

Abstract
Patient-to-patient variability in disease progression continues to complicate clinical decisions of treatment regimens for cardiovascular diseases, metastatic cancers and osteoporosis. Here, we investigated if monocytes, circulating white blood cells that enter tissues and contribute to disease progression by differentiating into macrophages or osteoclasts, could be useful in understanding this variability. Monocyte-derived macrophages and osteoclasts produce cysteine cathepsins, powerful extracellular matrix proteases which have been mechanistically linked to accelerated atherosclerotic, osteoporotic, and tumor progression. We hypothesized that multivariate analysis of temporal kinase activation states during monocyte differentiation could predict cathepsin proteolytic responses of monocyte-derived macrophages and osteoclasts in a patient-specific manner. Freshly isolated primary monocytes were differentiated with M-CSF or RANKL into macrophages or osteoclasts, respectively, and phosphorylation of ERK1/2, Akt, p38 MAPK, JNK, c-jun, and IκB-α were measured at days 1, 3, 6, and 9. In parallel, cell diameters and numbers of nuclei were measured, and multiplex cathepsin zymography was used to quantify cathepsins K, L, S, and V activity from cell extracts and conditioned media. There was extensive patient-to-patient variability in temporal kinase activation states, cell morphologies, and cathepsin K, L, S, and V proteolytic activity. Partial least squares regression models trained with temporal kinase activation states successfully predicted patient-specific morphological characteristics (mean cell diameter and number of nuclei) and patient-specific cathepsin proteolytic activity with predictability as high as 95%, even with the challenge of incorporating the complex, unknown cues from individual patients’ unique genetic and biochemical backgrounds. This personalized medicine approach considers patient variability in kinase signals to predict cathepsin activity. Such analyses may provide beneficial tools for personalized kinase and protease inhibitor therapies for tissue destructive diseases.
Introduction
Patient-to-patient variability in disease progression continues to complicate clinical decisions of treatment regimens for both cardiovascular diseases and cancer that originates at different sites and is diagnosed at different stages of progression. Monocytes are circulating white blood cells that participate in pathogenesis of cardiovascular disease and cancer in response to cues from cells or the environment by leaving the vasculature, entering the tissue, and differentiating into macrophages or osteoclasts. In atherosclerotic lesions, monocyte derived macrophages ingest lipids, become foam cells, and contribute to plaque growth and extracellular matrix (ECM) degradation and remodeling.1 In cancer, monocyte-derived tumor associated macrophages (TAMs) contribute to almost 50% of the tumor volume and promote tumor invasion, migration and metastasis.2, 3 Osteoclasts are the multinucleated cells that resorb bone by secreting high levels of cathepsin K to cleave type I collagen, the major structural protein in bone.4, 5 Osteoclasts have been implicated in later stages of atherosclerotic plaque calcification and shown to be differentiated from infiltrated monocytes.6–8 In cancer, osteoclasts are involved in positive feedback loops to develop osteolytic bone lesions with metastasized cancer cells.9, 10
Once differentiated from monocytes, both macrophages and osteoclasts contribute to tissue remodeling through the production and secretion of cysteine cathepsins, proteases that have been identified as the most potent mammalian collagenases and elastases that, upon secretion, locally degrade collagen, elastin, and other ECM substrates.11, 12 Cathepsins K, L, S, and V produced by macrophages and osteoclasts are highly implicated in atherosclerotic vascular remodeling as well as tumor associated tissue remodeling.13–17
Pathologically overactive osteoclasts generate elevated levels of cathepsin K and are the main etiological agents of osteoporosis and osteolytic lesions. Despite the number of pharmacological inhibitors being developed to block this activity, many are failing clinical trials.9, 18, 19 This may be due to variability in cathepsin activity among patients, altering pharmacokinetics, and in turn, increasing side effects due to under- or over-dosing. Studies have measured circulating cathepsin levels in patients with similar diseases and shown a wide range of variability, whether measured in plasma or serum, among healthy or diseased patients.6, 7, 20–22 Some of these studies showed higher cathepsin activity correlating with disease, but others were unable to obtain statistical significance due to high variability among patient cohorts.
Patient variability due to genetic polymorphisms has been investigated, but studies on patient variability at the cellular level of activity have been limited. At any given moment, cells are receiving multiple cues from their environment, and the internal milieu of one individual patient differs from another, presenting a unique biochemical environment and cocktail of stimuli to circulating monocytes in the peripheral blood. Any pre-existing conditions such as cardiovascular disease, anemia, obesity, diabetes, or a multitude of other conditions that alter blood biochemistry may stimulate or pre-condition the cells that are exposed to them. This inherent variability in patient-specific, in vivo environments adds another level of complexity and an additional unknown variable.
One tool that has been used successfully to extract important biological information by integrating unknown and measurable variables at a nexus, is the cue-signal-response paradigm. The cue-signal-response paradigm integrates multiple extracellular cues received by cells, measures changes in the induced signals, and interprets cell decisions to execute responses.23–26 In particular, computational analysis of dynamic changes in kinase activation has shown that kinases serve as integrators of stimuli from different soluble, cellular, and physical cues, to generate specific cellular responses.27–30 Prediction of monocyte differentiation and resultant proteolytic activity from an individual signaling pathway in isolation is problematic due to complex crosstalk in signaling networks, but is substantially improved when multiple pathways are considered.23, 26 We have previously used activation of just seven kinases in adult bone marrow-derived stem cells to show that osteogenic differentiation decisions were encoded in temporal kinase activation profiles, and analyzed them with the multivariate analytical technique partial least squares regression (PLSR) to predict terminal differentiation outcomes and phenotype.24
In the current study, we investigated if monocyte differentiation into macrophages or osteoclasts, and the resulting cathepsin activity of these cells were encoded in kinase activation profiles, and if data-driven, multivariate analysis models could predict patient-specific cell differentiation and proteolytic activity. We hypothesized that the temporal kinase activation states would be predictive even with the challenge of incorporating complex unknown cues provided by the genetic and biochemical background of each individual patient that leads to variability.
MATERIALS AND METHODS
THP-1 Cell Culture
Human THP-1 acute monocytic leukemia cells (American Type Culture Collection [ATCC]) were cultured in RPMI medium 1640 (Mediatech) containing 10% fetal bovine serum (FBS, Atlanta Biologicals), 0.05% β-mercaptoethanol, 1% L-glutamine, and 1% penicillin/streptomycin (Life Technologies). Cells were maintained with 5% CO2 at 37 °C. For macrophage differentiation, monocytes were incubated with 100 nM phorbol myristate acetate (PMA, Sigma-Aldrich) for 24 h, followed by incubation for an additional 11 days in growth medium, with media changed twice per week. For osteoclast differentiation, monocytes were incubated with 100 nM 1α,25-dihydroxyvitamin D3 (Alfa Aesar) for 12 days, with medium changed twice per week. For all differentiation, cells were seeded at 300,000 cells/cm2.
Primary monocyte isolation and differentiation
Heparinized venous blood from healthy volunteers was diluted 1:1 in sterile PBS, layered on Ficoll-Paque (GE healthcare), and centrifuged at 400g for 30 minutes. The buffy coat layer was isolated, red blood cells lysed, and peripheral blood mononuclear cells (PBMCs) were washed 3 times in PBS. Monocytes adhered overnight and all other non-adherent cells were removed. For macrophage differentiation, isolated monocytes were cultured in RPMI containing 10% male human serum and 30ng/μl macrophage colony stimulating factor (M-CSF, Peprotech). For osteoclast differentiation, isolated monocytes were cultured in alpha-MEM (Life Technologies) supplemented with 10% fetal bovine serum, 30ng/ml M-CSF, and 30ng/ml receptor activator of NFκB Ligand (RANKL) (Peprotech). Medium was replaced every 3 days.
