-, STAT3-, STAT5-deficient or WT control mice were cultured with anti-CD3 and anti-CD28 in the presence or absence of IL-9. Exogenous IL-9 failed to further enhance IL-17 production in these STAT1-, STAT3- knockout T cells, while IL-9 significantly upregulated IL-17 in control T cells (Fig. 5 B, C). In contrast, IL-9 increased IL-17 production both in WT littermates and STAT5-deficient T cells in response to anti-CD3 and anti-CD28 Abs stimulation (Fig. 5D). Addition of exogenous IL-9 further enhanced IL-17 production in STAT5-deficient cells (Fig. 5D). Taken together, our results indicate that STAT1 and STAT3, but not STAT5, regulate IL-9 mediated IL-17 production in T cells.
Discussion
In this study, we investigated the role of IL-9 in T cell development in EAE using IL-9 knockout mice. We show that IL-9−/− mice are protected from clinical disease, and the suppression of EAE in IL-9−/− mice was largely reversed when treated with rIL-9. Thus, IL-9 is required for the development and pathogenesis of EAE.
Several important conclusions can be drawn from the current study. First, null mutation of IL-9 results in a defect in antigen-specific T cell proliferation. Evidence supporting this notion includes the finding that IL-9−/− T cells exhibit a substantial reduction of proliferation in response to PLP peptide restimulation compared to WT littermates. Second, IL-9−/− T cells fail to differentiate and express a cytokine profile conducive to disease. Indeed, T cells from IL-9−/− mice produce significantly lower levels of IL-17, IFN-γ, TNF-α and IL-12p70 and higher levels of IL-10 in the CNS. Third, IL-9−/− T cells lack appropriate chemokine signals to migrate into the CNS after stimulation by antigen. In mice, CCR2 [32] and CCR6 [33] chemokines play an important role in eliciting migratory responses of IL-17+ T cells [34]. Thus, the decrease in chemokine receptors such as CCR2 and CCR6 on activated CD4+ T cells in IL-9−/− mice may explain, at least in part, the decreased migration of Th17 cells in the CNS.
Moreover, IL-9 deficiency reduces the number of inflammatory myeloid cells, including macrophages, mast cells and dendritic cells, in the CNS and dampens the levels of their cytokine production, which may contribute to the decrease in EAE in IL-9−/− mice. It has been documented that invasion of myeloid cells and production of inflammatory mediators are the driving forces behind CNS inflammation and demyelination, the pathological hallmarks of EAE and MS [35]. Published studies have demonstrated that the IL-9 receptor is expressed on hematopoietic progenitors [36], inflammatory macrophages [37, 38], mast cells [28], and dendritic cells [39], and the direct positive effect of IL-9 on myeloid cell function has been documented. Our data are consistent with these studies and extend their findings by demonstrating an IL-9 positive upregulation of inflammatory innate immunity in EAE, suggesting multiple actions of IL-9 on encephalitogenic T cells as well as non-T cells in immunopathology.
The fact that antigen-specific Th17+ T cells fail to induce EAE in IL-9−/− mice highlights the important role of IL-9 enriched microenvironment in Th17 cell-mediated pathology. Although mechanistically elusive, our results suggest that IL-9 in the periphery, such as the lymph nodes, affects Th17 cell survival and/or expansion. IL-9 in the microenvironment can be produced by effector Th17, Th9 [40] and other cells [28, 41]. It is unclear whether IL-9 also affects Th1 and Th2 cells in vivo, but analysis of the effects will help elucidate the role of IL-9 in autoimmune diseases.
The resistance to EAE in IL-9−/− animals may also result from an active immunoregulatory mechanism induced by IL-10 production. This notion was supported by our observation that IL-10 production was upregulated during the entire course of the study. IL-10 and IL-4 are required for IL-9 expression by T cells [42, 43]. However, production of IL-5 and IL-13 is required for IL-9 [44, 45]. The cascade of cytokine production that is required for IL-9 may defect in deficiency mice. The question whether CD4+ T cells are polarized towards Th2 in the absence of IL-9 needs to be further addressed. Although it remains to be elucidated which type of cells produce IL-10 in IL-9−/− mice resistant to EAE, CD4+CD25+ regulatory T cells are an important candidate, given that IL-10 producing regulatory CD4+CD25+ T-cells suppress the induction of EAE [46, 47]. Nevertheless, this finding indicates that IL-9 may be a negative regulator for IL-10 production in the CNS.
