

Abstract
Previous studies demonstrated that diet-induced obesity increased plasma angiotensin II concentrations and elevated systolic blood pressures in male mice. Adipocytes express angiotensinogen and secrete angiotensin peptides. We hypothesize that adipocyte-derived angiotensin II mediates obesity-induced increases in systolic blood pressure in male high fat-fed C57BL/6 mice. Systolic blood pressure was measured by radiotelemetry during week 16 of low fat or high fat feeding in Agtfl/fl and adipocyte-angiotensinogen deficient mice (AgtaP2). Adipocyte angiotensinogen deficiency had no effect on diet-induced obesity. Basal 24 hour systolic blood pressure was not different in low fat-fed Agtfl/fl compared to AgtaP2 mice (124 ± 3 vs. 128 ± 3 mmHg, respectively). In Agtfl/fl mice, high fat feeding significantly increased systolic blood pressure (24 hr; 134 ± 2 mmHg; P<0.05). In contrast, high fat-fed AgtaP2 mice did not exhibit an increase in systolic blood pressure (126 ± 2 mmHg). Plasma angiotensin II concentrations were increased by high fat-feeding in Agtfl/fl mice (low fat, 32 ± 14; high fat, 219 ± 58 pg/ml, P<0.05). In contrast, high fat-fed AgtaP2 mice did not exhibit elevated plasma angiotensin II concentrations (high fat, 18 ± 7 pg/ml). Similarly, adipose tissue concentrations of angiotensin II were significantly decreased in low fat and high fat-fed AgtaP2 mice compared to controls. In conclusion, adipocyte angiotensinogen deficiency prevented high fat-induced elevations in plasma angiotensin II concentrations and systolic blood pressure. These results suggest that adipose tissue serves as a major source of angiotensin II in the development of obesity-hypertension.
Keywords: obesity, angiotensinogen, angiotensin, hypertension, adipose
Introduction
The prevalence of hypertension has surged over the last 10 years consistent with an increasing incidence of obesity.1 In men, a rise in body mass index is the primary contributor to the increased prevalence of hypertension.1 Factors that consistently link obesity to hypertension include increased sodium reabsorption and activity of the sympathetic nervous system and the renin-angiotensin systems (RAS).2-4
The RAS is activated in human and experimental obesity.5-11 Previous studies demonstrated activation of the systemic RAS in experimental models of diet-induced obesity.5,6,12,13 Obesity prone rats fed a moderately high fat (HF) diet exhibited increased plasma angiotensin II (AngII) concentrations and elevated systolic blood pressures (SBP).5 Administration of an angiotensin type 1 receptor (AT1R) antagonist to obese rats normalized blood pressure.12 In humans, administration of inhibitors of the RAS is an effective therapy for the treatment of obesity-related hypertension.14 However, mechanisms for a stimulated RAS in the setting of obesity are not well defined.
Angiotensinogen (AGT) is the only known precursor to AngII. Early studies demonstrated a high level of AGT gene expression in rat adipose tissue.15,16 Moreover, AGT gene expression increased when stem cells were differentiated to mature white adipocytes.17,18 In addition to AGT, adipocytes express multiple components of the RAS required for the synthesis of AngII19,20, and release angiotensin peptides19,21,22. In mice with transgenic over-expression of AGT in adipocytes, plasma AGT concentrations increased and were associated with increased SBP.23 Conversely, adipocyte-specific AGT deficiency reduced plasma AGT concentrations and SBP in male and female mice fed standard diet.24 These results suggest that adipocyte-derived AGT modulates the systemic RAS and blood pressure control.
Expression of AGT in adipose tissue has been reported to either increase5,6,25-28, decrease11,29, or not change30 in human and/or experimental obesity. Differences in AGT gene expression in obese adipose tissue may arise from the source of tissue, species, or whether subjects exhibit obesity-related disorders. When corrected for adipose tissue mass, secretion of AGT from adipose tissue correlated positively to body mass index and SBP in obese humans and in HF-fed mice.30 Moreover, hypertrophied adipocytes, as well as other inflammatory cell types in obese adipose tissue express components of the RAS required for the conversion of AGT to AngII. Therefore, an expanded adipose tissue mass may serve as a source of systemic AngII with obesity. We hypothesized that adipocyte-derived AGT and/or AngII contribute to activation of the systemic RAS and blood pressure control in HF-fed male mice. In this study, we quantified effects of adipocyte-specific AGT deficiency on the development of obesity, activation of the adipose and systemic RAS, and on obesity-induced increases in blood pressure.
Methods
Animals
C57BL/6 mice with loxP sites flanking exon 2 of the AGT gene (1672 bps including exon 2), as described previously24, were bred to transgenic male C57BL/6 mice expressing aP2-Cre recombinase. For all studies, Agtfl/fl littermate controls were used for comparison to mice with adipocyte-AGT deficiency (AgtaP2). Diets, radiotelemetry monitoring of blood pressure, measurement of plasma and adipose parameters, gene expression and statistical analyses are described in online-only Data Supplement. Carotid artery catheters and radiotelemeters were implanted into anesthetized (isoflurane) mice during week 15 of diet feeding. At study endpoint, mice were anesthetized with ketamine/xylazine (100/10 mg/kg, ip) for exsanguination and tissue harvest. All procedures involving animals were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory animals, and were approved by the Institutional Animal Care and Use Committee at the University of Kentucky.
