

Abstract
Background
Pathogenesis of inflammatory bowel diseases (IBD), ulcerative colitis (UC) and Crohn’s disease (CD), involves interaction between environmental factors and inappropriate immune responses in the intestine of genetically predisposed individuals. Bile acids and their nuclear receptor, FXR, regulate inflammatory responses and barrier function in the intestinal tract.
Methods
We studied the association of five variants (rs3863377, rs7138843, rs56163822, rs35724, rs10860603) of the NR1H4 gene encoding FXR with IBD. 1138 individuals (591 non-IBD, 203 UC, 344 CD) were genotyped for five NR1H4 genetic variants with TaqMan SNP Genotyping Assays.
Results
We observed that the NR1H4 SNP rs3863377 is significantly less frequent in IBD cases than in non-IBD controls (allele frequencies: P = 0.004; wild-type vs. SNP carrier genotype frequencies: P = 0.008), whereas the variant rs56163822 is less prevalent in non-IBD controls (allele frequencies: P = 0.027; wild-type vs. SNP carrier genotype frequencies: P = 0.035). The global haplotype distribution between IBD and control patients was significantly different (P = 0.003). This also held true for the comparison between non-IBD and UC groups (P = 0.004), but not for the comparison between non-IBD and CD groups (P = 0.079).
Conclusions
We show that genetic variation in FXR is associated with IBD, further emphasizing the link between bile acid signaling and intestinal inflammation.
Keywords: Bile acid homeostasis, Crohn’s disease, Farnesoid X receptor, Inflammatory bowel disease, Single nucleotide polymorphisms, Ulcerative colitis
Background
Inflammatory bowel disease (IBD) is a chronic condition characterized by recurrent inflammation of intestinal mucosa, and results from aberrant regulation of the mucosal immune system in genetically susceptible individuals. The etiology of IBD involves a complex interaction of genetic, environmental, and immunomodulatory factors. Two major forms of chronic mucosal inflammation have been defined. In Crohn’s disease (CD), the whole gastrointestinal tract may be affected, although the most frequent site of inflammation is the terminal ileum, whereas in ulcerative colitis (UC) the mucosal inflammation typically affects the colon [1]. For CD pathogenesis, a strong genetic component has been suggested by the concordance of 63.6% in monozygotic twins, but only of 3.6% in dizygotic twins. The concordance of monozygotic twins is lower (6%) in UC, indicating that genetic susceptibility may play a somewhat smaller role in this disease [2].
Nuclear receptors are a large family of transcription factors that are involved in the regulation of numerous processes, including reproduction, development, and a wide range of metabolic pathways [3]. The ligand-dependent activation function at the carboxy-terminus of most nuclear receptors allows them to sense metabolic changes within cells, and orchestrate rapid transcriptional changes in response [4-6].
The farnesoid X receptor (FXR; gene symbol NR1H4) is a nuclear receptor that functions as the main sensor of intracellular bile acid levels [7-9]. The human NR1H4 gene is located on chromosome 12 and is composed of 11 exons and 10 introns [10]. The translation initiation codon of the NR1H4 gene lies at the 3′ end of exon 3, whereas exons 1 and 2, together with the 5′ region of exon 3, contain the 5′ untranslated region (5’-UTR). Multiple FXR isoforms can be generated via alternative promoter usage and alternative splicing, and these isoforms may have differential transactivation abilities on specific target promoters [11]. FXR typically acts by binding to FXR response elements within the target promoters as heterodimers with another member of the nuclear receptor family, retinoid X receptor-α (RXRα) [12]. In response to elevated levels of intracellular bile acids, activated FXR is well known to induce protective gene expression circuits against bile acid toxicity in the liver and intestine [13]. Expression of bile acid efflux systems in ileocytes (organic solute transporter α/β; OSTα/β) [14,15] and hepatocytes (bile salt export pump; BSEP) [16-18] is upregulated by bile acid-activated FXR, while the expression of the respective bile acid uptake systems apical sodium-dependent bile acid transporter (ASBT) [19] and Na+-taurocholate cotransporting polypeptide (NTCP) [20,21] is suppressed by it. FXR also represses transcription of three genes coding for bile acid synthesizing enzymes, namely cholesterol-7α-hydroxylase (CYP7A1), sterol-12α-hydroxylase (CYB8B1), and sterol-27-hydroxylase (CYP27A1) [22,23]. Thus, elevated levels of bile acids can suppress their own de novo production through a negative feedback loop involving FXR. In addition, FXR regulates several genes that can protect against intestinal inflammation and bacterial overgrowth [24-26]. Fxr-deficient mice have increased ileal concentrations of gut bacteria and exhibit defects in the integrity of the intestinal epithelial barrier. In agreement with this, the products of a number of genes that are regulated by Fxr in the ileum, including angiogenin (Ang1), inducible nitric oxide synthase (iNos), and interleukin-18 (Il-18), are known to have antimicrobial actions [26]. Furthermore, it has been reported that reduced expression of Fxr/FXR is associated with colon inflammation in rodent models of colitis and in CD patients [25]. Recently, FXR activation was shown to decrease NF-κB-mediated immune responses and intestinal permeability in mouse models of colitis [27]. It was subsequently shown that intestinal inflammation reduces FXR activation as well as the expression of FXR target genes such as intestinal bile acid-binding protein (IBABP) and fibroblast growth factor 15/19 (FGF15/19) [28]. In agreement with this, it has been proposed that FXR may contribute to the resistance of both human and mouse gastric epithelial cells against inflammation-induced injury [29]. The fact that FXR thus appears to play a role in the protection of the integrity of the intestinal epithelial barrier and its inverse correlation with the level of intestinal inflammation suggest a potential connection between FXR and the molecular pathogenesis of IBD. FXR variants have been previously studied in association with several liver diseases, such as gallstone disease [30], cholangiocarcinoma [31], intrahepatic cholestasis of pregnancy [32], and idiopathic infantile cholestasis [33]. Here, we have investigated five NR1H4 single nucleotide polymorphisms - two common SNPs and three rare variants - which have been previously studied in the context of human disease, in a well-sized IBD vs. non-IBD cohort, and report that two of these genetic variants are associated with IBD.
