

Abstract
The stable isotopes of hydrogen (δ2H) and oxygen (δ18O) in human urine are measured during studies of total energy expenditure by the doubly labeled water method, measurement of total body water, and measurement of insulin resistance by glucose disposal among other applications. An ultrasensitive laser absorption spectrometer based on off-axis integrated cavity output spectroscopy was demonstrated for simple and inexpensive measurement of stable isotopes in natural isotopic abundance and isotopically enriched human urine. Preparation of urine for analysis was simple and rapid (approx. 25 samples per hour), requiring no decolorizing or distillation steps. Analysis schemes were demonstrated to address sample-to-sample memory while still allowing analysis of 45 natural or 30 enriched urine samples per day. The instrument was linear over a wide range of water isotopes (δ2H = −454 to +1702 ‰ and δ18O= −58.3 to +265 ‰). Measurements of human urine were precise to better than 0.65 ‰ 1σ for δ2H and 0.09 ‰ 1σ for δ18O for natural urines, 1.1 ‰ 1σ for δ2H and 0.13 ‰ 1σ for δ18O for low enriched urines, and 1.0 ‰ 1σ for δ2H and 0.08 ‰ 1σ for δ18O for high enriched urines. Furthermore, the accuracy of the isotope measurements of human urines was verified to better than ±0.81 ‰ in δ2H and ±0.13 ‰ in δ18O (average deviation) against three independent IRMS laboratories. The ability to immediately and inexpensively measure the stable isotopes of water in human urine is expected to increase the number and variety of experiments which can be undertaken.
Introduction
Analysis of the stable isotopes of hydrogen (δ2H) and oxygen (δ18O) in human body water is used in a variety of biomedical applications including measurement of total energy expenditure (TEE) by the doubly labeled water (DLW) method1-3, measurement of total body water4, and measurement of insulin resistance by glucose disposal 5, 6 among other applications. Currently, the vast majority of studies use isotope-ratio mass spectrometry (IRMS) for analysis of δ2H and δ18O in body waters. For IRMS analysis, bodily fluids (e.g. urine) require either extensive purification, such as cryogenic distillation followed by decolorization7, or analysis by CO2 equilibration for 18O measurements and zinc or chromium reduction for 2H measurements8, 9. These preparation methods and IRMS analyses are labor-intensive, costly, and limited to only a few measurement laboratories worldwide. However, in order for the aforementioned biomedical applications to become widely available, measurements of a large number of samples must be completed quickly, accurately, and inexpensively, preferably at a location near the site of sample generation.
Ultrasensitive laser absorption spectroscopy, such as Off-Axis Integrated Cavity Output Spectroscopy (OA-ICOS) and cavity ring down spectroscopy (CRDS), provides the opportunity to measure δ2H and δ18O rapidly, accurately, and inexpensively 10-12. Furthermore, laser-based instrumentation does not require highly-trained operators and has a small footprint, allowing measurements to be made by researchers generating the samples. While studies have shown that laser-based instruments require corrections for organic contamination of samples 11, 13, 14, two laboratories have recently shown that the organic component of urine does not adversely affect laser-based isotope measurements7, 15. O’Grady et al. utilized CRDS to measure natural isotopic abundance human urines that had been either cryogenically distilled or decolorized with carbon black7. Thorsen et al. used CRDS to measure natural and enriched isotopic abundance human urines that had been decolorized, and found significant instrumental memory requiring a mathematical memory correction and careful ordering of samples15.
In this study, we demonstrate for the first time the accuracy, precision, speed, and simplicity of OA-ICOS technology for measurements of δ2H and δ18O in both natural and isotopically enriched human urine, without extensive pretreatment or purification. In addition, we detail simple methodologies, including the use of an internal control water and straightforward cleaning procedures, for ensuring that sample-to-sample memory effects are addressed while maintaining a high sample throughput (30 - 45 unknown urine samples per day). We also demonstrate the use of the Spectral Contamination Identifier to ensure that samples are free of measurement effects from organic components of urine. Finally, OA-ICOS results are compared with IRMS measurements from three independent laboratories to demonstrate the accuracy of the OA-ICOS technique.