Results
Adipocyte AGT deficiency had no significant effect on the development of diet-induced obesity
Genotyping demonstrated the presence of a 182 bp band in AgtaP2 mice (Figure S1A). AGT mRNA abundance was not significantly influenced by diet or genotype in liver or kidneys from Agtfl/fl compared to AgtaP2 mice (Figure S1B,C). In white adipose tissue, AGT mRNA abundance was modestly, but not significantly increased in HF-fed Agtfl/fl compared to LF-fed Agtfl/fl mice (Figure S1D). AGT mRNA abundance was significantly decreased in adipose tissue from AgtaP2 compared to Agtfl/fl mice (Figure S1D; P<0.05). In LF-fed mice, AgtaP2 mice had increased body weights compared to Agtfl/fl mice starting at week 5 (Figure 1A; P<0.05). With HF feeding, body weight increased in both genotypes compared to LF-fed mice (Figure 1A; P<0.05). In addition, fat mass increased significantly in HF-fed mice of each genotype compared to LF (Figure 1B; P<0.05). However, body weight (Figure 1A), fat and lean mass (Figure 1B, P<0.05) were not significantly different in HF-fed Agtfl/fl and AgtaP2 mice. HF-fed Agtfl/fl and AgtaP2 mice exhibited impaired glucose tolerance (Figure S2; P<0.05) compared to LF, with no differences between genotypes.
Figure 1.

Adipocyte AGT deficiency had no significant effect on obesity development . A, Weekly body weight in 16 week LF and HF-fed Agtfl/fl and AgtaP2 mice. Data are mean ± SEM from n = 15-17 (LF) or n = 21-32 (HF) mice/group. *, P < 0.05 compared to LF-fed Agtfl/fl. Fat mass (gm, B) and lean mass (gm, C) in LF and HF-fed Agtfl/fl and AgtaP2 mice. Data are mean ± SEM from n = 5 mice/group (LF) and n = 12-18 mice/group (HF). *, P<0.05 compared to LF within genotype.
Adipocyte AGT deficiency prevented obesity-induced increases in SBP
In LF-fed mice, adipocyte AGT deficiency had no significant effect on SBP (Figure 2A, 24 hr; Figure S3), diastolic blood pressures (DBP), pulse pressures, physical activity, or heart rate (Table 1). Moreover, both genotypes of LF-fed mice exhibited significantly lower SBP during the light compared to night cycle (Figure S3, P<0.05). In HF-fed mice of both genotypes, heart rate increased significantly, while physical activity was significantly decreased (Table 1; P<0.05). In HF-fed Agtfl/fl mice, SBP (24 hr) was increased significantly compared to LF (Figure 2A; P<0.05). In contrast, HF-fed AgtaP2 mice did not exhibit increased SBP compared to LF-fed mice of either genotype (Figure 2A). In addition, SBP was significantly decreased in HF-fed AgtaP2 compared to HF-fed Agtfl/fl mice (24 hrs; Figure 2A; P<0.05) during both the night and light cycle (Figure 2B; P<0.05).
Figure 2.

Adipocyte AGT deficiency prevented obesity-induced elevation in SBP. A, SBP (24 hour average for each mouse) in LF and HF-fed Agtfl/fl and AgtaP2 mice. *, P<0.05 compared to LF within genotype. **, P<0.05 compared to HF-fed Agtfl/fl. B, SBP during the night (12 hour average for each mouse) and light (12 hour average for each mouse) cycle in HF-fed Agtfl/fl and AgtaP2 mice. Data are mean ± SEM from n = 6-7 mice/group (LF) and n = 9 mice/group (HF). *, P<0.05 compared to LF within genotype. **, P<0.05 compared to Agtfl/fl within diet group.
Table 1.
Characteristics of LF and HF-fed Agtfl/fl and AgtaP2 mice.
| Agtfl/fl | AgtaP2 | |||
|---|---|---|---|---|
|
| ||||
| Parameter /group |
LF | HF | LF | HF |
| Mean arterial pressure (mmHg) |
109.3 ± 2.1 | 116.6 ± 1.1 | 112.5 ± 2.8 | 112.8 ± 2.4 |
| Diastolic blood pressure (mmHg) |
94.9 ± 2.9 | 100.2 ± 1.6 | 96.0 ± 3.3 | 98.0 ± 2.3 |
| Pulse pressure (mmHg) |
28.6 ± 3.7 | 32.5 ± 1.9 | 32.3 ± 1.3 | 28.4 ± 2.0 |
| Activity (counts/min) |
8.7 ± 1.3 | 6.5 ± 0.9* | 7.9 ± 1.5 | 5.3 ± 0.4* |
| Heart rate (beats/min) |
575 ± 7 | 589 ± 6* | 569 ± 9 | 599 ± 10* |
Data are mean ± SEM for 24 hour measurements from n = 6-7 mice/group in LF and 9 mice/group in HF.
P<0.05 compared to LF within genotype.
Adipocyte AGT deficiency prevented obesity-induced elevations in plasma AngII concentrations
In LF-fed AgtaP2 mice, plasma AGT concentrations were not significantly reduced compared to LF-fed Agtfl/fl (Figure 3A). Both genotypes of HF-fed mice exhibited a significant increase in plasma AGT concentrations compared to LF-fed controls (Figure 3A; P<0.05). Plasma AngII concentrations were markedly increased in HF-fed Agtfl/fl compared to LF-fed Agtfl/fl mice (Figure 3B; P<0.05). In contrast, plasma AngII concentrations were decreased in HF-fed AgtaP2 compared to HF-fed Agtfl/fl mice (Figure 3B; P<0.05), and were not significantly different from LF-fed controls of either genotype.
