

Abstract
Protein farnesyl transferase (FTase) catalyzes transfer of a 15-carbon farnesyl group from farnesyl diphosphate (FPP) to a conserved cysteine in the C-terminal Ca1a2X motif of a range of proteins, including the oncoprotein H-Ras (“C” refers to the cysteine, “a” to any aliphatic amino acid, and “X” to any amino acid) and the lipid chain interacts with, and forms part of the Ca1a2X peptide binding site. Previous studies have shown that H-Ras biological function is ablated when it is modified with lipids three-to-five orders of magnitude less hydrophobic than FPP. Here, we employed a library of anilinogeranyl diphosphate (AGPP) and phenoxygeranyl diphosphate (PGPP) derivatives with a range of polarity (logP (lipid alcohol) = 0.7–6.8, logP (farnesol) = 6.1) and shapes to examine whether FTase catalyzed transfer to peptide was dependent on the hydrophobicity of the lipid. Analysis of steady-state transfer kinetics for analogues to dansyl-GCVLS peptide revealed that the efficiency of lipid transfer was highly dependent on both the shape and size, but was independent of the polarity of the analogue. These observations indicate that hydrophobic features of isoprenoids critical for their association with membranes and/or protein receptors are not required for efficient transfer to Ca1a2X peptides by FTase. Furthermore, the results of these studies indicate that the role played by the farnesyl lipid in the FTase mechanism is primarily structural. We propose a model where the FTase active site stabilizes a membrane interface-like environment to explain these results.
Keywords: Protein Farnesyltransferase, Farnesyl diphosphate, Enzyme Catalysis, Lipophilicity, Terpenoids
Introduction
Protein farnesyl transferase (FTase) catalyzes transfer of a 15-carbon farnesyl group from farnesyl diphosphate (FPP, 1, Figure 1) to a conserved cysteine in the C-terminal Ca1a2X motif of a range of proteins, including the oncoprotein H-Ras (“C” refers to the cysteine, “a” to primarily aliphatic amino acids, and “X” to any amino acid). Farnesylation is obligatory for the proper biological function of H-Ras and a number of small molecule inhibitors of FTase (FTIs) have been developed as anti-cancer agents.[1–5] Several FTIs are currently in phase I, II and III clinical trials for the treatment of cancer,[2, 6–7] but the response in patients has not been significant.[1] An explanation for the lack of FTI clinical efficacy is the process of alternative prenylation where some FTase substrates can become geranylgeranylated by geranylgeranyl transferase type I (GGTase-I) when FTase activity is limiting.[1, 8–10] This has led to substantial interest in developing alternative lipids incapable of supporting normal prenyl group function.[11–13] Several studies have examined lipid features that influence the efficiency of isoprenoid transfer to Ca1a2X peptides by FTase.[13–19] These studies have focused on how the length of the isoprenoid affected transfer kinetics,[14] replacing the terminal isoprene with aryl substituents,[15, 18, 20–21] and altering the steric demands and electronic properties of the isoprenoid branched methyl groups.[13–14, 16, 22–23] Despite these efforts, information on the structural features that give rise to productive interactions of FPP analogues with the FTase active site remain limited. The reaction mechanism of FTase is unexpectedly complex (Figure 2). Product release is the rate determining step (kcat) for the FTase reaction, and an unusual feature of the FTase mechanism is that product dissociation is greatly enhanced by binding of either a new FPP or Ca1a2X peptide substrate. Remarkably, the hydrophobic thioether product has decreased affinity for the enzyme despite the non-polar amino acid resides lining the active site.[24] X-ray crystallographic analysis shows that the lipid chain interacts with, and forms a substantial part of the Ca1a2X peptide binding site throughout the course of the reaction.[25–26]
Figure 1.
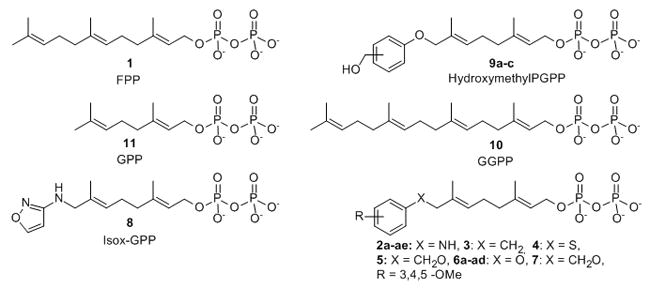
FPP, GPP, GGPP and FPP analogues.
Figure 2.
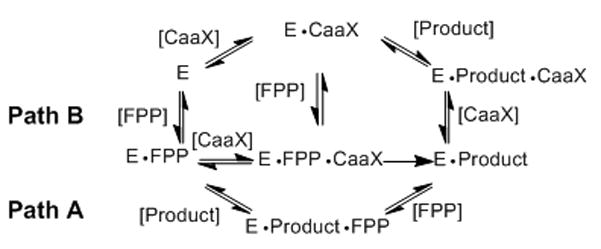
FTase reaction mechanism showing two pathways. Path A represents FPP stimulated product release, and Path B represents peptide stimulated product release. E is the FTase enzyme, E•FPP is the FTase•FPP complex, E•FPP•CaaX is the FTase•FPP•CaaX peptide complex, E•Product is the FTase bound product complex, E•Product•FPP is the FTase bound to both FPP and the reaction product, E•CaaX is the peptide bound FTase inhibitory complex, and E•Product•CaaX is the peptide bound enzyme product complex.
FPP analogues have been used to study the physical interactions between the lipid, FTase and the Ca1a2X peptide as well as the biological function of the modification.[12, 15–16, 18, 27] The Ca1a2X tetrapeptide is sufficient for prenyltransferase recognition, and the kinetics of FPP transfer to dns-GCVLS is identical to that of full length H-Ras.[28–29] The analogue 8-anilinogeranyl diphosphate (AGPP, 2a, Figure 1) is transferable to Ca1a2X substrates with apparent steady-state kinetics nearly identical to FPP, and the aniline moiety appears to act as an isostere for the FPP terminal isoprene.[15, 20] AGPP has been used to probe the endogenous modification of proteins by FTase and is competitive with FPP in vitro and in cell culture.[20, 30–31] We previously prepared and examined the FTase catalyzed lipid transfer of an AGPP analogue library to the dns-GCVLS peptide corresponding to the H-Ras Ca1a2X motif and found that reactivity depends on both size and shape of the lipid.[32–33] Small meta- and para-substitutions on the aniline ring increase reactivity with dns-GCVLS while others with ortho-substitutions were potent dns-GCVLS modification FTase inhibitors. In other work, investigation of ten FPP analogues showed that the normal biological function of H-Ras is blocked when modified with isoprenoids that are three-to-five orders of magnitude less hydrophobic than the farnesyl group.[12] H-Ras biological function appears to require a minimum lipophilicity of the prenyl group to allow important interactions downstream of the C-terminal processed H-Ras protein. These observations suggest that hydrophilic FPP analogues are prenyl function inhibitors (PFIs) that may serve as lead compounds for a unique class of potential anti-cancer therapeutics. However, the poor reactivity of the least polar FPP analogue Isox-GPP, 8 with H-Ras and Ca1a2X peptides raised the possibility that other polar FPP analogues might also be poor substrates for FTase.
The anti-Ras behavior of this small set of more polar FPP analogues prompted us to examine the relationship of isoprenoid hydrophobicity to the efficiency of FTase catalyzed lipid transfer to Ca1a2X peptides. Additional FPP analogues with a range of polarity and shapes were prepared and their transfer efficiency determined. We found that the efficiency of lipid transfer was highly dependent on both the shape and size, but was independent of the hydrophobicity of the analogue. The apparent catalytic efficiency (kcat/Kmpeptide) for transfer of several analogues to a dns-GCVLS (H-Ras Ca1a2X sequence) peptide was greater than that for the natural substrate FPP. These observations indicate that hydrophobic features of isoprenoids critical for their association with membranes and/or protein receptors are not required for efficient transfer to Ca1a2X peptides by FTase, and that hydrophobic interactions between the lipid and aromatic residues in the FTase active site do not drive binding of FPP to the enzyme. Furthermore, the results of these studies indicate that the role played by the farnesyl lipid in the FTase mechanism is primarily structural. We propose a model where the FTase active site stabilizes a membrane interface-like environment to explain these results.
Results and Discussion
Synthesis of FPP analogues where the AGPP amino group is replaced by other moieties
In order to examine whether the AGPP aniline nitrogen contributed specific interactions obligatory for efficient analogue transfer to Ca1a2X peptides, molecules 3, 4, 5, 6a and 7 with CH2, S, CH2O, O linkers between the terminal aryl group and the geranyl chain were synthesized. Methylene linked analogue 3 was prepared in five steps from 8-chlorogeranyl acetate using a modified procedure of Spencer and co-workers (Scheme 1).[34] THP ether 13 was obtained by protection of 8-chlorogeraniol 12 in quantitative yield. Coupling of benzyl magnesium chloride with chloride 13 followed by removal of the THP ether provided alcohol 15 which was converted to the corresponding bromide and diphosphorylated to give diphosphate 3.
Scheme 1.