Data presented herein are actually opposite to those previously reported by Elyaman W et al, using IL-9R KO mice [17]. The exact role of IL-9 in CNS inflammation will thus remain extremely controversial. Beriou G et al. demonstrated that higher frequency of memory CD4+ T cells can translate to IL-9+IL-17+ cells [48]. Whether IL-9 signals play a role can through IL-17 receptors remains to be elucidated. It is also possible there are additional ligands to IL-9R that could explain the difference in phenotype between the IL-9 KO and the IL-9R KO.
A role of STAT1 in IL-9 mediated Th17 differentiation provides a molecular mechanism for understanding IL-9 and Th17 in the pathogenesis of EAE [49]. Recently, chip-seq mapping of STAT1 binding revealed more than 11,000 sites in unstimulated human Hela cells and 40,000 sites following IFN-γ stimulation [50]. Indeed, STAT proteins bind to the IFN-γ activation site element, which is found to be necessary for IL-9-dependent activation of the promoter of this gene [49]. IFN-γ induces phosphorylation of STAT1 to a similar extent as human IL-9 but does not affect proliferation of transected murine lymphoma cells; however, IL-9 induces a sustained STAT1 phosphorylation, which affects cell growth associated with induction of a STAT3 target gene [51]. We show here that IL-9 affects Th17 cells not only through activation of STAT3 [52, 53], but also STAT1, suggesting that IL-9 triggers complex STAT signaling pathways.
We have provided evidence that IL-9 controls pathways central to the induction and development of EAE. Disruption of the IL-9 gene reduces inflammatory infiltrates in the CNS and attenuates clinical symptoms of EAE. Thus, our study has not only helped to further elucidate the molecular mechanism of IL-9 in inflammation and autoimmunity, but it has also suggested a potential target for human multiple sclerosis therapy.
Materials and Methods
Mice
STAT3−/−, STAT5−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME). STAT1−/−, mice were purchased from Taconic (Germantown, NY). Homozygous breeding pairs of IL-9−/− mice (N10 BALB/c), as previously described [23], were a kind gift from Dr. Noelle’s laboratories (Dartmouth Medical School, Lebanon, New Hampshire), and bred in the Thomas Jefferson University animal facility. For all experiments 8~10 week-old female mice were used. Throughout this study, both WT and heterozygous littermate mice were used for comparison with the deficient mice. Since no discrepancy was observed between the heterozygous and homozygous WT groups, hereafter they will both be referred to as WT littermates. All animals were housed under specific pathogen-free conditions and animal protocols were approved by the Thomas Jefferson University Animal Care and Use Committee. Paralyzed mice were afforded easy access to food and water.
Active EAE induction with myelin PLP180-199 peptide
EAE was induced with myelin PLP180-199 peptide (WTTCGSIAFPSKTSASIGSL; Louisville, KY) as described earlier [54]. Briefly, IL-9−/− mice and WT littermates (five to ten mice per group) were immunized subcutaneously (s.c) in the flanks with 200 μg of PLP180-199 peptide in 0.1 ml phosphate-buffered saline and 0.1 ml CFA containing 0.4 mg Mycobacterium tuberculosis (H37Ra; Difco Laboratories, Detroit, MI) and injected intravenously (i.v.) with 400 ng pertussis toxin (Sigma, St Louis, MO) on the day of immunization and 2 days later. EAE was scored as described previously: grade 1, piloerection; 2, limp tail or isolated weakness of gait without limp tail; 2.5, partial hind leg paralysis; grade 3, hind leg paralysis; grade 4, complete hind and fore limb paralysis; grade 5, moribund or death stage.
rIL-9 treatment
IL-9−/− and WT littermates immunized with PLP180-199 were injected intraperitoneally with 10 ng of rIL-9 (Akron Biotech, Philadelphia, PA) dissolved in 0.3 ml of PBS every day beginning on the day of PLP180-199 immunization until day 30. IL-9−/− and WT littermates receiving no treatment served as controls for these experiments.
Immunohistology
To assess the infiltration of immune cells in the CNS, IL-9−/− and WT littermates were euthanized 5 days after disease onset by CO2 asphyxiation. The lumbar region of spinal cords was removed on day 15 and stored in 10% buffered formalin. Paraffin-embedded 5 μm thick transverse sections of the spinal cord (six sections per mouse) were stained with haematoxylin and eosin (H&E) for infiltration of cells. Slides were assessed in a blind fashion for histology score, as documented previously [55]. Briefly, inflammation: 0, none; 1, a few inflammatory cells; 2, organization of perivascular infiltrates; and 3, increasing severity of perivascular cuffing with extension into the adjacent tissue.