Figure 3.

Adipocyte AGT deficiency had no significant effect on HF-induced elevations in plasma AGT concentrations, but ablated HF-induced elevations in plasma AngII concentrations. A, Plasma AGT concentrations in LF and HF-fed Agtfl/fl and AgtaP2 mice. B, Plasma AngII concentrations in LF and HF-fed Agtfl/fl and AgtaP2 mice. Data are mean ± SEM from n = 7-8 mice/group (LF) and n = 11-14 mice/group (HF). *, P<0.05 compared to LF within genotype. **, P<0.05 compared to Agtfl/fl within diet group.
Since plasma AGT concentrations were increased to a similar extent in HF-fed mice of each genotype (Figure 3A), but AngII concentrations differed markedly between genotypes (Figure 3B), we quantified plasma renin activity (PRA; angiotensin I generated by endogenous renin and AGT) and plasma renin concentration (PRC; angiotensin I generated by endogenous renin in the presence of exogenous AGT). PRA was not significantly different in LF or HF-fed mice of each genotype (Figure 4). When plasma from LF-fed mice of each genotype was incubated with exogenous AGT to quantify PRC, angiotensin I production increased markedly (Figure 4). Notably, as expected in HF-fed Agtfl/fl mice exhibiting high plasma concentrations of AngII and elicitation of negative feedback regulation of renin, PRC was significantly decreased compared to LF-fed Agtfl/fl mice (Figure 4; P<0.05). Unexpectedly, PRC was also decreased in HF-fed AgtaP2 mice exhibiting low plasma AngII concentrations compared to LF-fed Agtfl/fl. Since PRC quantified active renin in plasma, we quantified total plasma renin (renin+prorenin) concentrations in mice of each genotype. Total plasma renin concentrations were increased in HF-fed AgtaP2 compared to HF-fed Agtfl/lf mice (Figure S4; P<0.05).
Figure 4.
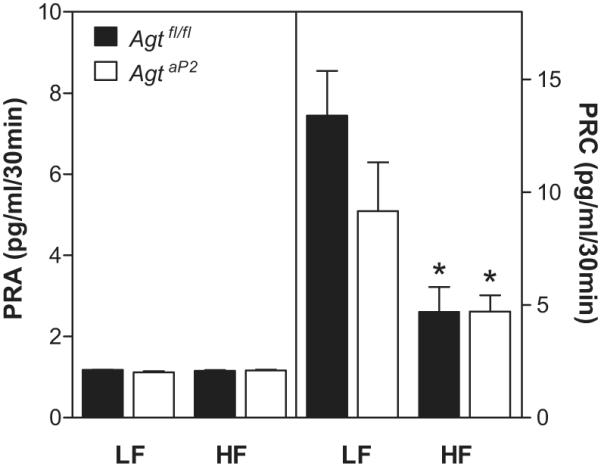
Plasma renin activity (PRA; left y-axis) and plasma renin concentration (PRC; right y-axis) in LF and HF-fed mice of each genotype. Data are mean ± SEM from n = 8 - 20 mice/group. *, P<0.05 compared to LF within genotype.
To define mechanisms for marked reductions in plasma AngII concentrations in HF-fed AgtaP2 mice, we quantified renin-like activity and AngII concentrations in adipose tissue. Renin-like activity, as reflected by levels of angiotensin I released from adipose explants, was increased in HF-fed Agtfl/fl compared to HF-fed AgtaP2 mice (Figure S5; P<0.05). Concentrations of AngII in adipose tissue (pg/mg tissue) were reduced markedly in both LF and HF-fed AgtaP2 compared to Agtfl/fl mice (Figure 5A; P<0.05). When AngII concentrations in adipose tissue were normalized for total fat mass of mice, HF-fed Agtfl/fl mice exhibited a tendency towards increased total adipose AngII content compared to LF-fed Agtfl/fl, which was significantly decreased in AgtaP2 mice (Figure 5B). In contrast to adipose, liver AngII concentrations were at the limits of detection (<0.1 pg/mg tissue), and were not influenced by genotype and/or diet (data not shown). To define mechanisms for differences in AngII concentrations between adipose and liver, we quantified mRNA abundance of the prorenin receptor and ACE. Adipose tissue from HF-fed mice of each genotype expressed relatively greater mRNA abundance for the prorenin receptor and ACE compared to liver (Figure 6A; P<0.05). Abundance of the prorenin receptor and ACE mRNA were modestly increased in adipose tissue from HF-fed Agtfl/fl mice compared to LF-fed Agtfl/fl controls (Figure 6B). While renin gene expression was below detectable limits in adipose tissue, cathepsin D and tonin, enzymes capable of processing AGT to angiotensin peptides, were also expressed in adipose tissue of mice in each group (Figure S6).
Figure 5.

Adipose AngII tissue concentrations are decreased in adipocyte AGT deficient mice. A, AngII concentrations (pg/mg tissue wet weight) in subcutaneous adipose tissue from LF and HF-fed Agtfl/fl and AgtaP2 mice. B, AngII concentrations from A corrected for total fat mass. Data are mean ± SEM from n = 4-6 mice/group (LF) and n = 5-11 mice/group (HF). *, P<0.05 compared to Agtfl/fl.