TRAP Histological Staining
On day 15, both macrophages and osteoclasts were stained for tartrate-resistant acid phosphatase (TRAP) activity according to the manufacturer’s instructions (Sigma-Aldrich). TRAP activity was visualized under a light microscope as brown and dark red areas. Multinucleated cells were deemed as those with three or more nuclei.
Flow Cytometry
TRAP activity was quantified using flow cytometry modified from an existing protocol31 and using Fast Red violet (Sigma Aldrich) instead of Fast Garnet GBC. Adherent cells were released using 2 mM ethylene diamine tetraacetate (EDTA), fixed with 4% paraformaldehyde and permeabilized using 0.02% Triton X-100. Cells were then incubated in 2X TRAP Staining Solution (8% of 12.5 mg/ml napthol-ASBI phosphate, 2% of 10 mg/ml Fast Red violet diluted in solution containing 50 mM MES, 50 mM Na Tartrate and pH at 6.3) at room temperature for 9 minutes. Reaction was ended using ice cold PBS. TRAP activity was detected at 488 nm excitation, 610 nm short pass dichroic and measured through a 675 ± 20 narrow bandpass filter. For CD68 labeling, adherent cells were released using 2 mM ethylene diamine tetraacetate (EDTA), fixed with 4% paraformaldehyde. Cells were incubated in mouse anti-CD68 antibody (Millipore) at 4 °C for 30 minutes. They were then incubated with donkey anti-mouse Alexafluor 488 (Life Technologies) at 4 °C for 30 minutes. Data were expressed as percent of the total cell population positive for CD68.
Cell morphology measurements
Cell diameter and number of nuclei measurements were used for cell morphology. For macrophages, 20 cells were measured per patient, and a single diameter was measured for each cell. For osteoclasts, the number of cells measured varied from 5 to 20 as only cells with more than 3 nuclei were included; due to their irregular shapes, the narrowest and widest parts of each cell were measured and averaged to approximate diameter.
Kinase phosphorylation analysis with Bioplex assays
Differentiating cells were lysed and total protein concentration was determined using microBCA assay (Pierce). Bioplex® bead kits (BioRad) were used according to manufacturer’s instructions with 5 μg protein from each sample. Phosphorylation of ERK1/2 (Thr202/Tyr204, Thr185/Tyr187), Akt (Ser473), p38 MAPK (Thr180/Tyr182), JNK (Thr183/Tyr185), c-jun (Ser63), NFκB p65 (Ser536) and IκB-α (Ser32/Ser36) were measured. Signal values for each phosphorylated kinase were normalized to the signal detected in a master lysate prepared in bulk from pre-stimulated monocytes that was used as a control for all assays. Signal values for each kinase were normalized between 0 and 1 by dividing by the maximum value over the entire 9 days for all treatments.
Partial least square regression (PLSR) analysis
M × N data matrix was generated with data from M patients and N kinase phosphorylation signals. Each column of the independent X matrix corresponds to a unique input or signal: phosphorylated kinase signal from days 1, 3, 6, and 9, and each column of the dependent Y matrix corresponds to unique outputs. Each row represents a unique patient and stimulation condition (i.e. patient 1- MCSF, patient 1-RANKL, patient 2- MCSF, etc.). All data was mean-centered and scaled to unit variance. SIMCA-P (UMetrics) was used to solve the PLSR problem with the nonlinear iterative partial least squares (NIPALS) algorithm.32
Multiplex cathepsin zymography
Media was replaced with serum-free media on day 14 and incubated overnight. This conditioned media was collected and concentrated using VivaSpin®500 Centrifugal Concentrator (Vivaproducts). Cellular protein was extracted in lysis buffer (20 nM Tris-HCl at pH 7.5, 5 mM EGTA, 150 mM NaCl, 20 mM β-glycerol-phosphate, 10 mM NaF, 1 mM sodium orthovanadate, 1% Triton X-100, 0.1% Tween-20) with 0.1 mM leupeptin freshly added. Cathepsin zymography was performed on cell extracts and on conditioned media as described previously.33 Briefly, equal amounts of protein in non-reducing loading buffer were separated on 12.5% SDS-polyacrylamide gels containing 0.2% gelatin at 4°C. Enzymes were renatured and then the gels were incubated overnight at 37°C in acetate buffer, pH 4 with 1 mM EDTA and freshly added 2mM dithiothreitol. Gels were then rinsed, stained with Coomassie blue, and imaged using an ImageQuant LAS 4000 (GE Healthcare). Densitometry was performed using ImageJ to quantify the intensity of the white cleared band of proteolytic activity. For cathepsin K inhibitor studies, gels were incubated overnight at 37 °C in the presence of 1μM cathepsin K inhibitor (1-(N-benzyloxycarbonyl-leucyl)-5-(N-Boc-phenylalanyl- leucyl) carbohydrazide [Z-L-NHNHCONHNH-LF-Boc], EMD Biosciences) or vehicle. 21
RESULTS
THP-1 monocyte differentiation into macrophages or osteoclasts is encoded in temporal kinase activation states
Clonal THP-1 monocyte cells were used to identify a set of kinases for predicting macrophage or osteoclast differentiation outcomes. These cells have been differentiated into macrophages with phorbol 12-myristate 13-acetate (PMA) and into osteoclasts with 1,25α vitamin D3 (1,25 D3).34–37 We first confirmed this in our hands by stimulating THP-1 cells with 100 nM PMA for 1 day, followed by 8 days of culture, or 100 nM 1,25α vitamin D3 for 12 days. After 12 days, osteoclast differentiation was confirmed by tartrate resistant acid phosphatase (TRAP) positive staining and multi-nucleation (n≥3) as indicated by Hoechst staining (Fig. 1A).
Figure 1. Multivariate analysis of kinase signals of THP-1 monocytes predicts and distinguishes macrophage and osteoclast differentiation outcomes.
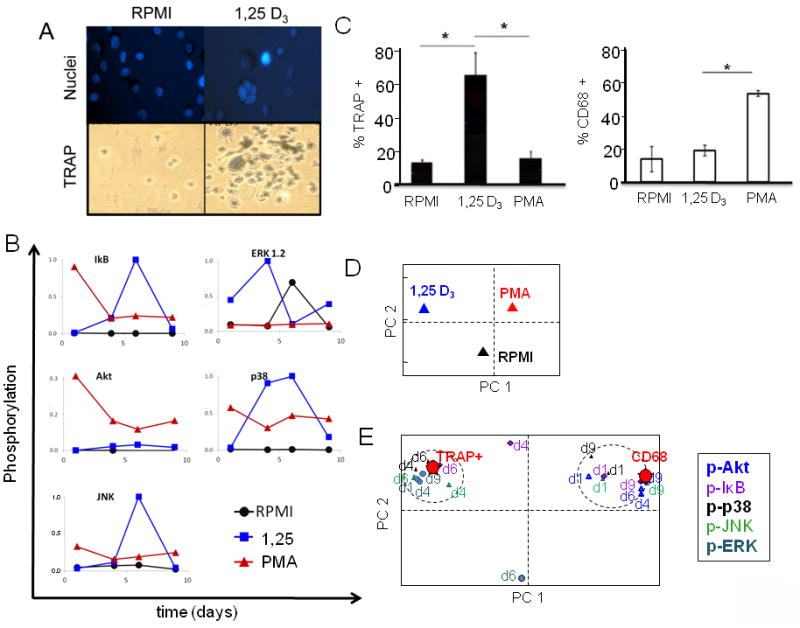
THP-1 monocytes were stimulated with 100nM PMA or 100nM 1,25 vitamin D3 (1,25 D3) for 12 days. A) Nuclei were stained with Hoechst for determining multi-nucleation (top row). Cells were fixed and stained with colorimetric TRAP activity assay (bottom row). B) On days 1, 4, 7 and 9 of differentiation, macrophages and osteoclasts were lysed and kinase signals were quantified using Bioplex technology and normalized to maximum signal over the time period. C) On day 12, flow cytometry for TRAP activity and CD68 expression was performed. D) PLSR analysis was performed using kinase signals and the scores plot shows polar separation of osteoclasts (1,25 D3) and macrophages (PMA) along principle component 1 (PC1). E) The loadings plot shows polar separation of temporal kinase phosphorylation and covariance with differentiation outcomes of osteoclasts (TRAP+) or macrophages (CD68).