Further, sections were stained with CD4 antibodies to determine the in vivo cytokine analysis in the CNS. Sections were incubated with CD4 (dilution 1:10) antibodies overnight at 4°C. The corresponding secondary horseradish peroxidase-labeled antibodies were added for 30 min at room temperature. Signals were amplified using a tyramide signal amplification kit per the manufacturer’s protocol.
Isolation of CNS mononuclear cells, ELISA, RT-PCR
MNCs from the CNS of PLP180-199-immunized mice were isolated by Percoll gradient centrifugation as previously described [55]. Briefly, mice were sacrificed and transcardially perfused with ice-cold GKN solution (2 g D−(+) glucose, 0.4 g KCl, 8 g NaCl, 3.56 g Na2 HPO4 12H20, and 0.78 g NaH2 PO4 2H20 in 1 L; pH 7.4) with 2 U/ml heparin (Sigma-Aldrich). Spinal cords were removed into GKN/0.02% BSA (w/v), mechanically dissociated through a 100-μmcell strainer, and enzymatically digested by incubation with 250 μg/ml collagenase/dispase and 250 μg/ml DNase I (Roche, Basel, Switzerland) at 37 °C for 20~30 min. The digested CNS preparation was washed with GKN/BSA, and the pellet was fractionated on a 70/37/30% Percoll gradient. Spinal cord homogenates from IL-9−/− mice and WT littermates from a subsequent EAE were analyzed by ELISA kit (R&D Systems) for TNF-α, IFN-γ,IL-12p70, IL-4, IL-10.
To determine IL-9, IL-10, CD11b, CD11c mRNA expression in the CNS, these cells were assayed using RT- PCR, with β-actin expression serving as control. Relative expression was calculated following the previously described protocol [55].
Recall responses
For evaluation of T-cell recall responses, lymph node cells from PLP180-199-immunized IL-9−/−, WT, IL-9−/− + rIL-9, or WT littermates + rIL-9 mice were harvested on day 10 p.i.; the cells were plated at a density of 5 ×105 cells/well in a 96-well plate in RPMI-complete in the presence or absence (background) of PLP180-199 (10 μg/ml) peptide or with Concanavalin A (Con A; 2 μg/ml). For proliferation, [3H] thymidine (1 μCi/well) was added at 60 hr and mean incorporation of thymidine in DNA was measured after 12 hr by 1450 Microbeta Wallac Trilux Liquid Scintillation Counter (Perkin Elmer Life Sciences, Foster City, CA).
Cytokine analysis
For cytokine assays, myelin PLP180-199-immunized lymph node cells (5 ×106/ml) were cultured in RPMI-complete with concentrations (10 μg/ml) of PLP180-199 peptide. Culture supernatants for IFN-γ were collected after 48hr, for IL-4 (eBioscience) and IL-17 were collected after 72 hr and for IL-10 at 96 hr of culture. Cytokines were measured by ELISA kit (R&D Systems) per the manufacturer’s instructions.
Chemokine receptor analysis
MNCs from lymph node cells of WT littermates and IL-9−/− mice were isolated at day 8 p.i. For flow cytometry, these cells were incubated for 20 mins in the dark at 4oC with Abs CD4, CD62L, CCR2 (Abcam, Cambridge, MA), CCR5, and CCR6 (BD PharMingen, San Diego, CA).
Adoptive EAE induction
Female 8~12 week-old IL-9−/− and WT littermates were immunized in the footpads with 100 μg of PLP180-199 in CFA containing 5 mg/ml Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI) on days 0 and 7. Lymph node cells were harvested on day 9 post-immunization (p.i) and cultured for 3 days in RPMI 1640 supplemented with 10% FCS, penicillin/streptomycin, L-glutamine, HEPES, sodium pyruvate and 2-ME. All cells were cultured in the presence of PLP180-199 (20 μg/ml), IL-23 (10 ng/ml) or PBS as a control. After 72 hr, cells were harvested, washed in PBS and transferred to naïve recipient mice (5×106 cells/mouse) via the tail vein. Mice were given two doses (400 ng/mouse) of pertussis toxin i.v. on days 0 and 2 and EAE disease induction was assessed as above.