Figure 6.

Adipose tissue has more abundant mRNA expression of the prorenin receptor and ACE compared to liver. A, Relative mRNA abundance of the prorenin receptor and ACE in subcutaneous adipose tissue (Subc) compared to livers from HF-fed mice of each genotype. B, Effects of diet and/or genotype on adipose tissue expression of the prorenin receptor and ACE. Data are mean ± SEM from n = 3-5 mice/group (LF) and n = 7-12 mice/group (HF). *, P<0.05 compared to subc within genotype. **, P<0.05 compared to prorenin receptor within tissue.
Discussion
In this study, we investigated the role of adipocyte-derived AGT and/or AngII in the development of obesity-hypertension. A major finding is that adipocyte-specific deficiency of AGT prevented obesity-induced increases in SBP. Notably, adipocyte AGT deficiency also prevented obesity-induced increases in plasma AngII concentrations, suggesting that adipose tissue is a significant contributor to systemic AngII and blood pressure control. In support, adipose tissue AngII concentrations tended to increase when normalized to total fat mass of obese mice, and were reduced in HF-fed adipocyte AGT deficient mice. In addition, adipose tissue expressed components (e.g., ACE, prorenin receptor, cathepsin D, tonin) that convert AGT to AngII, while liver had relatively lower abundance of these processing enzymes and undetectable levels of AngII. These results indicate that adipose tissue becomes a primary source of systemic AngII and contributes to obesity-induced increases in SBP.
It is well accepted that the liver is the primary source of systemic AGT in rodents and humans. Whole body deficiency of AGT markedly decreased plasma AGT concentrations and blood pressure in mice.31 By comparison, adipocyte AGT deficiency in mice fed standard diet reduced plasma AGT concentrations and SBP by 25%.24 In this study, AgtaP2 mice fed a LF diet did not exhibit significant reductions in plasma AGT concentrations and SBP, and had slightly higher body weights compared to Agtfl/fl mice. These results suggest that differences in diet composition (LF versus standard mouse diet24) may have influenced the body weight and blood pressure phenotype of adipocyte AGT deficiency. Results from this study demonstrate that plasma AGT concentrations were increased by obesity, and were not influenced in adipocyte AGT deficient mice. Thus, elevated systemic AGT concentrations in obese mice were not adipocyte-derived, and presumably came from liver. Moreover, modest elevations in AGT mRNA abundance in adipose tissue from HF-fed control mice, as demonstrated in this and previous studies5,6, most likely did not contribute to increased plasma AGT concentrations in obese mice.
A surprising finding was that despite elevations in plasma AGT concentrations in obese adipocyte AGT deficient mice, plasma AngII concentrations were markedly reduced. These results demonstrate dissociation of plasma AGT concentrations from plasma AngII levels, suggesting that elevated plasma AngII of obese Agtfl/fl mice was not derived from systemic AGT. In humans, even though changes in systemic AGT concentrations can influence the systemic RAS32-34, the high plasma AGT concentration in relation to the Km for renin35 supports renin as the rate-limiting step in AngII production. In contrast, previous investigators demonstrated that mice exhibit low PRA.36 In this study, despite a significant increase in plasma AGT concentrations in obese mice, generation of angiotensin I from endogenous renin in plasma was not different between obese and lean mice. However, upon addition of exogenous AGT, angiotensin I generation increased markedly, demonstrating that plasma AGT concentrations were rate limiting. However, obese mice could not effectively convert exogenous AGT to angiotensin I in plasma, suggesting obesity-induced alterations in renin. Mechanisms for obesity-induced reductions in active PRC are unclear. However, previous studies demonstrated that PRA was reduced in obese hypertensive, but not normotensive women.37 Moreover, regional adiposity may influence renin, as central obesity was associated with increased PRA, while patients with peripheral obesity did not exhibit changes in renin activity.38 Interestingly, in HF-fed adipocyte AGT deficient mice exhibiting low plasma AngII concentrations, the total renin pool (including prorenin) increased in plasma. These results suggest that obese adipocyte AGT deficient mice respond to low systemic AngII concentrations to increase prorenin production. Further studies are required to investigate changes in plasma renin with obesity. However, the rate limiting nature of AGT in plasma of lean and/or obese mice, coupled with obesity-induced reductions in PRC, suggest that circulating concentrations of AngII were not derived from blood-borne AGT.
In this study reduced adipose tissue renin-like activity and AngII concentrations paralleled reductions in plasma AngII concentrations and SBP in HF-fed adipocyte AGT deficient mice. These results suggest that adipose tissue converts AGT to AngII, which is then released into the systemic circulation. In support, similar to previous findings suggesting a complete RAS in adipose tissue (for review see39, 40), mouse adipose tissue expressed ACE, prorenin receptor, cathepsin D and tonin mRNA. Expression levels of the prorenin receptor and ACE were modestly increased by HF feeding, suggesting nutritional regulation of these adipose RAS components may have contributed to increased conversion of AGT to AngII in adipose tissue. In addition, expression of several of these synthetic RAS components, as well as AngII concentrations, were more abundant in adipose compared to liver. Conversion of AGT to angiotensin peptides through tonin and/or cathepsin may have contributed to increased angiotensin I release from adipose tissue explants. Alternatively, the adipocyte prorenin receptor may have interacted with circulating prorenin to increase production of angiotensins in adipose tissue of obese mice. Adipose tissue expands markedly with obesity; by comparison, the mass of liver or other AngII-producing tissues (e.g.,kidney) did not increase appreciably with obesity. These results suggest that the expanded adipose mass with obesity becomes an important source of systemic AngII in the control of blood pressure.