In parallel, lysates were collected on days 1, 4, 6, and 9 of differentiation for Bioplex phosphorylated kinase assays. Six μg of total protein was used to quantify phosphorylation of Akt, ERK1/2, p38, IκB, and JNK at each of these time-points in cultures stimulated with either PMA or 1,25 D3. These kinases were chosen as they have all been implicated during monocyte differentiation into either macrophages, osteoclasts, or both.34, 36, 38–40 Kinase phosphorylation signals were normalized to the maximum activation across all conditions for all time points and treatment conditions (Fig 1B). The plots illustrate the difficulty in identifying patterns to link the cues, signals, and responses of differentiating cells based on an individual pathway.
Quantitative measurements of macrophage and osteoclast differentiation were necessary to populate a response matrix for the partial least squares regression (PLSR) model, and establish a mathematical relationship between kinase phosphorylation signatures and monocyte differentiation responses. To quantify macrophage differentiation, flow cytometry was performed after labeling the cells with an anti-CD68 antibody. For osteoclasts, traditional, qualitative colorimetric TRAP staining was modified with a fluorogenic phosphatase substrate to enable fluorescent TRAP activity quantification by flow cytometry.31 As expected, there was a significantly greater number of TRAP+ cells after stimulation with 1,25 D3 compared to the other two conditions, while treatment with PMA significantly increased the percentage of cells positive for CD68 expression (Fig. 1C, n=3, p<.01).
With quantitative signals from kinase phosphorylation measurements (Fig. 1B) and quantified differentiation responses from flow cytometry (Fig. 1C), we were then able to determine if these kinase activation states could be used to predict macrophage or osteoclast differentiation responses. Based on the principles of eigenvectors and orthogonal transformation, PLSR algorithm groups signals with covarying responses, and reduces dimensionality of the data by plotting them along principal component (PC) axes that capture maximal variance. The PLSR algorithm then computes a linear solution in principal component space based on the proposed relationship between the independent variables X (signals) and dependent variables Y (responses) to calculate a coefficient matrix such that Y = F (X) in principal component space. Methods to determine significance of principal components is described in supplemental methods. The kinase signals that co-vary the greatest with the dependent variable (macrophage or osteoclast differentiation responses in this case) are weighted more heavily in the solution function that will be used to predict the responses (Y) from the given input data matrix (X). When analyzing biological measurements, these principal component axes can be assigned to biological phenomena such as differentiation, proliferation or apoptosis.
In the scores plot shown in figure 1D, treatment conditions are plotted onto principal component axes according to their covariance. In other words, similar cellular responses to treatments are grouped together when projected onto the principal components. PMA and 1,25 D3 stimulated kinase signatures were segregated along the first principle component (PC 1). Undifferentiated THP-1 monocytes cultured only in RPMI media were segregated along PC 2 from the differentiated cells. This suggested that the first principal component could be defined as macrophage/osteoclast differentiation axis, and the second as the proliferation axis. The loadings plot is shown in figure 1E and depicts the contribution of an individual kinase’s activation at a specific time, to macrophage or osteoclast differentiation according to the calculated weighted coefficients, and plots them onto weighted principal components. A clear polarization of signals with either CD68 or TRAP responses is depicted.
Goodness of prediction was tested using a bootstrapping approach; cross-validation was performed by omitting an observation, then using the calculated weighted coefficient matrix to predict response values without those removed observations. This procedure was repeated until every observation had been excluded exactly once. Then predictability was determined using root mean square error between predicted and experimentally observed values. Using Akt, ERK1/2, p38, IκB, and JNK phosphorylation, greater than 99% predictability of CD68 expression and TRAP positive staining was achieved suggesting that these kinases would be useful in predicting monocyte differentiation responses.
Primary monocyte-derived macrophages and osteoclasts from healthy donors exhibit extensive morphological variability
The cell line was useful for proof-of-principle that a key set of kinase activation signatures could be predictive of monocyte differentiation decisions. The next step was to apply this methodology to primary monocytes isolated from different individuals’ peripheral blood. Monocytes were isolated from healthy donors and stimulated for macrophage differentiation with M-CSF for 14 days, or for osteoclast differentiation with M-CSF and RANKL for 14 days. Morphological differences in diameter and number of nuclei among the donors’ differentiated macrophages and osteoclasts were the first indicator of variability among donors. Representative images of differentiated cells are shown with patient matched macrophages and osteoclasts, by column (Fig. 2A). Osteoclast differentiation was confirmed by multi-nucleation (n ≥3) and TRAP+ histological staining (Fig. 2B).
Figure 2. Phenotypic variability of macrophages and osteoclasts derived from peripheral blood monocytes.
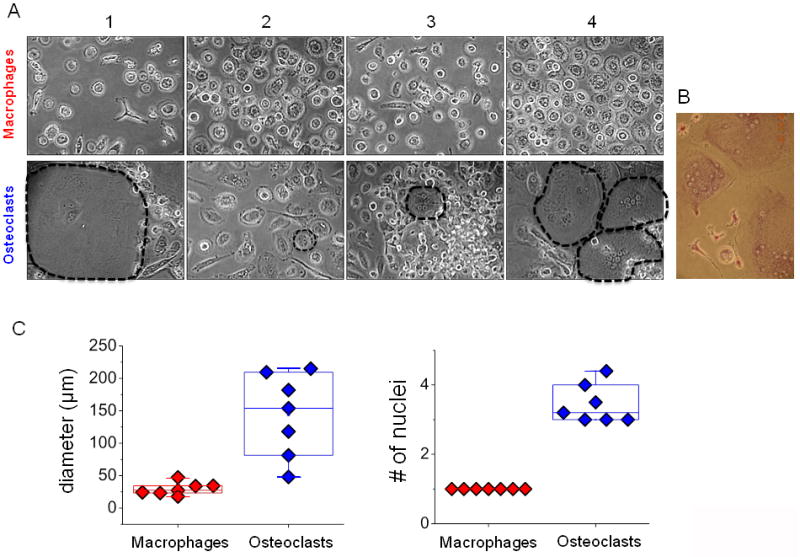
Monocytes isolated from peripheral blood were cultured for 14 days and treated with 30ng/μl M-CSF alone to differentiate them into macrophages or with 30ng/μl M-CSF and 30ng/μl sRANKL to drive osteoclastic differentiation and cultured. A) Representative pictures of monocyte derived macrophages and monocyte derived osteoclasts after 12 days of differentiation are shown. Dotted lines outline osteoclasts. B) On day 15, multinucleated cells in the culture were stained for TRAP activity to confirm osteoclastic differentiation. A representative image of colorimetric assay staining is shown. C) Mean diameter and average number of nuclei per patient were measured (n=7).