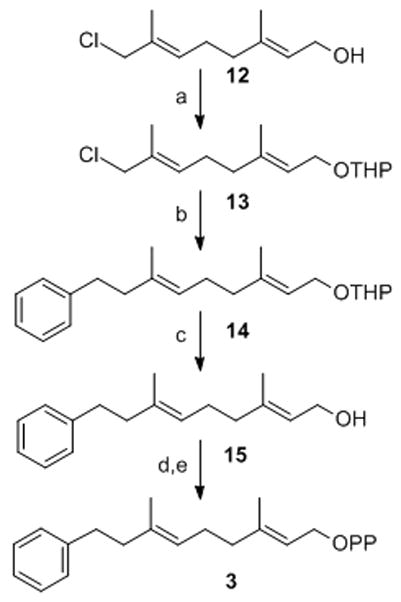
Synthesis of (2E,6E)-3,7-Dimethyl-9-phenyl-nona-2,6-dien-1-diphosphate: a) PPTS, DHP, DCE; b) PhCH2MgCl, Et2O, 0°C; c) PPTS, MeOH; d) Ph3PBr2, CH3CN; e) (Bu4N)3HP2O7, CH3CN.
The key step in the preparation of analogues 4, 5 and 7 was alkylation of the appropriate, thiophenol or benzylalcohol with 8-bromogeranyl acetate 16 (Scheme 2). The desired diphosphates 4, 5 and 7 were then obtained by saponification of acetates 18a–c to give alcohols 19a–c which were converted to the corresponding bromides followed by diphosphorylation with (Bu4N)3HP2O7 in CH3CN. Compound 6a was prepared by solid state Mitsunobu reaction as described below.
Scheme 2.
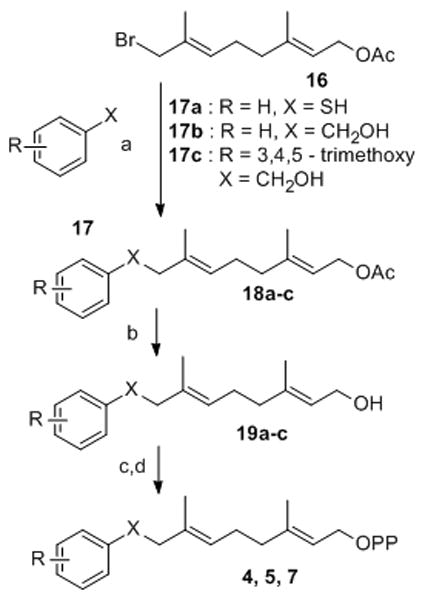
Synthesis of diphosphates 4, 5, 7: a) NaH, THF; b) K2CO3, MeOH/H2O; c) Ph3PBr2, CH3CN; d) (Bu4N)3HP2O7, CH3CN.
Analogue linker atom has a moderate effect on reactivity with Ca1a2X peptide
The kinetic parameters appkcat, and appKmpeptide for analogue transfer to dns-GCVLS were measured utilizing a continuous fluorescence assay. The apparent kcat/Km peptide is the catalytic efficiency of FTase which measures the ability of the enzyme to catalyze a reaction at low peptide substrate concentrations. We found that the linker appears to have only a modest effect on the transfer efficiency, suggesting that the aniline nitrogen is not critical for efficient transfer of the unsubstituted parent molecules. Consistent with previous observations, transfer of the bulky 3,4,5-trimethoxy substituted analogue 7 was not detected. All reactions between FPP analogues and dns-GCVLS peptide were analyzed by HPLC with detection of the resulting components by dansyl fluorescence. No product detected by HPLC where we observe no increase in dansyl fluorescence. Although single turnover reactions for the ”non-reactive” analogue diphosphates can not be excluded by this method, the lack of steady state turnover indicates that product release is impaired.
Synthesis of phenoxygeranyl diphosphate FPP analogue library
A library of 25 transferable AGPP derivatives had been previously prepared.[35–36] The number of analogues to be examined was increased by preparing a directed library of 33 ether linked phenoxygeranyl diphosphates (PGPP) 6a–ad and 9a–c (Scheme 3 & 4). The PGPP ether linkage removes the aniline H-bond donor, alters the conformational preference of the lipid and allows for the straightforward introduction of polar functional groups. A focused phenoxygeranyl diphosphate library was prepared by a mixed solid-phase organic synthesis (SPOS)-solution phase route (Scheme 3). Reduction of the previously described resin-bound aldehyde 20 with NaBH4 in DCE/EtOH (1:1) furnished the corresponding alcohol 21 in 82% yield. Diversity was introduced into the library by solid phase Mitsunobu reaction of alcohol 21 with 5 equivalents of the appropriately substituted phenols in DCE at room temperature to provide the corresponding resin bound ethers 23a–ad. The resin bound THP ethers 23a–ad were released from the resin as the corresponding allylic bromides by agitation with two equivalents of Ph3PBr2 in CH2Cl2 for 4 h and the bromides were then trapped in situ with 6 equivalents of (Bu4N)3HP2O7 in CH3CN to give the desired phenoxygeranyl diphosphates 6a–ad. The crude diphosphates were converted to the NH4+ form by ion exchange chromatography and then purified by RP-HPLC. Release of the THP resin linked lipids as the corresponding allylic bromides provides a traceless linker pathway to the desired FPP analogues 6a–ad. The phenoxygeraniols 24a–ad were made by cleavage of ethers 23a–ad from the resin by treatment with DCE/MeOH/PPTS at 80°C followed by silica gel column chromatography (Table 2).
Scheme 3.
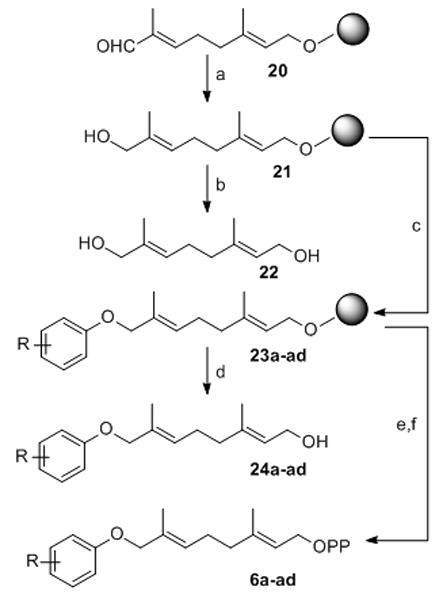
Synthesis of phenoxygeranyl diphosphates 6a–6ad: a) NaBH4, DCE/EtOH; b) PPTS, MeOH/DCE, reflux c) Ph3P, DEAD, Phenols, DCE; d) PPTS, MeOH/DCE, reflux; e) Ph3PBr2, CH2Cl2; f) (Bu4N)3HP2O7, CH3CN
Scheme 4.
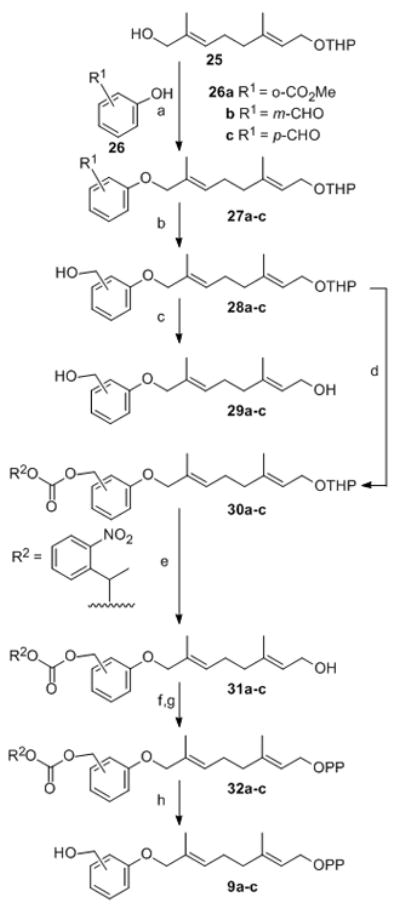
Synthesis of Hydroxymethylphenoxygeranyl diphosphate: a) DEAD, Ph3P, THF; b) NaBH4, EtOH; c) PPTS, M e O H ; d )αα-methyl-o-nitrobenzyl chloroformate,3 pyridine/CH2Cl2; e) PPTS, MeOH; f) Ph3PBr2, CH3CN; g) (Bu4N)3HP2O7, CH3CN; h) NH4HCO3/H2O, hγ, 0°C.
Table 2.