An important finding of this study was that obesity-induced increases in SBP were not observed in adipocyte AGT deficient mice. The RAS is generally considered to be activated in obese rodents and humans (for review see41). AngII activates the sympathetic nervous system42,43 and increases sodium retention, two mechanisms that have been consistently linked to obesity-associated hypertension2-4. Given the marked reductions in plasma AngII concentrations in adipocyte AGT deficient mice, it is likely that diminished AngII-mediated regulation of sodium reabsorption and sympathetic nervous system activity contributed to the pronounced effects of adipocyte AGT deficiency to prevent obesity-associated hypertension.
In conclusion, results demonstrate that adipocyte-derived AngII is a primary contributor to obesity-induced elevations in plasma AngII concentrations and SBP. Reductions in plasma AngII concentrations were paralleled by decreased adipose tissue AngII concentrations in HF-fed adipocyte AGT deficient mice. Adipose tissue expressed relatively greater abundance of AngII synthetic processing enzymes and AngII concentrations than liver. These results demonstrate that adipose tissue is a significant contributor to an activated systemic RAS in the development of obesity-associated hypertension.
Perspectives
Evidence supports activation of the RAS in obesity-related hypertension. Adipose tissue expresses several RAS components that may link obesity to hypertension. We demonstrate that adipocyte AGT deficiency prevents obesity-induced increases in plasma AngII concentrations and SBP. Adipose tissue AngII concentrations were markedly reduced in adipocyte AGT deficient obese mice. Our results suggest that adipose-derived AngII contributes to the systemic RAS and blood pressure control in obese mice. These results support use of inhibitors of the RAS in the treatment of obesity-hypertension. Moreover, interventions that could mitigate activation of the adipose RAS may provide novel areas of treatment for obesity-related hypertension.
Supplementary Material
Figure S1. A. Genotyping of tails from LF and HF-fed Agtfl/fl and AgtaP2 mice. Primers were used to detect Cre+ (5′-ACCTGAAGATGTTCGCGATT and 5′-CGGCATCAACGTTTTCTTTT), IL-2 gene (5′-CTAGGCCACAGAATTGAAAGATCT and 5′-GTAGGTGGAAATTCTAGCATCATCC). Cre+ mice were identified by a product of 182bp. B. AGT mRNA abundance in liver; C. AGT mRNA abundance in kidney; D. AGT mRNA abundance in white adipose tissue. Data are mean ± SEM from n = 6-10 mice/group. *, P<0.05 compared to Agtfl/fl within diet group.
Figure S2. Glucose tolerance tests in LF and HF-fed Agtfl/fl and AgtaP2 mice. A, Blood glucose concentrations at several time points after administration of glucose (2 mg/kg body weight). B, Area under the curve (AUC) for data in A, above. Data are mean ± SEM from n = 7-10 mice/group. *, P<0.05 compared to LF within genotype.
Figure S3. SBP (12 hour averages for each mouse) of LF-fed Agtfl/fl and AgtaP2 mice during the night and light cycle. Carotid artery catheters and radiotelemeters were implanted at week 15 of LF, mice were allowed one week to recover, and SBP was recorded for 3 - 5 days. Data are mean ± SEM from n = 6-7 mice/group (LF) over the 3 - 5 days of recording. *, P<0.05 compared to night cycle within genotype.
Figure S4. Total renin (renin+prorenin) in plasma of LF and HF-fed Agtfl/fl and AgtaP2 mice. Data are mean ± SEM from n = 5-6 mice/group. *, P<0.05 compared to LF within genotype.
Figure S5. Renin-like activity, as evidenced by generation of angiotensin I, in explants of adipose tissue from LF and HF-fed Agtfl/fl and AgtaP2 mice. Data are mean ± SEM from n = 3-4 mice/group. *, P<0.05 compared to LF within genotype. **, P<0.05 compared to Agtfl/fl within diet group.
Figure S6. Cathepsin D (A) and tonin (B) mRNA abundance in adipose tissue from LF and HF-fed Agtfl/fl and AgtaP2 mice. Data are mean ± SEM from n = 5-7 mice/group. P = 0.06 genotype by diet interaction.
Novelty and Significance.
- What is New?
- Demonstration for the first time that adipocyte AGT deficiency reduces plasma AngII concentrations and prevents the development of obesity-associated hypertension.
- Demonstration of dissociation of systemic concentrations of AGT from AngII in the setting of obesity.
- What is Relevant?
- Since obesity accounts for almost the entire increased prevalence of hypertension in men in the US, this is highly relevant to target novel therapies for hypertension.
- This study demonstrates that adipose tissue can be a major source of systemic AngII in obesity-induced hypertension.
- Summary
- Adipocyte AGT deficiency prevents obesity-induced increases in SBP.
- Adipocyte AGT deficiency markedly reduces plasma and adipose tissue AngII concentrations in obese mice.
- The expanded fat mass from obesity becomes a significant source of systemic AngII in the development of hypertension.
