

Abstract
Modulating molecular chaperones is emerging as an attractive approach to treat neurodegenerative diseases associated with protein aggregation, DPN (diabetic peripheral neuropathy) and possibly, demyelinating neuropathies. KU-32 [N-(7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyl-tetrahydro-2H-pyran-2-yloxy)-8-methyl-2-oxo-2H-chromen-3-yl)acetamide] is a small molecule inhibitor of Hsp90 (heat shock protein 90) and reverses sensory deficits associated with myelinated fibre dysfunction in DPN. Additionally, KU-32 prevented the loss of myelinated internodes induced by treating myelinated SC (Schwann cell)-DRG (dorsal root ganglia) sensory neuron co-cultures with NRG1 (neuregulin-1 Type 1). Since KU-32 decreased NRG1-induced demyelination in an Hsp70-dependent manner, the goal of the current study was to clarify how Hsp70 may be mechanistically linked to preventing demyelination. The activation of p42/p44 MAPK (mitogen-activated protein kinase) and induction of the transcription factor c-Jun serve as negative regulators of myelination. NRG1 activated MAPK, induced c-Jun expression and promoted a loss of myelin segments in DRG explants isolated from both WT (wild-type) and Hsp70 KO (knockout) mice. Although KU-32 did not block the activation of MAPK, it blocked c-Jun induction and protected against a loss of myelinated segments in WT mice. In contrast, KU-32 did not prevent the NRG1-dependent induction of c-Jun and loss of myelin segments in explants from Hsp70 KO mice. Overexpression of Hsp70 in myelinated DRG explants prepared from WT or Hsp70 KO mice was sufficient to block the induction of c-Jun and the loss of myelin segments induced by NRG1. Lastly, inhibiting the proteasome prevented KU-32 from decreasing c-Jun levels. Collectively, these data support that Hsp70 induction is sufficient to prevent NRG1-induced demyelination by enhancing the proteasomal degradation of c-Jun.
Keywords: diabetic neuropathy, molecular chaperones, myelin basic protein, Schwann cells, sensory neurons
Abbreviations: CHIP, C-terminus Hsp70-interacting protein; CMT1, Charcot–Marie–Tooth disease type 1; DAPI, 4,6-diamidino-2-phenylindole; DMEM, Dulbecco's modified Eagle's medium; DPN, diabetic peripheral neuropathy; DRG, dorsal root ganglia; FCS, fetal calf serum; HRP, horseradish peroxidise; HS, heat shock; Hsc70, heat-shock cognate 70 stress protein; HSF1, heat shock factor 1; Hsp, heat shock protein; HSR, heat shock response; JNK, c-Jun N-terminal kinase; KO, knockout; KU-32, N-(7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyl-tetrahydro-2H-pyran-2-yloxy)-8-methyl-2-oxo-2H-chromen-3-yl)acetamide; mAb, monoclonal antibody; MAPK, mitogen-activated protein kinase; MBP, myelin basic protein; NRG1, neuregulin-1 Type 1; pAb, polyclonal antibody; PBST, phosphate-buffered saline containing 0.1% Tween 20; PGP9.5, protein gene product 9.5; phospho-c-Jun, phosphorylated c-Jun; PMP22, peripheral myelin protein 22; SC, Schwann cell; WT, wild-type
INTRODUCTION
Molecular chaperones, such as Hsp90 (heat shock protein 90) and Hsp70, are essential for the folding of nascent polypeptides (client proteins) into their biologically active structures. The binding of ATP to the N-terminal nucleotide-binding domain of Hsp90 promotes dimerization of the N-termini and clamping around the bound client protein. Subsequent nucleotide hydrolysis by the chaperone's intrinsic ATPase activity provides the energy necessary for conformational changes that facilitate folding and maturation of the client (Hartl et al., 2011). N-terminal Hsp90 inhibitors function by blocking ATP-mediated dimerization, which destabilizes the complex and results in client protein degradation via the ubiquitin–proteasome pathway, producing toxicity. However, induction of client protein degradation and cytotoxicity can occur at drug concentrations that also activate an antagonistic aspect of Hsp90 biology, induction of the cytoprotective HSR (heat shock response).
Chaperones are also essential for the refolding of aggregated, damaged and denatured proteins. Under normal conditions, Hsp90 binds HSF1 (heat shock factor 1) and this complex renders HSF1 inactive. However, upon exposure to stress, the Hsp90–HSF1 complex can disassemble and release HSF1 (Neef et al., 2011). Phosphorylation of monomeric HSF1 stimulates its trimerization and the transcriptional up-regulation of the HSR, characterized in part by increased expression of Hsp27, Hsp40, Hsp70, Hsp90 and antioxidant genes. Similar to disruption of the Hsp90–HSF1 complex by cellular stress, small molecule inhibitors of Hsp90 can mimic this dissociation and also induce a HSR (Blagg and Kerr, 2006).
The ability of Hsp90 inhibitors to promote Hsp70 expression and decrease protein aggregation has been proposed as a potential approach to treat neurodegenerative diseases associated with protein mis-folding. In this regard, N-terminal Hsp90 inhibitors can decrease protein aggregation in Alzheimer's (Dou et al., 2003; Dickey et al., 2007a), Parkinson's (Shen et al., 2005) and Huntington's disease models (Fujikake et al., 2008), as well as improve motor function in spinal and bulbar muscular atrophy (Waza et al., 2005). Similarly, an N-terminal Hsp90 inhibitor increased Hsp70 expression, improved the processing of aggregated peripheral myelin protein 22 and increased myelination of DRG (dorsal root ganglia) explants prepared from a mouse model of CMT1 (Charcot–Marie–Tooth disease type 1), a prevalent demyelinating neuropathy (Rangaraju et al., 2008). Unfortunately, a limitation of many N-terminal Hsp90 inhibitors is their narrow therapeutic window that dissociates the cytotoxic effects of client protein degradation from the cytoprotective effects of chaperone induction (Rangaraju et al., 2008).
Hsp90 also contains a C-terminal nucleotide-binding domain that can be weakly inhibited with novobiocin. Similar to N-terminal inhibitors, structural modification of novobiocin has resulted in a class of inhibitors that also promote cytotoxicity (Donnelly and Blagg, 2008; Samadi et al., 2011). However, we have developed a novel class of novobiocin-based C-terminal Hsp90 inhibitors (novologues) which circumvent this issue and exhibit robust neuroprotection of primary neurons in the absence of cytotoxicity (Ansar et al., 2007; Kusuma et al., 2012). KU-32 [N-(7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyl-tetrahydro-2H-pyran-2-yloxy)-8-methyl-2-oxo-2H-chromen-3-yl) acetamide] is a second generation novologue that also shows efficacy in ameliorating neurodegeneration associated with DPN (diabetic peripheral neuropathy) (Urban et al., 2010, 2012). Mechanistically, this protection is linked to induction of Hsp70 since KU-32 was ineffective in reversing clinically relevant indices of DPN in mice lacking the inducible forms of Hsp70 (Hsp70.1 and Hsp70.3). Thus, modulating molecular chaperones such as Hsp90 and Hsp70 offers a powerful approach to ameliorate neurodegenerative disease, even if its onset, i.e., DPN, is not causally linked to any one specific mis-folded or aggregated protein.
DPN affects both non-myelinated and myelinated sensory nerves (Farmer et al., 2012) and modulating Hsp90 with KU-32 reversed the loss and improved the innervation of unmyelinated intra-epidermal nerve fibres in diabetic mice (Urban et al., 2012). KU-32 also protected unmyelinated, embryonic sensory neurons from glucose-induced death (Urban et al., 2010; Kusuma et al., 2012). Moreover, modulating chaperone expression seems sufficient to aid myelinated fibres since KU-32 improved motor nerve conduction velocity and prevented neuregulin-induced demyelination (Urban et al., 2010).