Results
Study population
The study population was European and consisted of more women (806) than men (332). Detailed demographic data is given in Table1.
Table 1.
Demographic data of the population included in the analysis
| Characteristics | non-IBD | IBD | CD | UC |
|---|---|---|---|---|
|
Population |
591 (51.9%) |
547 (48.1%) |
344 (30.2%) |
203 (17.8%) |
|
Mean age (± SD) |
55.7 (± 12.6) |
42.6 (± 15.1) |
40.9 (± 14.9) |
45.4 (± 14.9) |
|
Median age |
59 |
41 |
39 |
44 |
|
Minimum age |
20 |
16 |
16 |
18 |
| Maximum age | 81 | 82 | 79 | 82 |
NR1H4 sequence variability
All five NR1H4 variants selected for the study are single nucleotide substitutions, previously identified within the NR1H4 gene (http://www.ncbi.nlm.nih.gov/snp/). Three of these SNPs can be considered as rare variants: rs3863377, rs56163822, rs7138843, with reported minor allele frequencies (MAF) of 4%, 2.2%, and 0.9%, respectively. The other two variants, rs10860603 and rs35724, are common SNPs, with minor allele frequencies of 20.5% and 40.8% (http://www.ncbi.nlm.nih.gov/snp/) in European individuals. The genotype frequencies in all groups were in Hardy-Weinberg equilibrium (represented by the χ2 values in Table2). The obtained allele and genotype frequencies are given in Tables 3 and 2, respectively.
Table 2.
Genotype frequencies of the NR1H4 variants in the study population and the genotype association analysis
|
SNP rs3863377(G→ A) | ||||||||
| |
WT [GG] |
Het [GA] |
Hom [AA] (%) |
P |
P’ |
OR |
CI |
χ2 |
| non-IBD |
506 (94.4) |
28 (5.2) |
2 (0.4) |
|
|
|
|
0.14 |
| IBD |
438 (97.8) |
10 (2.2) |
0 |
0.009** † |
0.022* |
0.39 |
0.19-0.80 |
0.01 |
| CD |
271 (97.8) |
6 (2.2) |
0 |
0.030* † |
0.031* |
0.37 |
0.15-0.191 |
0.01 |
| UC |
167 (97.7) |
4 (2.3) |
0 |
0.100 † |
0.066 |
0.40 |
0.14-1.16 |
0.01 |
|
SNP rs56163822(−1G → T) | ||||||||
| |
WT [GG] |
Het [GT] |
Horn [TT] (%) |
P |
P’ |
OR |
CI |
χ2 |
| non-IBD |
560 (96.6) |
20 (3.4) |
0 |
|
|
|
|
0.04 |
| IBD |
505 (93.9) |
33 (6.1) |
1 (0.09) |
0.035* |
0.451 |
1.83 |
1.04-3.23 |
0.00 |
| CD |
319 (94.4) |
19 (5.6) |
1 (0.09) |
0.115 |
0.846 |
1.67 |
0.88-3. 17 |
0.00 |
| UC |
186 (93.0) |
14 (7.0) |
0 |
0.034* |
0.154 |
2.11 |
1.04-4.26 |
0.13 |
|
SNP rs7138843(A → T) | ||||||||
| |
WT [AA] |
Het [AT] |
Horn [TT] (%) |
P |
P’ |
OR |
CI |
χ2 |
| non-IBD |
495 (96.7) |
17 (3.3) |
0 |
|
|
|
|
0.03 |
| IBD |
417 (95.2) |
21 (4.8) |
0 |
0.248 |
0.772 |
147 |
0.76-2.82 |
0.06 |
| CD |
261 (96.0) |
11 (4.0) |
0 |
0.369 † |
0.612 |
1.23 |
0.57-2.66 |
0.04 |
| UC |
156 (94.0) |
10 (6.0) |
0 |
0.097 † |
0.981 |
1.87 |
0.84-4.16 |
0.10 |
|
SNP rs10860603(G → A) | ||||||||
| |
WT [GG] |
Het [GA] |
Horn [AA] (%) |
P |
P’ |
OR |
CI |
χ2 |
| non-IBD |
424 (72.8) |
143 (24.6) |
15 (2.6) |
|
|
|
|
049 |
| IBD |
407 (75.8) |
121 (22.5) |
9 (1.7) |
0.261 |
0.384 |
0.86 |
0.66-1.12 |
0.00 |
| CD |
254 (75.4) |
76 (22.5) |
7 (2.1) |
0.403 |
0.611 |
0.88 |
0.65-1.19 |
0.22 |
| tiC |
153 (76.5) |
45 (22.5) |
2 (1.0) |
0.312 |
0.521 |
0.82 |
0.57-1.19 |
0.43 |
|
SNP rs35724(G→ C) | ||||||||
| |
WT [GG] |
Het [GC] |
Horn [CC] (%) |
P |
P’ |
OR |
CI |
χ2 |
| non-IBD |
217 (37.3) |
264 (454) |
101 (17.3) |
|
|
|
|
1.78 |
| IBD |
177 (32.9) |
251 (46.7 |
110 (20.4) |
0.125 |
0.209 |
1.213 |
0.95-1.55 |
1.47 |
| CD |
111 (32.8) |
155 (45.9) |
72 (21.3) |
0.175 |
0.279 |
1.216 |
0.92-1.61 |
1.68 |
| UC | 66 (33.0) | 96 (48.0) | 38 (19.0) | 0.277 | 0.233 | 1.207 | 0.86-1.69 | 0.09 |
Table 3.