Acknowledgments
We thank Eboni Lewis for skilled surgical implantation of radiotelemeters in mice, Michael Karounos for assistance with the development and maintenance of the breeding colony, and Victoria English for quantification of the RAS.
Sources of Funding These studies were supported by National Heart, Lung and Blood Institute (HL73085, LAC; HL62846, AD; T32HL091812, FY) and by the National Center for Research Resources (P20RR021954; LAC).
Footnotes
Conflict of Interest/Disclosure(s) Statement None
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cutler JA, Sorlie PD, Wolz M, Thom T, Fields LE, Roccella EJ. Trends in hypertension prevalence, awareness, treatment, and control rates in United States adults between 1988-1994 and 1999-2004. Hypertension. 2008;52:818–827. doi: 10.1161/HYPERTENSIONAHA.108.113357. [DOI] [PubMed] [Google Scholar]
- 2.Hamm LL, Chen J. Fat chance for hypertension and chronic kidney disease. Hypertension. 2011;58:756–757. doi: 10.1161/HYPERTENSIONAHA.111.180414. [DOI] [PubMed] [Google Scholar]
- 3.Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, Smith G, Stec DE. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285:17271–17276. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reisin E, Jack AV. Obesity and hypertension: mechanisms, cardio-renal consequences, and therapeutic approaches. Med Clin North Am. 2009;93:733–751. doi: 10.1016/j.mcna.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Boustany CM, Bharadwaj K, Daugherty A, Brown DR, Randall DC, Cassis LA. Activation of the systemic and adipose renin-angiotensin system in rats with diet-induced obesity and hypertension. Am J Physiol Regul Integr Comp Physiol. 2004;287:R943–R949. doi: 10.1152/ajpregu.00265.2004. [DOI] [PubMed] [Google Scholar]
- 6.Gupte MN, Boustany-Kari CM, Bharadwaj K, Police S, Thatcher S, Gong MC, English VL, Cassis LA. Ace2 is expressed in mouse adipocytes and regulated by a high fat diet. Am J Physiol Regul Integr Comp Physiol. 2008;295:R781–788. doi: 10.1152/ajpregu.00183.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engeli S, Schling P, Gorzelniak K, Boschmann M, Janke J, Ailhaud G, Teboul M, Massiera F, Sharma AM. The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? Int J Biochem Cell Biol. 2003;35:807–825. doi: 10.1016/s1357-2725(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 8.Bloem LJ, Manatunga AK, Tewksbury DA, Pratt JH. The serum angiotensinogen concentration and variants of the angiotensinogen gene in white and black children. J Clin Invest. 1995;95:948–953. doi: 10.1172/JCI117803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Umemura S, Nyui N, Tamura K, Hibi K, Yamaguchi S, Nakamaru M, Ishigami T, Yabana M, Kihara M, Inoue S, Ishii M. Plasma angiotensinogen concentrations in obese patients. Am J Hypertens. 1997;10:629–633. doi: 10.1016/s0895-7061(97)00053-8. [DOI] [PubMed] [Google Scholar]
- 10.Licata G, Scaglione R, Ganguzza A, Corrao S, Donatelli M, Parrinello G, Dichiara MA, Merlino G, Cecala MG. Central obesity and hypertension. Relationship between fasting serum insulin, plasma renin activity, and diastolic blood pressure in young obese subjects. Am J Hypertens. 1994;7:314–320. doi: 10.1093/ajh/7.4.314. [DOI] [PubMed] [Google Scholar]
- 11.Engeli S, Bohnke J, Gorzelniak K, Janke J, Schling P, Bader M, Luft FC, Sharma AM. Weight loss and the renin-angiotensin-aldosterone system. Hypertension. 2005;45:356–362. doi: 10.1161/01.HYP.0000154361.47683.d3. [DOI] [PubMed] [Google Scholar]
- 12.Boustany CM, Brown DR, Randall DC, Cassis LA. AT1-receptor antagonism reverses the blood pressure elevation associated with diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2005;289:R181–186. doi: 10.1152/ajpregu.00507.2004. [DOI] [PubMed] [Google Scholar]
- 13.Dobrian AD, Davies MJ, Prewitt RL, Lauterio TJ. Development of hypertension in a rat model of diet-induced obesity. Hypertension. 2000;35:1009–1015. doi: 10.1161/01.hyp.35.4.1009. [DOI] [PubMed] [Google Scholar]
- 14.Iacobellis G, Gao YJ, Sharma AM. Do cardiac and perivascular adipose tissue play a role in atherosclerosis? Curr Diab Rep. 2008;8:20–24. doi: 10.1007/s11892-008-0005-2. [DOI] [PubMed] [Google Scholar]
- 15.Cassis LA, Lynch KR, Peach MJ. Localization of angiotensinogen messenger RNA in rat aorta. Circ Res. 1988;62:1259–1262. doi: 10.1161/01.res.62.6.1259. [DOI] [PubMed] [Google Scholar]
- 16.Cassis LA, Saye J, Peach MJ. Location and regulation of rat angiotensinogen messenger RNA. Hypertension. 1988;11:591–596. doi: 10.1161/01.hyp.11.6.591. [DOI] [PubMed] [Google Scholar]