Cell diameter and number of nuclei of monocyte derived macrophages and monocyte-derived osteoclasts were quantified and shown in the box and whisker plots (Fig. 2C). Mean diameter of the macrophages was 29.2 ± 12 μm, 41% deviation in diameter, among seven patients, here on referred to as Group I, and with one nucleus. Osteoclasts exhibited greater range of donor variability with a mean diameter of 143.5 ± 85μm, a 59% deviation, and median number of nuclei of 3 (Fig. 2C) with one osteoclast from a particular donor contained as many as 9 nuclei, although the median number of nuclei among all counted osteoclasts for that donor was 5. TRAP and CD68 expression were initially measured for first sets of samples using flow cytometry. However, changes in forward and side scatter (FSC and SSC) due to these morphology differences made it difficult to reliably gate and quantify appreciable shifts in fluorescence for these groups. Due to this, cell size and number of nuclei were used as quantitative morphological measurements.
Patient-to-patient variability in cell morphology is encoded in temporal kinase activation states of the differentiating monocytes
We tested the hypothesis that a coefficient matrix could still be calculated from multivariate kinase activation to predict donor monocyte differentiation responses despite the donor-specific morphological variability. Cell lysates were collected on days 1, 3, 6 and 9 of primary monocyte differentiation and temporal kinase phosphorylation signatures were measured using Bioplex assays as described earlier. These kinase activation signatures were variable for Group I, as indicated by color variations in the heat map, even for the same kinase, treatment condition, and time (Fig. 3A). The input matrix (X) of kinase phosphorylation and dependent response matrix (Y) of cell morphology data of diameter and number of nuclei were used to calculate the weighted coefficient matrix that would link the initiating cues to the differentiated cell phenotype responses using the kinases’ signals. Separate models were made for macrophages and osteoclast differentiation cues, with the inherent, but unknown patient-specific factors that stimulate monocytes in vivo, affecting kinase signatures of both datasets.
Figure 3. Patient-to-patient variability in macrophage and osteoclast differentiation outcomes of cell diameter and number of nuclei is captured in kinase activation state of differentiating monocytes.
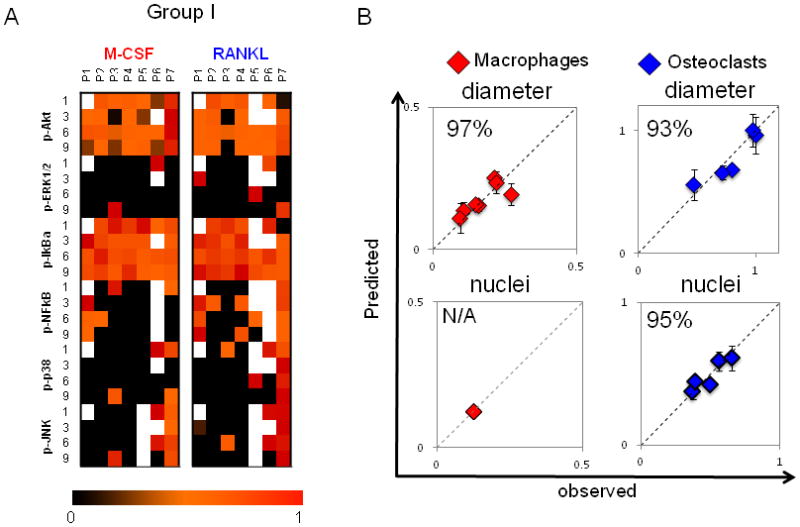
A) On days 1, 3, 6 and 9 of differentiation, macrophages and osteoclasts were lysed, and kinase signals were quantified using Bioplex technology. A compendium of time-dependent kinase signals of patients 1–7 (group I) is shown. Signals are normalized to the highest value within a given time point and a kinase. White boxes correspond to missing measurements. B) A PLSR model was generated with kinase signals of differentiating macrophages as inputs and cell diameter as an output (R2Y = 0.734 Q2= 0.061, 1 significant PC). A separate PLSR model was generated for differentiating osteoclasts with their kinase signals in input matrix and their cell size and number of nuclei in output matrix (R2Y = 0.815, Q2 = 0.248, 2 significant PCs). Prediction was made with cross-validation and jack-knifing approaches, and predictability was calculated based on RMSEE. Plots of predicted vs. observed are shown, with blue diamonds for osteoclasts and red diamonds for macrophages. Predictability for cell diameter was 96% for macrophages and 89% for osteoclasts. Predictability for number of nuclei was 94% for osteoclasts. With only one nucleus, no variation can be predicted for macrophage.
PLSR analysis also can identify the most important kinase signals and time points for a differentiation outcome by calculating the variable importance for projection (VIP) using a weighted sum of squares of the coefficients calculated for a signal, such that those signals projecting strongly either positively or negatively with a differentiation response are highly ranked. If the VIP value was greater than 1, then the kinase signals were regarded as significant. JNK activation on days 1, 3 and 6 were important for determining cell morphological responses for both macrophages and osteoclasts (Supp. Table 1). Interestingly, the effects of JNK activation on macrophage and osteoclasts were opposite; with a positive correlation for macrophage diameter but a negative correlation for osteoclast diameter and number of nuclei. Using the bootstrapping method described earlier, goodness of prediction was calculated. Predictability was 97% for cell diameter of macrophages, and 93% for osteoclasts. Predictability was 95% for number of nuclei for osteoclasts (Fig. 3B). With only one nucleus, no variation could be predicted for monocyte-derived macrophages.
To test this predictability a priori, this trained model was applied to 7 additional donors, denoted as Group II, with kinase activation heat map and variable signals shown (Fig. 4A). Cell diameter was predicted at 90% and 92% for macrophages and osteoclasts, respectively, but osteoclast nuclei predictability dropped to 71% (Fig 4B). Quantification of these experimental measurements for Group II is shown in box and whisker plots with mean diameter of the macrophages as 34.4 ± 10 μm (29% deviation) among 7 donors (Group II) and osteoclasts at a mean diameter of 173.9 ± 54μm (31% deviation). Median number of nuclei for these osteoclasts was 4, one higher than training Group I, but two donors from Group II had higher average numbers of nuclei (>6) which could have lowered the nuclei predictability (Fig. 4C).
Figure 4. A priori predictions of differentiated morphologies.