NRG1 (neuregulin-1 Type 1) forms a family of EGF (epidermal growth factor)-like ligands that signal through Erb B receptors (Esper et al., 2006), which localize primarily to SCs (Schwann cells) in peripheral nerve (Grinspan et al., 1996). NRG1 isoforms have complex effects on SC biology and are critical for promoting peripheral nerve myelination (Taveggia et al., 2005; Nave and Salzer, 2006). However, the pro-myelinating effects of neuregulins are influenced by their concentration and neuregulins may also contribute to demyelination if their expression becomes enhanced under pathological conditions (Zanazzi et al., 2001; Syed et al., 2010). Indeed, pathological activation of Erb B2 can increase demyelination (Guertin et al., 2005; Tapinos et al., 2006) and inhibiting Erb B2 can improve some features of DPN modelled in diabetic mice (McGuire et al., 2009). Additionally, hyperglycaemia can increase the extent of NRG1-induced demyelination (Yu et al., 2008). Although it remains unclear if increased Erb B2 activation contributes to demyelination in human DPN and other neuropathies, compounds which interfere with common downstream components that contribute to demyelination may be of potential benefit in treating diseases that degrade the integrity of the myelin sheath in peripheral nerve.
Our previous work demonstrated that KU-32 decreased NRG1-induced demyelination in an Hsp70-dependent manner (Urban et al., 2010). However, it remained unclear how Hsp70 induction may be mechanistically linked to preventing demyelination. The activation of p42/p44 MAPK (mitogen-activated protein kinase) (Harrisingh et al., 2004; Ogata et al., 2004; Syed et al., 2010; Napoli et al., 2012) and induction of the transcription factor c-Jun (Parkinson et al., 2008) function as negative regulators of myelination (Jessen and Mirsky, 2008). The data herein provide evidence that the induction of Hsp70 by KU-32 is necessary to prevent NRG1-induced demyelination by blocking c-Jun expression and phosphorylation. Re-expression of Hsp70 on the null background was sufficient to block the induction of c-Jun following NRG1 treatment and inhibiting the proteasome negated the protective effect of KU-32 on preventing c-Jun induction. Given the role of altered signalling through the NRG1–Erb B2 ligand/receptor pair in promoting demyelination, these data suggest that molecular chaperones may antagonize negative regulators of myelination and be of potential benefit in treating demyelinating neuropathies.
MATERIALS AND METHODS
DMEM (Dulbecco's modified Eagle's medium) was obtained from Mediatech. MG132 was purchased from Tocris. Ascorbic acid and collagen were obtained from Sigma–Aldrich. KU-32 was synthesized and structural purity verified (>95%) as described (Burlison et al., 2006; Donnelly and Blagg, 2008). Mouse mAbs (monoclonal antibodies) against Hsp70, Hsc70 (heat-shock cognate 70 stress protein), Hsp90 as well as rabbit pAb (polyclonal antibody) for Hsp40 were purchased from Stressgen (Enzo Life Sciences). Hsp27 goat pAb, c-Jun rabbit pAb, JNK (c-Jun N-terminal kinase) mouse pAb, phospho-JNK mouse mAb and all HRP (horseradish peroxidase)-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology. Phospho-c-Jun (phosphorylated c-Jun) rabbit mAb was from Cell Signaling Technology. β-actin mouse mAb was from MP Biologicals. S100β rabbit pAb was purchased from Dako Cytomation. Mouse mAb against MBP (myelin basic protein) (SMI-94R) was from Covance and rabbit pAb detecting PGP9.5 (protein gene product 9.5) was purchased from Chemicon. Alexa Fluor®-conjugated secondary antibodies were all obtained from Molecular Probes.
Preparation of neonatal sensory neuron/SC cultures
Breeding colonies of WT (wild-type) C57Bl/6 and Hsp70.1/70.3 double KO (knockout; Hsp70 KO) mice on a C57Bl/6 background (Hunt et al., 2004) were maintained using mice initially purchased from Harlan Laboratories and the Mutant Mouse Resource Center respectively. Absence of Hsp70.1 and 70.3 was confirmed by genotyping of genomic DNA and corroborated by lack of inducible Hsp70 protein expression as determined by immunoblot analysis. Hsp70 primers (forward: GTACACTTTAAACTCCCTCC; reverse: CTGCTTCTCTTGTCTTCG) amplified a 450 bp band while primers against the neo cassette (forward: ATGGGATCGGCC-ATTGAACAAG; reverse: ACTCGTCAAGAAGGCGATAGAAGG) amplified a 650 bp band. The PCR conditions were (94°C for 5 min; 35 cycles of 94°C for 40 s; 65°C for 1 min; 72°C for 40 s; 72°C for 5 min) using KlenTaq polymerase (DNA Polymerase Technology) and 200–300 ng of genomic DNA template.
DRG were dissected from neonatal (P0–P1) C57Bl/6 or Hsp70 KO pups and placed into L15 medium (Yu et al., 2008). Following dissociation of tissues using 0.25% trypsin and 0.5% collagenase at 37°C for 30 min, cells were collected by centrifugation for 5 min at 1000 g and resuspended in DMEM containing 25 mM glucose, 10% FCS (fetal calf serum; Atlas Biologicals). The cells were triturated with a fire polished glass pipette and 6–7×104 cells were seeded on to collagen-coated glass coverslips or dishes. The cultures were maintained in DMEM maintenance medium containing 25 mM glucose, 10% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin, 50 μM gentamycin (MP Biologicals) and 50 ng/ml nerve growth factor (Harlan Biosciences). Fibroblasts were removed by treating the cells with 10 μM cytosine β-d-arabinoside for 2 days and the cultures then maintained in maintenance medium for 1 week to allow SC proliferation and association with axons.
Myelination was initiated by adding freshly prepared ascorbic acid (50 μg/ml in maintenance medium) to induce basal lamina formation. Myelination progressed for 18–21 days with the medium being changed every 2–3 days and fresh ascorbate added. Demyelination was induced by the addition of 150–200 ng/ml neuregulin-1-β1 epidermal growth factor domain (amino acids 176–246) (NRG1, R&D Systems) for 48–72 h. To examine the effect of KU-32 on preventing demyelination, the cells were incubated for 16 h with 0.05% DMSO (vehicle) or 1 μM KU-32 prior to adding NRG1; demyelination was assessed 48–72 h after adding NRG1.
HS (heat shock) treatment and immunoblot analyses
For HS treatment, cell culture plates were sealed and placed into a 43–44°C water bath for 30 min. Depending on the treatment paradigm, cells were either immediately collected or returned to a 37°C incubator to recover before cell lysis. Cells were scraped into lysis buffer containing 50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P40, 1% deoxycholate, 0.1% SDS, 0.5 mM sodium orthovandate, 40 mM NaF, 10 mM β-glycerophosphate and 1×Complete Protease Inhibitors (Roche Diagnostics) and homogenized by sonication. The crude cell lysates were centrifuged at 10000 g for 10 min at 4°C and the total protein concentration of the supernatant was determined using a Bio-Rad protein assay and BSA as the standard. Approximately 30–35 μg of protein was separated by SDS/PAGE and transferred to nitrocellulose for immunoblot analyses.
The membranes were incubated with 5% non-fat dry milk in PBST (phosphate-buffered saline containing 0.1% Tween 20) for 1–2 h at room temperature (25°C) and probed with primary antibodies recognizing Hsp70, Hsc70, Hsp90, Hsp40, Hsp27, c-Jun, JNK or β-actin at 4°C overnight. For detection of the phosphorylated proteins, 5% non-fat dry milk was substituted with 5% BSA. After primary antibody incubation, membranes were washed with PBST and incubated with HRP-conjugated anti-mouse, anti-rabbit, anti-chicken or anti-goat secondary antibodies. Immunoreactivity for each protein was visualized using an enhanced chemiluminescence detection kit (GE Healthcare Life Sciences). The films were digitally scanned and densitometrically analysed using Image J (National Institutes of Health) software.