Experimental
Preparation of Enriched Urine Test Samples
Urine test samples for the study were prepared according to the following procedure. A single sample (> 151 mL) of urine was collected into a sterile cup and well mixed. Three aliquots of 50 mL each were pipetted into sterile 50 mL conical vials for preparation of three urine test samples, one each of natural isotopic abundance, low-enriched, and high-enriched. An additional 1mL aliquot of the original urine was prepared for immediate isotopic analysis as described below to ascertain the native 2H/1H and 18O/16O isotope ratios of the urine prior to isotopic enrichment. The low-enriched urine sample was prepared by adding 0.8 μL of 2H2O (Sigma Aldrich, St. Louis, MO) and 1.0 μL of 98 atom percent excess (APE) H 182O (ICON Isotopes, Summit, NJ) to one 50 mL aliquot. The high-enriched urine sample was prepared by adding 3.4 μL of 2H2O and 4.9 μL of 98 APE H 182O to one 50 mL aliquot. The third 50 ml “natural” sample was used as collected. A small aliquot of each of the low-enriched and high-enriched urine samples was analyzed to ensure that the target enrichments, which were chosen to approximate enrichments frequently found in DLW experiments16, were roughly achieved. Finally, the 50 ml samples of each of the natural, low-enriched, and high-enriched urines were divided into 1mL aliquots and frozen for storage. This procedure ensured a large quantity of urine with identical isotope ratios for use in this validation study. Additional enriched urine samples, from a different urine collection, were prepared using the same basic procedure to create five urine samples of varying enrichment for the comparison study with IRMS. Additional urine samples from multiple individuals (>30) were collected and used without modification for optimization of the sample preparation and OA-ICOS analysis procedures.
Off-Axis ICOS Instrumentation
We utilized a commercially-available, Off-Axis Integrated Cavity Output Spectroscopy (OA-ICOS) laser absorption spectrometer (Los Gatos Research (LGR) Liquid Water Isotope Analyzer (LWIA-24d)) for analysis of the 2H/1H and 18O/16O stable isotopes in liquid water. The OA-ICOS instrument employed near-infrared tunable diode laser absorption spectroscopy with the laser coupled off-axis to a high-finesse optical cavity17 to provide highly accurate quantification of δ18O and δ2H in injected water samples in a reasonably compact and very robust instrument12. Samples were introduced into the OA-ICOS instrument via a LC PAL autoinjector (CTC Analytics) equipped with a heated injector block (LGR), where the water samples were evaporated for isotope analysis. Liquid samples were injected into the injector block using a Hamilton 1.2 μL, zero dead volume syringe (P/N: 203185/01). Simultaneous measurements of both δ18O and δ2H were completed at a speed of 1080 injections per day, or 80 seconds per measurement of an individual injection. The number of injections per sample was contingent upon the type of sample (e.g. natural water or urine) and level of isotopic enrichment as described below. Data from the instrument were analyzed using LGR’s commercially-available Post Analysis Software (LGR, version 2.2.0.12), which utilized inter-run standard measurements to automatically calibrate isotope measurements. The data were checked for the presence of any organic contamination using the commercially-available Spectral Contamination Identifier (SCI) (LGR, version 1.0.0.69)18. No contamination was found in any of the urine utilized for this study. Subsequent urine analysis did find a few (< 1%) urines with small but detectable contamination that can be corrected for using the SCI 14, 18.
Off-Axis ICOS Analysis of Urine Samples
Prior to each analysis, frozen urine samples were thawed, vortexed for 5 seconds, and centrifuged at 6000 rpm for 10 - 30 minutes. One hundred fifty (150) microliters of supernatant was micropipetted into an autosampler vial with a total volume of 350 microliters (National Scientific, Rockwood, TN). For the precision tests, a larger sample of urine was required, so 750 microliters of supernatant was micropipetted into a 2 mL autosampler vial (Microanalytical Analysis Supplies, Suwanee, GA). The urine samples were then analyzed for δ18O and δ2H on the OA-ICOS instrument without further preparation. No distillation or decolorizing steps were undertaken, reducing the probability of sample-handling induced errors. Using the procedure described above, approximately 25 urines could be prepared per hour, limited in our laboratories by the number of samples we could concurrently centrifuge.