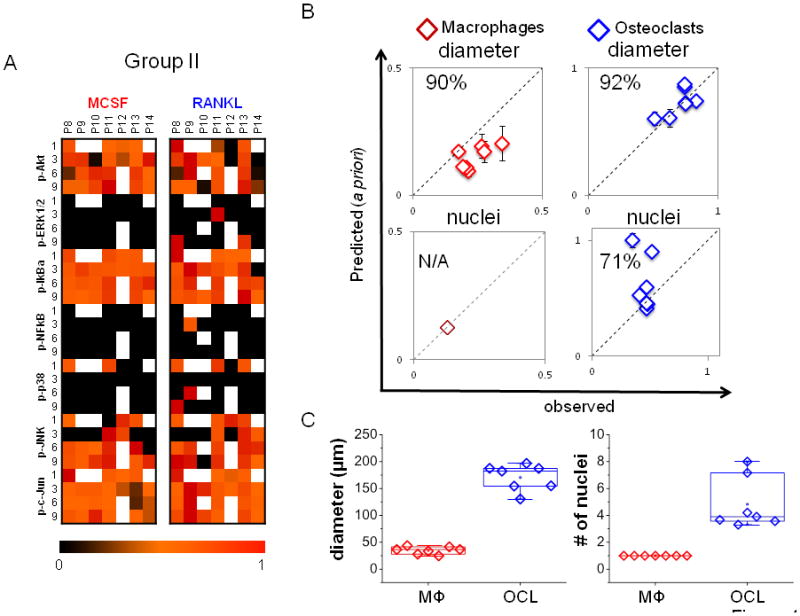
A) A compendium of time-dependent kinase signals of patients 8–14 (group II) used for a priori prediction is shown. B) The trained models for macrophages and osteoclasts generated with Group I data were used to predict cell diameter and number of nuclei from Group II kinase data. The model was effective in predicting cell diameter, with 90% predictability for macrophages and 91% predictability for osteoclasts, but only 71% for osteoclast nuclei. With only one nucleus, no variation can be predicted for macrophage. C) Experimental/observed quantification of mean diameter and average number of nuclei per patient (n=7).
Cathepsin proteolytic activity of differentiated macrophages and osteoclasts reflect patient-to-patient variability
With some predictability of morphology, we tested if behavioral or functional activity of the differentiated macrophages and osteoclasts could also be predicted for individual patients. Cathepsin activity was this metric. After the 14 day differentiation period during which kinase phosphorylation data was collected, conditioned media and cell extracts were collected and assayed for cathepsin activity from a separate group of cells seeded from Group II donors. Multiplex cathepsin zymography was used. By this assay, active cathepsins K, L, S, and V produce cleared white bands of degraded gelatin on a Coomassie stained gelatin-polyacrylamide gel. Intensity correlates to level of proteolytic activity and this can be quantified with densitometry. An added benefit of this assay is that cathepsins K, L, S, and V all produce a detectable signal on the same gel but appear at distinct, expected electrophoretic migration distances of 37 kD for cathepsin K, 35 kD for cathepsin V, 25 kD for cathepsin S, and 20 kD for cathepsin L.41 It is also more sensitive for cathepsin K than Western blotting.42
Cathepsin activity from cell extracts (Fig. 5A,B) and conditioned media (Fig. 5C,D) was quantified, and patient variability is shown in the box and whisker plots with representative gels of four donors’ zymograms shown. Zymograms shown in figure 5 were incubated at pH 4 for maximal cathepsin V and L signals, and were also incubated at pH 6 for maximal cathepsin K signal33 (Supp. Fig. 1). From these zymogram results, there were similar cathepsin activity profiles across donors within a cell type, but distinctive between osteoclasts and macrophages. A 75 kD cathepsin activity band consistently appeared in osteoclast lysates and conditioned media, suggestive of it being cathepsin K activity. However, its electrophoretic migration distance differed from the expected 37 kD distance for cathepsin K.21 To verify its identity, osteoclast lysates were loaded for cathepsin zymography and incubated in the presence or absence of 1μM cathepsin K inhibitor, which blocked the appearance of the band in question after staining the zymogram, confirming its identity as cathepsin K (Fig. 5E, F). Cathepsins K and V activity was higher in both conditioned media and cell extracts of osteoclasts compared to macrophages. This was expected for cathepsin K since it is the key enzyme used by osteoclasts for bone resorption.43 Cathepsin V, however, was more unexpected since its tissue localization has been reported as being restricted to thymus, testis, cornea, and macrophages.11 Macrophage cathepsin L activity was high in both conditioned media and cell extracts supporting reports of its expression by macrophages in atherosclerosis.13 Cathepsin L was not secreted by osteoclasts, however, although it was active intracellularly. Cathepsin L also showed the greatest patient-to-patient variability.
Figure 5. Cathepsin proteolytic profiles of differentiated macrophages and osteoclasts reflect patient-to-patient variability.
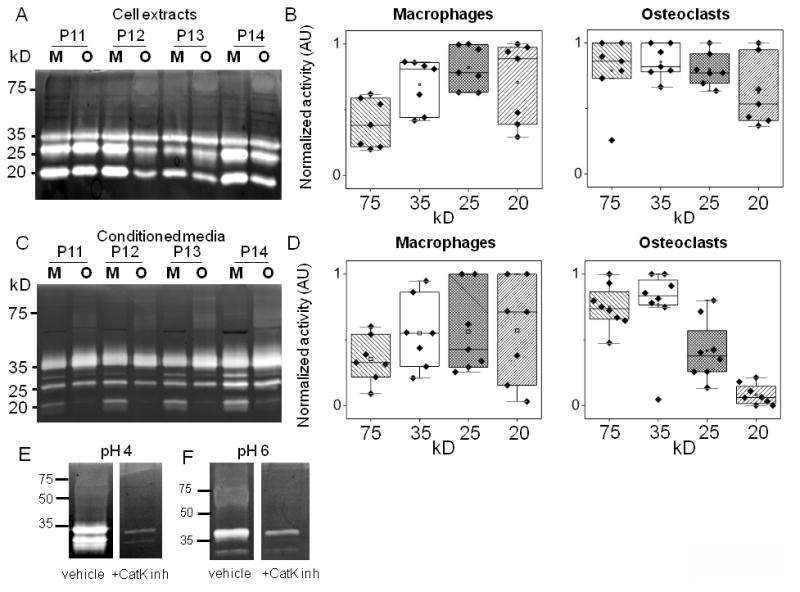
Multiplex cathepsin zymography (assay buffer, pH 4) and quantification of (A, B) cell extracts or (C, D) conditioned media for macrophage and osteoclast differentiation from patient monocytes. Quantification of cathepsin activity and patient variability is represented in the box and whisker plots. To confirm identity of 75kD band as cathepsin K, osteoclast lysates were loaded for zymography and 1μM of cathepsin K inhibitor (1-(N-benzyloxycarbonyl-leucyl)-5-(N-Boc-phenylalanyl- leucyl) carbohydrazide [Z-L-NHNHCONHNH-LF-Boc] was incubated with the zymogram during the overnight incubation at pH 4 (E) or pH 6 (F). The 75kD active band no longer appears.
Patient-specific cathepsin proteolytic activity of monocyte-derived macrophages and osteoclasts can be predicted
To test predictability of cathepsins K, L, S and V activity in differentiated macrophages and osteoclasts, cell-type specific coefficient matrices were calculated using the previously collected kinase signatures from Group II. Predictability was calculated as before with cell diameter and nuclei data, but this time for cathepsin activity in cell extracts. Plots of predicted values versus experimentally observed values are shown in Figure 6. Predictability for all cathepsins was greater than or equal to 90%, and this was true for both macrophage and osteoclast outcomes. For macrophages, predictability for cathepsin K was 90%, cathepsin V was 95%, cathepsin S was 94%, and cathepsin L was 93% (Fig. 6A). For osteoclasts, predictability for cathepsin K was 90%, cathepsin V was 90%, cathepsin S was 95%, and cathepsin L was 90% (Fig. 6B). Predictions for cathepsins secreted into the conditioned medium were generally lower than 90% and considered not to be predictive (Supp. Fig. 2). Analysis of the VIPs for predicting cathepsin activity identified c-jun phosphorylation as important for cathepsin activity of monocyte-derived macrophages and osteoclasts (Supp Table 2), and this has been implicated previously by us41.
Figure 6. Multivariate analysis of kinase activation successfully predicted patient-specific cathepsin proteolytic activity of monocyte derived macrophages and osteoclasts.