Immunofluorescence analysis
Myelinated DRG explants grown on glass coverslips were rinsed with PBS and fixed with fresh 4% (w/v) paraformaldehyde for 20 min at room temperature. The cells were permeabilized by incubating with −20°C methanol for 15 min, then blocked with 10% normal goat serum (Invitrogen) containing 0.3% Triton X-100 for 15 min at room temperature. Primary antibodies against MBP (SMI-94R, 1:500), PGP9.5 (1:500), Hsp70 (1:80) and S100β (1:1000) were diluted in blocking buffer (10% goat serum in PBS) and incubated with the cells overnight at 4°C in an humidified chamber. The cells were washed with PBS and incubated with Alexa Fluor® 568, Alexa Fluor® 488 or Alexa Fluor® 647-conjugated secondary antibodies. Coverslips were counterstained with DAPI (4,6-diamidino-2-phenylindole) to visualize nuclei and mounted on slides using the Prolong Antifade kit. Slides were imaged on an Olympus/3I Spinning Disk Confocal/TIRF Inverted Microscope and 6–8 random fields per coverslip were captured using the imaging software, SlideBook 5.0 (Intelligent Imaging Innovations Inc.).
The extent of demyelination was quantified as previously described (Urban et al. 2010). MBP-positive segments were counted as internodes and the percentage of broken versus total internodes was calculated and expressed as a percent of degenerated segments for each picture frame. Changes in internode length were quantified utilizing an open source imaging software-CellProfiler (http://www.cellprofiler.org). Individual myelin internodes with a length within 20–200 μm were identified through Otsu's method (Otsu, 1979) for thresholding and segmentation. Throughout image processing, visual inspection and manual editing were performed during segment identification in the case of errors or regions where segments intersected or touched the border. Major axis lengths for each identified segment were then computed to give the length of the internodes. For immunofluorescent quantification of Hsp70 expression in premyelinating cultures, intensity was set as the threshold factor instead of length. Hsp70 protein expression was computed as area×intensity in fluorescence units. Co-localization of fluorescent channels was achieved using Image J.
Preparation of Hsp70 adenovirus and Hsp70-promoter-luciferase reporter
The cDNA sequence of human Hsp70 (HSPA1A) was amplified by PCR with a forward primer containing a BamHI site (AGCTTGGATCCGAATTCACCAT-GGCCAAAGCCGCGGCG), and a reverse primer containing a SalI site (AAGTCGACATCTACCTCCTCAATGGTGGGGCCTG). The PCR product was subsequently digested and cloned into the p-Shuttle-IRES-hrGFP-1 vector (Agilent Technologies) between the BglII and SalI site to add an in-frame C-terminal FLAG tag. The integrity of the sequence was verified by DNA sequencing and recombinant adenovirus was generated using the pAdEasy kit as per the manufacturer's directions. To infect myelinated neuronal cultures, concentrated viral particles were diluted in maintenance medium and 16 h after infection, the medium was replaced by fresh non-viral medium prior to further treatment. Recombinant expression of Hsp70 was confirmed by antibodies against Hsp70 or the C-terminal FLAG epitope tag.
To prepare the luciferase reporter, a 1.5 kb region upstream of the start codon of the human HSPA1A gene and containing 5′ KpnI and 3′ SacI sites to direct cloning into a pGL3 basic luciferase reporter plasmid was synthesized by GeneArt (Life Technologies). The integrity of the promoter sequence and the presence of two HS elements were verified by DNA sequencing. 50B11 cells (Chen et al., 2007) were grown in 10 cm dishes in DMEM containing 25 mM glucose, 10% FCS and 5 μg/ml blasticidin. The cells were transfected using Lipofectamine™ and after 24 h, were re-seeded into 24 well plates at a density of 2×105 cells per well. After 6 h, the cells were treated with the indicated concentrations of KU-32 for 16 h, luciferase activity was assessed and normalized to the total protein concentration of each well. Results shown are from triplicate wells obtained in three separate experiments.
Statistical analysis
Datasets are presented as means±S.E.M. Equality of variances was verified and a one-way ANOVA or Kruskal–Wallis non-parametric test was performed. Differences among treatment groups were determined using Tukey's or Dunn's post-hoc tests.
RESULTS
KU-32 induces Hsp70 expression in DRG explant cultures
We have previously shown that KU-32 inhibited NRG1-induced demyelination in an Hsp70-dependent manner but it remained unclear if neuroprotection may also be associated with the induction of other chaperones. Immunoblot analysis of unmyelinated DRG explant cultures treated with 1 μM KU-32 for 4–24 h indicated that Hsp70 was the primary chaperone up-regulated by KU-32 (Figures 1A and 1B). Although Hsp90 and Hsp40 can be induced in response to HS, KU-32 did not significantly increase their expression. Similarly, the drug did not alter the level of the constitutively expressed Hsp70 paralog, Hsc70 (see below), and had no effect on the resident chaperones of the endoplasmic reticulum, Grp78 and Grp94 (data not shown). Although KU-32 did tend to increase the expression of Hsp27, a small Hsp that may be involved in transiently stabilizing mis-folded or damaged proteins until their interaction with Hsp70/Hsp40 (Muchowski and Wacker, 2005), this did not quite reach significance. Consistent with the induction of Hsp70 protein, KU-32 also dose-dependently increased the expression of a luciferase reporter that was driven by the human Hsp70 promoter (Figure 1C). Since primary sensory neurons transfect poorly, an immortalized sensory neuron cell line (50B11 cells) was used for the transfection (Chen et al., 2007). Importantly, 50B11 cells have a very low basal level of Hsp70 expression, similar to primary sensory neurons. However, KU-32 is a weak activator since it was not as effective as geldanamycin, a prototypical Hsp90 N-terminal inhibitor that robustly activates the HS elements within the Hsp70 promoter (Calamini et al., 2012).
Figure 1. KU-32 induces Hsp70 in mouse DRG explants.

(A) Primary DRG explants from C57Bl/6 mice were isolated and grown in culture for 1 week. Cells were treated with vehicle (0.05% DMSO, 24 h) or 1 μM KU-32 for 4, 8 or 24 h. Cell lysates were collected and immunoblotted for Hsp90, Hsp70, Hsp40 and Hsp27. (B) Hsp levels were normalized to β-actin for each time point and changes were expressed as the fold control (n = 3–6 at each time point) **P<0.01 versus control. (C) 50B11 cells were transfected with a luciferase reporter driven by the human Hsp70 promoter and treated with vehicle or the indicated concentrations of KU-32 for 16 h. Cell lysates were prepared and luciferase activity was assessed. Results are expressed as a percent of the vehicle treated cells and are from three experiments performed in triplicate. Geldanamycin (250 nM) was used as a positive control and it activated the reporter by 4.5±0.29-fold compared with the untreated control. ***P<0.001 versus control; ζP<0.001 versus 10 nM; ˆP<0.01 versus 100 nM.