Subsequent to sample preparation, urine samples were immediately analyzed on the OA-ICOS instrument. The instrument was calibrated using deionized internal working standards that had been previously calibrated by OA-ICOS against the VSMOW2 and SLAP2 international standards 19. For each OA-ICOS instrumental session, working standards were chosen such that their isotope ratios bracketed the expected isotope ratios of the urine samples while minimizing, as much as possible, the total span of isotope ratios. Samples and working standards were interleaved throughout each analysis to ensure high accuracy by frequent intra-run calibration. Interleaving of standards and samples had the additional benefit of prolonging the syringe lifetime by effectively rinsing urine solutes from the syringe on a regular basis. In addition, an internal control water of known isotopic composition within the range of the isotope ratios of the working standards was measured periodically throughout each analysis to ensure the quality of the data collected (e.g. Internal Control 1, δ18O = −7.08 ± 0.08 ‰ and δ2H = −43.6 ± 0.28 ‰ was used for natural isotope abundance measurements).
Inter-sample memory effects are well known in water isotope analysis 3, including analyses made with laser absorption spectroscopy instruments 10, 15, which have intrinsic, instrumental memory effects, most likely due to adsorption of water onto the internal surfaces of the instrument and mixing of water in the syringe. Instrumental memory is routinely addressed by injecting water samples multiple times and ignoring the results from the first few injections 10. Analyses of urine have additional memory effects which worsen over time due to the accumulation of urine solutes in the injector block. In order to address both the instrumental and the additional solute memory between successive samples, the following analysis schemes were optimized. For unenriched, natural urine samples, the instrument was programmed to inject each sample and working standard 8 times. The first four injections were discarded on account of memory while the last four injections were averaged to provide an individual analysis of the urine sample. Since each injection cycle required 80 s, this analysis scheme allowed for a maximum of 45 unknown unenriched urines to be analyzed per day, in addition to the associated working standards and internal controls. For isotopically-enriched urine samples (δ2H > +100 ‰), the instrument was programmed to inject each sample and working standard 12 times. The first 9 injections were discarded on account of memory while the last three injections were averaged to provide an individual analysis of the urine sample. This analysis scheme allowed for a maximum of 30 unknown enriched urines to be analyzed per day, in addition to the associated working standards and internal controls.
As expected, during analyses of urine samples the memory between successive samples was found to increase as solutes from the urine accumulated in the injector block of the instrument. The rate of increase depended strongly on the urine sample; samples of higher specific gravity (i.e. higher solute concentration) increased the memory effect much faster than those of low specific gravity. An internal control water of known isotope ratio was measured periodically throughout the run and used to determine when the solute build up had reached a point at which the above schemes were unable to fully ameliorate the sample to sample memory. For this study, a measurement of the internal control that was more than ± 1.4 ‰ for δ2H or ±0.2 ‰ for δ18O away from the known value was used to indicate that the injector block required cleaning. In every case after injector block cleaning, the memory between successive samples returned to the level of the instrumental memory effect (i.e. agreed with the known values of the internal control to better than the manufacturer’s stated precision of ± 0.6 ‰ for δ2H and ± 0.2 ‰ for δ18O).
At the conclusion of each analysis, the injector block, the connector to the transfer tube, and the septum support were thoroughly cleaned by ultrasonication in a soap solution for 1 hour, ultrasonication in tap water for 1 hour, and finally ultrasonication in a fresh aliquot of tap water for 1 hour. The injector block was then thoroughly rinsed in deionized water and the inside blown dry using a duster-type air canister. The injector block was reattached to the autosampler and allowed to heat up for at least 20 minutes before beginning a new analysis. High-throughput analysis was facilitated by utilizing two injector blocks, so that one could be cleaned while the second was in use. The Teflon transfer tube was replaced when deposits were visible within the tube or the sample-to-sample memory was seen to be increasing. Regular maintenance of the instrument, including deliming of the injector block, was performed according to the user manual. The syringe was cleaned daily using N-methylpyrrolidone (NMP) to remove solute buildup and condition the syringe. The syringe was rinsed with deionized water before use.