Allele frequencies of the NR1H4 variants in the study population and the allele association analysis
|
SNP rs3863377(G → A) | |||||
|
Case |
MAF (%) |
P |
OR |
CI |
Reported MAFs1 |
| non-IBD |
32 (2.9) |
|
|
|
4.0% |
| IBD |
10 (1.1) |
0.004** † |
0.37 |
0.18-0-75 |
|
| CD |
6 (1.1) |
0.015* † |
0.36 |
0.15-0.86 |
|
| UC |
4 (1.2) |
0.075 † |
0.38 |
0.14-1.10 |
|
|
SNP rs56163822, (G →T) | |||||
|
Case |
MAF (%) |
P |
OR |
CI |
Reported MAFs1 |
| non-IBD |
20 (1.7) |
|
|
|
2.2% |
| IBD |
34 (3.2) |
0.027* |
1.86 |
1.06-3.25 |
|
| CD |
20 (2.9) |
0.081 |
1.74 |
0.93-3.25 |
|
| UC |
14 (3.5) |
0.036* |
2.07 |
1.03-4.13 |
|
|
SNP rs7138843, (A → T) | |||||
|
Case |
MAF (%) |
P |
|
|
Reported MAFs1 |
| non-IBD |
17 (1.7) |
|
|
|
0.9% |
| IBD |
21 (2.4) |
ns |
|
|
|
| CD |
11 (2.0) |
ns |
|
|
|
| UC |
10 (3.0) |
ns |
† |
|
|
|
SNP rs10860603, (G → A) | |||||
|
Case |
MAF (%) |
|
P |
|
Reported MAFs1 |
| non-IBD |
173 (14.9) |
|
|
|
20.5% |
| IBD |
139 (12.9) |
ns |
|
|
|
| CD |
90 (13.4) |
ns |
|
|
|
| UC |
49 (12.3) |
ns |
|
|
|
|
SNP rs35724, (G → C) | |||||
|
Case |
MAF (%) |
P |
|
|
Reported MAFs1 |
| non-IBD |
466 (40.0) |
|
|
|
40.8% |
| IBD |
471 (43.8) |
ns |
|
|
|
| CD |
299 (44.4) |
ns |
|
|
|
| UC | 172 (43.0) | ns | |||
Genetic variation in the NR1H4 gene and IBD
The NR1H4 SNP variants rs3863377 and rs56163822 were found to be significantly associated with IBD when considering an uncorrected significance level of p<0.05. Upon statistical analysis of the allele (Table3) and genotype (Table2) frequencies, we observed that the NR1H4 variant rs3863377 is significantly less frequent in IBD cases than in non-IBD controls (allele frequencies: P = 0.004; wild-type vs. SNP carrier genotype frequencies: P = 0.008) even when considering an Bonferroni-corrected significance level of P<0.01. Upon subgrouping the IBD patients, the significance of the inverse association of the rs3863377 SNP remained for the CD patients when considering an uncorrected significance level of P<0.05 (allele frequencies: P = 0.015; wild-type vs. SNP carrier genotype frequencies: P = 0.024), but not for the UC group (allele frequencies: P = 0.075; wild-type vs. SNP carrier genotype frequencies: P = 0.083). Conversely, the variant rs56163822 is less prevalent in non-IBD subjects than in IBD patients (allele frequencies: P = 0.027; wild-type vs. SNP carrier genotype frequencies: P = 0.035); an observation, which is, however, not significant, when considering a corrected significance level of P<0.01. Upon subgrouping the patient cohort according to IBD subtypes, the uncorrected association remained only significant for the UC group (allele frequencies: P = 0.036; wild-type vs. SNP carrier genotype frequencies: P = 0.034). Upon adjustment for age and gender, the uncorrected significance (P<0.05) of genotype frequency association with IBD remained for rs3863377, but was reduced to P>0.05 for rs56163822. There were no significant differences in the allele frequency distribution between the subject groups for NR1H4 variants rs7138843, rs10860603, and rs35724.
NR1H4 haplotype analysis
All individuals, for whom genotype determination could be performed for all five NR1H4 SNPs under study were included in the haplotype prediction analyses. Four hundred and eighty-six non-IBD cases, along with 243 CD patients and 150 UC patients were thus haplotyped. Twenty haplotypes and up to thirty-nine diplotypes were predicted by the software FAMHAP to exist in the studied cohort. NR1H4 haplotypes were significantly differentially distributed in the IBD and control groups (Table4, P = 0.003) upon global haplotype distribution analysis. This observation held partially true upon stratification according to disease subtype. Here, the haplotype frequencies differ significantly between the UC patients and the non-IBD control group (Table5, P = 0.004), but not between the CD patients and the control subjects (Table6, P = 0.079). We particularly note that the haplotype 14, GTTGC, is predicted to occur significantly (P = 0.005) more frequently in the UC group in comparison with the non-IBD cohort. This haplotype harbours the more frequent allele G at the first SNP position rs3863377, which was shown to be significantly associated with IBD even after Bonferroni correction. It is thus a possible risk haplotype for the development of IBD, although we note that the overall frequency for this haplotype is rather low. Upon best reconstruction analysis the significance of the association of the haplotype 14, GTTGC, with the UC group was, however, lost (P=0.012) (Table5, italicized section). No significant associations were observed for the predicted diplotype patterns and IBD (data not shown). As shown in the LD plot (Figure1), there was no significant linkage disequilibrium between any of the five NR1H4 SNPs studied.
