

Abstract
A palladium-catalyzed reaction is presented for the synthesis of highly substituted indoles involving three independent components in a one-pot reaction. Two distinct palladium catalyzed coupling reactions occur using a single catalytic system: a Buchwald-Hartwig reaction and an arene-alkene coupling. Quantum chemical computations provide insight into the mechanism of the latter coupling step.
Keywords: indole methodology, multicomponent, palladium catalysis
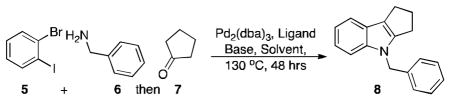
Methods development for the synthesis of heterocycles[1] continues to enable many areas of chemical research and new organometallic catalytic methods[2] are providing synthetic routes to important targets previously unavailable to the synthetic chemist. Indoles, which are ubiquitous in pharmaceuticals and in natural products,[3] have inspired dozens of methods for their construction.[4] The search for easy, diversity oriented routes to indoles is of importance to both the synthetic and biological communities.[5] Some of these strategies have employed the use of palladium catalysis and there are reports of indole syntheses using Heck and Buchwald-Hartwig reactions.[6] There are also reports of multicomponent assembly methods to form indoles.[7] We report here a general palladium catalyzed multicomponent method for indole synthesis incorporating both a Buchwald-Hartwig[8] reaction and an arene-alkene coupling[9] reaction using a single catalyst-ligand system. This method employs three components and allows for easy diversification from readily available starting materials. Multicomponent assembly reactions are advantageous in that they provide expedient methods for constructing complex structures, minimize synthetic steps, and use relatively simple starting materials.[10] The overall one-pot – three step multicomponent assembly reported here is illustrated in Scheme 1.
Scheme 1.

Three-reactant palladium catalyzed indole synthesis.
Reaction optimization established that use of the DPPF ligand [1,1′-bis(diphenylphosphanyl)ferrocene; Figure 1] and Cs2CO3 as base (Table 1, Entry 4) gave the best yields. Xphos, tBuXphos, and Xantphos (Entries 1, 2, and 3, respectively) gave product albeit in lower yields, while PPh3 (Entry 5) and PtBu3 (Entry 6) did not promote the reaction. KOtBu gave lower yields (Entry 7) – possibly because of enolate formation from the ketone or aldehyde, which reduces the likelihood of an amine-carbonyl condensation event. K3PO4 gave no product (Entry 8). The addition of MgSO4 favors enamine formation by effectively excluding water from the carbonyl + amine ⇆ enamine + H2O equilibrium reaction. Attempts at using a palladium(II) catalyst as a source of palladium(0) gave no product (Entry 11); this may indicate that reduction of Pd(II) to Pd(0) is too slow for the Pd(II) species to be an effective precatalyst for this process.
Figure 1.

Ligands screened.
Table 1.
Optimization of reaction conditions.
| ||||
|---|---|---|---|---|
| Entry | Ligand | Base | Solvent | Yield |
| 1 | Xphos | Cs2CO3 | Toluene | 55% |
| 2 | tBuXphos | Cs2CO3 | Toluene | 49% |
| 3 | Xantphos | Cs2CO3 | Toluene | 61% |
| 4 | DPPF | Cs2CO3 | Toluene | 68% |
| 5 | PPh3 | Cs2CO3 | Toluene | 0% |
| 6 | PtBu3 | Cs2CO3 | Toluene | 0% |
| 7 | DPPF | KOtBu | Toluene | 51% |
| 8 | DPPF | K3PO4 | Toluene | 0% |
| 9 | DPPF | Cs2CO3 | Dioxane | trace |
| 10 | DPPF | Cs2CO3 | DMF | 14% |
| 11a | DPPF | Cs2CO3 | Toluene | 0% |
Pd2dba3 was replaced with Pd(OAc)2.