IRMS Analysis of Urine Samples
Hubert Curien Multidisciplinary Institute (IPHC)
For IPHC IRMS analysis only, water from urine was extracted by cryogenic distillation under vacuum for 15 min and placed in an inert glass tube (Chromacol). The on-line determination of hydrogen and oxygen isotope ratios was performed using a high-temperature conversion elemental analyzer (TC/EA) coupled with a Delta V Plus Isotope-Ratio Mass spectrometer and a Conflo III interface (THERMO, Brêmen, Germany) 20, 21. The elemental analyzer was equipped with a bottom feed connector and a glassy carbon tube heated to 1400 °C. After pyrolysis, H2 and CO were separated with a GC column at 90 °C and measured during the same injection in magnetic jump mode. High purity hydrogen (N60) and carbon monoxide (N47) from Linde Gaz (France) were used as reference gases. Urine standards, prepared with enriched waters from Euriso-top (Saint Aubin, France) mixed with pooled human urine, and urine quality controls, included to validate the measurement results, were made with different levels of enrichments and normalized against VSMOW2 and SLAP2.
University of Colorado Anschutz Medical Campus
For UC IRMS analysis only, urine was prepared by decolorization with activated charcoal followed by filtration. The determination of hydrogen and oxygen isotope ratios was performed using a Thermo Fisher Delta V Advantage IRMS22. For hydrogen analysis, the sample was injected into a chromium metal reactor at 850°C, reducing water in the sample to form hydrogen gas, whose isotope ratio was measured by the IRMS. For oxygen, sample urine was transferred into an Exetainer tube and the headspace atmosphere was replaced with 0.3% CO2 in helium. After incubating at ambient temperature for 48 hours, the headspace CO2 isotope ratio was measured on the IRMS.
University of Aberdeen, Scotland
For Aberdeen IRMS analysis only, urine was equilibrated with CO2 gas using the small sample equilibration technique for analysis of 18O 23. Preweighed Vacutainers were injected with 10 uL of urine and reweighed (0.0001 g), to account for differences in the amount of urine added. Subsequently, the Vacutainers with the samples were injected with 0.5 mL CO2 with a known oxygen isotopic enrichment and left to equilibrate at 60C for 16 h. For analysis of 18O/16O ratios, equilibrated CO2 samples were admitted to an ISOCHROM mGAS system (Micromass, UK), which uses a gas chromatograph column to separate nitrogen and CO2 in a stream of helium before analysis by IRMS24. All samples were run adjacent to three working standards that had been characterized against VSMOW and SLAP and all data were normalized to the accepted values for these international reference materials.
Results and Discussion
Linearity
The range of δ18O and δ2H values used in biomedical applications is far beyond the natural abundance range measured in environmental studies; for example enrichments of more than 700 ‰ in δ2H are common for DLW experiments in humans 25. It is thus critical to determine the linearity of the instrument over a very wide range of δ18O and δ2H values. Water samples of known isotope ratio were obtained ranging from −454 to +1702 ‰ in δ2H and −58.3 to +265 ‰ in δ18O. Figure 1a shows the excellent linearity of the instrument over the entire range of δ18O values. Figure 1b shows the excellent linearity in δ2H. In Figure 1b, the most enriched sample in δ2H (open circle) has not been included in the regression line since the uncertainty in the “known” IRMS value (±4.92 ‰, 1σ) is significantly higher than the uncertainty of OA-ICOS measurements. Nevertheless, for the most enriched sample, the residual of the measured value from the regression line (−9.54 ‰) is less than two standard deviations from the “known” IRMS value.
Figure 1.

Linearity of OA-ICOS demonstrated by the excellent linear correlations between the known and measured δ18O (a) and δ2H (b) values. In Figure 1b, the most enriched sample in δ2H (open circle) has not been included in the regression line since the uncertainty in the “known” IRMS value (±4.92 ‰, 1σ) is significantly higher than the uncertainty of OA-ICOS measurements.
Accuracy
The accuracy of the OA-ICOS instrument for measurements of urine samples was determined by measuring aliquots of the same urine samples by OA-ICOS and by three separate IRMS laboratories. Urine samples were prepared for OA-ICOS measurement as described in the experimental section above. Urine samples were prepared for IRMS analysis by each laboratory according to standard practice for that laboratory as described above. The working calibration standards used in these analyses were measured by all four labs and the data corrected to the same standard values. Figure 2 shows the excellent agreement between the OA-ICOS and IRMS analyses. Panels (a) and (b) show, for δ18O and δ2H respectively, the agreement between the measured OA-ICOS values and the mean of the IRMS values. Panels (c) and (d) show, for δ18O and δ2H respectively, the amount that each of the individual measurements deviates from the mean of the IRMS values. The error bars represent the standard deviation of the mean IRMS value. The data show that the OA-ICOS isotope measurements of human urines are accurate to better than ±0.81 ‰ in δ2H and ±0.13 ‰ in δ18O (average deviations) against three independent IRMS laboratories and in every case within two standard deviations of the IRMS mean value. Unfortunately, one of the IRMS for analysis of δ2H was in need of repair, so those data are not available.