In vitro Th17 cell differentiation
Lymph nodes were removed from IL-9−/−, STAT1−/−, STAT3−/−, STAT5−/−, WT littermates and passed through a 100μm cell strainer. Erythrocytes were lysed and cell numbers were determined. Naïve CD4+ T cells were purified using magnetic bead separation kits (Miltenyi Biotec, CA) and sorted by flow cytometry. Cells were cultured with anti-CD3 (1 μg/ml), anti-CD28 (1 μg/ml) (BD Pharmingen, San Jose, CA) and TGF-β (3 ng /ml) (Invitrogen) in a serumfree medium (X-VIVO-20; Lonza) in the presence or absence of recombinant mouse IL-9 (10 ng/ml) (Akron Biotech, Philadelphia, PA). Th17 cells were generated with TGF-β (3 ng /ml), IL-6 (30 ng/ml) and anti-IL-4 (10 μg/ml) (all from Invitrogen).
Statistics
Data sets were tested for statistical significance using unpaired, 2-tailed, Student’s t tests (for parametric data) or Mann-Whitney tests (for non-parametric data). Differences were considered significant if p<0.05. Data were analyzed and represented graphically using Prism software (GraphPad Software Inc., San Diego, CA).
Supplementary Material
Acknowledgments
The authors thank Randolph J. Noelle for providing IL-9−/− mice and Katherine Regan for editorial assistance.
This work was supported by grants from the National Institutes of Health and the National Multiple Sclerosis Society.
Abbreviations
- PLP
myelin proteolipid protein
- CCR
C-C chemokine receptors
- STAT
signal transducer and activator of transcription
- MNC
monoclear cells
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol. 2007;19:353–361. doi: 10.1016/j.smim.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinman L. A rush to judgment on Th17. J Exp Med. 2008;205:1517–1522. doi: 10.1084/jem.20072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 6.Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81:1258–1268. doi: 10.1189/jlb.1006610. [DOI] [PubMed] [Google Scholar]
- 7.Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, Duquette P, Prat A. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol. 2009;66:390–402. doi: 10.1002/ana.21748. [DOI] [PubMed] [Google Scholar]
- 8.Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montes M, Zhang X, Berthelot L, Laplaud DA, Brouard S, Jin J, Rogan S, Armao D, Jewells V, Soulillou JP, Markovic-Plese S. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin Immunol. 2009;130:133–144. doi: 10.1016/j.clim.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 12.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 13.Dong C. Differentiation and function of pro-inflammatory Th17 cells. Microbes Infect. 2009;11:584–588. doi: 10.1016/j.micinf.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 15.Zhou L, Littman DR. Transcriptional regulatory networks in Th17 cell differentiation. Curr Opin Immunol. 2009;21:146–152. doi: 10.1016/j.coi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Quinn DB, Palmer MT, Lee YK, Weaver CT. Emergence of the Th17 pathway and its role in host defense. Adv Immunol. 2008;99:115–163. doi: 10.1016/S0065-2776(08)00605-6. [DOI] [PubMed] [Google Scholar]
- 17.Elyaman W, Bradshaw EM, Uyttenhove C, Dardalhon V, Awasthi A, Imitola J, Bettelli E, Oukka M, van Snick J, Renauld JC, Kuchroo VK, Khoury SJ. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0812530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nowak EC, Weaver CT, Turner H, Begum-Haque S, Becher B, Schreiner B, Coyle AJ, Kasper LH, Noelle RJ. IL-9 as a mediator of Th17-driven inflammatory disease. J Exp Med. 2009;206:1653–1660. doi: 10.1084/jem.20090246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adamson AS, Collins K, Laurence A, O’Shea JJ. The Current STATus of lymphocyte signaling: new roles for old players. Curr Opin Immunol. 2009;21:161–166. doi: 10.1016/j.coi.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, Housseau F, Yu H, Pardoll DM, Drake CG. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Rostami A. IL-9: Basic Biology, Signaling Pathways in CD4+ T Cells and Implications for Autoimmunity. J Neuroimmune Pharmacol. 2009 doi: 10.1007/s11481-009-9186-y. [DOI] [PubMed] [Google Scholar]