Our optimized conditions work effectively with an array of substrates (Chart 1) and proceed with a catalyst loading of 2 mol% Pd2(dba)3 (4% Pd).[11] A wide range of carbonyls are suitable as substrates for this process. Cyclic (8, 14, 17, 18, 21) and acyclic ketone (15, 20, 22) variants were used and both gave similar results. Aldehydes (16 and 19) also successfully form indoles using our optimized conditions. A variety of primary amines can be used and the aryl component can be a carbocycle, an anisole, or a pyridine. For all of the indoles prepared, the appropriate o-bromoiodo-arene was used as a reactant. A larger scale reaction (5 mmol rather than the typical 1 mmol) was also conducted for the synthesis of 14. A substantial change in yield was not observed for this larger scale reaction (e.g., there was only a 2% increase in yield for the 5 mmol reaction). The optimized yield for the synthesis of 8 is 68%, which equates to each of the three steps in our one-pot reaction sequence (Buchwald-Hartwig coupling, condensation, and arene-alkene coupling) proceeding in 88% yield. Indeed, the yield of our one-pot transformation is comparable to the overall yield of Urabe and co-worker’s recently reported three-step three-pot indole synthesis.[12]
Chart 1.

Three-component indole products and yields. Conditions: 1 mmol o-bromoiodoarene, 1 mmol primary amine, 2 mmol ketone/aldehyde, 2 mol% Pd2dba3, 5 mol% DPPF, 2.2 mmol Cs2CO3, 10 mmol MgSO4, toluene (0.5 M), 130 °C. Yields of isolated product after purification by column chromatography.
Three bonds are formed in this transformation: an N-aryl bond by a Buchwald-Hartwig coupling, an N-vinyl bond by condensation of a nitrogen nucleophile onto an aldehyde or ketone giving an enamine, and a C-C bond by an arene-alkene coupling. We envisioned that several reaction sequences could be used to construct the indole core from these three components. As shown in Scheme 2, several experiments were carried out to probe the order of events in our reaction. Experiments 1–3 bear on the intermediacy of enolates.[13] 1) The use of a strong base (NaHMDS) favors enolate formation; although Buchwald-Hartwig coupling succeeded (to form 9), subsequent enolate coupling (to form 10) failed. 2) Initial enolate generation and coupling succeeded but subsequent amine addition did not give the Buchwald-Hartwig product (10). 3) Attempted enolate coupling using optimized conditions (i.e., with a weaker base) gave only trace amount of intended product; the major product formed arose from dimerization of cyclopentanone (12). On the basis of these results, we conclude that enolates do not play a significant role in the transformation described herein. 4) Preformation of the enamine (13) yielded only small amounts of product using optimized conditions, indicating that such a process likely does not initiate the catalytic cycle. 5) Initial Buchwald-Hartwig coupling, followed by addition of the ketone, did produce the indole product in 68% yield. In light of the results of experiments 1–5, we propose the catalytic cycle shown in Scheme 3.
Scheme 2.

Indole synthesis order of events examined.
Scheme 3.

Proposed multicomponent assembly catalytic mechanism.DPPF ligand omitted for clarity.
Several mechanisms for indole ring formation from an N-arylenamine were examined using density functional theory calculations (B3LYP/6-31G(d)[C,H,N],LANL2DZ [Pd] with the simplified model system shown in Figure 2).[14] No transition state structures corresponding to the alkene insertion step of a traditional Heck reaction were found. Instead, a two-step alkene insertion process via an intermediate metallacycle (A→B→C) was located. Simple insertion (i.e., A→B, as in the Heck reaction) is likely disfavored due to the strain associated with endo-cyclization and the fact that intermediate cation B is stabilized by imine resonance. Formation of B can be formulated as the attack of an enamine on an electrophilic metal center (with or without prior Br− loss). Intermediate C, which displays a significant agostic interaction (Pd---H distance of 2.0 Å and elongated C–H distance of 1.15 Å), can readily undergo β-hydride elimination to form indole D. Although this elimination requires a cis [Pd] and H relationship, either methylene hydrogen can be removed, since B can undergo a conformational change (from one half-chair to another) with a low barrier (B→B′, Figure 2); note that this would, in principle, allow β-hydride elimination to occur for any substrate with at least one hydrogen at the nucleophilic enamine carbon, regardless of alkene configuration.