Table 4.
Haplotype analysis of IBD patients and non-IBD controls
| Order | Haplotype a) b) c) d) e) | Cases (%) | Controls (%) | OR (C.I.) | P | Order | Haplotype a) b) c) d) e) | Cases (%) | Controls (%) | OR (C.I.) | P |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
G G A G G |
405.9 (52.2) |
510.1 (52.6) |
0.98 (0.81-1.19) |
|
1 |
G G A G G |
417 (53.6) |
530 (54.6) |
0.96 (0.79-1.16) |
a)ns |
| 2 |
G G A G C |
243.9 (31.3) |
261 (26.9) |
1.24 (1.01-1.53) |
0.064 |
2 |
G G A G C |
235 (30.2) |
241 (24.7) |
1.31 (1.06-1.62) |
a)0.012 |
| 3 |
G G A A C |
55.9 (7.2) |
91.3 (9.4) |
0.75 (0.53-1.05) |
0.091 |
3 |
G G A A C |
63 (8.1) |
108 (11.1) |
0.7 (0.51-0.97) |
a)0.03 |
| 4 |
G G A A G |
27.4 (3.5) |
47.5 (4.9) |
0.71 (0.44-1.14) |
|
4 |
G G A A G |
18 (2.3) |
31 (3.2) |
0.72 (0.40-1.29) |
a)ns |
| 5 |
A G A G G |
2.7 (0.4) |
13.4 (1.4) |
0.25 (0.07-0.93) |
0.017 |
5 |
A G A G G |
2 (0.3) |
13 (1.3) |
0.19 (0.04-0.84) |
0.017 |
| 6 |
A G A G C |
6.6 (0.8) |
12 (1.2) |
0.68 (0.26-1.76) |
|
6 |
A G A G C |
8 (1.0) |
14 (1.4) |
0.71 (0.30-1.70) |
ns |
| 7 |
G G T G C |
5 (0.6) |
9.1 (0.9) |
0.68 (0.23-2.04) |
|
7 |
G G T G C |
4 (0.5) |
9 (0.9) |
0.55 (0.17-1.80) |
ns |
| 8 |
G T A G G |
3.8 (0.5) |
7.5 (0.8) |
0.63 (0.18-2.17) |
NA |
8 |
G T A G G |
2 (0.3) |
7 (0.7) |
0.35 (0.07-1.71) |
ns |
| 9 |
G T A G C |
10.4 (1.3) |
5.7 (0.6) |
2.27 (0.82-6.32) |
0.090 |
9 |
G T A G C |
12 (1.5) |
6 (0.6) |
2.52 (0.94-6.74) |
ns |
| 10 |
G G T G G |
2 (0.3) |
3.1 (0.3) |
0.79 (0.13-4.70) |
NA |
10 |
G G T G G |
1 (0.1) |
2 (0.2) |
0.62 (0.06-6.89) |
ns |
| 11 |
G T A A G |
0.8 (0.1) |
1.8 (0.2) |
NA |
NA |
11 |
G T A A G |
1 (0.1) |
2 (0.2) |
0.63 (0.06-6.89) |
ns |
| 12 |
A G A A G |
0.2 (0) |
2.0 (0.2) |
0.13 (0–11.93) |
NA |
12 |
A G A A G |
0 |
1 (0.1) |
NA |
ns |
| 13 |
G G T A C |
2.4 (0.3) |
2.3 (0.2) |
1.29 (0.21-7.99) |
NA |
13 |
G G T A C |
4 (0.5) |
4 (0.4) |
1.25 (0.31-5.00) |
ns |
| 14 |
G T T G C |
7.4 (1) |
1.0 (0.1) |
NA |
NA |
14 |
G T T G C |
7 (0.9) |
1 (0.1) |
8.80 (1.08-71.70) |
0.026 |
| 15 |
G G T A G |
1.1 (0.1) |
0.5 (0.1) |
NA |
NA |
15 |
G G T A G |
1 (0.1) |
0 |
NA |
ns |
| 16 |
G T T A G |
1.2 (0.2) |
1.0 (0.1) |
NA |
NA |
16 |
G T T A G |
3 (0.4) |
1 (0.1) |
3.75 (0.39-36.15) |
ns |
| 17 |
A G A A C |
0.1 (0) |
0.6 (0.1) |
NA |
NA |
17 |
A G A A C |
0 |
0 |
NA |
NA |
| 18 |
A T A G C |
0.4 (0.1) |
0 (0) |
NA |
NA |
18 |
A T A G C |
0 |
0 |
NA |
NA |
| 19 | G T T A C | 1 (0.1) | 0 (0) | NA | NA | 19 | G T T A C | 0 | 0 | NA | NA |
389 cases (778 haplotypes) and 485 controls (970 haplotypes) were included in the analysis.
Global haplotype distribution: P = 0.003.
Left table side: Predicted haplotypes obtained with Monte Carlo simulations using FAMHAP. The calculation of odds ratios and a global P value for haplotype distribution was performed on these results using FAMHAP. The haplotype order follows a priority ranking within the control group. Nineteen haplotypes were predicted. The haplotype base positions correspond to a) rs3863377, b) rs56163822, c) rs7138843, d) rs10860603, and e) rs35724. As FAMHAP tested all haplotypes with a frequency >0.01, the frequency distribution of eight haplotypes (1–7, 9) was included. Relevant P-values for four haplotypes were displayed by FAMHAP. None of the individual haplotypes are significantly differentially distributed between the IBD and control groups.