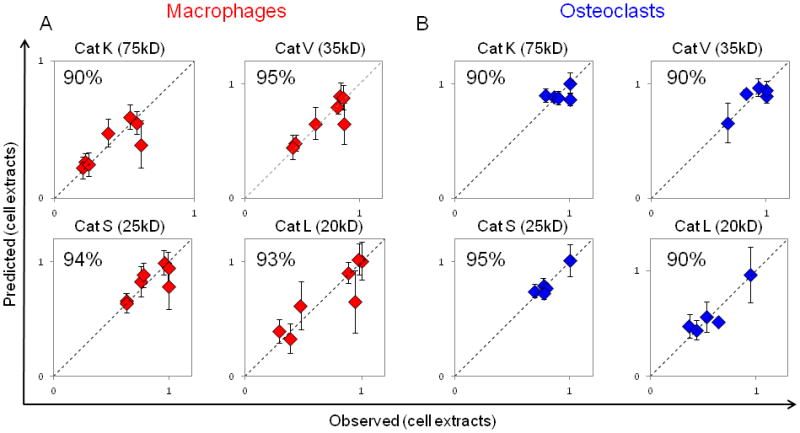
Predictability of cathepsins K, L, S, and V activity in cell extracts using a PLSR model trained with kinase measurements of differentiating macrophages (A) (R2Y = 0.837, Q2 = 0.618, 1 significant PC) or of differentiating osteoclasts (B) (R2Y = 0.564, Q2 = −0.0823, 1 significant PC).
Discussion
Variability in disease may be due to a number of factors. From these results, we submit that each person’s individual biochemical milieu of cytokines, growth factors, and other stimuli, contain a bevy of cues that explicitly and acutely pre-condition cells for specific responses to induce the variability in response to treatment or in progression of disease. Cathepsins are proteases expressed by macrophages and osteoclasts that are also biomarkers and mediators of tissue destructive diseases. By using a systems biology approach to link cell differentiation cues and responses through integration of signals at the kinase level, where integration of ubiquitous information is processed intelligently by the differentiating cell, we were able to mathematically predict relative amounts of cathepsin activity and distinguish which donors would have greater cathepsin activity compared to others.
Despite stimulation with identical cues, in vitro, patient-to-patient variability was evident in kinase activation signatures, differentiated cell morphology, and cathepsin activity. This suggests that there were additional cues that pre-conditioned the circulating monocytes from unique, patient-specific milieus prior to isolation. Along with a patient’s genetic background, there are many bioactive molecules circulating in the blood that control macrophage and osteoclast differentiation, such as monocyte chemoattractant protein-1, RANKL, tumor necrosis factor-α, parathyroid hormone, and calcitonin, among others, that differ from patient to patient. The complex question of how to account for the influence of these undetermined and unmeasured cues may be answered using kinase signatures, as demonstrated by this study. Kinases are up- and downstream of growth factor binding to receptors, cytokine stimulation, transcription factor binding, gene transcription, and protein translation. Therefore, by measuring kinase activation as this nexus of inputs that precede outputs, predictive information of cell fate prior to synthesis of differentiated cell-specific proteins and behavioral responses is provided. Although there was variability in magnitude and duration of kinase activation signals among the seven original patients of this study, a weighted coefficient matrix could be used to calculate and predict the responses of cell diameter and number of nuclei with as high as 97% predictability (Fig 3). Osteoclasts’ larger cell diameters and multi-nucleation are distinct characteristics from macrophages, although they differentiate from the same progenitor cell. By using these metrics, PLSR was able to predict differentiation outcomes in terms of cell diameter and number of nuclei. Even further, it was predictive of the patient-specific differences in these outcomes, which varied by as much as 59% for osteoclasts (Figs 3 and 4).
This was tested a priori on a second group of patients with predictability remaining above 90% for cell diameter, but an additional functional outcome of cathepsin activity was measured to link the patient variability in differentiated macrophages and osteoclasts to cell function; cathepsin activity from cell extracts was also highly predictive using only temporal kinase signatures (Fig 6), and macrophage models were more predictive than osteoclasts. This is a limitation of the model, but it makes sense considering the wider range of values for osteoclasts and the fact that they are multi-nucleated cells formed from fusion of pre-osteoclasts. Another limitation is that it was difficult to predict secreted cathepsin activity levels in a patient specific manner (Supp. Fig 2). As an explanation, cathepsins are highly regulated at multiple stages from transcription to translation to secretion, and are susceptible to external influences such as degradation, oxidation, denaturation, and inhibition.44 Additionally, we have recently shown that cannibalism occurs between cathepsins in the extracellular space serving to degrade each other,45 and the time frame at which conditioned media is collected could allow this additional behavior to occur and reduce the amount of secreted cathepsin present. These factors cannot be accounted for by changes in intracellular kinases. The multivariate analysis was able to provide clues to identify cathepsin K despite its altered electrophoretic migration. Cathepsin K is post-translationally modified in several ways that can affect its electrophoretic migration under non-reducing conditions: 1) its glycosylation can lead to altered targeting and secretion,44 2) it binds to chondroitin sulfates to form large oligomeric complexes,46–48 and 3) there are also reports of cathepsin K assuming either a tensed or relaxed state depending on ionic conditions at physiological pH that may be altering its electrophoretic migration.49
Development of cathepsins as biomarkers of disease is a growing field, but the lack of predictability for secreted cathepsins shown in this study, may have translated to the difficulties of others that have used ELISAs to measure cathepsins in plasma or serum and correlated their levels with disease. There have been varied successes in cardiovascular disease, osteoporosis, cancer, and osteolytic bone metastases.6, 7, 20–22 Serum cathepsin K measurements have been controversial; one study determined statistically significant elevated cathepsin K in postmenopausal women with osteoporosis compared to healthy age matched women, even though the standard deviation of the measurements was greater than the mean.7 A different study reported the opposite; serum cathepsin K levels could not be used to identify pre- and post-menopausal women with osteoporosis or osteopenia.7 In cancer biomarker studies, serum cathepsin levels have been measured for prostate, breast, and lung cancer, and all have yielded wide range of patient to patient variability and inconclusive findings regarding their correlation with disease.7, 50, 51 By studying mononuclear cells from the blood and the proteases they will produce, inferences can be made about local proteolytic activity contributing to the focal disease and matrix degradation, whereas the circulating levels in the blood could be attributed to any number of cells from different regions of the body.
Those large cohort studies motivate the need for personalized medicine approaches for individualized assessment incorporating the indefinable, therefore non-quantifiable patient-inherent factors that provide cues to elicit cellular responses; these factors may explain why some individuals have greater propensity to make proteolytic enzymes over others (Fig. 5). In this study, monocytes were specifically targeted as they are the “effector” cells that enter tissue, differentiate, and advance disease. Recently, the effects of some inherent, circulating patient-specific factor (or a group of factors) that increased protease production by monocytes was demonstrated in a comparative study between individuals with and without sickle cell disease.41 In that study, we showed that the chronic inflammatory milieu of sickle cell disease activated monocytes to induce greater cathepsin proteolytic activity after binding to endothelial cells compared to those without the disease. This could be an example of patient-inherent factors that precondition monocytes for elevated proteolytic activity. Although the donors of this current study did not have sickle cell disease, they still exhibited wide range of variability in cathepsin activity, perhaps due to each person’s unique biochemical milieu; yet kinase activation signals could predict these responses.