- 22.Zhu YX, Sun HB, Tsang ML, McMahel J, Grigsby S, Yin T, Yang YC. Critical cytoplasmic domains of human interleukin-9 receptor alpha chain in interleukin-9-mediated cell proliferation and signal transduction. J Biol Chem. 1997;272:21334–21340. doi: 10.1074/jbc.272.34.21334. [DOI] [PubMed] [Google Scholar]
- 23.McMillan SJ, Bishop B, Townsend MJ, McKenzie AN, Lloyd CM. The absence of interleukin 9 does not affect the development of allergen-induced pulmonary inflammation nor airway hyperreactivity. J Exp Med. 2002;195:51–57. doi: 10.1084/jem.20011732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, Sakaguchi N, Sakaguchi S. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204:2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loetscher P, Uguccioni M, Bordoli L, Baggiolini M, Moser B, Chizzolini C, Dayer JM. CCR5 is characteristic of Th1 lymphocytes. Nature. 1998;391:344–345. doi: 10.1038/34814. [DOI] [PubMed] [Google Scholar]
- 27.Sato W, Aranami T, Yamamura T. Cutting edge: Human Th17 cells are identified as bearing CCR2+CCR5- phenotype. J Immunol. 2007;178:7525–7529. doi: 10.4049/jimmunol.178.12.7525. [DOI] [PubMed] [Google Scholar]
- 28.Hultner L, Kolsch S, Stassen M, Kaspers U, Kremer JP, Mailhammer R, Moeller J, Broszeit H, Schmitt E. In activated mast cells, IL-1 up-regulates the production of several Th2-related cytokines including IL-9. J Immunol. 2000;164:5556–5563. doi: 10.4049/jimmunol.164.11.5556. [DOI] [PubMed] [Google Scholar]
- 29.Renauld JC, Goethals A, Houssiau F, Merz H, Van Roost E, Van Snick J. Human P40/IL-9. Expression in activated CD4+ T cells, genomic organization, and comparison with the mouse gene. J Immunol. 1990;144:4235–4241. [PubMed] [Google Scholar]
- 30.Uyttenhove C, Coulie PG, Van Snick J. T cell growth and differentiation induced by interleukin-HP1/IL-6, the murine hybridoma/plasmacytoma growth factor. J Exp Med. 1988;167:1417–1427. doi: 10.1084/jem.167.4.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demoulin JB, Van Roost E, Stevens M, Groner B, Renauld JC. Distinct roles for STAT1, STAT3, and STAT5 in differentiation gene induction and apoptosis inhibition by interleukin-9. J Biol Chem. 1999;274:25855–25861. doi: 10.1074/jbc.274.36.25855. [DOI] [PubMed] [Google Scholar]
- 32.Aranami T, Yamamura T. Th17 Cells and autoimmune encephalomyelitis (EAE/MS) Allergol Int. 2008;57:115–120. doi: 10.2332/allergolint.R-07-159. [DOI] [PubMed] [Google Scholar]
- 33.Singh SP, Zhang HH, Foley JF, Hedrick MN, Farber JM. Human T cells that are able to produce IL-17 express the chemokine receptor CCR6. J Immunol. 2008;180:214–221. doi: 10.4049/jimmunol.180.1.214. [DOI] [PubMed] [Google Scholar]
- 34.Webb A, Johnson A, Fortunato M, Platt A, Crabbe T, Christie MI, Watt GF, Ward SG, Jopling LA. Evidence for PI-3K-dependent migration of Th17-polarized cells in response to CCR2 and CCR6 agonists. J Leukoc Biol. 2008;84:1202–1212. doi: 10.1189/jlb.0408234. [DOI] [PubMed] [Google Scholar]
- 35.El Behi M, Dubucquoi S, Lefranc D, Zephir H, De Seze J, Vermersch P, Prin L. New insights into cell responses involved in experimental autoimmune encephalomyelitis and multiple sclerosis. Immunol Lett. 2005;96:11–26. doi: 10.1016/j.imlet.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 36.Holbrook ST, Ohls RK, Schibler KR, Yang YC, Christensen RD. Effect of interleukin-9 on clonogenic maturation and cell-cycle status of fetal and adult hematopoietic progenitors. Blood. 1991;77:2129–2134. [PubMed] [Google Scholar]
- 37.Druez C, Coulie P, Uyttenhove C, Van Snick J. Functional and biochemical characterization of mouse P40/IL-9 recetors. J Immunol. 1990;145:2494–2499. [PubMed] [Google Scholar]