Although these data indicate that Hsp70 expression is modulated by KU-32, immunoblot analysis of the unmyelinated DRG explants did not allow us to assess whether induction of Hsp70 was occurring within sensory neurons and/or SCs. Unmyelinated DRG explants from C57Bl/6 mice were treated with either vehicle or KU-32 for 24 h and processed for immunostaining. A 30-min HS followed by 1 h recovery was applied to a parallel set of cultures and served as a positive control. To characterize Hsp70 expression in the mixed culture, cells were double immunostained with antibodies against Hsp70 and neuronal (PGP9.5) or SC (S100β) markers. As shown in Figure 2(A), a basal level of Hsp70 fluorescence co-localized with PGP9.5 that was limited to the cell body and was not observed within axons. Short-term HS increased Hsp70 expression in neurons as well as PGP9.5-negative cells (arrows). Similarly, the increase in the Hsp70 signal after KU-32 treatment was also evident in both PGP9.5-positive and PGP9.5-negative cells. These data suggest that KU-32 may increase Hsp70 within SCs and co-staining of Hsp70 and S100β in the explants verified a prominent expression of Hsp70 in cells co-labelled with S100β, a SC marker (Figure 2B). Thus, KU-32 can induce expression of Hsp70 in neuronal cell bodies and SCs.
Figure 2. Hsp70 is induced in neurons and SCs.

DRG explant cultures were treated with vehicle, 1 μM KU-32 for 24 h, or subjected to 30 min HS plus a 1 h recovery (HS+R). Localization of Hsp70 expression (red) in neurons or SCs were examined using double-fluorescence-labelling with antibodies against PGP9.5 (A) (green) or S100β (blue) (B), respectively. Confocal images were taken and co-localization of Hsp70 with either PGP9.5 or S100β was performed using Image J. Arrows indicate increased Hsp70 in SCs. Scale bar, 15 μm.
Hsp70 is necessary for drug efficacy and sufficient to prevent NRG1-induced demyelination
To gain insight into the mechanism by which Hsp70 induction contributes to preventing demyelination, myelinated DRG explants were prepared from neonatal WT C57Bl/6 and Hsp70 KO mice. Deletion of the Hsp70 gene was verified by genotyping and the lack of Hsp70 induction by KU-32 and HS (Figure 3A). However, no difference existed in the expression of Hsc70 between WT and Hsp70 KOs and Hsc70 levels were not altered by KU-32 or HS (Figure 3B).
Figure 3. Hsp70 is necessary for protecting against NRG1-induced demyelination by KU-32.

(A) One-week-old mouse DRG explants from WT or Hsp70 KO mice were treated with vehicle or 1 μM KU-32 for 24 h in the absence or presence of HS (30 min at 42°C). Heat shocked cultures were then switched to 37°C for 0–4 h of recovery time. Cell lysates were prepared and the expression of Hsp70 was determined by immunoblot analysis. (B) DRG explants from WT or Hsp70 KO mice were treated with vehicle or 1 μM KU-32 for 24 h in the absence or presence of HS (30 min at 42°C and 4 h recovery at 37°C). The level of Hsc70 was determined by immunoblot.
Treating myelinated WT explants with 200 ng/ml NRG for 72 h led to a marked degeneration of the myelinated internodes as indicated by the fragmented, vesicular appearance of the MBP staining (Figure 4A). Importantly, this degeneration was not a consequence of impaired axonal integrity as no decrease or irregularity in PGP9.5 staining was seen, as we have previously reported (Urban et al., 2010). The extent of myelin degeneration was quantified by imaging random fields from each treatment group and calculating the percentage of damaged versus total myelin segments. While there was approximately 8% basal demyelination in control cultures, the number of degenerated myelin internodes in WT cultures treated with 200 ng/ml NRG1 was ∼7-fold greater (Figure 4B). However, incubation with 1 μM KU-32 prior to NRG1 treatment prevented this increase since the number of damaged segments remained close to the control level. KU-32 had no effect on the number of damaged myelin segments in the absence of NRG1 treatment. As we have shown previously (Urban et al., 2010), NRG1 treatment also decreased the expression of myelin protein zero as determined by immunoblot analysis and this effect was attenuated by pretreating the co-cultures with KU-32 (Figure 4C).
Figure 4. KU-32 requires Hsp70 to block NRG1-induced demyelination.

DRG explants were established from C57Bl/6 (A, B) or Hsp70 KO (D, E) mice and myelinated in vitro for 3 weeks. The cultures were treated with vehicle or 1 μM KU-32 for 16 h prior to inducing demyelination with 200 ng/ml NRG1 for 3 days. Myelin internodes were labelled via MBP staining and nuclei visualized with a DAPI stain. Five to eight images were taken for each individual culture and the number of total and degraded myelin segments was counted per frame. Data shown are means±S.E.M. from three preparations per genotype. ***P<0.001 versus control; ˆP<0.01 versus NRG1 minus KU-32. (C) Explant cultures from WT mice were treated as above and lysates were prepared for immunoblot analysis of myelin protein zero (P0).
Myelinated cultures prepared from the Hsp70 KO mice showed a similar basal level (∼6%) of damaged segments as was seen in the WT cultures. However, NRG1 stimulated approximately a 6-fold increase in degenerated segments (Figure 4D). The reason for this slight resistance to NRG1 is not known, but might be due to a generally higher basal level of myelination in Hsp70 KO cultures. Since Hsp70 has not been characterized as having an inhibitory role in myelination, the reason for the increased number of myelinated segments that is reproducibly observed in explant cultures from the Hsp70 KO mice is unknown. However, this may be related to the slightly lower basal expression of c-Jun that was observed in the myelinated cultures from the Hsp70 KO mice (see below and Figures 6 and 7). Nevertheless, pretreatment with KU-32 failed to significantly reduce the percentage of damaged myelin internodes in these cultures (Figure 4E). These findings are consistent with our previous in vivo observation that Hsp70 is central for the neuroprotective efficacy of KU-32 (Urban et al., 2010).
Figure 6. KU-32 inhibited NRG1-induced expression of c-Jun in WT but not Hsp70 deficient co-cultures.

(A) NRG1 treatment induces c-Jun in SCs. Fully myelinated WT DRG explants were treated with 100 ng/ml NRG1 for 16 h and stained for MBP and c-Jun levels. DAPI staining was used to visualize nuclei, scale bar, 10 μM. (B) Fully myelinated DRG explants from WT or Hsp70 KO mice were treated with vehicle or 1 μM KU-32 overnight then stimulated with 200 ng/ml NRG for 16 h. Cell lysates were prepared and the level of phospho-c-Jun and c-Jun were determined by immunoblot. The level of phospho-c-Jun (C) and c-Jun (D) were quantified using Image J and the data shown are means±S.E.M. from five experiments. **P<0.01 versus control in each genotype; ˆP<0.05 versus NRG1 in WT mice only.
Figure 7. Ectopic expression of Hsp70 is sufficient to block NRG1-induced demyelination in WT and Hsp70 KO neurons.

Fully myelinated WT (A) or Hsp70 KO (B) DRG explants were either uninfected or transduced with blank or Hsp70-FLAG adenoviruses for 16 h. The cultures were treated with vehicle or 150–200 ng/ml NRG for 3 days and phospho-c-Jun and c-Jun were detected by immunoblot analysis. The levels of phospho-c-Jun (C) and c-Jun (D) were quantified using Image J and the data shown are mean and the range from two experiments.
While the above results support that Hsp70 is necessary for the efficacy of KU-32 in preventing NRG1-induced demyelination, it remains unclear whether the neuroprotection arises from a direct effect of Hsp70. To determine whether induction of Hsp70 was sufficient to protect against NRG1-induced demyelination, a recombinant adenovirus expressing Hsp70 with a C-terminal FLAG-tag (Hsp70-FLAG) was generated to genetically overexpress Hsp70. Fully myelinated DRG explants from WT and Hsp70 KO mice were infected with a blank or Hsp70-FLAG adenovirus for 16 h, the cells were treated with NRG1 for 72 h and the cultures stained for MBP. In order to maintain myelin damage at a comparable level between WT and Hsp70 KO cultures, WT cells were treated with 150 ng/ml NRG1, while the Hsp70 KO cultures were treated with 200 ng/ml NRG1. In uninfected cultures, this approach resulted in approximately 45% of the total segments being degenerated in the WT cultures whereas approximately 50% of the total segments showed degeneration in the Hsp70 KO cultures (Figures 5A and 5B). In myelinated WT and Hsp70 KO cultures, infection with blank virus did not decrease the magnitude of NRG1-induced demyelination. In contrast, infection of either the WT or Hsp70 KO cultures with the Hsp70-FLAG adenovirus led to a significant decrease in the extent of damaged myelin segments induced by NRG1 treatment. This decrease correlated with the ectopic expression of the epitope-tagged Hsp70 which was similar in both WT and Hsp70 KO cultures (see Figure 7).