OR, odds ratio; NA, not applicable, FAMHAP did not calculate an odds ratio on these results because of a low haplotype frequency value predicted.
Right table side (italics): Most likely occurring haplotype frequencies (haplotype in best reconstruction) predicted by FAMHAP. Based on these predictions significance tests were performed. P-values were calculated using the Fisher’s exact test or (if marked with a) ) using the Chi-Square test. A Bonferroni-corrected P-value of 0.003 (19 tests) was taken as significance level. According to this, none of the haplotypes appeard to be significantly differently distributed between the case and control groups.
OR, odds ratio; C.I., confidence interval; NA, OR not applicable because of at least one cell count with the value of null; ns, not significant and P >0.05.
Table 5.
Haplotype analysis of UC patients and non-IBD controls
| Order | Haplotype a) b) c) d) e) | Cases (%) | Controls (%) | OR (C.I.) | P | Order | Haplotype a) b) c) d) e) | Cases (%) | Controls (%) | OR (C.I.) | P |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
G G A G G |
146.6 (49.5) |
507.5 (52.3) |
0.89 (0.69-1.16) |
|
1 |
G G A G G |
153 (51.7) |
530 (54.6) |
0.89 (0.68-1.15) |
a)ns |
| 2 |
G G A G C |
98.8 (33.4) |
263.9 (27.2) |
1.34 (1.01-1.77) |
0.022 |
2 |
G G A G C |
94 (31.8) |
239 (24.6) |
1.42 (1.07-1.89) |
a)0.015 |
| 3 |
G G A A C |
16.9 (5.7) |
89.5 (9.2) |
0.59 (0.35-1.02) |
0.010 |
3 |
G G A A C |
21 (7.1) |
111 (11.4) |
0.59 (0.36-0.96) |
a)0.038 |
| 4 |
G G A A G |
14.2 (4.8) |
49 (5.1) |
0.95 (0.52-1.73) |
|
4 |
G G A A G |
9 (3) |
30 (3.1) |
0.98 (0.46-2.09) |
a)ns |
| 5 |
A G A G G |
1.2 (0.4) |
14.1 (1.5) |
0.28 (0.04-1.79) |
0.110 |
5 |
A G A G G |
1 (0.3) |
13 (1.3) |
0.25 (0.03-1.91) |
ns |
| 6 |
A G A G C |
1.7 (0.6) |
11 (1.1) |
0.5 (0.10-2.53) |
|
6 |
A G A G C |
2 (0.7) |
14 (1.4) |
0.46 (0.10-2.06) |
ns |
| 7 |
G G T G C |
1.6 (0.5) |
9.4 (1) |
0.54 (0.10-2.97) |
|
7 |
G G T G C |
1 (0.3) |
11 (1.1) |
0.30 (0.04-2.30) |
ns |
| 8 |
G T A G G |
2.2 (0.7) |
7.8 (0.8) |
0.92 (0.21-4.13) |
NA |
8 |
G T A G G |
1 (0.3) |
7 (0.7) |
0.47 (0.06-3.81) |
ns |
| 9 |
G T A G C |
2.6 (0.9) |
5.4 (0.6) |
1.58 (0.36-6.99) |
NA |
9 |
G T A G C |
3 (1) |
6 (0.6) |
1.65 (0.41-6.62) |
ns |
| 10 |
G G T G G |
0.3 (0.1) |
3.0 (0.3) |
0.37 (0.01-13.09) |
NA |
10 |
G G T G G |
0 |
2 (0.2) |
NA |
ns |
| 11 |
G T A A G |
0.8 (0.3) |
1.8 (0.2) |
NA |
NA |
11 |
G T A A G |
1 (0.3) |
2 (0.2) |
1.64 (0.15-18.17) |
ns |
| 12 |
A G A A G |
0.2 (0.1) |
2.0 (0.2) |
0.33 (0-30.58) |
NA |
12 |
A G A A G |
0 |
1 (0.1) |
NA |
ns |
| 13 |
G G T A C |
1.1 (0.4) |
2.0 (0.2) |
1.83 (0.18-18.47) |
NA |
13 |
G G T A C |
2 (0.7) |
1 (0.1) |
6.59 (0.60-73.0) |
ns |
| 14 |
G T T G C |
4.4 (1.5) |
1.0 (0.1) |
NA |
0.005* |
14 |
G T T G C |
4 (1.4) |
1 (0.1) |
13.27 (1.48-119.30) |
0.012 |
| 15 |
G G T A G |
1.3 (0.4) |
0.7 (0.1) |
NA |
NA |
15 |
G G T A G |
1 (0.3) |
1 (0.1) |
3.29 (0.20-52.71) |
ns |
| 16 |
G T T A G |
1.2 (0.4) |
1.0 (0.1) |
NA |
NA |
16 |
G T T A G |
2 (0.7) |
1 (0.1) |
6.59 (0.60-73.00) |
ns |
| 17 |
A G A A C |
0.2 (0.1) |
0.9 (0.1) |
NA |
NA |
17 |
A G A A C |
0 |
0 |
NA |
NA |
| 18 |
A T A G C |
0.7 (0.2) |
0 |
NA |
NA |
18 |
A T A G C |
1 (0.3) |
0 |
NA |
ns |
| 19 | G T T A C | 0.2 (0.1) | 0 | NA | NA | 19 | G T T A C | 0 | 0 | NA | NA |
148 cases (296 haplotypes) and controls 485 (970 haplotypes) were included in the analysis.