Figure 2.

Computed mechanisms for arene-alkene coupling. Computed relative free energies (B3LYP/6-31G(d)[C,H,N], LANL2DZ[Pd], all relative to the energy of A, except for the E→D barrier) for structures involved in indole ring formation. Energies of minima are shown next to each structure; energies of transition state structures are shown over arrows.12 *Energy for [HPd(PH3)2-N-methylindole]+ complex.
An alternative to this “interrupted Heck” reaction was also examined. Deprotonation of one of the acidic methylene protons of B would lead to E, which can undergo reductive elimination to form indole D with a low predicted barrier (~5 kcal/mol). Although both mechanisms (A→B/B′→C/C′→D/D′ and A→B→E→D) are consistent with our experimental results, we favor the latter on energetic grounds, assuming the reaction conditions allow for rapid deprotonation of B.[14] This mechanistic proposal differs from the traditional Heck mechanism proposed (or implied) in previous studies on N-arylenamine cyclizations of this type.[6c,15]
In conclusion, we have developed a versatile one-pot – three step multicomponent assembly route to highly substituted indoles using a palladium catalyzed reaction employing three independent components. Successful indole formation can be accomplished using suitable reaction conditions that favor in situ generation of an aniline, condensation to an arylenamine, and a subsequent arene-alkene coupling reaction to close the ring. A single palladium catalyst-ligand system mediates both coupling reactions. We have also presented a detailed mechanism for the palladium catalyzed ring-closing step. The mechanism described herein is supported by quantum chemical calculations and offers an alternative to direct Heck coupling as previously suggested. This work significantly extends indole construction and diversification strategies.
Supplementary Material
Acknowledgments
We thank the National Institute of Health (GM08153), the National Science Foundation (CHE-0910870 and CHE-030089 with the Pittsburgh Supercomputer Center) for their generous financial support. We also thank Professor Annaliese Franz (UC Davis) and Professor Tomislav Rovis (Colorado State) for helpful discussions.
Footnotes
We thank the National Institute of Health (GM08153), the National Science Foundation (CHE-0910870 and CHE-030089 with the Pittsburgh Supercomputer Center) for their generous financial support. We also thank Professor Annaliese Franz (UC Davis) and Professor Tomislav Rovis (Colorado State) for helpful discussions.
Experimental, spectral and computational data is available free of charge via the Internet at http://pubs.acs.org.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author
References
- 1.Recent examples: Kramer S, Skrydstrup T. Angew Chem Int Ed. 2012;51:4681–4684. doi: 10.1002/anie.201200307.Yao B, Wang Q, Zhu J. Angew Chem Int Ed. 2012;51:5170–5174. doi: 10.1002/anie.201201640.Ball CJ, Gilmore J, Willis MC. Angew Chem Int Ed. 2012;51:5718–5722. doi: 10.1002/anie.201201529.Gati W, Rammah MM, Rammah MB, Couty F, Evano G. J Am Chem Soc. 2012;134:9078– 9081. doi: 10.1021/ja303002a.
- 2.Recent examples: Huang C, Doyle AG. J Am Chem Soc. 2012;134:9541–9544. doi: 10.1021/ja3013825.Brazeau JF, Zhang S, Colomer I, Corkey BK, Toste FD. J Am Chem Soc. 2012;134:2742–2749. doi: 10.1021/ja210388g.Li B, Wang H, Zhu Q, Shi Z. Angew Chem Int Ed. 2012;51:3948–3952. doi: 10.1002/anie.201200271.Chen Y, Chen M, Liu Y. Angew Chem Int Ed. 2012;51:6493– 6497. doi: 10.1002/anie.201201799.