Figure 5. Hsp70 induction is sufficient to block NRG1-induced demyelination.
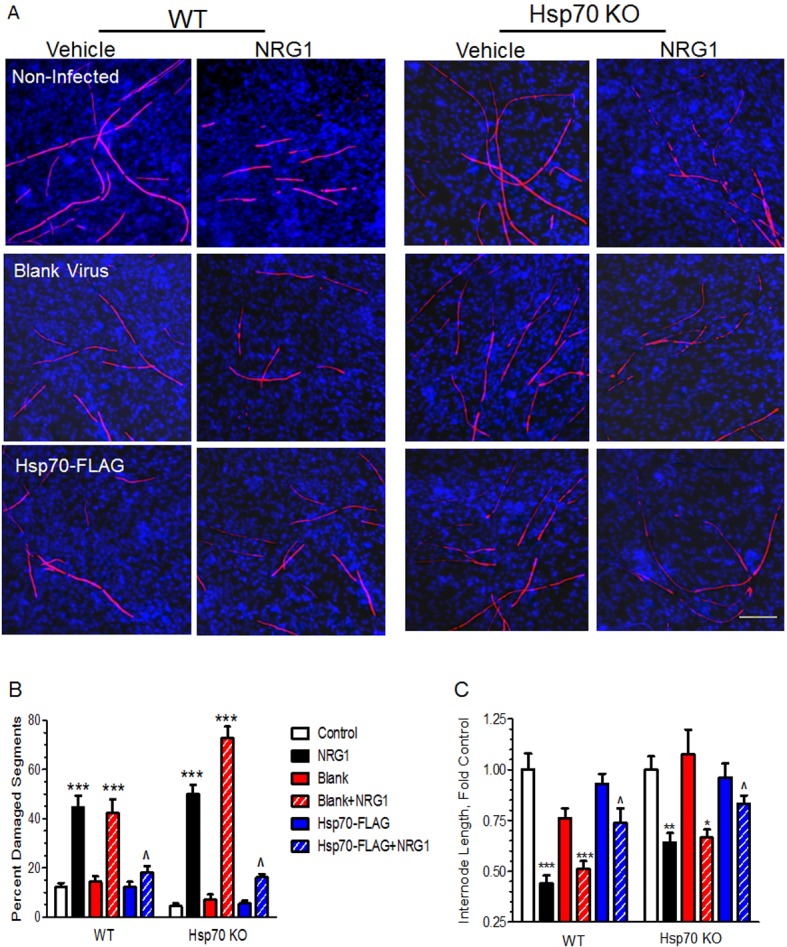
Fully myelinated WT or Hsp70 KO DRG explants were either uninfected or transduced with blank or Hsp70-FLAG adenoviruses for 16 h. The cultures were treated with vehicle or 200 ng/ml NRG for 3 days, stained for MBP and DAPI (A). The percentage of degenerated segments (B) and segment internode length (C) was quantified. Data shown are means±S.E.M. from three experiments per genotype. *P<0.05, **P<0.01, ***P<0.001 versus respective control; ˆP<0.05 versus blank+NRG1. Scale bar, 100 μm.
Since the percentage of damaged myelin segments only takes into account the gross number of ‘broken’ segments, it does not reflect the severity of the ongoing degeneration of the myelinated internodes. To further characterize the integrity of the myelin internodes, average internode length from each treatment was also assessed using CellProfiler. Briefly, linear segments of continuous MBP immunoreactivity that fell into the range of 20–200 μm were identified by the program and major axis length was computed for each segment. The results were expressed as fold of the untreated control and this analysis indicated that NRG1 decreased the average length of myelinated internodes by approximately 50% in either the uninfected or blank virus infected WT cultures (Figure 5C). However, overexpression of Hsp70 significantly attenuated the decrease in internode length. In the Hsp70 KO cultures, NRG1 decreased the internode length cultures by approximately 35% and expression of Hsp70-FLAG led to a similar extent of recovery that was observed in the WT cultures. These results support that Hsp70 induction is sufficient to improve myelination and recapitulate the neuroprotection seen with KU-32.
KU-32 blocks c-Jun induction in an Hsp70-dependent manner
c-Jun is an established negative regulator of myelination and has been proposed to mediate NRG1-induced demyelination (Parkinson et al., 2008; Syed et al., 2010). In addition, whereas c-Jun is minimally expressed in healthy nerves, it is significantly up-regulated in DPN and a number of human demyelinating neuropathies (Hur et al., 2011; Hutton et al., 2011). Similarly, we observed a low level of c-Jun expression in myelinated explant cultures from WT mice and the addition of NRG1 led to a strong induction of c-Jun (Figure 6A), largely in SCs as previously noted (Parkinson et al., 2008). We therefore evaluated whether the efficacy of myelin protection by KU-32 correlated with inhibition of c-Jun. Myelinated DRG explants from WT or Hsp70 KO mice were incubated with 1 μM KU-32 overnight prior to treating with NRG1 for 16 h, which was identified in preliminary experiments as a time point for essentially maximal c-Jun induction under our culture conditions. Cell lysates were prepared and the levels of phospho-c-Jun and total c-Jun (Figure 6B) were determined by immunoblot analysis. NRG1 induced a 2–2.5-fold increase in c-Jun and phospho-c-Jun expression in WT and a similar induction was observed in myelinated Hsp70 KO cultures treated with NRG1 (Figures 6C and 6D). Thus, Hsp70 is not necessary for the induction of c-Jun by NRG1. Notably, pretreatment with KU-32 abolished NRG1-induced c-Jun expression and phosphorylation in the WT cultures which correlates with the protection of myelin integrity. In contrast, KU-32 was unable to prevent the induction and phosphorylation of c-Jun in the Hsp70 KO neurons.
To determine if expression of Hsp70 was sufficient to decrease the induction of c-Jun, WT (Figure 7A) and Hsp70 KO (Figure 7B) cultures were infected with the Hsp70-FLAG adenovirus. Consistent with the ability of Hsp70 to protect against NRG1-induced demyelination (Figure 4), ectopic expression of Hsp70 in the WT cultures recapitulated the effect of KU-32 in preventing the increase in phosphorylated and total c-Jun after NRG1 treatment (Figures 7C and 7D). Similarly, re-expression of Hsp70-FLAG in the Hsp70 KO cultures was sufficient to block the NRG1-induced increase in c-Jun expression and phosphorylation (Figures 7C and 7D). Collectively, the data in Figures 4–6 strongly support that induction of Hsp70 is sufficient to prevent c-Jun induction and phosphorylation and that the efficacy of KU-32 in preventing NRG1-induced demyelination is linked to a necessary intersection between Hsp70 and c-Jun signalling.
KU-32 does not alter JNK or p42/p44 MAPK activation
It is well recognized that JNK serves as an upstream regulator of c-Jun by phosphorylating the protein at its N-terminus. However, neither NRG1 nor KU-32 had any effect on increasing JNK expression. Similarly, neither treatment increased JNK activation, as determined by a lack of change in the expression of phospho-JNK (Figure 8A). Alternatively, the activation of p42/p44 MAPK by NRG1 is a critical upstream signal for promoting demyelination (Harrisingh et al., 2004; Ogata et al., 2004; Napoli et al., 2012) and inhibiting the activation of p42/p44 MAPK blocked c-Jun induction by NRG1 (Syed et al., 2010). Although the level of phosphorylated MAPK was clearly increased in myelinated DRG explants stimulated with NRG1 for 45 min, pretreatment with KU-32 also had no effect on NRG1-induced MAPK activation (Figure 8B).