Global haplotype distribution: P=0.004.
Left table side: Predicted haplotype frequencies obtained with Monte Carlo simulations using FAMHAP: The calculation of odds ratios and a global P-value for haplotype distribution was performed on these results using FAMHAP. The haplotype order follows a priority ranking within the control group. Nineteen haplotypes were predicted. The haplotype base positions correspond to a) rs3863377, b) rs56163822, c) rs7138843, d) rs10860603, and e) rs35724. As FAMHAP tested all haplotypes with a frequency >0.01, the frequency distribution of eight haplotypes (1-7, 14) was included. Relevant P-values for four haplotypes were displayed by FAMHAP. One single haplotype (14) was significantly differentially distributed between the UC and control groups. OR, odds ratio; NA, not applicable, FAMHAP did not calculate an odds ratio on these results because of a low haplotype frequency value predicted.
Right table side (italics): Most likely occurring haplotype frequencies (haplotypes in best reconstruction) predicted by FAMHAP. Based on these predictions significance tests were performed. P-values were calculated using the Fisher’s exact test or (if marked with a) ) the Chi-Square-test. A Bonferroni-corrected P-value of 0.003 (19 tests) was taken as significance level. According to this, none of the haplotypes appeared to be significantly different distributed between the UC and control groups. OR, odds ratio; C.I., confidence interval; NA, OR not applicable because of at least one cell count with the value of null; ns, not significant and P > 0.05.
Table 6.
Haplotype analysis of CD patients and non-IBD controls
| Order | Haplotype a) b) c) d) e) | Cases (%) | Controls (%) | OR (C.I.) | P | Order | Haplotype a) b) c) d) e) | Cases (%) | Controls (%) | OR (C.I.) | P |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
G G A G G |
258.6 (53.6) |
511.2 (52.7) |
1.04 (0.83-1.29) |
|
1 |
G G A G G |
265 (55.5) |
530 (55.3) |
1.01 (0.81-1.26) |
a)ns |
| 2 |
G G A G C |
146.8 (30.5) |
259.0 (26.7) |
1.20 (0.95-1.53) |
|
2 |
G G A G C |
140 (29.4) |
239 (24.9) |
1.25 (0.98-1.60) |
a)ns |
| 3 |
G G A A C |
39.3 (8.2) |
93.9 (9.7) |
0.83 (0.56-1.22) |
|
3 |
G G A A C |
44 (9.2) |
110 (11.5) |
0.79 (0.54-1.14) |
a)ns |
| 4 |
G G A A G |
13 (2.7) |
45.9 (4.7) |
0.56 (0.30-1.04) |
0.062 |
4 |
G G A A G |
9 (1.9) |
31 (3.2) |
0.58 (0.27-1.22) |
ns |
| 5 |
A G A G G |
2.1 (0.4) |
14.4 (1.5) |
0.29 (0.07-1.24) |
0.053 |
5 |
A G A G G |
1 (0.2) |
13 (1.4) |
0.15 (0.02-1.17) |
0.044 |
| 6 |
A G A G C |
3.9 (0.8) |
11.6 (1.2) |
0.68 (0.22-2.15) |
|
6 |
A G A G C |
5 (1) |
14 (1.5) |
0.72 (0.26-2.00) |
ns |
| 7 |
G G T G C |
3.8 (0.8) |
9.4 (1.0) |
0.81 (0.24-2.68) |
|
7 |
G G T G C |
5 |
11 |
0.91 (0.32-2.65) |
ns |
| 8 |
G T A G G |
2.1 (0.4) |
7.8 (0.8) |
0.53 (0.11-2.49) |
NA |
8 |
G T A G G |
1 (0.2) |
7 (0.7) |
0.29 (0.04-2.33) |
ns |
| 9 |
G T A G C |
6.1 (1.3) |
5.7 (0.6) |
2.19 (0.70-6.91) |
0.122 |
9 |
G T A G C |
7 (1.5) |
6 (0.6) |
2.37 (0.79-7.09) |
ns |
| 10 |
G G T G G |
1.5 (0.3) |
3.1 (0.3) |
0.98 (0.14-6.86) |
NA |
10 |
G G T G G |
1 (0.2) |
2 (0.2) |
1.01 (0.09-11.13) |
ns |
| 11 |
G T A A G |
0 |
1.6 (0.2) |
NA |
NA |
11 |
G T A A G |
0 |
2 (0.2) |
NA |
ns |
| 12 |
A G A A G |
0 |
2.1 (0.2) |
NA |
NA |
12 |
A G A A G |
0 |
1 (0.1) |
NA |
ns |
| 13 |
G G T A C |
0.9 (0.2) |
2.5 (0.3) |
0.73 (0.07-7.9) |
NA |
13 |
G G T A C |
0 |
2 (0.2) |
NA |
ns |
| 14 |
G T T G C |
3.1 (0.6) |
0.9 (0.1) |
NA |
NA |
14 |
G T T G C |
3 (0.6) |
1 (0.1) |
6.07 (0.63-58.53) |
ns |
| 15 |
G G T A G |
NA |
NA |
NA |
NA |
15 |
G G T A G |
0 |
0 |
NA |
NA |
| 16 |
G T T A G |
0.7 (0.1) |
1.0 (0.1) |
NA |
NA |
16 |
G T T A G |
1 (0.2) |
1 (0.1) |
2.02 (0.13-32.30) |
ns |
| 17 |
A G A A C |
NA |
NA |
NA |
NA |
17 |
A G A A C |
0 |
0 |
NA |
NA |
| 18 |
A T A G C |
NA |
NA |
NA |
NA |
18 |
A T A G C |
0 |
0 |
NA |
NA |
| 19 | G T T A C | NA | NA | NA | NA | 19 | G T T A C | 0 | 0 | NA | NA |
241 cases (482 haplotypes) and 485 controls (970 haplotypes) were included in the analysis.