Figure 2.

Accuracy of OA-ICOS for urine analyses demonstrated by the excellent agreement between OA-ICOS and the mean of IRMS measurements for δ18O (a) (three IRMS measurements) and δ2H (b) (two IRMS measurements). Deviation of each individual measurement from the IRMS mean for δ18O (c) and δ2H (d) for measurements by OA-ICOS (blue dots), TC/EA IRMS (red squares), and Cr reduction/CO2 equilibration IRMS (magenta and green triangles).
Precision
The precision of the OA-ICOS technique for measuring natural and enriched urine samples was determined by making repeated analyses on urine from a single vial. This test was performed on three different days over a period of months for each of the three prepared urine samples, natural, low-enriched, and high-enriched. An example of the excellent precision obtained for the natural urine sample can be seen in Figure 3, where the δ2H average is −42.25 ± 0.33 ‰ (1σ) and the δ18O average is −4.38 ± 0.03 ‰ (1σ). Each point reports the results of one complete urine analysis (i.e. the average of the last four of eight injections, calibrated using intra-run, interleaved working standards). The complete results from this series of precision tests are shown in Table 1. The average lines in the table contain the averages and standard deviations of all the data taken during three analytical sessions spanning a period of months, demonstrating that the long-term inter-run precision is equivalent to the intra-run precision. The data clearly show that the precision of OA-ICOS is comparable to the precision obtained using IRMS16.
Figure 3.
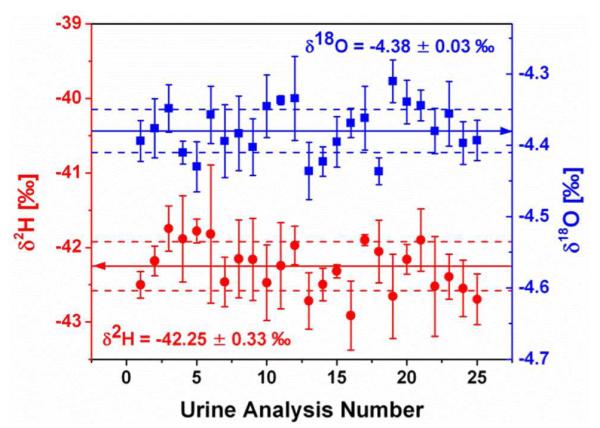
Precision of OA-ICOS for urine analysis demonstrated by making 25 repeated analyses from a single vial of a 750 μl natural urine sample. Error bars indicate the standard deviation of the four averaged injections that make up a single analysis. The δ2H (left axis, red circles) average (solid line) is −42.25 ± 0.33 ‰ (1σ, dashed lines) and the δ18O (right axis, blue squares) average (solid line) is −4.38 ± 0.03 ‰ (1σ, dashed lines).
Table 1.
Precision of OA-ICOS for repeated analyses of natural, low-enriched, and high-enriched urines. The average lines contain the averages and standard deviations of all the data taken during three analytical sessions spanning a period of months, demonstrating that the long-term inter-run precision is equivalent to the intra-run precision.