Figure 8. KU-32 does not inhibit JNK or p42/p44 MAPK.

Myelinated WT cultures were treated with vehicle or 1 μM KU-32 and stimulated with 200 ng/ml NRG. Cell lysates were prepared and the levels of phospho JNK and total JNK (A) or phospho p42/p44 MAPK and total MAPK (B) were determined by immunoblot analysis. Cells were stimulated with NRG1 for 16 h or 45 min prior to preparing cell lysates to determine JNK and MAPK levels respectively. Gels shown are representative of results obtained in five experiments.
Reduction of c-Jun expression by KU-32 is proteasome-dependent
As NRG1-induced myelin degeneration is not associated with the accumulation of a particular mis-folded protein or protein aggregate, Hsp70-mediated assistance in protein refolding is an unlikely mechanism of myelin protection by KU-32. However, Hsp70 may intersect with cellular signal transduction through proteasome-mediated degradation of signalling proteins. For example, the anti-apoptotic effect of Hsp70 and its co-chaperone, CHIP (C-terminus Hsp70-interacting protein), have been linked to their facilitating ubiquitination and proteasomal disposal of ASK1, an upstream JNK kinase (Hwang et al., 2005). To determine whether KU-32 eliminates c-Jun through protein degradation, myelinated DRG explant cultures were pre-treated overnight with KU-32. Subsequently, the proteasome inhibitor MG132 was added coincident with NRG1 and c-Jun expression was examined by immunoblot analysis 16 h after this treatment (Figures 9A and 9B). As in earlier experiments, NRG1 increased c-Jun/phospho-c-Jun (upper band) expression and KU-32 significantly blocked this induction. However, addition of 2 μM MG132 abrogated the block to c-Jun induction that was observed in cultures treated with KU-32 and NRG1. As expected, MG132 modestly increased the level of c-Jun and phospho-c-Jun in cultures treated with only NRG1 due to inhibition of the proteasome. Collectively, these data support that KU-32 reduces the expression of c-Jun and phospho-c-Jun by enhancing their proteasomal clearance in an Hsp70-dependent manner.
Figure 9. KU-32 promotes proteasomal degradation of c-Jun.

Fully myelinated DRG explants from WT mice were treated with 1 μM KU-32 for 16 h and then treated with 200 ng/ml NRG1 in the absence or presence of 2 μM MG132 for an additional 16 h. Cell lysates were prepared the level of c-Jun was determined by immunoblot analysis (A) and quantified (B) using Image J. Results are means±S.E.M. from five independent experiments. *P<0.05 versus control; ˆP<0.05 versus NRG1 only; #P<0.002 versus NRG1+KU-32.
DISCUSSION
Hsp70 is a cytosolic chaperone that has demonstrated prominent neuroprotection in models of cerebral ischaemia and a variety of neurodegenerative disorders associated with aberrant protein aggregates (Li and Dobrowsky, 2012). For example, transgenic overexpression of PMP22 (peripheral myelin protein 22) results in formation of PMP22 aggregates and is a model of CMT1A (Robertson et al., 2002). N-terminal Hsp90 inhibitors that promoted a robust induction of Hsp70 decreased the formation of PMP22 aggregates (Fortun et al., 2007) and increased myelination using in vitro cultures prepared from the C22 mouse model of CMT1A (Rangaraju et al., 2008). Although KU-32 can reverse DPN and prevent NRG1-induced demyelination in an Hsp70-dependent manner, neither model has an aetiology related to the production of protein aggregates (Urban et al., 2010). Thus, Hsp70 may also decrease neurodegeneration through mechanisms independent of its role as a chaperone that can aid protein refolding (Koren et al., 2009).
c-Jun is a basic leucine zipper transcription factor of the AP-1 (activator protein 1) family and is well recognized for its role in promoting apoptosis of sympathetic neurons following growth factor withdrawal (Palmada et al., 2002). This pro-apoptotic effect of c-Jun was attenuated by genetic overexpression of Hsp70 in sympathetic neurons, which suppressed c-Jun induction and phosphorylation upon NGF (nerve growth factor) withdrawal (Bienemann et al., 2008). Previous evidence indicates that c-Jun also functions as a negative regulator of myelination and drives dedifferentiation of myelinated fibres upon pathologic expression (Parkinson et al., 2008). Our data support the premise that an intersection between Hsp70 and c-Jun signalling is critical to preventing demyelination since KU-32 is ineffective at attenuating NRG1-induced demyelination and c-Jun induction in explant cultures from Hsp70 KO mice. Further, that protection against demyelination was recapitulated following re-expression of Hsp70 in cultures prepared from the Hsp70 KO mice also supports this conclusion. However, one shortcoming of our study is that we cannot conclude that the induction of Hsp70 specifically in SCs is solely responsible for the protection. This may be most clearly demonstrated in a tissue-specific Hsp70 KO mouse and additional work will examine the cell autonomous roles of chaperone signalling in neurons versus peripheral glia in contributing to drug efficacy.
Mechanistically, Hsp70 is known to inhibit JNK (Gabai et al., 1998) and inactivation of JNK/c-Jun signalling by Hsp70 has been suggested to prevent neuronal apoptosis (Salehi et al., 2006). However, KU-32 treatment did not decrease JNK expression or phosphorylation in a time course that preceded the significant reduction in c-Jun levels. A possible explanation is that the level of Hsp70 induction by KU-32 may be too transient or of insufficient magnitude to markedly impact JNK activity. Alternatively, the necessity of JNK in affecting downstream biologies related to c-Jun induction may be context dependent. Indeed, JNK phosphorylation of c-Jun is not required for dedifferentiation of myelinated SCs (Parkinson et al., 2008). Thus, it is unlikely that an interaction between Hsp70 and JNK contributed to the efficacy of KU-32 in preventing demyelination. In contrast, recent work has shown that activation of p42/p44 MAPK by NRG1 is necessary for c-Jun induction and demyelination (Syed et al., 2010; Napoli et al., 2012). Although NRG1 increased MAPK activation, KU-32 pretreatment did not decrease MAPK phosphorylation following stimulation of the co-cultures with NRG1. These results support that the ability of KU-32 to prevent c-Jun induction is also not due to a block in Erb B2 activation and an inhibition of early signalling events.
The ability of KU-32 to decrease c-Jun levels appears associated with its proteasomal degradation since incubating the cultures with the proteasome inhibitor MG132 negated the effect of KU-32 on blunting c-Jun induction. Although c-Jun is not recognized as forming protein aggregates, these results are consistent with the proteasomal clearance of aggregated tau by Hsp70 and other chaperones (Dickey et al., 2007a; Koren et al., 2009). Since the clearance of aggregated protein by Hsp70 often enlists CHIP (a CHIP that also binds to the 26S proteasome) to promote ubiquitination and degradation (Dickey et al., 2007b), it will be important to determine if CHIP is also involved in c-Jun degradation by KU-32 treatment.
One caveat to our approach is that the proteasomal inhibitor MG132 can increase JNK activation and apoptosis (Meriin et al., 1998; Nakayama et al., 2001). Although we did not observe substantial cell death at the MG132 concentration used in the present study, we observed a small increase in c-Jun levels by MG132 in cells treated with NRG1. This result would suggest that MG132 may increase the extent of NRG1-induced demyelination. However, inhibiting the proteasome with MG132 blocked SC dedifferentiation during Wallerian degeneration of sciatic nerve explants (Lee et al., 2009). Although our work and the study of Lee et al. used different models, these data suggest that any block to SC degeneration by MG132 must be downstream of c-Jun induction since MG132 enhanced the level of c-Jun induced by NRG1. Alternatively, inhibiting the proteasome with MG132 can actually stimulate HSF1 and increase chaperone expression (Neef et al., 2011), which may account for the observed effect in delaying Wallerian degeneration (Lee et al., 2009).