- 3.a) Mason JS, Morize I, Menard PR, Cheney DL, Hulme C, Labaudiniere RF. J Med Chem. 1999;42:3251–3264. doi: 10.1021/jm9806998. [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Pfefferkorn JA, Roecker AJ, Cao G, Barluenga S, Mitchell HJ. J Am Chem Soc. 2000;122:9939–9953. [Google Scholar]; c) Pindur U, Lemster T. Curr Med Chem. 2001;8:1681–1698. doi: 10.2174/0929867013371941. [DOI] [PubMed] [Google Scholar]; d) Knolker H, Reddy KR. Chem Rev. 2002;102:4303–4427. doi: 10.1021/cr020059j. [DOI] [PubMed] [Google Scholar]; e) Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; f) Taber DF, Tirunahari PK. Tetrahedron. 2011;67:7195– 7210. doi: 10.1016/j.tet.2011.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Fischer E, Jourdan F. Ber. 1883;16:2241–2245. [Google Scholar]; b) Fischer E, Hess O. Ber. 1884;17:559–568. [Google Scholar]; c) Bartoli G, Leardini R, Medici A, Rosini G. J Chem Soc. 1978:692–696. [Google Scholar]; d) Bartoli G, Palmieri F, Bosco M, Dalpozzo R. Tet Lett. 1989;30:2129–2132. [Google Scholar]; e) Bartoli G, Bosco M, Dalpozzo R. J Chem Soc. 1991:2757–2761. [Google Scholar]; f) Madelung W. Ber. 1912;45:1128–1134. [Google Scholar]; g) Nenitzescu CD. Bull Soc Chim Romania. 1929;11:37–43. [Google Scholar]; h) Larock RC, Yum EK. J Am Chem Soc. 1991;113:6689– 6690. [Google Scholar]
- 5.Shiri M. Chem Rev. 2012;122:3508–3549. doi: 10.1021/cr2003954. [DOI] [PubMed] [Google Scholar]
- 6.a) Willis MC, Brace GN, Holmes IP. Angew Chem Int Ed. 2005;44:403–406. doi: 10.1002/anie.200461598. [DOI] [PubMed] [Google Scholar]; b) Jensen T, Pedersen H, Bang-Andersen B, Madsen R, Jørgensen M. Angew Chem Int Ed. 2008;47:888–890. doi: 10.1002/anie.200703763. [DOI] [PubMed] [Google Scholar]; c) Nazare M, Schneider C, Lindenschmidt A, Will DW. Angew Chem Int Ed. 2004;43:4526–4528. doi: 10.1002/anie.200460122. [DOI] [PubMed] [Google Scholar]; d) Chen C, Lieberman DR, Larsen RD, Verhoeven TR, Reider PJ. J Org Chem. 1997;62:2676–2677. doi: 10.1021/jo970278i. [DOI] [PubMed] [Google Scholar]; e) Newman SG, Lautens M. J Am Chem Soc. 2010;132:11416–11417. doi: 10.1021/ja1052335. [DOI] [PubMed] [Google Scholar]; f) Jia Y, Zhu J. J Org Chem. 2006;71:7826–7834. doi: 10.1021/jo061471s. [DOI] [PubMed] [Google Scholar]; g) Fang Y, Lautens M. Org Lett. 2005;7:3549–3552. doi: 10.1021/ol051286l. [DOI] [PubMed] [Google Scholar]; h) Fayol A, Fang Y, Lautens M. Org Lett. 2006;8:4203–4206. doi: 10.1021/ol061374l. [DOI] [PubMed] [Google Scholar]; i) Edmondson SD, Mastracchio A, Parmee EM. Org Lett. 2000;2:1109–1112. doi: 10.1021/ol000031z. [DOI] [PubMed] [Google Scholar]; j) Wagaw S, Yang BH, Buchwald SL. J Am Chem Soc. 1999;121:10251–10263. [Google Scholar]