Hydrophobicity and steady-state kinetic properties of isoprenoids for reaction with dns-GCVLS
| R-group and position on ring [a] | Compound number | Apparent logP [b] | apparent kcat[c] (s−1)•10−2 | apparent Km dns-GCVLS [c] (μM) | apparent kcat/Km dns-GCVLS [c] (μM−1•s−1)•10−2 |
|---|---|---|---|---|---|
| GPP | 11 | 3.6 | 0.4 ± 0.1 | 27 ± 9 | 1.5 ± 0.7 |
| Isox-GPP | 8 | 0.7 | 4 ± 1 | 1.3 ± 0.4 | 2.8 ± 0.2 |
| FPP | 1 | 6.1 | 14 ± 2 | 0.8 ± 0.1 | 18 ± 3 |
| H | 6a | 4.1 | 30 ± 2 | 2.0 ± 0.2 | 15 ± 2 |
| H | 2a | 3.6 | 12 ± 2 [d] | 0.5 ± 0.1 [d] | 24 ± 6 [d] |
| o-F | 6b | 4.3 | 19 ± 1 | 1.3 ± 0.1 | 15 ± 3 |
| m-F | 6c | 4.8 | 0.4 ± 0.04 | 0.1 ± 0.015 | 8.5 ± 1.6 |
| p-F | 6d | 3.7 | 58 ± 5 | 4.5 ± 0.5 | 13 ± 3 |
| o-F | 2b | 4.1 | 15 ± 3 [d] | 0.9 ± 0.2 [d] | 16 ± 1 [d] |
| m-F | 2c | 3.7 | 10 ± 1 [d] | 0.34 ± 0.08 [d] | 26 ± 3 [d] |
| p-F | 2d | 3.5 | 9.5 ± 0.4 [d] | 0.26 ± 0.04 [d] | 35 ± 4 [d] |
| 2,6-di-F | 6e | 4.4 | 167 ± 3 | 16.5 ± 0.6 | 10 ± 2 |
| 3,4-di-F | 6f | 4.9 | 11 ± 0. 4 | 0.7 ± 0.05 | 16 ± 4 |
| 2,3-di-F | 6g | 4.4 | 23 ± 0. 5 | 1.3 ± 0.7 | 19 ± 1 |
| 3,5-di-F | 6h | 4.8 | 10 ± 0. 2 | 0.5 ± 0.02 | 18 ± 2 |
| 2,3,5,6-tetra-F | 6i | 4.6 | 310 ± 2 | 51.2 ± 3.9 | 6 ± 1 |
| m-CN | 6j | 3.9 | 4 ± 0.1 | 0.42 ± 0.02 | 10 ± 2 |
| p-CN | 6k | 2.7 | 193 ± 9 | 33 ± 2.3 | 6 ± 2 |
| m-CN | 2h | 3.1 | 13 ± 1 [d] | 1.0 ± 0.1 [d] | 11.5 ± 0.3 [d] |
| p-CN | 2i | 2.9 | 70 ± 3 [d] | 10.2 ± 0.6 [d] | 6.8 ± 0.1 [d] |
| p-NO2 | 6l | 4.2 | 55 ± 2 | 6.3 ± 0.4 | 8 ± 1 |
| p-NO2 | 2j | 3.1 | 160 ± 20 [d] | 15 ± 2 [d] | 10.7 ± 0.2 [d] |
| o-Br | 6m | 5.1 | 30 ± 3 | 3.1 ± 0.34 | 10 ± 1 |
| m-Br | 6n | 6.0 | 6 ± 0.4 | 70 ± 0.1 | 8 ± 2 |
| p-Br | 6o | 5.6 | 50 ± 1 | 18.7 ± 0.8 | 3 ± 1 |
| o-Br | 2k | 5 | 13 ± 1 [d] | 1.0 ± 0.2 [d] | 13 ± 3 [d] |
| p-Br | 2l | 4.6 | 27 ± 8 [d] | 1.7 ± 0.5 [d] | 16 ± 2 [d] |
| o-CH3 | 2m | 4.1 | 16.6 ± 0.9 [d] | 4.0 ± 0.4 [d] | 4.2 ± 0.5 [d] |
| m-CH3 | 2n | 3.3 | 6.9 ± 0.5 [d] | 0.17 ± 0.06 [d] | 38 ± 8 [d] |
| p-CH3 | 2o | 3.4 | 60 ± 20 [d] | 4 ± 1 [d] | 16 ± 1 [d] |
| o-CF3 | 2p | 4 | 30 ± 10 [d] | >10 [d] | 0.9 ± 0.1 [d] |
| m-CF3 | 6p | 6.0 | 13 ± 0.5 | 1.1 ± 0.1 | 12 ± 2 |
| 3,5-di-Cl | 6q | 5.2 | 19 ± 0.7 | 1.3 ± 0.1 | 15 ± 2 |
| 3,4-di-Cl | 6r | 5.1 | 24 ± 1 | 2.1 ± 0.13 | 12 ± 2 |
| o-I | 6s | 4.4 | 11 ± 0.5 | 3.2 ± 0.20 | 4 ± 1 |
| m-I | 6t | 3.7 | 7 ± 0.2 | 0.4 ± 0.02 | 17 ± 2 |
| p-I | 6u | 3.6 | 27 ± 1 | 2.3 ± 0.2 | 12 ± 1 |
| m-I | 2r | 4.8 | 24 ± 2 [d] | 0.9 ± 0.1 [d] | 26 ± 3 [d] |
| p-I | 2s | 4.9 | 17 ± 3 [d] | 1.4 ± 0.4 [d] | 12 ± 4 [d] |
| o-CH2OH | 9a | 1.0 | NR[f] | NR [f] | NR[f] |
| m-CH2OH | 9b | 0.9 | 44 ± 1 | 4.0 ± 0.2 | 10 ± 2 |
| p-CH2OH | 9c | 0.8 | 115 ± 6 | 8.0 ± 0.6 | 14 ± 3 |
| o-MeO | 2t | 4.1 | 8.8 ± 0.9 [d] | 0.9 ± 0.2 [d] | 9.3 ± 0.6 [d] |
| m-MeO | 2u | 3.3 | 53 ± 5 [d] | 4.3 ± 0.6 [d] | 12 ± 2 [d] |
| p-MeO | 2v | 3 | 140 ± 40 [d] | 30 ± 10 [d] | 5 ± 2 [d] |
| o-Et | 6v | 4.0 | 11 ± 1 | 2.5 ± 0.4 | 4 ± 1 |
| m-Et | 6w | 5.5 | 17 ± 0.7 | 2.1 ± 0.2 | 8 ± 1 |
| p-Et | 6x | 5.1 | 28 ± 0.8 | 4.4 ± 0.3 | 6 ± 1 |
| o-Et | 2w | 4.8 | 17 ± 2 [d] | 7 ± 1 [d] | 2.4 ± 0.6 [d] |
| m-Et | 2x | 4.6 | 8 ± 2 [d] | 3.8 ± 0.8 [d] | 2.1 ± 0.1 [d] |
| m-CF3O | 6y | 5.1 | 3 ± 0.2 | 0.3 ± 0.01 | 11 ± 2 |
| p-CF3O | 6z | 5.1 | 16 ± 0.3 | 1.3 ± 0.1 | 12 ± 2 |
| m-CF3O | 2aa | 4.9 | 40 ± 10 [d] | 2.3 ± 0.6 [d] | 19 ± 1 [d] |
| p-CF3O | 2ab | 4.9 | 27 ± 6 [d] | 3.1 ± 0.9 [d] | 8.7 ± 0.3 [d] |
| m-iPr | 6aa | 6.7 | 21 ± 0.5 | 1.9 ± 0.1 | 11 ± 1 |
| p-iPr | 6ab | 6.8 | 44 ± 5 | 21.4 ± 2.9 | 2 ± 1 |
| o-Ph | 6ac | Nde [e] | NR [f] | NR [f] | NR [f] |
| p-Bn | 6ad | 6.5 | 30 ± 0.6 | 2.3 ± 0.1 | 13 ± 3 |
The analogues are listed in order of increasing substituent surface area.
LogP measurements of the corresponding alcohol.
app kcat,, appKmpeptide and apparent kcat/Kmpeptide were determined using a Michaelis-Menton analysis as described.[32]
From Troutman et al.[32]
Nde indicates not determined.
NR indicates no reaction determined by RP-HPLC product analysis.
Three additional hydroxymethyl PGPP derivatives 9a–c were prepared by solution methods (Scheme 4) to test the hypothesis that the size and shape of the lipid rather than the absolute polarity are essential for efficient transfer by FTase to peptide. AGPP analogues with methoxy, trifluoromethoxy and ethyl groups are transferred to dns-GCVLS by FTase[32] and the polar hydroxymethyl group is roughly isosteric with these moieties (Scheme 4). Preparation of these polar PGPP analogues required masking of the reactive hydroxymethyl group during synthesis. The similar reactivity of the benzylic and allylic alcohol functions as well as the acid and base sensitive diphosphate in these amphipathic molecules narrowly constrain suitable protecting groups that can be used. Consequently, we employed the photolabile α-methyl-o-nitrobenzyl carbonate group to protect the hydroxymethyl group of analogues 28a–c during THP cleavage, subsequent diphosphorylation, ion exchange and purification. Alcohols 29a–c were obtained by Mitsunobu reaction of 8-hydroxy-OTHP protected geraniol 25 with either 3- or 4-hydroxybenzaldehyde or methylsalicylate followed by NaBH4 reduction. The THP protected alcohols 28a–c were acylated with αα-methyl-o-nitrobenzyl chloroformate to give carbonates 30a–c. The corresponding isoprenoid alcohols 31a–c were obtained in quantitative yield by cleavage of THP ethers 30a–c with PPTS in MeOH. The nitrobenzyl protected diphosphates 32a and 32c were obtained by treating the alcohols 31a–c with Ph3PBr2 to give the corresponding bromide which were then trapped in situ by (Bu4N)3HP2O7 in CH3CN. Carbonate 32c was not isolated, rather, p-hydroxymethyl PGPP 9c was purified directly from the diphosphorylation reaction.