Patient variability in kinase activation signatures and cathepsin activity profiles may provide insight into proper dosing and efficiency of therapeutic small-molecule kinase inhibitors and cathepsin inhibitors currently in the pharmaceutical pipeline. Variability among the patients’ kinase activation (Fig 3A, 4A) also suggests that a one-size-fits-all approach of administering kinase inhibitors may not be the best strategy for all patients. Certainly the varying levels of active cathepsins among patients can be a confounding factor when prescribing doses of cathepsin inhibitors and cause severe side effects in some patients, that have prematurely ended many cathepsin S and K inhibitor clinical trials.18
Conclusions
Kinases are signal integrators between environmental cues and cellular responses, and analysis of multiple kinase pathways yielded high predictability for monocyte differentiation into macrophages and osteoclasts, described by cell morphology and cathepsin activity. Although there are a myriad of patient-specific factors that cannot be accounted for, we suggest that the wide range of patient-to-patient variability in proteolytic expression from monocyte-derived macrophages and osteoclast shown in our study may provide clues to wide range of disease progression and responses to therapy observed between patients. Lastly, using data-driven, multivariate analysis model of kinases, we could predict patient-specific cathepsin activity profiles which may provide beneficial tools for personalized protease inhibitor therapies.
Supplementary Material
Supplemental figure 1. Cathepsin proteolytic profiles of differentiated macrophages and osteoclasts in assay buffer, pH 6.
Multiplex cathepsin zymography (assay buffer, pH 6) and quantification of (A, B) cell extracts or (C, D) conditioned media for macrophage and osteoclast differentiation from patient monocytes. Quantification of cathepsin activity and patient variability is represented in the box and whisker plots. As with zymograms incubated in pH 4, the 75kD cathepsin activity was apparent in osteoclasts and cathepsin V activity was higher in osteoclasts as well. Cathepsin L activity was higher in cell extracts of macrophages.
Supplemental figure 2. Multivariate analysis of kinase activation was not highly predictive of secreted cathepsin proteolytic activity of patient monocyte derived macrophages and osteoclasts.
Predictability of secreted cathepsins K, L, S, and V activity using a PLSR model trained with kinase measurements of differentiating macrophages (A) (R2Y = 0.693, Q2 = 0.541, 1 significant PC) or of differentiating osteoclasts (B) (R2Y = 0.478, Q2 = 0.073, 1 significant PC).
Supplemental table 1. Variable importance of projection (VIP) for macrophage diameter and osteoclast diameter and number of nuclei.
Kinase signals with significant VIP values for the PLSR model predictive of macrophage diameter, and osteoclast diameter and number of nuclei. Kinase signals were regarded as significant if the VIP value was greater than 1. Kinase signals with significant VIP values for both macrophages and osteoclasts were highlighted.
Supplemental table 2. VIPs for cell-associated cathepsin activity of macrophages and osteoclasts.
Kinase signals with significant VIP values for the PLSR model predictive of cell-associated cathepsin activity of monocyte-derived macrophages and for monocyte-derived osteoclasts.
Insight, Innovation and Integration.
Cathepsin proteases contribute to development and progression of atherosclerosis, cancer, and osteoporosis, and insight gained from differences in cathepsin proteolytic activity may be responsible for patient-to-patient variability in rates of disease progression and pharmacologic responses to small molecule kinase and protease inhibitors. Using data-driven, multivariate analysis models based on kinase activation signatures and quantitative measurements of cathepsin activity, we successfully predicted patient-specific proteolytic activity of monocyte-derived macrophages and osteoclasts. Integrating computational methods with biological problems supported this innovative hypothesis that kinase activation profiles which encode cellular proteolytic activity, was useful to predict cathepsin levels of distinct patients. Further, these models could account for unknown patient-inherent factors that influence disease susceptibility by integration at the kinase level.
Acknowledgments
This work was funded in some part by the Georgia Cancer Coalition, Regenerative Engineering and Medicine (REM) Seed Grant, Parker H. Petit Institute for Bioengineering and Biosciences, the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000454, and NIH New Innovator grant Award Number 1DP2OD007433-01 from the Office of the Director, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Office of the Director, National Institutes of Health or the National Institutes of Health.
References
- 1.Ross R. N Engl J Med. 1999:340, 115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, Berman T, Joyce JA. Genes Dev. 24:241–255. doi: 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, Peters C, Hanahan D, Joyce JA. Genes Dev. 2006;20:543–556. doi: 10.1101/gad.1407406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi GP, Chapman HA, Bhairi SM, DeLeeuw C, Reddy VY, Weiss SJ. FEBS Lett. 1995;357:129–134. doi: 10.1016/0014-5793(94)01349-6. [DOI] [PubMed] [Google Scholar]
- 5.Bromme D, Okamoto K. Biological Chemistry Hoppe-Seyler. 1995;376:379–384. doi: 10.1515/bchm3.1995.376.6.379. [DOI] [PubMed] [Google Scholar]
- 6.Aaronson DS, Horvath CM. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 7.Lancet. 2010;376:1874. [Google Scholar]
- 8.Micheletti RG, Fishbein GA, Currier JS, Singer EJ, Fishbein MC. Mod Pathol. 2008;21:1019–1028. doi: 10.1038/modpathol.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Gall C, Bonnelye E, Clezardin P. Curr Opin Support Palliat Care. 2008;2:218–222. doi: 10.1097/SPC.0b013e32830baea9. [DOI] [PubMed] [Google Scholar]
- 10.Mundy GR. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 11.Yasuda Y, Li Z, Greenbaum D, Bogyo M, Weber E, Bromme D. J Biol Chem. 2004;279:36761–36770. doi: 10.1074/jbc.M403986200. [DOI] [PubMed] [Google Scholar]
- 12.Garnero P, Borel O, Byrjalsen I, Ferreras M, Drake FH, McQueney MS, Foged NT, Delmas PD, Delaisse JM. J Biol Chem. 1998;273:32347–32352. doi: 10.1074/jbc.273.48.32347. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, Sukhova GK, Yang JT, Sun J, Ma L, Ren A, Xu WH, Fu H, Dolganov GM, Hu C, Libby P, Shi GP. Atherosclerosis. 2006;184:302–311. doi: 10.1016/j.atherosclerosis.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 14.Lutgens E, Lutgens SP, Faber BC, Heeneman S, Gijbels MM, de Winther MP, Frederik P, van der Made I, Daugherty A, Sijbers AM, Fisher A, Long CJ, Saftig P, Black D, Daemen MJ, Cleutjens KB. Circulation. 2006;113:98–107. doi: 10.1161/CIRCULATIONAHA.105.561449. [DOI] [PubMed] [Google Scholar]
- 15.Platt MO, Ankeny RF, Jo H. Arterioscler Thromb Vasc Biol. 2006;26:1784–1790. doi: 10.1161/01.ATV.0000227470.72109.2b. [DOI] [PubMed] [Google Scholar]