- 17.Saye JA, Cassis LA, Sturgill TW, Lynch KR, Peach MJ. Angiotensinogen gene expression in 3T3-L1 cells. Am J Physiol. 1989;256:C448–C451. doi: 10.1152/ajpcell.1989.256.2.C448. [DOI] [PubMed] [Google Scholar]
- 18.Matsushita K, Wu Y, Okamoto Y, Pratt RE, Dzau VJ. Local renin angiotensin expression regulates human mesenchymal stem cell differentiation to adipocytes. Hypertension. 2006;48:1095–1102. doi: 10.1161/01.HYP.0000248211.82232.a7. [DOI] [PubMed] [Google Scholar]
- 19.Saye JA, Ragsdale NV, Carey RM, Peach MJ. Localization of angiotensin peptide-forming enzymes of 3T3-F442A adipocytes. Am J Physiol. 1993;264:C1570–C1576. doi: 10.1152/ajpcell.1993.264.6.C1570. [DOI] [PubMed] [Google Scholar]
- 20.Shenoy U, Cassis L. Characterization of renin activity in brown adipose tissue. Am J Physiol. 1997;272:C989–999. doi: 10.1152/ajpcell.1997.272.3.C989. [DOI] [PubMed] [Google Scholar]
- 21.Cassis LA, Dwoskin LP. Presynaptic modulation of neurotransmitter release by endogenous angiotensin II in brown adipose tissue. J Neural Transm Suppl. 1991;34:129–137. doi: 10.1007/978-3-7091-9175-0_17. [DOI] [PubMed] [Google Scholar]
- 22.Schling P, Schafer T. Human adipose tissue cells keep tight control on the angiotensin II levels in their vicinity. J Biol Chem. 2002;277:48066–48075. doi: 10.1074/jbc.M204058200. [DOI] [PubMed] [Google Scholar]
- 23.Massiera F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, Quignard-Boulange A, Negrel R, Ailhaud G, Seydoux J, Meneton P, Teboul M. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J. 2001;15:2727–2729. doi: 10.1096/fj.01-0457fje. [DOI] [PubMed] [Google Scholar]
- 24.Yiannikouris F, Karounos M, Charnigo R, English VL, Rateri DL, Daugherty A, Cassis LA. Adipocyte-specific deficiency of angiotensinogen decreases plasma angiotensinogen concentration and systolic blood pressure in mice. Am J Physiol Regul Integr Comp Physiol. 2012;302:R244–251. doi: 10.1152/ajpregu.00323.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rahmouni K, Mark AL, Haynes WG, Sigmund CD. Adipose depot-specific modulation of angiotensinogen gene expression in diet-induced obesity. Am J Physiol Endocrinol Metab. 2004;286:E891–895. doi: 10.1152/ajpendo.00551.2003. [DOI] [PubMed] [Google Scholar]
- 26.Frederich RC, Jr, Kahn BB, Peach MJ, Flier JS. Tissue-specific nutritional regulation of angiotensinogen in adipose tissue. Hypertension. 1992;19:339–344. doi: 10.1161/01.hyp.19.4.339. [DOI] [PubMed] [Google Scholar]
- 27.Giacchetti G, Faloia E, Sardu C, Camilloni MA, Mariniello B, Gatti C, Garrapa GG, Guerrieri M, Mantero F. Gene expression of angiotensinogen in adipose tissue of obese patients. Int J Obes Relat Metab Disord. 2000;24(Suppl 2):S142–143. doi: 10.1038/sj.ijo.0801305. [DOI] [PubMed] [Google Scholar]
- 28.Van Harmelen V, Ariapart P, Hoffstedt J, Lundkvist I, Bringman S, Arner P. Increased adipose angiotensinogen gene expression in human obesity. Obes Res. 2000;8:337–341. doi: 10.1038/oby.2000.40. [DOI] [PubMed] [Google Scholar]
- 29.Okada S, Kozuka C, Masuzaki H, Yasue S, Ishii-Yonemoto T, Tanaka T, Yamamoto Y, Noguchi M, Kusakabe T, Tomita T, Fujikura J, Ebihara K, Hosoda K, Sakaue H, Kobori H, Ham M, Lee YS, Kim JB, Saito Y, Nakao K. Adipose tissue-specific dysregulation of angiotensinogen by oxidative stress in obesity. Metabolism. 2010;59:1241–1251. doi: 10.1016/j.metabol.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yasue S, Masuzaki H, Okada S, Ishii T, Kozuka C, Tanaka T, Fujikura J, Ebihara K, Hosoda K, Katsurada A, Ohashi N, Urushihara M, Kobori H, Morimoto N, Kawazoe T, Naitoh M, Okada M, Sakaue H, Suzuki S, Nakao K. Adipose tissue-specific regulation of angiotensinogen in obese humans and mice: impact of nutritional status and adipocyte hypertrophy. Am J Hypertens. 2010;23:425–431. doi: 10.1038/ajh.2009.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanimoto K, Sugiyama F, Goto Y, Ishida J, Takimoto E, Yagami K, Fukamizu A, Murakami K. Angiotensinogen-deficient mice with hypotension. J Biol Chem. 1994;269:31334–31337. [PubMed] [Google Scholar]
- 32.Bohlender J, Menard J, Ganten D, Luft FC. Angiotensinogen concentrations and renin clearance : implications for blood pressure regulation. Hypertension. 2000;35:780–786. doi: 10.1161/01.hyp.35.3.780. [DOI] [PubMed] [Google Scholar]
- 33.Menard J, el Amrani AI, Savoie F, Bouhnik J. Angiotensinogen: an attractive and underrated participant in hypertension and inflammation. Hypertension. 1991;18:705–707. doi: 10.1161/01.hyp.18.5.705. [DOI] [PubMed] [Google Scholar]