The other two hydroxymethyl PGPP derivatives 9a and 9b were obtained in quantitative yield by photolysis of the RP-HPLC purified carbonates 32a and 32b with Pyrex filtered UV light for 5–10 min in aqueous ammonium bicarbonate at 0°C. The advantage of photo-deprotection is that the nitroso byproducts are separated from the pure analogue diphosphates 9a and 9b by CH2Cl2 extraction, avoiding additional chromatography (Scheme 4).
Analogue hydrophobicity depends on terminal aromatic moiety substituents
The lipophilicity of the anilinogeranyl diphosphates 2a–ae and phenoxygeranyl diphosphates 6a–ad and 9a–c is correlated with the apparent logP (logPapp) of the parent alcohols (Table 2) and was determined from RP-HPLC capacity factors. LogP is the logarithm of the partition coefficient between water saturated octanol and octanol saturated water and is a useful metric of hydrophobicity and the ability of compounds to associate with membranes. The incorporation of aromatic rings and heteroatoms into the analogues decrease their hydrophobicity relative to farnesol.
FTase catalysis depends on isoprenoid size and shape, not hydrophobicity
The kinetic parameters appkcat, appKmpeptide and apparent kcat/Kmpeptide were measured for transfer of each PGPP analogue to dns-GCVLS utilizing a continuous fluorescence assay (Table 2). Thirty one of the 33 PGPP analogues 6a–ad and 9a–c were detectably transferred to dns-GCVLS peptide by FTase (Table 2). The aryl substituents of 20 PGPP molecules were identical to previously reported AGPP analogues.[32] In general, transfer kinetics and hydrophobicity of PGPP and AGPP analogues with identical aryl moieties were similar. The apparent logP of PGPP analogues varied within ±1.3 units of the corresponding AGPP (Table 2) and apparent kcat/Kmpeptide were within a factor of ±5.3. However, there are a number of important differences in reactivity between the two different classes of FPP analogues. The p-Et-PGPP 6x, p-iPr-PGPP 6ab, m-iPr-PGPP 6aa, p-Bn-PGPP 6ad and o-I-PGPP 6s analogues were efficient substrates, whereas corresponding AGPP analogues were not transferred to the dns-GCVLS peptide.[32] Larger substituents in the transferable PGPP series may be productively accommodated in the FTase active site because the ether linkage is conformationally less restricted than the aniline linkage in the corresponding AGPP analogues. We found that the meta- and para-hydroxymethyl PGPP 9b and 9c were efficiently transferred by FTase while the ortho- isomer 9a was not, further reinforcing the observation that reactivity depends on substituent position and size. Remarkably, the para-hydroxymethyl PGPP 9b was almost as efficient a substrate as FPP, indicating that FTase activity is not necessarily decreased by hydrophilic lipid analogues.
FPP analogue transfer efficiency does not correlate with lipid hydrophobicity
The apparent kcat/Kmpeptide, appKmpeptide and appkcat for the library of 54 transferable PGPP and AGPP analogues as well as GPP and FPP was plotted against apparent logP to determine if there was a simple relationship between FTase catalytic activity and the lipid shape, size and hydrophobicity (Figure 3).
Figure 3.

Plot of apparent logP vs apparent kcat/Kmpeptide for FPP, GPP and 54 transferable AGPP and PGPP analogues with dns-GCVLS. There are 39 different aryl structures present in the analogue library, where 16 are unique to the PG series, 8 unique to the AG series and 15 represented in both. The open diamonds ◆ are FPP, GPP, AGPP and PGPP as indicated, the solid squares ■ are ortho-, solid triangles ▲ are meta-, solid circles ● are para- substituted analogues and the open circles ○ are Isox-GPP and multi-substituted PG analogues. The underlying data is from table 2.
Thirty nine different substituted aryl groups are represented in the library, where 15 substituents are identical in both the anilinogeranyl and phenoxygeranyl series, as well as eight unique AGPP and 16 unique PGPP structures. The hydrophobicity of the compounds in the library spanned six orders of magnitude and included molecules with both greater (p-iPr-PGPP 6ab, logP = 6.8) and substantially lower (Isox-GPP 8, logP = 0.7) apparent logP than FPP (logP = 6.1). Surprisingly, we found no discernable relationship between apparent kcat/Kmpeptide and apparent logP of the transferable analogues. Similar plots of appkcat vs. apparent logP and appKmdns-GCVLS vs. apparent logP also show no obvious relationship between the measured properties.
The pattern of analogue reactivity with dns-GCVLS leads to the surprising conclusion that isoprenoid transfer efficiency does not depend on lipid hydrophobicity. Rather, isoprenoid size and shape appear to be the most important lipid physical property for FTase catalyzed transfer to Ca1a2X peptides. This result is even more startling in light of the extensive contacts between the farnesyl hydrocarbon and the predominantly hydrophobic aromatic amino acid side chains in the enzyme active site revealed in X-ray crystal structures of the binary FTase•FPP and ternary FTase•FPP•CaaX and FTase•product complexes.[26, 37–44] These observations are important because they indicate that the biological functions of the isoprenoid that depend on hydrophobic association with membranes and/or protein receptors are not critical for efficient transfer to Ca1a2X peptides by FTase. Furthermore, the results of these studies indicate that the role played by the farnesyl lipid in the FTase mechanism is primarily structural. Several data provide evidence to support these conclusions.
There are multiple points in the FTase mechanism where lipid binding is structurally important (Figure 1).[26] Mutagenesis and computational studies suggest that the primary source of free energy for FPP binding to FTase are electrostatic interactions between the negatively charged diphosphate group and the positively charged amino acid residues at the upper rim of the FTase active site.[15, 45] The lipid binds to one wall of the FTase active site in an extended conformation in the E•FPP complex and forms a substantial part of the peptide binding site in the E•FPP•CaaX complex.[40–41] Lipids such as GPP that are too small to adequately fill the farnesyl lipid binding site, as well as molecules that are substantially larger than FPP such as GGPP, are poor substrates for FTase.[14–15, 46] Larger molecules physically interfere with the transfer reaction by occluding the peptide binding site.[14, 18] The poor transfer kinetics for GPP (logP = 3.6) relative to FPP are not due to reduced lipophilicity of the geranyl group, as AGPP is almost identical in size to FPP, but has the same logP as GPP and is transferred to Ca1a2X peptides more efficiently than FPP.[32] Additionally, a number of other analogues with a range of lipophilicities are transferred more efficiently to dns-GCVLS than FPP (Table 2), reinforcing the observation that the size and shape of the isoprenoid are critical determinants of transferability.[32, 47] In particular, the divergent transfer efficiencies of the four most polar analogues, Isox-GPP 8 and o-, m-, p-hydroxymethyl PGPP 9a–c, illustrate this point (Table 2).
The Ca1a2X peptide adopts a single extended conformation in the E•FPP•CaaX, E•Product and E•FPP•Product complexes.[26] Notably, Ca1a2X peptides with a wide range of hydrophobicity and amino acid residues in the a1, a2 and X positions are productively accommodated in the active site in essentially the same conformation.[26, 48] The third isoprene of the lipid diphosphate makes intimate contacts with the a2 residue and the peptide backbone of the Ca1a2X peptide co-substrate in the E • FPP • CaaX, E-product and E•FPP•Product complexes.[26] In contrast, contacts between the first and second isoprenes and FTase active site amino acid side chains are disrupted upon product formation and new interactions form between the lipid and the Ca1a2X peptide in the E•Product complex.[26] It is clear that substantial changes in the structure, electronics and hydrophobicity of the terminal isoprene and its interactions with the Ca1a2X peptide do not interfere with achieving the transition state and product formation (Tables 1 & 2).[32] The dissociation constant (KDpeptide) for four Ca1a2X peptides with the FTase•FPP complex are very different from each other and are not correlated with kcat/Kmpeptide.[16] Furthermore, changes in the structure of the first and second isoprene can also yield transferable analogues.[16, 49–52] However, there is no simple relationship between the structures of the lipid donor and peptide acceptor and their reactivity.
Table 1.
Steady-state kinetic properties of FPP diphosphate analogues for reaction with dns-GCVLS
| R-group and position on ring [a] | Compound number | Apparent logP [b] | apparent kcat [c] (s−1)•10−2 | apparent Kmdns-GCVLS[c] (μM) | apparent kcat/Kmdns-GCVLS[c] (μM−1•s−1)•10−2 |
|---|---|---|---|---|---|
| NH | 2a | 3.6 | 10 ± 3 | 0.4 ± 0 | 20 ± 4 |
| CH2 | 3 | 4.8 | 40 ± 2 | 3.8 ± 0.3 | 11 ± 8 |
| S | 4 | 4.8 | 17 ± 0.5 | 2.0 ± 0.1 | 8 ± 2 |
| CH2O | 5 | 4.0 | 17 ± 0.3 | 1 ± 0.4 | 17 ± 10 |
| O | 6a | 4.1 | 30 ± 2 | 2.0 ± 0.2 | 16 ± 8 |
| CH2O,X=3,4,5- Trimethoxy | 7 | 3.2 | Nde [d] | Nde [d] | Nde [d] |
The analogues are listed in order of increasing substituent surface area.