| Urine Sample | Date | n | δ2H ± 1σ | δ18O ± 1σ |
|---|---|---|---|---|
| Natural | 06/02/11 | 25 | −42.15 ± 0.70 | −4.30 ± 0.14 |
| Natural | 06/03/11 | 25 | −42.71 ± 0.71 | −4.37 ± 0.06 |
| Natural | 02/06/12 | 25 | −42.27 ± 0.33 | −4.38 ± 0.04 |
|
| ||||
| Natural Average | 75 | −42.37 ± 0.65 | −4.35 ± 0.09 | |
|
| ||||
| Low-Enriched | 08/09/11 | 25 | 134.15 ± 1.0 | 12.94 ± 0.15 |
| Low-Enriched | 10/25/11 | 25 | 134.97 ± 0.87 | 13.08 ± 0.05 |
| Low-Enriched | 01/05/12 | 25 | 133.77 ± 1.0 | 12.95 ± 0.09 |
|
| ||||
| Low-Enriched Average | 75 | 134.29 ± 1.1 | 12.99 ± 0.13 | |
|
| ||||
| High-Enriched | 03/15/12 | 23 | 837.0 ± 0.80 | 92.83 ± 0.07 |
| High-Enriched | 04/21/12 | 19 | 836.1 ± 0.97 | 92.87 ± 0.10 |
| High-Enriched | 04/22/12 | 16 | 837.5 ± 0.82 | 92.84 ± 0.04 |
|
| ||||
| High-Enriched Average | 58 | 836.8 ± 1.0 | 92.84 ± 0.08 | |
Repeatability and Ruggedness
The repeatability of the urine preparation technique was determined by preparing 9 separate aliquots of the same urine sample into 9 separate vials for sequential measurement on the instrument. This test was performed for each of the three prepared urine samples. The analytical results were identical to within the instrument precision for multiple preparations of the same sample. The ruggedness of the urine preparation technique was determined by measuring aliquots of the same sample which had been prepared by two different scientists. This test was performed for all three of the prepared urine samples and no difference was found for any of the samples or preparations of samples to within the instrument precision (intraclass correlation coefficient > 0.99999).
Conclusions
An ultrasensitive laser-absorption spectrometer, based on off-axis integrated cavity output spectroscopy, was utilized to measure the stable isotopes of hydrogen (δ2H) and oxygen (δ18O) in natural isotopic abundance and isotopically enriched human urine. The analyzer had a small footprint and simple, inexpensive operation, allowing measurements to be made quickly by researchers generating the samples, rather than by a select few measurement laboratories. Unlike previously reported analyses, preparation of urine was shown to be simple and rapid (approx. 25 samples per hour), requiring no decolorizing or distillation steps, thus reducing the probability of sample-handling induced errors. Analysis schemes were demonstrated which utilize multiple injections of each sample as well as inclusion of an internal control water of known isotope ratio to address sample-to-sample memory while still allowing analysis of 45 natural or 30 enriched urine samples per day. The instrument was shown to be linear over a wide range of water isotopes (−454 to +1702 ‰ for δ2H and −58.3 to +265 ‰ for δ18O). Intra-run and inter-run precision for measurements of human urine with natural and enriched isotopic abundances were shown to be better than 0.65 ‰ 1σ for δ2H and 0.09 ‰ 1σ for δ18O for natural urines, 1.1 ‰ 1σ for δ2H and 0.13 ‰ 1σ for δ18O for low enriched urines, and 1.0 ‰ 1σ for δ2H and 0.08 ‰ 1σ for δ18O for high enriched urines. The simple urine preparation technique was shown to be repeatable and rugged (no significant difference between preparations made by different scientists) to within the instrument precision. Furthermore, the accuracy of the isotope measurements of human urines was verified to better than ±0.81 ‰ in δ2H and ±0.13 ‰ in δ18O (average deviations) against three independent IRMS laboratories. The ability to immediately and inexpensively measure the stable isotopes of water in human urine is expected to increase the number and variety of experiments which can be undertaken in the areas of measurement of total energy expenditure by the doubly labeled water method, measurement of total body water, and measurement of insulin resistance by glucose disposal among other applications.
Acknowledgements
This work was supported by NIH SBIR Grant # 2R44RR023231-02A1. The Mass Spec Lab at the University of Colorado Anschutz Medical Campus is supported by NIH Grant #P30DK048520.