Global haplotype distribution: P=0.079.
Left table side: Predicted haplotype frequencies obtained with Monte Carlo simulations using FAMHAP: The calculation of odds ratios and a global P-value for haplotype distribution was performed on these results using FAMHAP. The haplotype order follows a priority ranking within the control group. Nineteen haplotypes were predicted. The haplotype base positions correspond to a) rs3863377, b) rs56163822, c) rs7138843, d) rs10860603, and e) rs35724. As FAMHAP tested all haplotypes with a frequency >0.01, the frequency distribution of eight haplotypes (1-7, 9) was included. Relevant P-values for three haplotypes were displayed by FAMHAP. None of the individual haplotypes are significantly differentially distributed between the CD and control groups. OR, odds ratio; NA, not applicable, FAMHAP did not calculate an odds ratio on these results because of a low haplotype frequency value predicted.
Right table side (italics): Most likely occurring haplotype frequencies (haplotypes in best reconstruction) predicted by FAMHAP. Based on these predictions significance tests were performed.
P-values were calculated using the Fisher’s exact test or (if marked with a) ) the Chi-Square-test. A Bonferroni-corrected P-value of 0.003 (19 tests) was taken as significance level. According to this, none of the haplotypes appeared to be significantly different distributed between the CD and control groups.
OR, odds ratio; C.I., confidence interval; NA, OR not applicable because of at least one cell count with the value of null; ns, not significant and P > 0.05.
Figure 1.
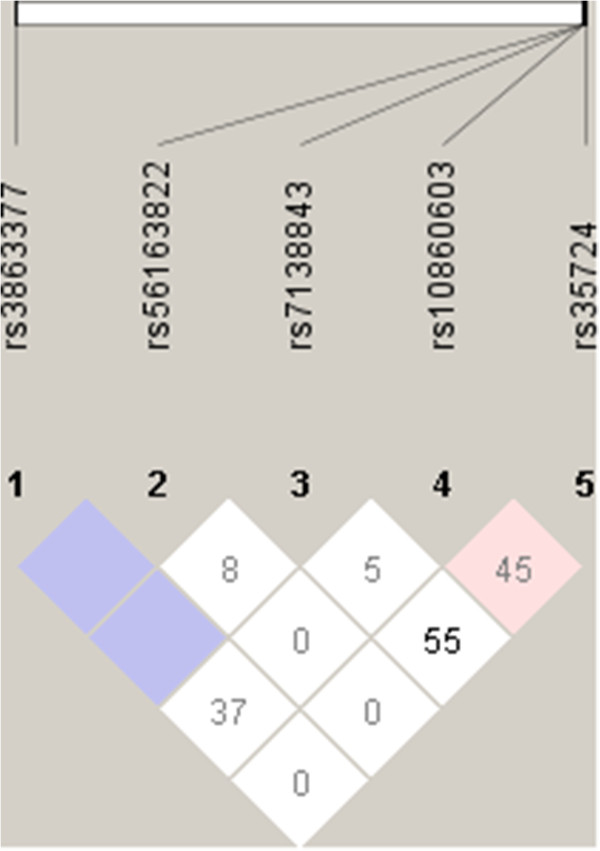
Pairwise linkage disequilibrium calculations between the five NR1H4 SNPs under study in the non-IBD population. A plot with D’ values; Colour scheme: D’ < 1 and LOD < 2, white, D’ = 1 and LOD < 2, blue, D’ < 1 and LOD ≥ 2, pink.
Discussion
The complex pathophysiology of IBD still remains largely unelucidated, although multiple factors, both genetic and environmental, are clearly involved. SNPs and mutations within several genes have been proposed to be associated with the risk to develop IBD. Prior studies have revealed more than 70 genes that are potentially associated with IBD [34,35]. The region on chromosome 16q11-12 named IBD1 was identified in 1996, and the fine mapping of this region led to the identification of the NOD2 (nucleotide-binding oligomerization domain 2)/CARD15 (caspase activation recruitment domain 15) genes [36,37], and a member of a family of pattern recognition receptors (PRRs) that recognizes microbial components and modifies inflammatory responses to bacterial triggers such as lipopolysaccharides (LPS), through the activation of NF-κB [38,39]. Furthermore, genes that play roles in immunological cell-cell interactions and signaling, such as the tumor necrosis factor receptor 1 (TNFR1) [40], the interleukin-23 receptor (IL23R) [41], and other genes that are involved in immune response to bacteria, such as the toll-like receptor 4 (TLR4) [42,43], have been proposed to be associated with IBD. In addition, regulatory genes, such as the protein tyrosine phosphatase N2 (PTPN2) [44,45] and the anti-inflammatory nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ) [46], as well as genes encoding membrane transporters multidrug resistance gene 1 (MDR1)[47-49] and the organic cation transporter 1/2 (OCTN1/2) [47,50] have been proposed to be associated with the risk of chronic mucosal inflammation.