LogP measurements of the corresponding alcohol.
appkcat,, appKmpeptide and apparent kcat/Kmpeptide were determined using a Michaelis-Menton analysis as described. [32]
Nde indicates not determined.
The thioether product is substantially more hydrophobic than either substrate and product release depends on binding of either a new lipid diphosphate or Ca1a2X peptide (Figure 1).[40, 35] Product release stimulated by lipid diphosphate binding results in formation of the E•Product•FPP complex where two lipid moieties interact with the enzyme. The structure of this complex revealed by x-ray crystallographic analysis shows that the new FPP and alkylated peptide are in the active site while the farnesyl thioether is flipped out into an exit groove.[26, 40, 53–54] Presumably, the association of the FPP and displacement of the alkylated lipid is driven by electrostatic interaction of the diphosphate moiety with the enzyme. Displacement of the alkylated peptide product by Ca1a2X peptide is consistent with the free energy of E•Product being higher than E•CaaX and the observation that farnesylation of competitive Ca1a2X peptides decreases their affinity for FTase.[24] We have previously observed that AGPP may be less efficient than FPP at stimulating product release due to the decreased hydrophobicity of AGPP.[32, 35] However, AGPP is transferred more efficiently to dns-GCVLS than is FPP, indicating that there may be greater flux through the peptide stimulated release pathway for AGPP compared with FPP at comparable isoprenoid concentrations. It is possible that analogues more hydrophilic than AGPP are even less efficient at stimulating product release from the E•Product complex due to their relatively stronger interactions with the bulk solvent water. However, the efficiency of peptide stimulated release for some of the hydrophilic analogues (notably p-hydroxymethyl PGPP 9c and p-CN-PGPP 6k) must increase correspondingly, as their overall efficiency of transfer is comparable or significantly better than for FPP (Table 2).
The FTase active site stabilizes a membrane interface-like environment
The FTase active site is predominantly lined with tryptophan and tyrosine residues.[26] Partition of tryptophan and tyrosine side chains into membrane interfaces is strongly favored while their partition into the membrane alkyl phase is disfavored.[55] Zwitterionic membrane interfaces are about 15 Å thick and consist of a complex and thermally disordered mixture of water, charged lipid headgroups and methylenes from the edges of the hydrocarbon core.[55] The highly favorable free energy for partition of both tryptophan and tyrosine side-chains into membrane interfaces suggests that the FTase active site lining stabilizes a membrane interface like volume. Similar to membrane interfaces, the FTase active site contains a large number of water molecules as well as a variety of charged groups. The Arg 202β, Glu 198β and Asp 200β side chains are at the bottom, and the highly polar diphosphate binding and Zn+2 coordinating residues are at the upper rim of the active site. Small N-acyl peptides (1–6 residues) composed of non-polar or aromatic residues and charged C-termini are unstructured and partition almost exclusively into palmitoyloleoylphosphatidylcholine (POPC) membrane interfaces and are virtually insoluble in the membrane alkyl phase.[55] The dns-GCVLS peptide as well as the C-termini of H-, K- and N-Ras are unstructured in solution and bind to FTase in an extended conformation.[56] X-ray crystallographic analysis shows that Ca1a2X peptide substrate binding is mediated by interactions through ordered water molecules.[39]
FPP and the transferable analogues are intrinsically flexible and adopt a number of interconverting conformations in solution.[57] The conformational space accessible to the lipid chain in these molecules is significantly reduced upon binding to FTase.[57] The loss of conformational entropy is proposed to be partially compensated for by contacts with the binding site as well as by displacement of waters hydrating the active site and lipid. Structural studies reveal that the terminal FPP isoprene is buried in a pocket formed from W102β, Y154β, Y205β, C254β, W303β and the dns-GCVLS leucine Ca1a2X a2 side chain. Efficient transfer of the substantially less hydrophobic analogues implies that the free energy for lipid diphosphate association does not depend strongly on the lipid chain hydophobicity. The FTase dissociation constant (KD) for FPP and a series of four transferable FPP analogues with a range of hydrophobicities are similar to each other and are not correlated with kca or Kmanalogue for transfer to dansyl-GCVLS.[58] This is particularly important as farnesol preferentially partitions into alkyl phases relative to the aqueous phase (Table 2). The free energy change for moving amino acid side chains from water into a membrane interface is about 1/2 that of moving the same residue into the membrane alkyl phase.[55] A membrane interface like active site would act to level both favorable and unfavorable changes in free energy for transfer of the substrates and products into and out of the FTase active site. Therefore, one function of the active site aromatic residues is to reduce the affinity of FTase for alkyl phases in order to facilitate release of the hydrophobic product once the diphosphate has been displaced. This is consistent with the observation that farnesol is a poor inhibitor of FTase. Interaction of the FPP lipid chain with the active site residues is important for orienting and tethering the charged diphosphate moiety in a conformation most conducive to binding and activation as a leaving group. Furthermore, the leveling effect allows the PGPP and AGPP series lipid chains to function as a structural anchor for the diphosphate leaving group despite their reduced hydrophobicity. Previously, Gibbs and co-workers developed a pharmacophore model for FPP analogues where hydrophobic elements were placed at the lipid C3- and C11-methyl groups to account for critical interactions observed in a variety of analogues.[33, 51–52] Our observations that efficiency of isoprenoid transfer does not depend on the lipid hydrophobicity, but does depend on the lipid moiety occupying an appropriate volume of the active site are consistent with this model.
Conclusion
These observations are consistent with an FTase active site that has evolved to either provide no net stabilization or which slightly destabilizes the association of alkyl groups (and phases) while stabilizing the binding of Ca1a2X peptides with open hydrogen bonds. The conclusion that interactions between the FTase active site and alkyl phases are destabilized provides part of the explanation for why the substantially more hydrophobic farnesylated thioether product can be ejected from the active site by an incoming Ca1a2X peptide. Loss of the diphosphate upon thioether formation removes much of the free energy driving binding of FPP to FTase, allowing displacement of the product by either a new lipid diphosphate or new Ca1a2X peptide. These results also show that analogues with large changes in hydrophobicity engineered into the isoprenoid structure retain activity as FTase substrates, which may be important for the development of prenylated protein function inhibitors.
Experimental Section
All the reactions except for resin preparation were performed in PTFE tubes using a Quest 210 apparatus manufactured by Argonaut Technologies. All RP-HPLC was performed on an Agilent 1100 HPLC system equipped with a microplate autosampler, diode array and fluorescence detector. N-dansyl-GCVLS was purchased from Peptidogenics (San Jose, CA, USA). Spectrofluorometric analyses were performed in 96-well flat bottom, non-binding surface, black polystyrene plates (Corning, Excitation wavelength, 340 nm; emission wavelength 505 nm with a 10 nm cutoff) with a SpectraMax GEMINI XPS fluorescence well-plate reader. HPLC analysis of peptide reactions utilized a microsorb C18 column with 0.01% TFA in water (A) and 0.01% TFA CH3CN (B) as the mobile phase as described [32]. Absorbance readings were determined using a Cary UV/Vis spectrophotometer. All assays were performed at minimum in triplicate where the average values are reported with a one standard of deviation error. Recombinant mammalian protein farnesyl transferase was a gift from Dr. Carol Fierke (University of Michigan). Reaction temperature refers to the external bath. All solvents and reagents were purchased from VWR (EM Science-Omnisolv high purity) and Aldrich respectively and used as received. Merrifield-Cl resin was purchased from Argonaut technologies. Synthetic products were purified by silica gel flash chromatography (EtOAc/hexane) unless otherwise noted. RP-HPLC purification of lipid diphosphates were carried out using a Varian Dynamax, 10 μm, 300 Å, C-18 (10 mm × 250 mm) column and eluted with a gradient mobile phase and flow rate of 4 mL/min: 90% of A and 10% of B linear increase to 100% of B and retained in the same ratio for two more minutes and brought back to 90% of A and 10% of B over 5 minutes and monitored at 254 nm & 210 nm. A is 25mM aqueous ammonium acetate, B is CH3CN. 1H NMR and 13C NMR spectra of alcohols were obtained in CDCl3 and 1H and 31P of diphosphates in D2O with a Varian Inova spectrometer operating at 400 MHz (1H), 100.6 MHz (13C) and 161.8 MHz (31P). Chemical shifts are reported in ppm from CDCl3 internal peak at 7.27 ppm for 1H and 77.4 ppm for 13C; D2O (TSP, 0 ppm for 1H; H3PO4 as an external reference, 0 ppm for 31P). ESI-MS were performed at the University of Kentucky Mass Spectra Facility. Positive and negative ion electrospray ionization (ESI) mass spectra were obtained on a Thermofinnigan LCQ with sample introduction by direct infusion. High resolution impact (EI) ionization mass spectra were recorded at 25eV on a JEOL JMS-700T MSstation (magnetic sector instrument) at a resolution of greater than 10000. Samples were introduced via heatable direct probe inlet. Perfluorokerosine (pfk) was used to produce reference masses. Spectral data for all new molecules are reported in supplementary data.