The neuroprotection elicited by Hsp70 in the context of a degenerative signal suggests that induction of c-Jun by NRG1 may be akin to a form of transient proteotoxic stress, similar to demyelination promoted by the maladaptive activation of the unfolded protein response in transgenic mice expressing a deletion of Ser63 in myelin protein zero (Pennuto et al., 2008; Gow and Wrabetz, 2009). Notably, the induction of c-Jun may play a fundamental role in the onset of human demyelinating neuropathies (Hutton et al., 2011) and at first sight, the ability of Hsp70 to attenuate c-Jun expression and prevent demyelination would seem primarily beneficial in the context of treating demyelinating disorders. However, the expression of c-Jun in SCs is also necessary for the expression of glial-derived neurotrophic factor and artemin, which are critical paracrine factors that activate axonal Ret receptors to promote regeneration following nerve damage (Fontana et al., 2012). Given the essential role of nerve regeneration in recovery of sensation in neuropathies such as DPN (Zochodne, 2012), it is conceivable that prolonged inhibition of c-Jun levels in SCs may impinge on the regenerative capacity of myelinated axons.
In summary, these proof-of-principle studies suggest it will be important to determine if the in vitro efficacy of KU-32 in decreasing demyelination can be recapitulated in models of chemical-induced demyelinating neuropathy or a rodent model of DPN that superimposes diabetes on genetically hypertensive rats to enhance myelin thinning (Gregory et al., 2012). As degenerative changes in myelinated SCs is a substantial feature in human DPN, understanding how molecular chaperones modulate signalling events underlying peripheral nerve demyelination may open new translational avenues for clinical management of DPN and/or other human neuropathies.
Footnotes
This work was supported the Juvenile Diabetes Research Foundation and The National Institutes of Health [grant numbers NS054847 (to R.T.D.), CA120458 and CA109265 (to B.S.J.B.) and NS075311 (to B.S.J.B. and R.T.D.)].
REFERENCES
- Ansar S, Burlison JA, Hadden MK, Yu XM, Desino KE, Bean J, Neckers L, Audus KL, Michaelis ML, Blagg BS. A non-toxic Hsp90 inhibitor protects neurons from Abeta-induced toxicity. Bioorg Med Chem Lett. 2007;17:1984–1990. doi: 10.1016/j.bmcl.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Bienemann AS, Lee YB, Howarth J, Uney JB. Hsp70 suppresses apoptosis in sympathetic neurones by preventing the activation of c-Jun. J Neurochem. 2008;104:271–278. doi: 10.1111/j.1471-4159.2007.05006.x. [DOI] [PubMed] [Google Scholar]
- Blagg BS, Kerr TD. Hsp90 inhibitors: small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med Res Rev. 2006;26:310–338. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BS. Novobiocin: redesigning a DNA gyrase inhibitor for selective inhibition of Hsp90. J Am Chem Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- Calamini B, Silva MC, Madoux F, Hutt DM, Khanna S, Chalfant MA, Saldanha SA, Hodder P, Tait BD, Garza D, Balch WE, Morimoto RI. Small-molecule proteostasis regulators for protein conformational diseases. Nat Chem Biol. 2012;8:185–196. doi: 10.1038/nchembio.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Mi R, Haughey N, Oz M, Höke A. Immortalization and characterization of a nociceptive dorsal root ganglion sensory neuronal line. J Periph Nerv Syst. 2007;12:121–130. doi: 10.1111/j.1529-8027.2007.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS Jr, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90–CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007a;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Patterson C, Dickson D, Petrucelli L. Brain CHIP: removing the culprits in neurodegenerative disease. Trends Mol Med. 2007b;13:32–38. doi: 10.1016/j.molmed.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Donnelly A, Blagg BS. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem. 2008;15:2702–2717. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci USA. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esper RM, Pankonin MS, Loeb JA. Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res Brain Res Rev. 2006;51:161–175. doi: 10.1016/j.brainresrev.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Farmer KL, Li C, Dobrowsky RT. Diabetic peripheral neuropathy: should a chaperone accompany our therapeutic approach? Pharmacol Rev. 2012;64:880–900. doi: 10.1124/pr.111.005314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana X, Hristova M, Da Costa C, Patodia S, Thei L, Makwana M, Spencer-Dene B, Latouche M, Mirsky R, Jessen KR, Klein R, Raivich G, Behrens A. c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J Cell Biol. 2012;198:127–141. doi: 10.1083/jcb.201205025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J, Verrier JD, Go JC, Madorsky I, Dunn WA, Notterpek L. The formation of peripheral myelin protein 22 aggregates is hindered by the enhancement of autophagy and expression of cytoplasmic chaperones. Neurobiol Dis. 2007;25:252–265. doi: 10.1016/j.nbd.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem. 2008;283:26188–26197. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabai VL, Meriin AB, Yaglom JA, Volloch VZ, Sherman MY. Role of Hsp70 in regulation of stress-kinase JNK: implications in apoptosis and aging. FEBS Lett. 1998;438:1–4. doi: 10.1016/s0014-5793(98)01242-3. [DOI] [PubMed] [Google Scholar]
- Gow A, Wrabetz L. CHOP and the endoplasmic reticulum stress response in myelinating glia. Curr Opin Neurobiol. 2009;19:505–510. doi: 10.1016/j.conb.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory J, Jolivalt C, Goor J, Mizisin A, Calcutt N. Hypertension-induced peripheral neuropathy and the combined effects of hypertension and diabetes on nerve structure and function in rats. Acta Neuropathol. 2012;124:561–573. doi: 10.1007/s00401-012-1012-6. [DOI] [PubMed] [Google Scholar]
- Grinspan JB, Marchionni MA, Reeves M, Coulaloglou M, Scherer SS. Axonal interactions regulate Schwann cell apoptosis in developing peripheral nerve: neuregulin receptors and the role of neuregulins. J Neurosci. 1996;16:6107–6118. doi: 10.1523/JNEUROSCI.16-19-06107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin AD, Zhang DP, Mak KS, Alberta JA, Kim HA. Microanatomy of axon/glial signaling during Wallerian degeneration. J Neurosci. 2005;25:3478–3487. doi: 10.1523/JNEUROSCI.3766-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrisingh MC, Perez-Nadales E, Parkinson DB, Malcolm DS, Mudge AW, Lloyd AC. The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. 2004;23:3061–3071. doi: 10.1038/sj.emboj.7600309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Hunt CR, Dix DJ, Sharma GG, Pandita RK, Gupta A, Funk M, Pandita TK. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol Cell Biol. 2004;24:899–911. doi: 10.1128/MCB.24.2.899-911.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J, Sullivan KA, Pande M, Hong Y, Sima AAF, Jagadish HV, Kretzler M, Feldman EL. The identification of gene expression profiles associated with progression of human diabetic neuropathy. Brain. 2011;134:3222–3235. doi: 10.1093/brain/awr228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton EJ, Carty L, Laurá M, Houlden H, Lunn MPT, Brandner S, Mirsky R, Jessen K, Reilly MM. c-Jun expression in human neuropathies: a pilot study. J Periph Nerv Syst. 2011;16:295–303. doi: 10.1111/j.1529-8027.2011.00360.x. [DOI] [PubMed] [Google Scholar]