- 7.a) Lu BZ, Zhao W, Wei H, Dufour M, Farina V, Senanayake CH. Org Lett. 2006;8:3271–3274. doi: 10.1021/ol061136q. [DOI] [PubMed] [Google Scholar]; b) Ohta Y, Chiba H, Oishi S, Fujii N, Ohno H. J Org Chem. 2009;74:7052–7058. doi: 10.1021/jo901328q. [DOI] [PubMed] [Google Scholar]; c) Kaspar LT, Ackermann L. Tetrahedron. 2005;61:11311–11316. [Google Scholar]; d) Leogane O, Lebel H. Angew Chem Int Ed. 2008;47:350–352. doi: 10.1002/anie.200703671. [DOI] [PubMed] [Google Scholar]; e) Barluenga J, Jimenez-Aquino A, Valdes C, Aznar F. Angew Chem Int Ed. 2007;46:1529–1532. doi: 10.1002/anie.200604407. [DOI] [PubMed] [Google Scholar]; f) Barluenga J, Jimenez-Aquino A, Aznar F, Valdes C. J Am Chem Soc. 2009;131:4031–4041. doi: 10.1021/ja808652a. [DOI] [PubMed] [Google Scholar]; g) Bouyssi D, Monteiro N, Balme G. Beilstein J Org Chem. 2011;7:1387–1406. doi: 10.3762/bjoc.7.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Kosugi M, Kameyama M, Migita T. Chem Lett. 1983:927–928. [Google Scholar]; b) Guram AS, Buchwald SL. J Am Chem Soc. 1994;116:7901–7902. [Google Scholar]; c) Paul F, Patt J, Hartwig JF. J Am Chem Soc. 1994;116:5969–5970. [Google Scholar]; d) Wolfe JP, Wagaw S, Buchwald SL. J Am Chem Soc. 1996;118:7215– 7216. [Google Scholar]
- 9.a) Heck RF. J Am Chem Soc. 1968;90:5518–5526. [Google Scholar]; b) Mizoroki T, Mori K, Ozaki A. Bull Chem Soc Jpn. 1971;44:581. [Google Scholar]; c) Heck RF, Nolley JP. J Org Chem. 1972;37:2320–2322. [Google Scholar]; d) Dieck HA, Heck RF. J Am Chem Soc. 1974;96:1133– 1136. [Google Scholar]
- 10.a) Boger DL, Desharnais J, Capps K. Angew Chem Int Ed. 2003;42:4138–4176. doi: 10.1002/anie.200300574. [DOI] [PubMed] [Google Scholar]; b) Trost BM. Angew Chem Int Ed. 1995;34:259–281. [Google Scholar]; c) Noyori R. Nat Chem. 2009;1:5–6. doi: 10.1038/nchem.143. [DOI] [PubMed] [Google Scholar]; d) Bienayme H, Hulme C, Oddon G, Schmitt P. Chem Eur J. 2000;6:3321–3329. doi: 10.1002/1521-3765(20000915)6:18<3321::aid-chem3321>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]; e) Posner GH. Chem Rev. 1986;86:831–844. [Google Scholar]; f) Biggs-Houck JE, Younai A, Shaw JT. Curr Opin Chem Bio. 2010;14:371–382. doi: 10.1016/j.cbpa.2010.03.003. [DOI] [PubMed] [Google Scholar]; g) Hardy S, Martin SF. Org Lett. 2011;13:3102–3105. doi: 10.1021/ol201010s. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) D’Souza DM, Mueller TJJ. Chem Soc Rev. 2007;36:1095–1108. doi: 10.1039/b608235c. [DOI] [PubMed] [Google Scholar]
- 11.For recent reviews on palladium catalysis in indole synthesis see: Patil S, Buolamwini JK. Curr Org Synth. 2006;3:477–498.Cacci S, Fabrizi G. Chem Rev. 2005;105:2873–2930. doi: 10.1021/cr040639b.Cacchi S, Fabrizi G. Chem Rev. 2005;105:2873–2920. doi: 10.1021/cr040639b.