Synthesis of Farnesyl diphosphate (FPP, 1), Geranyldiphosphate (GPP, 11) and Anilinogeranyl diphosphates (2a–ae)
FPP 1 and GPP 11 were prepared as described by Davisson et al.[59] Anilinogeranyl analogues 2a–ae were prepared on solid support or in solution as previously described by Subramanian and Chehade et al.[20, 36]
2-((2E,6E)-8-chloro-3,7-dimethyl-octa-2,6-dienyloxy)-tetrahydro-pyran (13)
Chloride 12 [60] (2 g, 1.06 mmol), dihydropyran (1.07 g, 1.27 mmol) and PPTS (50 mg) in dry methanol (10 mL) were stirred overnight at room temperature. The reaction mixture was concentrated, extracted into CH2Cl2, the organic phase was washed with sat. NaHCO3, water, brine, dried (MgSO4), filtered and concentrated. Chromatographic purification of the crude product gave chloride 13 in quantitative yield. The spectral data were consistent with previous reports.[61]
2-((2E,6E)-3,7-Dimethyl-9-phenyl-nona-2,6-dienyloxy)-tetrahydro-pyran (14)
Benzyl magnesium chloride (3.68 mL, 1.0 M solution in Et2O, 3.68 mmol) was added dropwise to a solution of 13 (1 g, 3.68 mmol) in Et2O (20 mL) at 0 °C and stirred for 3 h. After allowing the reaction to warm to room temperature and stir overnight, it was diluted with sat. NH4Cl (5 mL) and extracted with Et2O (2X). The organic extracts were dried (MgSO4), filtered and evaporated. Chromatographic purification of the oily residue gave ether 14 (994 mg, 83%).
(2E,6E)-3,7-Dimethyl-9-phenyl-nona-2,6-dien-1-ol (15)
Compound 14 (990 mg, 0.3 mmol) and PPTS (50 mg) were stirred in dry MeOH (5 mL) overnight. The reaction mixture was concentrated, extracted with ethyl acetate, washed with sat. NaHCO3, brine, dried (MgSO4), filtered and evaporated. Chromatographic purification of the residue gave alcohol 15 (670 mg, 91%).
(2E,6E)-3,7-Dimethyl-9-phenyl-nona-2,6-dien-1-diphosphate (3)
Ph3PBr2 (103 mg, 0.246 mmol) in CH3CN (2 mL) was added dropwise to a cooled (0°C) solution of alcohol 15 (30 mg, 0.123 mmol) in CH3CN (5 mL) and stirred for 2 h. ((n-Bu)4N)3HP2O7 (480 mg, 0.492 mmol) in CH3CN (2 mL) was then added and the solution allowed to warm to room temperature over 30 min. The reaction mixture was concentrated and washed with Et2O. The organic extracts were discarded and the residue suspended in 2 mL ion exchange buffer (25 mM NH4HCO3 in 2% (v/v) i-PrOH/water). The resultant white solution was loaded onto a preequlibrated 4×30 cm column of Dowex AG 50W-X8 (100–200 mesh) cation-exchange resin (NH4+ form). The flask was washed with buffer (2 × 2 mL) and loaded onto the column before eluting with 100 mL of ion exchange buffer. The eluent was lyophilized to yield a white solid. This solid was dissolved in 25 mM solution of NH4HCO3 buffer (4 mL), purified by RP-HPLC (retention time about 7 min.) and lyophilized to give 3 (18 mg, 32%) as a white powder.
General procedure for synthesis of alcohols 19a–c
Compound 17 (either 17a–c: 0.45 mmol) in THF (2 mL) was added dropwise to a stirred suspension of NaH (182 mg, 0.45 mmol) in THF (5 mL) at 0°C and allowed to stir for 1 h. Bromide 16 (1.25 g, 0.45 mmol ) in THF (5 mL) was added and stirred at 0°C for 1 h. After the reaction was allowed to warm to room temperature and stirred overnight, then diluted by slow addition of water, concentrated and extracted with CH2Cl2. The organics were washed with water, brine, dried (MgSO4), filtered and concentrated. The residue was dissolved in MeOH (5 mL) and stirred at room temperature overnight with K2CO3 (1.9 g, 1.35 mmol). The reaction mixture was concentrated, extracted with ethyl acetate. The organics were washed with water, brine and concentrated in vacuo. Chromatography of the residue gave 19a–c (0.988 g of 19a: 83%; 0.886 g of 19b: 75%; 1.16 g of 19c: 73%).
General procedure for synthesis of diphosphates 4, 5 and 7
Alcohol 19 (either 19a–c, 30 mg each) was stirred with Ph3PCl2 (2 equiv.) in dry CH3CN (3 mL) at 0°C and allowed to warm to room temperature over 1h and stirred at the same temperature for 10 h. ((n-Bu)4N)3HP2O7 (480 mg, 0.492 mmol) in CH3CN (2 mL) was then added and stirred for 3 h at room temperature. The lipid diphosphate was isolated as described above for compound 3 (14 mg of 4: 26%, 21 mg of 5: 38%, 23 mg of 7: 47%).
Resin-bound alcohol 21
Resin 20 (11.62 g, 1.87 mmol/g, 21.7mmol) was agitated with DCE/EtOH (200 mL, 1:1) for 20 min followed by addition of NaBH4 (1.10 g, 30 mmol) in small portions. The resultant mixture was agitated for 3 h at room temperature and then heated to 50°C for 12 h. The reaction mixture was cooled to room temperature and filtered. The resin was thoroughly washed with 1:1 THF/H2O (3X), 1:1 MeOH/H2O (3X), THF (3X), 1:1 MeOH/CH2Cl2 (3X) and CH2Cl2 and dried (11.68 gm).
General Procedure for resin cleavage
Resin 21 (500 mg, 0.935 mmol) in 10 mL of DCE/MeOH (1:1 v/v) and PPTS (50 mg, 0.199 mmol) was heated at reflux for 12 h. The cooled resin was filtered, washed with CH2Cl2 (3X) and the combined filtrate was concentrated. The residue was extracted with ethyl acetate, washed with sat. NaHCO3, water, dried (MgSO4), filtered and concentrated. Chromatographic purification of the crude product gave diol 22 (128 mg, 81%). The spectral data were consistent with previous reports.[62]
General Procedure for Alcohols 24a–ad (Mitsunobu reaction)
Four equivalent of DEAD (0.217 mL, 0.608 mmol, 40% solution in toluene) was added to a suspension of resin 21 (100 mg, 1.52 mmol/g, 0.152 mmol), Ph3P (159 mg, 0.608 mmol) and appropriate phenol (4 equiv.) in DCE (4 mL) and agitated overnight. Product resin 23a–ad was washed with CH2Cl2 (5X) and THF (5X) and dried under vacuum. The alcohols 24a–ad were cleaved from the dried resin as for 22 above.
General Procedure for the Synthesis of Phenoxygeranyldiphosphates (6a–ad)
Ph3PBr2 (71 mg, 0.168 mmol) was added to resin 23a–ad (1.352 mmol/g) pre-swollen in dry CH2Cl2 and agitated for 3 h under N2. ((n-Bu)4N)3HP2O7 (760 mg, 0.775 mmol) in dry CH3CN (3 mL) was then added and agitated for 3 h at room temperature. The resultant heterogeneous mixture was filtered and the solid washed twice with dry CH3CN. The lipid diphosphate 6a–ad was isolated from the combined filtrate as described above for compound 3.
General Procedure for THP ethers 27a–c
DEAD (1.9 mL, 40% in toluene, 4.25 mmol) was added dropwise to a stirred solution of 25 (900 mg, 3.5 mmol), phenol 26 (518 mg for 26b and 26c, 646 mg in case of 26a, 4.25 mmol), Ph3P (1.11 g 4.25 mmol) in THF (10 mL) at 0°C and stirred for 1 h. After allowing the reaction to warm to room temperature and stir overnight, it was diluted with sat. NaHCO3, concentrated, and extracted with CH2Cl2 (2X). The organic extracts were dried (MgSO4), filtered and concentrated. Chromatographic purification of the oily residue gave 27a–c (912 mg of 27a : 66%, 1.05 g of 27b : 83%, 702 mg of 27c : 55%).
General Procedure for hydroxymethyl-phenoxygeranyl-THP ethers 28a–c
NaBH4 (104 mg, 2.6 mmol) was added to ether 27 (1.3 mmol of 27a–c) in EtOH (10 mL) at 0°C and stirred for 3 h. The mixture was diluted with water and extracted with CH2Cl2 (2X). The organic extracts were dried (MgSO4), filtered and evaporated. Chromatographic purification of the oily residue gave 28a–c in quantitative yield.