References
- (1).Lifson N, Gordan GB, McClintock R. Journal of Applied Physiology. 1955;7:704–710. doi: 10.1152/jappl.1955.7.6.704. [DOI] [PubMed] [Google Scholar]
- (2).Schoeller D, Van Santen E. Journal of Applied Physiology. 1982;53:955–959. doi: 10.1152/jappl.1982.53.4.955. [DOI] [PubMed] [Google Scholar]
- (3).Speakman J. Doubly4labelled water: theory and practice. Kluwer Academic publishers; New York: 1997. [Google Scholar]
- (4).Moore FD. Science. 1946;104:157–160. [PubMed] [Google Scholar]
- (5).DeFronzo RA, Tobin JD, Andres R. American Journal of Physiology. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- (6).Beysen C, Murphy EJ, McLaughlin T, Riiff T, Lamendola C, Turner HC, Awada M, Turner SM, Reaven G, Hellerstein MK. Diabetes Care. 2007;30:1143–1149. doi: 10.2337/dc06-1809. [DOI] [PubMed] [Google Scholar]
- (7).O’Grady SPO, Enright LE, Barnette JE, Cerling TE, Ehleringer JR. Isotopes in Envrionmental and Health Studies. 2010;46:476–483. doi: 10.1080/10256016.2010.536229. [DOI] [PubMed] [Google Scholar]
- (8).Wong WW, Lee LS, Klein PD. American Journal of Clinical Nutrition. 1987;45:905–913. doi: 10.1093/ajcn/45.5.905. [DOI] [PubMed] [Google Scholar]
- (9).Schoeller DA, Colligan AS, Shriver T, Avak H, Bartok-Olson C. Journal of Mass Spectrometry. 2000;35:1128–1132. doi: 10.1002/1096-9888(200009)35:9<1128::AID-JMS41>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- (10).Lis G, Wassenaar L, Hendry M. Analytical Chemistry. 2008;80:287–293. doi: 10.1021/ac701716q. [DOI] [PubMed] [Google Scholar]
- (11).Brand WA, Geilmann H, Crosson ER, Rella CW. Rapid Communications in Mass Spectrometry. 2009;23:1879–1884. doi: 10.1002/rcm.4083. [DOI] [PubMed] [Google Scholar]
- (12).Berman E, Gupta M, Gabriell C, Garland T, McDonnell J. Water Resources Research. 2009;45:W10201. [Google Scholar]
- (13).West AG, Goldsmith GR, Brooks PD, Dawson TE. Rapid Communications in Mass Spectrometry. 2010;24:1948. doi: 10.1002/rcm.4597. [DOI] [PubMed] [Google Scholar]
- (14).Schultz NM, Griffis TJ, Lee X, Baker JM. Rapid Communications in Mass Spectrometry. 2011;25:3360–3368. doi: 10.1002/rcm.5236. [DOI] [PubMed] [Google Scholar]
- (15).Thorsen T, Shriver T, Racine N, Richman BA, Schoeller D. Rapid Communications in Mass Spectrometry. 2011;25:3–8. doi: 10.1002/rcm.4795. [DOI] [PubMed] [Google Scholar]
- (16).Schoeller D. American Journal of Clinical Nutrition. 1983;38:999–1005. doi: 10.1093/ajcn/38.6.999. [DOI] [PubMed] [Google Scholar]
- (17).Baer DS, Paul JB, Gupta M, O’Keefe A. Applied Physics B. 2002;75:261–265. [Google Scholar]
- (18).Leen JB, Berman E, Liebson L, Gupta M. Review of Scientific Instruments. 2012;83:044305. doi: 10.1063/1.4704843. [DOI] [PubMed] [Google Scholar]
- (19).Certificate of Analysis. Los Gatos Research; Mountain View, CA: 2010. [Google Scholar]
- (20).Gehre M, Geilmann H, Richter J, Werner RA, Brand WA. Rapid Communications in Mass Spectrometry. 2004;18:2650–2660. doi: 10.1002/rcm.1672. [DOI] [PubMed] [Google Scholar]
- (21).Ripoche N, Ferchaud-Roucher V, Krempf M, Ritz P. Journal of Mass Spectrometry. 2006;41:1212–1218. doi: 10.1002/jms.1093. [DOI] [PubMed] [Google Scholar]
- (22).Sonko BJ, Miller LV, Jones RH, Donnelly JE, Jacobsen DJ, Hill JO, Fennessey PV. Analytical Biochemistry. 2003;323:211–217. doi: 10.1016/j.ab.2003.09.011. [DOI] [PubMed] [Google Scholar]
- (23).Speakman J, Nagy K, Masman D, Mook W, Poppitt S, Strathearn G, Racey P. Analytical Chemistry. 1990;62:703–708. [Google Scholar]
- (24).Speakman J, Krol E. Physiological and Biochemical Zoology. 2005;78:650–667. doi: 10.1086/430234. [DOI] [PubMed] [Google Scholar]
- (25).Schoeller D, Ravussin E, Schutz Y, Acheson K, Baertschi P, Jequier E. American Journal of Physiology. 1986;250:R823–830. doi: 10.1152/ajpregu.1986.250.5.R823. [DOI] [PubMed] [Google Scholar]