- 12.Yamagishi M, Nishigai K, Ishii A, Hata T, Urabe H. Angew Chem Int Ed. 2012;51:6471–6474. doi: 10.1002/anie.201201024. [DOI] [PubMed] [Google Scholar]
- 13.For a recent review of enolate coupling, see: Culkin DA, Hartwig JF. Acc Chem Res. 2003;36:234–245. doi: 10.1021/ar0201106.
-
14.a) A, the A→B transition state structure, B, and the B→C transition state structure were recomputed using P(CH3)3 ligands instead of PH3 ligands, but no significant mechanistic differences were observed. Energies (kcal/ mol) relative to that of A: +5.6 (A→B), −17.2 (B), +14.7 (B→C). The predicted E→D barrier using P(CH3)3 ligands is 6.0 kcal/mol. b) A, the A→B transition state structure, and B were recomputed using a reactant with a carbomethoxy group on the internal alkene carbon of the enamine, a model of a system that did not work experimentally (i.e., which formed only the aryl amine; see below). Energies (kcal/mol) relative to that of A: +11.0 (A→B), −12.5 (B). This larger barrier and smaller exergonicity for the A→B reaction are consistent with reduced reactivity of a less nucleophilic enamine.
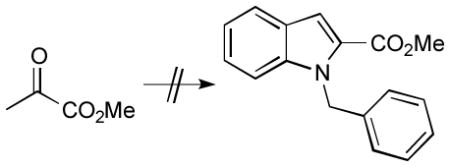 c) A, the A→B transition state structure, and B were recomputed using a reactant with a carbomethoxy group on the terminal alkene carbon of the enamine and a methyl group on the internal carbon (see below), a model of a system that formed the aryl amine as the major product and only trace amounts of the indole product. Energies (kcal/mol) relative to that of A: +6.7 (A→B), −18.1 (B). These results suggest that the problem with this reaction is not an inherent electronic deactivation of the enamine (also borne out by calculations with the terminal ester but no internal methyl group; see Supporting Information); interactions between the ester group and the full-size di-phosphine ligand or inherent acidity of the β-ketoester may therefore be responsible for the experimental problems.
c) A, the A→B transition state structure, and B were recomputed using a reactant with a carbomethoxy group on the terminal alkene carbon of the enamine and a methyl group on the internal carbon (see below), a model of a system that formed the aryl amine as the major product and only trace amounts of the indole product. Energies (kcal/mol) relative to that of A: +6.7 (A→B), −18.1 (B). These results suggest that the problem with this reaction is not an inherent electronic deactivation of the enamine (also borne out by calculations with the terminal ester but no internal methyl group; see Supporting Information); interactions between the ester group and the full-size di-phosphine ligand or inherent acidity of the β-ketoester may therefore be responsible for the experimental problems.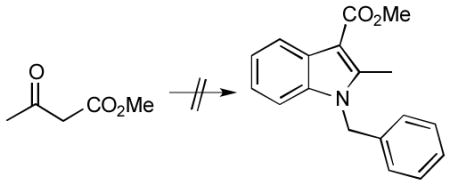
- 15.a) Koerber-Ple K, Massiot G. Synlett. 1994:759–760. [Google Scholar]; b) Lachance N, April M, Joly M. Synthesis. 2005;15:2571–2577. [Google Scholar]; c) Yamazaki K, Nakamura Y, Kondo Y. J Org Chem. 2003;68:6011–6019. doi: 10.1021/jo0340307. [DOI] [PubMed] [Google Scholar]; d) Chen C, Lieberman DR, Larsen RD, Verhoeven TR, Reider PJ. J Org Chem. 1997;62:2676– 2677. doi: 10.1021/jo970278i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.