General Procedure for diols 29a–c
Ether 28 (28a–c, 100 mg, 0.27 mmol) was stirred with PPTS (20 mg) in dry CH3OH (3 mL) overnight at room temperature. The solvent was evaporated and the residue extracted with ethyl acetate (2 × 20 mL). The organic extracts were washed with sat. NaHCO3, brine, dried (MgSO4), filtered and evaporated. Chromatographic purification of the oily residue gave 29a–c in quantitative yield.
General Procedure for carbonates 30a, 30b and 30c
[Caution! a-methyl-o-nitrobenzyl chloroformate[63] was prepared in-situ using the highly toxic phosgene.[64] The reaction and subsequent work up must be carried out in an efficient fume hood!] Ether 28 (28a–c, 150 mg, 0.41 mmoles) in 1:3 pyridine/CH2Cl2 (4 mL) was added dropwise to a solution of α-methyl-o-nitrobenzyl chloroformate (165 mg, 0.41 mmoles) in dry CH2Cl2 (3 mL) at 0°C. The reaction was allowed to warm to room temperature and stirred for 24 h. The solvent was removed under vacuum and the residue was dissolved in CH2Cl2 (20 mL), washed with 1M NaHSO3 (2X), brine, dried (MgSO4), filtered and evaporated. Chromatographic purification of the oily residue gave 158mg of 30a: 69%, 174 mg of 30b: 76%, 168mg of 30c: 73%).
General Procedure for compounds 31a–c
Compounds 31a–c were prepared from ethers 30a–c (100 mg, 0.18 mmol) by the same method as 29a–c above (Yield, 72mg of 31a: 85%, 69mg of 31b: 82%, 70mg of 31c: 83%).
General Procedure for hydroxymethyl diphosphates 32a–c
Ph3PBr2 (45 mg, 0.106 mmol) in CH3CN (3 mL) was added dropwise to a cooled (0°C) solution of alcohol 31 (31a–c, 50 mg, 0.106 mmol) in CH3CN (2 mL) and stirred for 3 h. ((n-Bu)4N)3HP2O7 (417 mg, 0.424 mmol) in CH3CN (2 mL) was then added and the solution allowed to warm to room temperature over 1 h. The reaction mixture was worked up and diphosphates 32a–b isolated as for 3 above (31 mg of 32a: 31%, 28 mg of 32b: 39%, 18 mg of 9c: 35%). The compound 32c was unstable and obtained as 9c.
Preparation of compounds 9a and 9b
Compound 31a or 31b (10 mg) in 25 mM solution of NH4HCO3 (1 mL) was irradiated with pyrex filtered UV light for 5 min at 0°C in a Rayonet device. The yellow solution was extracted with CH2Cl2, the organics discarded and aqueous layer lyophilized to obtain 9a/9b in quantitative yield.
Steady-state peptide kinetics
The kinetic constants appkcat, appKmpeptide and apparent kcat/Kmpeptide for transfer of isoprenoids 1, 6a–6ad by FTase to peptide were determined using a continuous spectrofluorometric assay originally developed by Pompliano et al. [65] and modified for a 96-well plate format as described.[32] Analogue transfer to peptide was analyzed using reverse phase high performance liquid chromatography (RP-HPLC) as described.[32]
LogP Determination
The apparent logP values for the corresponding alcohols 24a–ad were estimated from the capacity factors (k′) using reverse phase high performance liquid chromatography (RP-HPLC).[12]
Acknowledgments
We thank Dr. Carol Fierke, Heather Hartman, Katherine Hicks and Jennifer Pickett, University of Michigan, for the gift of mammalian protein farnesyl transferase and Dr. Trevor Creamer, University of Kentucky, for helpful discussions.
Abbreviations
- FTase
protein farnesyltransferase
- FPP
farnesyl diphosphate
- GGTase-I
protein geranylgeranyltransferase type 1
- GPP
geranyl diphosphate
- GGPP
geranylgeranyl diphosphate
- FTI
protein farnesyltransferase inhibitor
- PFI
prenyl function inhibitor
- CaaX
tetrapeptide sequence cysteine-aliphatic amino acid-aliphatic amino acid-X (serine, glutamine, or methionine for FTase)
- dns
dansylated
- RP-HPLC
reverse-phase high-performance liquid chromatography
- H-Ras
Harvey-Ras
- K-Ras
Kirsten-Ras
- N-Ras
neuronal-Ras
- AGPP
8-anilinogeranyl diphosphate
- PGPP
8-phenoxygeranyl diphosphate
- appkcat
apparent turnover number
- appKmpeptide
apparent Michaelis-Menten constant for peptide
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
Supporting Information: Spectral data for 3, 4, 5, 6a–6ad, 7, 14, 15, 18a–18c, 19a–19c, 24a–24ad, 27a–27c, 28a–28c, 29a–29c, 30a–30c, 31a–31c, 32a–32c. 1H spectra of 3, 15,19a–d, 24a–ad, 1H, 31P and LRMS spectra of 4, 5, 6a–6ad, 7, 9a–9c are available.
References
- 1.Basso AD, Kirschmeier P, Bishop WR. J Lipid Res. 2006;47:15–31. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Gotlib J. Curr Hematol Rep. 2005;4:77–84. [PubMed] [Google Scholar]
- 3.Isobe T, Herbst RS, Onn A. Semin Oncol. 2005;32:315–328. doi: 10.1053/j.seminoncol.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 4.Khuri FR. Clin Lung Cancer. 2003;5(Suppl 1):S36–40. doi: 10.3816/clc.2003.s.014. [DOI] [PubMed] [Google Scholar]
- 5.Mitsch A, Bergemann S, Gust R, Sattler I, Schlitzer M. Arch Pharm (Weinheim) 2003;336:242–250. doi: 10.1002/ardp.200300758. [DOI] [PubMed] [Google Scholar]
- 6.Doll RJ, Kirschmeier P, Bishop WR. Curr Opin Drug Discov Devel. 2004;7:478–486. [PubMed] [Google Scholar]
- 7.Woo JT, Nakagawa H, Krecic AM, Nagai K, Hamilton AD, Sebti SM, Stern PH. Biochem Pharmacol. 2005;69:87–95. doi: 10.1016/j.bcp.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 8.James GL, Brown MS, Goldstein JL. Methods Enzymol. 1995;255:38–46. doi: 10.1016/s0076-6879(95)55007-0. [DOI] [PubMed] [Google Scholar]
- 9.Cox AD, Der CJ. Biochim Biophys Acta. 1997;1333:F51–71. doi: 10.1016/s0304-419x(97)00011-5. [DOI] [PubMed] [Google Scholar]
- 10.Sebti SM. Oncologist. 2003;8:30–38. doi: 10.1634/theoncologist.8-suppl_3-30. [DOI] [PubMed] [Google Scholar]
- 11.Gibbs BS, Zahn TJ, Mu Y, Sebolt-Leopold JS, Gibbs RA. J Med Chem. 1999;42:3800–3808. doi: 10.1021/jm9902786. [DOI] [PubMed] [Google Scholar]
- 12.Roberts MJ, Troutman JM, Chehade KA, Cha HC, Kao JP, Huang X, Zhan CG, Peterson YK, Subramanian T, Kamalakkannan S, Andres DA, Spielmann HP. Biochemistry. 2006;45:15862–15872. doi: 10.1021/bi061704+. [DOI] [PubMed] [Google Scholar]
- 13.Dudler T, Gelb MH. Biochemistry. 1997;36:12434–12441. doi: 10.1021/bi971054x. [DOI] [PubMed] [Google Scholar]
- 14.Micali E, Chehade KA, Isaacs RJ, Andres DA, Spielmann HP. Biochemistry. 2001;40:12254–12265. doi: 10.1021/bi011133f. [DOI] [PubMed] [Google Scholar]
- 15.Chehade KA, Kiegiel K, Isaacs RJ, Pickett JS, Bowers KE, Fierke CA, Andres DA, Spielmann HP. J Am Chem Soc. 2002;124:8206–8219. doi: 10.1021/ja0124717. [DOI] [PubMed] [Google Scholar]
- 16.Reigard SA, Zahn TJ, Haworth KB, Hicks KA, Fierke CA, Gibbs RA. Biochemistry. 2005;44:11214–11223. doi: 10.1021/bi050725l. [DOI] [PubMed] [Google Scholar]
- 17.Kale TA, Hsieh SA, Rose MW, Distefano MD. Curr Top Med Chem. 2003;3:1043–1074. doi: 10.2174/1568026033452087. [DOI] [PubMed] [Google Scholar]
- 18.Turek-Etienne TC, Strickland CL, Distefano MD. Biochemistry. 2003;42:3716–3724. doi: 10.1021/bi0266838. [DOI] [PubMed] [Google Scholar]