- 34.Arnal JF, Cudek P, Plouin PF, Guyenne TT, Michel JB, Corvol P. Low angiotensinogen levels are related to the severity and liver dysfunction of congestive heart failure: implications for renin measurements. Am J Med. 1991;90:17–22. doi: 10.1016/0002-9343(91)90501-n. [DOI] [PubMed] [Google Scholar]
- 35.Jeunemaitre X, Inoue I, Williams C, Charru A, Tichet J, Powers M, Sharma AM, Gimenez-Roqueplo AP, Hata A, Corvol P, Lalouel JM. Haplotypes of angiotensinogen in essential hypertension. Am J Hum Genet. 1997;60:1448–1460. doi: 10.1086/515452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hansen PB, Yang T, Huang Y, Mizel D, Briggs J, Schnermann J. Plasma renin in mice with one or two renin genes. Acta Physiol Scand. 2004;181:431–437. doi: 10.1111/j.1365-201X.2004.01315.x. [DOI] [PubMed] [Google Scholar]
- 37.Lamounier-Zepter V, Ehrhart-Bornstein M, Bornstein SR. Insulin resistance in hypertension and cardiovascular disease. Best Pract Res Clin Endocrinol Metab. 2006;20:355–367. doi: 10.1016/j.beem.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Ruano M, Silvestre V, Castro R, Garcia-Lescun MC, Rodriguez A, Marco A, Garcia-Blanch G. Morbid obesity, hypertensive disease and the renin-angiotensin-aldosterone axis. Obes Surg. 2005;15:670–676. doi: 10.1381/0960892053923734. [DOI] [PubMed] [Google Scholar]
- 39.Thatcher S, Yiannikouris F, Gupte M, Cassis L. The adipose renin-angiotensin system: role in cardiovascular disease. Mol Cell Endocrinol. 2009;302:111–117. doi: 10.1016/j.mce.2009.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cassis LA, Police SB, Yiannikouris F, Thatcher SE. Local adipose tissue renin-angiotensin system. Curr Hypertens Rep. 2008;10:93–98. doi: 10.1007/s11906-008-0019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Putnam K, Shoemaker R, Yiannikouris F, Cassis LA. The renin angiotensin system: A target of and contributor to dyslipidemias, altered glucose homeostasis and hypertension of the metabolic syndrome. Am J Physiol Heart Circ Physiol. 2012;302:H1219–30. doi: 10.1152/ajpheart.00796.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keim KL, Sigg EB. Activation of central sympathetic neurons by angiotensin II. Life Sci I. 1971;10:565–574. doi: 10.1016/0024-3205(71)90042-7. [DOI] [PubMed] [Google Scholar]
- 43.Falcon JC, 2nd, Phillips MI, Hoffman WE, Brody MJ. Effects of intraventricular angiotensin II mediated by the sympathetic nervous system. Am J Physiol. 1978;235:H392–399. doi: 10.1152/ajpheart.1978.235.4.H392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A. Genotyping of tails from LF and HF-fed Agtfl/fl and AgtaP2 mice. Primers were used to detect Cre+ (5′-ACCTGAAGATGTTCGCGATT and 5′-CGGCATCAACGTTTTCTTTT), IL-2 gene (5′-CTAGGCCACAGAATTGAAAGATCT and 5′-GTAGGTGGAAATTCTAGCATCATCC). Cre+ mice were identified by a product of 182bp. B. AGT mRNA abundance in liver; C. AGT mRNA abundance in kidney; D. AGT mRNA abundance in white adipose tissue. Data are mean ± SEM from n = 6-10 mice/group. *, P<0.05 compared to Agtfl/fl within diet group.
Figure S2. Glucose tolerance tests in LF and HF-fed Agtfl/fl and AgtaP2 mice. A, Blood glucose concentrations at several time points after administration of glucose (2 mg/kg body weight). B, Area under the curve (AUC) for data in A, above. Data are mean ± SEM from n = 7-10 mice/group. *, P<0.05 compared to LF within genotype.
Figure S3. SBP (12 hour averages for each mouse) of LF-fed Agtfl/fl and AgtaP2 mice during the night and light cycle. Carotid artery catheters and radiotelemeters were implanted at week 15 of LF, mice were allowed one week to recover, and SBP was recorded for 3 - 5 days. Data are mean ± SEM from n = 6-7 mice/group (LF) over the 3 - 5 days of recording. *, P<0.05 compared to night cycle within genotype.
Figure S4. Total renin (renin+prorenin) in plasma of LF and HF-fed Agtfl/fl and AgtaP2 mice. Data are mean ± SEM from n = 5-6 mice/group. *, P<0.05 compared to LF within genotype.
Figure S5. Renin-like activity, as evidenced by generation of angiotensin I, in explants of adipose tissue from LF and HF-fed Agtfl/fl and AgtaP2 mice. Data are mean ± SEM from n = 3-4 mice/group. *, P<0.05 compared to LF within genotype. **, P<0.05 compared to Agtfl/fl within diet group.
Figure S6. Cathepsin D (A) and tonin (B) mRNA abundance in adipose tissue from LF and HF-fed Agtfl/fl and AgtaP2 mice. Data are mean ± SEM from n = 5-7 mice/group. P = 0.06 genotype by diet interaction.