- 16.Platt MO, Ankeny RF, Shi GP, Weiss D, Vega JD, Taylor WR, Jo H. American journal of physiology. Heart and circulatory physiology. 2007;292:H1479–1486. doi: 10.1152/ajpheart.00954.2006. [DOI] [PubMed] [Google Scholar]
- 17.Sukhova GKSG, Simon DI, Chapman HA, Libby P. The Journal of clinical investigation. 1998;102:576–583. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bromme D, Lecaille F. Expert Opin Investig Drugs. 2009;18:585–600. doi: 10.1517/13543780902832661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desmarais S, Masse F, Percival MD. Biol Chem. 2009;390:941–948. doi: 10.1515/BC.2009.092. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Ma L, Yang J, Ren A, Sun Z, Yan G, Sun J, Fu H, Xu W, Hu C, Shi GP. Atherosclerosis. 2006;186:411–419. doi: 10.1016/j.atherosclerosis.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Kundu AK, Putnam AJ. Biochem Biophys Res Commun. 2006;347:347–357. doi: 10.1016/j.bbrc.2006.06.110. [DOI] [PubMed] [Google Scholar]
- 22.Kerschan-Schindl K, Hawa G, Kudlacek S, Woloszczuk W, Pietschmann P. Experimental Gerontology. 2005;40:532–535. doi: 10.1016/j.exger.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Janes KA, Kelly JR, Gaudet S, Albeck JG, Sorger PK, Lauffenburger DA. J Comput Biol. 2004;11:544–561. doi: 10.1089/cmb.2004.11.544. [DOI] [PubMed] [Google Scholar]
- 24.Platt MO, Wilder CL, Wells A, Griffith LG, Lauffenburger DA. Stem Cells. 2009;27:2804–2814. doi: 10.1002/stem.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffenburger DA, Yaffe MB. Science. 2005;310:1646–1653. doi: 10.1126/science.1116598. [DOI] [PubMed] [Google Scholar]
- 26.Miller-Jensen K, Janes KA, Brugge JS, Lauffenburger DA. Nature. 2007;448:604–608. doi: 10.1038/nature06001. [DOI] [PubMed] [Google Scholar]
- 27.Jordan JD, Landau EM, Iyengar R. Cell. 2000;103:193–200. doi: 10.1016/s0092-8674(00)00112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 29.Lee FS, Hagler J, Chen ZJ, Maniatis T. Cell. 1997;88:213–222. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- 30.Su B, Jacinto E, Hibi M, Kallunki T, Karin M, Ben-Neriah Y. Cell. 1994;77:727–736. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 31.Filgueira L. J Histochem Cytochem. 2004;52:411–414. doi: 10.1177/002215540405200312. [DOI] [PubMed] [Google Scholar]
- 32.Geladi P. Analytica Chimica Acta. 1986;185:1–17. [Google Scholar]
- 33.Wilder CL, Park KY, Keegan PM, Platt MO. Archives of biochemistry and biophysics. 2011;516:52–57. doi: 10.1016/j.abb.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwende H, Fitzke E, Ambs P, Dieter P. Journal of leukocyte biology. 1996;59:555–561. [PubMed] [Google Scholar]
- 35.Inada M, Tsukamoto K, Hirata M, Takita M, Nagasawa K, Miyaura C. Biochem Biophys Res Commun. 2008;372:434–439. doi: 10.1016/j.bbrc.2008.05.041. [DOI] [PubMed] [Google Scholar]
- 36.Nerenz RD, Martowicz ML, Pike JW. Mol Endocrinol. 2008;22:1044–1056. doi: 10.1210/me.2007-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitazawa R, Kitazawa S. Biochem Biophys Res Commun. 2002;290:650–655. doi: 10.1006/bbrc.2001.6251. [DOI] [PubMed] [Google Scholar]
- 38.Roodman GD. Ann N Y Acad Sci. 2006;1068:100–109. doi: 10.1196/annals.1346.013. [DOI] [PubMed] [Google Scholar]
- 39.Boyle WJ, Simonet WS, Lacey DL. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 40.Teitelbaum SL. J Clin Invest. 2004;114:463–465. doi: 10.1172/JCI22644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keegan PM, Surapaneni S, Platt MO. Anemia. 2012;2012:201781. doi: 10.1155/2012/201781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li WA, Barry ZT, Cohen JD, Wilder CL, Deeds RJ, Keegan PM, Platt MO. Anal Biochem. 2010;401:91–98. doi: 10.1016/j.ab.2010.02.035. [DOI] [PubMed] [Google Scholar]
- 43.Chapman HA, Riese RJ, Shi GP. Annu Rev Physiol. 1997;59:63–88. doi: 10.1146/annurev.physiol.59.1.63. [DOI] [PubMed] [Google Scholar]
- 44.Brix K, Dunkhorst A, Mayer K, Jordans S. Biochimie. 2008;90:194–207. doi: 10.1016/j.biochi.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 45.Barry ZT, Platt MO. J Biol Chem. 2012 doi: 10.1074/jbc.M111.332684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z, Kienetz M, Cherney MM, James MN, Bromme D. J Mol Biol. 2008;383:78–91. doi: 10.1016/j.jmb.2008.07.038. [DOI] [PubMed] [Google Scholar]
- 47.Li Z, Hou WS, Escalante-Torres CR, Gelb BD, Bromme D. J Biol Chem. 2002;277:28669–28676. doi: 10.1074/jbc.M204004200. [DOI] [PubMed] [Google Scholar]
- 48.Li Z, Yasuda Y, Li W, Bogyo M, Katz N, Gordon RE, Fields GB, Bromme D. J Biol Chem. 2004;279:5470–5479. doi: 10.1074/jbc.M310349200. [DOI] [PubMed] [Google Scholar]
- 49.Novinec M, Kovacic L, Lenarcic B, Baici A. Biochem J. 2010;429:379–389. doi: 10.1042/BJ20100337. [DOI] [PubMed] [Google Scholar]
- 50.Tumminello FM, Flandina C, Crescimanno M, Leto G. Biomed Pharmacother. 2008;62:130–135. doi: 10.1016/j.biopha.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Naumnik W, Niklinska W, Ossolinska M, Chyczewska E. Folia Histochem Cytobiol. 2009;47:207–213. doi: 10.2478/v10042-009-0024-0. [DOI] [PubMed] [Google Scholar]
- 52.Eriksson L, Johansson E, Kettaneh-Wold N, Trygg J. Multivariate and Megavariate Data Analysis Basic Principles and Applications (Part I) 2. Umetrics AB; Umea, Sweden: 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1. Cathepsin proteolytic profiles of differentiated macrophages and osteoclasts in assay buffer, pH 6.
Multiplex cathepsin zymography (assay buffer, pH 6) and quantification of (A, B) cell extracts or (C, D) conditioned media for macrophage and osteoclast differentiation from patient monocytes. Quantification of cathepsin activity and patient variability is represented in the box and whisker plots. As with zymograms incubated in pH 4, the 75kD cathepsin activity was apparent in osteoclasts and cathepsin V activity was higher in osteoclasts as well. Cathepsin L activity was higher in cell extracts of macrophages.
Supplemental figure 2. Multivariate analysis of kinase activation was not highly predictive of secreted cathepsin proteolytic activity of patient monocyte derived macrophages and osteoclasts.
Predictability of secreted cathepsins K, L, S, and V activity using a PLSR model trained with kinase measurements of differentiating macrophages (A) (R2Y = 0.693, Q2 = 0.541, 1 significant PC) or of differentiating osteoclasts (B) (R2Y = 0.478, Q2 = 0.073, 1 significant PC).
Supplemental table 1. Variable importance of projection (VIP) for macrophage diameter and osteoclast diameter and number of nuclei.
Kinase signals with significant VIP values for the PLSR model predictive of macrophage diameter, and osteoclast diameter and number of nuclei. Kinase signals were regarded as significant if the VIP value was greater than 1. Kinase signals with significant VIP values for both macrophages and osteoclasts were highlighted.
Supplemental table 2. VIPs for cell-associated cathepsin activity of macrophages and osteoclasts.
Kinase signals with significant VIP values for the PLSR model predictive of cell-associated cathepsin activity of monocyte-derived macrophages and for monocyte-derived osteoclasts.