- 38.Pilette C, Ouadrhiri Y, Van Snick J, Renauld JC, Staquet P, Vaerman JP, Sibille Y. Oxidative burst in lipopolysaccharide-activated human alveolar macrophages is inhibited by interleukin-9. Eur Respir J. 2002;20:1198–1205. doi: 10.1183/09031936.02.00005402. [DOI] [PubMed] [Google Scholar]
- 39.Mohamadzadeh M, Ariizumi K, Sugamura K, Bergstresser PR, Takashima A. Expression of the common cytokine receptor gamma chain by murine dendritic cells including epidermal Langerhans cells. Eur J Immunol. 1996;26:156–160. doi: 10.1002/eji.1830260124. [DOI] [PubMed] [Google Scholar]
- 40.Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, Khoury S, Oukka M, Kuchroo VK. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(−) effector T cells. Nat Immunol. 2008;9:1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soussi-Gounni A, Kontolemos M, Hamid Q. Role of IL-9 in the pathophysiology of allergic diseases. J Allergy Clin Immunol. 2001;107:575–582. doi: 10.1067/mai.2001.114238. [DOI] [PubMed] [Google Scholar]
- 42.Houssiau FA, Schandene L, Stevens M, Cambiaso C, Goldman M, van Snick J, Renauld JC. A cascade of cytokines is responsible for IL-9 expression in human T cells. Involvement of IL-2, IL-4, and IL-10. J Immunol. 1995;154:2624–2630. [PubMed] [Google Scholar]
- 43.Monteyne P, Renauld JC, Van Broeck J, Dunne DW, Brombacher F, Coutelier JP. IL-4-independent regulation of in vivo IL-9 expression. J Immunol. 1997;159:2616–2623. [PubMed] [Google Scholar]
- 44.Poulin LF, Habran C, Stordeur P, Goldman M, McKenzie A, Van Snick J, Renauld JC, Braun MY. Interleukin-9 stimulates the production of interleukin-5 in CD4+ T cells. Eur Cytokine Netw. 2005;16:233–239. [PubMed] [Google Scholar]
- 45.Steenwinckel V, Louahed J, Lemaire MM, Sommereyns C, Warnier G, McKenzie A, Brombacher F, Van Snick J, Renauld JC. IL-9 promotes IL-13-dependent paneth cell hyperplasia and up-regulation of innate immunity mediators in intestinal mucosa. J Immunol. 2009;182:4737–4743. doi: 10.4049/jimmunol.0801941. [DOI] [PubMed] [Google Scholar]
- 46.Burkhart C, Liu GY, Anderton SM, Metzler B, Wraith DC. Peptide-induced T cell regulation of experimental autoimmune encephalomyelitis: a role for IL-10. Int Immunol. 1999;11:1625–1634. doi: 10.1093/intimm/11.10.1625. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, Weiner HL. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 48.Beriou G, Bradshaw EM, Lozano E, Costantino CM, Hastings WD, Orban T, Elyaman W, Khoury SJ, Kuchroo VK, Baecher-Allan C, Hafler DA. TGF-beta induces IL-9 production from human Th17 cells. J Immunol. 185:46–54. doi: 10.4049/jimmunol.1000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Demoulin JB, Uyttenhove C, Van Roost E, DeLestre B, Donckers D, Van Snick J, Renauld JC. A single tyrosine of the interleukin-9 (IL-9) receptor is required for STAT activation, antiapoptotic activity, and growth regulation by IL-9. Mol Cell Biol. 1996;16:4710–4716. doi: 10.1128/mcb.16.9.4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 51.Demoulin JB, Van Snick J, Renauld JC. Interleukin-9 (IL-9) induces cell growth arrest associated with sustained signal transducer and activator of transcription activation in lymphoma cells overexpressing the IL-9 receptor. Cell Growth Differ. 2001;12:169–174. [PubMed] [Google Scholar]
- 52.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 53.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 54.Lyons JA, Ramsbottom MJ, Trotter JL, Cross AH. Identification of the encephalitogenic epitopes of CNS proteolipid protein in BALB/c mice. J Autoimmun. 2002;19:195–201. doi: 10.1006/jaut.2002.0619. [DOI] [PubMed] [Google Scholar]
- 55.Li H, Zhang GX, Chen Y, Xu H, Fitzgerald DC, Zhao Z, Rostami A. CD11c+CD11b+ dendritic cells play an important role in intravenous tolerance and the suppression of experimental autoimmune encephalomyelitis. J Immunol. 2008;181:2483–2493. doi: 10.4049/jimmunol.181.4.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.