- 19.Lepre CA, Peng J, Fejzo J, Abdul-Manan N, Pocas J, Jacobs M, Xie X, Moore JM. Comb Chem High Throughput Screen. 2002;5:583–590. doi: 10.2174/1386207023329950. [DOI] [PubMed] [Google Scholar]
- 20.Chehade KA, Andres DA, Morimoto H, Spielmann HP. J Org Chem. 2000;65:3027–3033. doi: 10.1021/jo991735t. [DOI] [PubMed] [Google Scholar]
- 21.Gaon I, Turek TC, Weller VA, Edelstein RL, Singh SK, Distefano MD. J Org Chem. 1996;61:7738–7745. doi: 10.1021/jo9602736. [DOI] [PubMed] [Google Scholar]
- 22.Rawat DS, Gibbs RA. Org Lett. 2002;4:3027–3030. doi: 10.1021/ol026176i. [DOI] [PubMed] [Google Scholar]
- 23.Stremler KE, Poulter CD. J Am Chem Soc. 1987;109:5542–5544. [Google Scholar]
- 24.Moores SL, Schaber MD, Mosser SD, Rands E, O’Hara MB, Garsky VM, Marshall MS, Pompliano DL, Gibbs JB. J Biol Chem. 1991;266:14603–14610. [PubMed] [Google Scholar]
- 25.Furfine ES, Leban JJ, Landavazo A, Moomaw JF, Casey PJ. Biochemistry. 1995;34:6857–6862. doi: 10.1021/bi00020a032. [DOI] [PubMed] [Google Scholar]
- 26.Long SB, Casey PJ, Beese LS. Nature. 2002;125419:645–650. doi: 10.1038/nature00986. [DOI] [PubMed] [Google Scholar]
- 27.Dolence JM, Poulter CD. Proc Natl Acad Sci U S A. 1995;92:5008–5011. doi: 10.1073/pnas.92.11.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang C, Hightower KE, Fierke CA. Biochemistry. 2000;39:2593–2602. doi: 10.1021/bi992356x. [DOI] [PubMed] [Google Scholar]
- 29.Pompliano DL, Rands E, Schaber MD, Mosser SD, Anthony NJ, Gibbs JB. Biochemistry. 1992;31:3800–3807. doi: 10.1021/bi00130a010. [DOI] [PubMed] [Google Scholar]
- 30.Troutman JM, Roberts MJ, Andres DA, Spielmann HP. Bioconjug Chem. 2005;16:1209–1217. doi: 10.1021/bc050068+. [DOI] [PubMed] [Google Scholar]
- 31.Coffinier C, Hudon SE, Lee R, Farber EA, Nobumori C, Miner JH, Andres DA, Spielmann HP, Hrycyna CA, Fong LG, Young SG. J Biol Chem. 2008;283:9797–9804. doi: 10.1074/jbc.M709629200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Troutman JM, Subramanian T, Andres DA, Spielmann HP. Biochemistry. 2007;46:11310–11321. doi: 10.1021/bi700516m. [DOI] [PubMed] [Google Scholar]
- 33.Henriksen BS, Zahn TJ, Evanseck JD, Firestine SM, Gibbs RA. J Chem Inf Model. 2005;45:1047–1052. doi: 10.1021/ci0496550. [DOI] [PubMed] [Google Scholar]
- 34.Spencer TA, Onofrey TJ, Cann RO, Russel JS, Lee LE, Blanchard DE, Castro A, Gu P, Jiang GJ, Shechter I. J Org Chem. 1999;64:807–818. doi: 10.1021/jo981617q. [DOI] [PubMed] [Google Scholar]
- 35.Troutman JM, Andres DA, Spielmann HP. Biochemistry. 2007;46:11299–11309. doi: 10.1021/bi700513n. [DOI] [PubMed] [Google Scholar]
- 36.Subramanian T, Wang Z, Troutman JM, Andres DA, Spielmann HP. Org Lett. 2005;7:2109–2112. doi: 10.1021/ol050386o. [DOI] [PubMed] [Google Scholar]
- 37.Long SB, Hancock PJ, Kral AM, Hellinga HW, Beese LS. Proc Natl Acad Sci U S A. 2001;98:12948–12953. doi: 10.1073/pnas.241407898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Long SB, Casey PJ, Beese LS. Structure. 2000;8:209–222. doi: 10.1016/s0969-2126(00)00096-4. [DOI] [PubMed] [Google Scholar]
- 39.Strickland CL, Windsor WT, Syto R, Wang L, Bond R, Wu Z, Schwartz J, Le HV, Beese LS, Weber PC. Biochemistry. 1998;37:16601–16611. doi: 10.1021/bi981197z. [DOI] [PubMed] [Google Scholar]
- 40.Long SB, Casey PJ, Beese LS. Biochemistry. 1998;37:9612–9618. doi: 10.1021/bi980708e. [DOI] [PubMed] [Google Scholar]
- 41.Dunten P, Kammlott U, Crowther R, Weber D, Palermo R, Birktoft J. Biochemistry. 1998;37:7907–7912. doi: 10.1021/bi980531o. [DOI] [PubMed] [Google Scholar]
- 42.Park HW, Boduluri SR, Moomaw JF, Casey PJ, Beese LS. Science. 1997;275:1800–1804. doi: 10.1126/science.275.5307.1800. [DOI] [PubMed] [Google Scholar]
- 43.Hightower KE, De S, Weinbaum C, Spence RA, Casey PJ. Biochem J. 2001;360:625–631. doi: 10.1042/0264-6021:3600625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Z, Demma M, Strickland CL, Radisky ES, Poulter CD, Le HV, Windsor WT. Biochemistry. 1999;38:11239–11249. doi: 10.1021/bi990583t. [DOI] [PubMed] [Google Scholar]
- 45.Cui G, Wang B, Merz KM., Jr Biochemistry. 2005;44:16513–16523. doi: 10.1021/bi051020m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reiss Y, Seabra MC, Armstrong SA, Slaughter CA, Goldstein JL, Brown MS. J Biol Chem. 1991;266:10672–10677. [PubMed] [Google Scholar]
- 47.Turek TC, Gaon I, Distefano MD, Strickland CL. J Org Chem. 2001;66:3253–3264. doi: 10.1021/jo991130x. [DOI] [PubMed] [Google Scholar]
- 48.Krzysiak AJ, Scott SA, Hicks KA, Fierke CA, Gibbs RA. Bioorg Med Chem Lett. 2007;17:5548–5551. doi: 10.1016/j.bmcl.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mu Y, Eubanks LM, Poulter CD, Gibbs RA. Bioorg Med Chem. 2002;10:1207–1219. doi: 10.1016/s0968-0896(01)00390-x. [DOI] [PubMed] [Google Scholar]
- 50.Zahn TJ, Weinbaum C, Gibbs RA. Bioorg Med Chem Lett. 2000;10:1763–1766. doi: 10.1016/s0960-894x(00)00337-1. [DOI] [PubMed] [Google Scholar]
- 51.Shao Y, Eummer JT, Gibbs RA. Org Lett. 1999;1:627–630. doi: 10.1021/ol990714i. [DOI] [PubMed] [Google Scholar]
- 52.Mu Y, Gibbs RA, Eubanks LM, Poulter CD. J Org Chem. 1996;61:8010–8015. doi: 10.1021/jo9614203. [DOI] [PubMed] [Google Scholar]
- 53.Wlodarczyk N, Gilleron P, Millet R, Houssin R, Goossens JF, Lemoine A, Pommery N, Wei MX, Henichart JP. Oncol Res. 2005;16:107–118. doi: 10.3727/000000006783981170. [DOI] [PubMed] [Google Scholar]
- 54.Reid TS, Terry KL, Casey PJ, Beese LS. J Mol Biol. 2004;343:417–433. doi: 10.1016/j.jmb.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 55.Wimley WC, White SH. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 56.Thapar R, Williams JG, Campbell SL. J Mol Biol. 2004;343:1391–1408. doi: 10.1016/j.jmb.2004.08.106. [DOI] [PubMed] [Google Scholar]
- 57.Cui G, Merz KM., Jr Biochemistry. 2007;46:12375–12381. doi: 10.1021/bi701324t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nguyen UTT, Crammer J, Gomis J, Reents R, Gutierrez-Rodriguez M, Goody RS, Alexandrov K, Waldmann H. ChemBiochem. 2007;8:408–423. doi: 10.1002/cbic.200600440. [DOI] [PubMed] [Google Scholar]
- 59.Davisson VJ, Woodside AB, Neal TR, Stremler KE, Muehlbacher M, Poulter CD. J Org Chem. 1986;51:4768–4779. [Google Scholar]
- 60.Marshall JA, Lebreton J. J Org Chem. 1988;53:4108–4112. [Google Scholar]
- 61.Nishitani K, Konomi T, Mimaki Y, Tsunoda T, Yamakawa K. Heterocycles. 1993;36:1957–1960. [Google Scholar]
- 62.Williams JR, Lin C, Chodosh DF. J Org Chem. 1985;50:5815–5822. [Google Scholar]
- 63.Hasan A, Stengele KP, Giegrich H, Cornwell P, Isham KR, Sachleben RA, Pfleiderer W, Foote RS. Tetrahedron. 1997;53:4247–4264. [Google Scholar]
- 64.Li S, Bowerman D, Marthandan N, Klyza S, Luebke KJ, Garner HR, Kodadek T. J Am Chem Soc. 2004;126:4088–4089. doi: 10.1021/ja039565w. [DOI] [PubMed] [Google Scholar]
- 65.Pompliano DL, Gomez RP, Anthony NJ. J Am Chem Soc. 1992;114:7945–7946. [Google Scholar]