- Hwang JR, Zhang C, Patterson C. C-terminus of heat shock protein 70-interacting protein facilitates degradation of apoptosis signal-regulating kinase 1 and inhibits apoptosis signal-regulating kinase 1-dependent apoptosis. Cell Stress Chaperones. 2005;10:147–156. doi: 10.1379/CSC-90R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. Negative regulation of myelination: relevance for development, injury, and demyelinating disease. Glia. 2008;56:1552–1565. doi: 10.1002/glia.20761. [DOI] [PubMed] [Google Scholar]
- Koren J III, Jinwal UK, Lee DC, Jones JR, Shults CL, Johnson AG, Anderson LJ, Dickey CA. Chaperone signalling complexes in Alzheimer's disease. J Cell Mol Med. 2009;13:619–630. doi: 10.1111/j.1582-4934.2008.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuma BR, Zhang L, Sundstrom T, Peterson LB, Dobrowsky RT, Blagg BSJ. Synthesis and evaluation of novologues as C-Terminal Hsp90 inhibitors with cytoprotective activity against sensory neuron glucotoxicity. J Med Chem. 2012;55:5797–5812. doi: 10.1021/jm300544c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Shin YK, Jung J, Seo S-Y, Baek S-Y, Park HT. Proteasome inhibition suppresses Schwann cell dedifferentiation in vitro and in vivo. Glia. 2009;57:1825–1834. doi: 10.1002/glia.20894. [DOI] [PubMed] [Google Scholar]
- Li C, Dobrowsky RT. Targeting molecular chaperones in diabetic peripheral neuropathy. In: Hayat G, editor. In Peripheral Neuropathy – Advances in Diagnostic and Therapeutic Approaches. Rijeka, Croatia: InTech; 2012. pp. 39–62. [Google Scholar]
- McGuire JF, Rouen S, Siegfreid E, Wright DE, Dobrowsky RT. Caveolin-1 and altered neuregulin signaling contribute to the pathophysiological progression of diabetic peripheral neuropathy. Diabetes. 2009;58:2677–2686. doi: 10.2337/db09-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriin AB, Gabai VL, Yaglom J, Shifrin VI, Sherman MY. Proteasome inhibitors activate stress kinases and induce Hsp72. Diverse effects on apoptosis. J Biol Chem. 1998;273:6373–6379. doi: 10.1074/jbc.273.11.6373. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Furusu A, Xu Q, Konta T, Kitamura M. Unexpected transcriptional induction of monocyte chemoattractant protein 1 by proteasome inhibition: involvement of the c-Jun N-terminal kinase-activator protein 1 pathway. J Immunol. 2001;167:1145–1150. doi: 10.4049/jimmunol.167.3.1145. [DOI] [PubMed] [Google Scholar]
- Napoli I, Noon Luke A, Ribeiro S, Kerai Ajay P, Parrinello S, Rosenberg Laura H, Collins Melissa J, Harrisingh Marie C, White Ian J, Woodhoo A, Lloyd Alison C. A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron. 2012;73:729–742. doi: 10.1016/j.neuron.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Nave KA, Salzer JL. Axonal regulation of myelination by neuregulin 1. Curr Opin Neurobiol. 2006;16:492–500. doi: 10.1016/j.conb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Neef DW, Jaeger AM, Thiele DJ. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov. 2011;10:930–944. doi: 10.1038/nrd3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T, Iijima S, Hoshikawa S, Miura T, Yamamoto S, Oda H, Nakamura K, Tanaka S. Opposing extracellular signal-regulated kinase and Akt pathways control schwann cell myelination. J Neurosci. 2004;24:6724–6732. doi: 10.1523/JNEUROSCI.5520-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu N. A threshold selection method from gray-level histograms. IEEE Trans Syst Man Cybernectics. 1979;9:62–66. [Google Scholar]
- Palmada M, Kanwal S, Rutkoski NJ, Gufstafson-Brown C, Johnson RS, Wisdom R, Carter BD. c-Jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J Cell Biol. 2002;158:453–461. doi: 10.1083/jcb.200112129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R, Jessen KR. c-Jun is a negative regulator of myelination. J Cell Biol. 2008;181:625–637. doi: 10.1083/jcb.200803013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennuto M, Tinelli E, Malaguti M, Del Carro U, D'Antonio M, Ron D, Quattrini A, Feltri ML, Wrabetz L. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot–Marie–Tooth 1B mice. Neuron. 2008;57:393–405. doi: 10.1016/j.neuron.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju S, Madorsky I, Pileggi JG, Kamal A, Notterpek L. Pharmacological induction of the heat shock response improves myelination in a neuropathic model. Neurobiol Dis. 2008;32:105–115. doi: 10.1016/j.nbd.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AM, Perea J, McGuigan A, King RHM, Muddle JR, Gabreëls-Festen AA, Thomas PK, Huxley C. Comparison of a new PMP22 transgenic mouse line with other mouse models and human patients with CMT1A*. J Anat. 2002;200:377–390. doi: 10.1046/j.1469-7580.2002.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi AH, Morris SJ, Ho WC, Dickson KM, Doucet G, Milutinovic S, Durkin J, Gillard JW, Barker PA. AEG3482 is an antiapoptotic compound that inhibits Jun kinase activity and cell death through induced expression of heat shock protein 70. Chem Biol. 2006;13:213–223. doi: 10.1016/j.chembiol.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Samadi AK, Zhang X, Mukerji R, Donnelly AC, Blagg BS, Cohen MS. A novel C-terminal HSP90 inhibitor KU135 induces apoptosis and cell cycle arrest in melanoma cells. Cancer Lett. 2011;312:158–167. doi: 10.1016/j.canlet.2011.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H-Y, He J-C, Wang Y, Huang Q-Y, Chen J-F. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem. 2005;280:39962–39969. doi: 10.1074/jbc.M505524200. [DOI] [PubMed] [Google Scholar]
- Syed N, Reddy K, Yang DP, Taveggia C, Salzer JL, Maurel P, Kim HA. Soluble neuregulin-1 has bifunctional, concentration-dependent effects on schwann cell myelination. J Neurosci. 2010;30:6122–6131. doi: 10.1523/JNEUROSCI.1681-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapinos N, Ohnishi M, Rambukkana A. Erb B2 receptor tyrosine kinase signaling mediates early demyelination induced by leprosy bacilli. Nature Med. 2006;12:961–966. doi: 10.1038/nm1433. [DOI] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, Chao MV, Falls DL, Role L, Salzer JL. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 2005;47:681–694. doi: 10.1016/j.neuron.2005.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban MJ, Li C, Yu C, Lu Y, Krise JM, McIntosh MP, Rajewski RA, Blagg BSJ, Dobrowsky RT. Inhibiting heat shock protein 90 reverses sensory hypoalgesia in diabetic mice. ASN Neuro. 2010;2:e00040. doi: 10.1042/AN20100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban MJ, Pan P, Farmer KL, Zhao H, Blagg BS, Dobrowsky RT. Modulating molecular chaperones improves sensory fiber recovery and mitochondrial function in diabetic peripheral neuropathy. Exp Neurol. 2012;235:388–396. doi: 10.1016/j.expneurol.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- Yu C, Rouen S, Dobrowsky RT. Hyperglycemia and downregulation of caveolin-1 enhance neuregulin-induced demyelination. Glia. 2008;56:877–887. doi: 10.1002/glia.20662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanazzi G, Einheber S, Westreich R, Hannocks MJ, Bedell-Hogan D, Marchionni MA, Salzer JL. Glial growth factor/neuregulin inhibits Schwann cell myelination and induces demyelination. J Cell Biol. 2001;152:1289–1299. doi: 10.1083/jcb.152.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zochodne DW. The challenges and beauty of peripheral nerve regrowth. J Periph Nerv Syst. 2012;17:1–18. doi: 10.1111/j.1529-8027.2012.00378.x. [DOI] [PubMed] [Google Scholar]