In this report, we describe the identification of single nucleotide polymorphisms associated with the diagnosis of IBD within the NR1H4 gene, encoding the nuclear receptor for bile acids, FXR, in a well-sized European cohort. Five NR1H4 SNPs were analyzed, all of which have previously been studied in the context of other human disease conditions: rs3863377, rs7138843, rs56163822, rs35724, and rs10860603. The NR1H4 variants rs7138843 and rs56163822 have been previously shown to be inversely associated with cholelithiasis in a Mexican population and may thus play a protective role in gallstone disease, while the variant rs3863377 showed no association with cholelithiasis [51]. The variant rs56163822 was found to be more common in a British control group than in patients with intrahepatic cholestasis of pregnancy (ICP), although this difference did not reach statistical significance [32]. NR1H4 variants rs35724 and rs10860603 have been previously shown to be significantly associated with elevated body mass index and obesity [52]. IBD is often associated with hepatobiliary manifestations, [53,54] implying that the etiology of the diseases affecting the two organs, intestine and liver, may have common factors, also supported by our findings that the same NR1H4 genetic variants may be associated with both.
The variant rs3863377 is located in the 5’ region of the NR1H4 gene, whereas the rs7138843 lies within the NR1H4 intron 7 and variants rs35724 and rs10860603 within NR1H4 intron 9. In none of these cases is it known, how the presence of the SNP may affect the expression and/or molecular function of FXR. As the SNP rs3863377 is located within the 5’ region, it may alter a binding site for a transcription factor and may thus affect NR1H4 gene expression. The intronic SNPs rs7138843, rs10860603, and rs35724 could potentially influence splicing of the FXR mRNA. The substitution -1 G > T in rs56163822 lies in the base position adjacent to the translation initiation site, and was shown to lead to reduced FXR protein expression and decreased level of FXR-dependent promoter activation in human embryonic kidney cells. In another study the functional activity of the -1 G > T variant also appeared to be compromised, although transcriptional and translational efficiencies of the variant appeared comparable to the wild-type in cell-free assays and in HeLa cells [55]. Interestingly, the mRNA expression levels of the FXR target genes SHP and OATP1B3 are significantly reduced in the livers of the carriers of the rs56163822 allele, while the FXR mRNA expression level remains comparable, further indicating that this polymorphism may rather lead to weakened function than to reduced expression level of FXR.
In our current genotyping analysis we have found that for the NR1H4 variant rs3863377, the IBD population has a significantly lower frequency of carriers of the rarer allele than the healthy population, suggesting that this 5’ region SNP may confer a protective effect against the disease. In the case of the rs56163822 NR1H4 variant, the rare allele is significantly more prevalent in the IBD population, suggesting that previously reported reduced FXR function exhibited by this variant may contribute to IBD pathogenesis. In the case of the rare NR1H4 variant under study, rs7138843, and the common SNPs rs10860603 and rs35724, no significant differences between the study populations were observed. In agreement with the associations observed for two of the five single SNP variants, the predicted global haplotype pattern was significantly different in IBD patients and non-IBD controls.
In our study, five NR1H4 SNPs were investigated. During the preparation of our manuscript, Nijmeijer et al.[56] published a study showing that mRNA expression of FXR and its target gene SHP are decreased in the ileum of Crohn’s disease patients, in further support of the importance of the role for FXR in IBD. These authors also studied potential association of nine NR1H4 SNPs with IBD in a Dutch population, but did not discover any associations that remained significant upon correction for multiple testing. We note that in their analysis Nijmeijer et al. did not include the SNP rs3863377, the inverse association by which with IBD remained significant even after Bonferroni correction in the current study. As numerous further polymorphisms are known to exist in the NR1H4 gene (http://www.ncbi.nlm.nih.gov/snp/), our report, as well as that by Nijmeijer et al., serve as initial characterizations of the role of FXR genetic variants in IBD. Furthermore, these findings warrant further studies into genetic variants in the NR1H4 gene in the context of other inflammatory conditions affecting further tissues that express FXR.
FXR ligands, such as the hydrophilic bile acid ursodeoxycholic acid, have been proposed as attractive options for the therapy of liver diseases, such as cholestatic disease and non-alcoholic fatty liver disease [57]. Our finding that FXR genetic variants are associated with IBD, together with prior observations on FXR expression being altered in Crohn’s disease [56] and on FXR promoting intestinal barrier integrity [27] and antibacterial defence [26], further emphasizes the potential benefits of FXR ligand administration also in IBD. We further speculate that testing for genetic variation in the NR1H4 gene may contribute to the early IBD diagnosis and prediction of therapy response in the future.
Conclusions
In conclusion, our results further support the role for FXR as a modulator of intestinal inflammation and as an important player in enteroprotection. The link between the bile acid receptor FXR and IBD also further emphasizes the potential importance of bile acid homeostasis and metabolism in the pathogenesis of IBD.
Methods
Study subjects
The study population was European, and comprised of 591 healthy subjects and 547 IBD patients, from which 203 were diagnosed to suffer from UC and 334 from CD. The IBD subjects were recruited at the centers participating in the Swiss Inflammatory Bowel Disease Cohort Study (SIBDCS) [58]. For the IBD patients, the diagnosis of UC or CD was confirmed by the study investigators based on clinical presentation, endoscopic findings, and histology. Non-IBD controls were recruited from gastroenterological patients undergoing surveillance colonoscopy, and showed no symptoms of IBD. History of colorectal cancer was used as an exclusion criterion for both IBD patients and non-IBD controls. All subjects provided their written informed consent to be included in the study. Ethical approvals were obtained from the local medical ethical committees of all study sites involved in the study: 1) The Swiss IBD Cohort Study (SIBDCS) (the participating centers are listed in http://ibdcohort.ch/index.php?id = 94&L = 2; ethical license EK-1316). 2) The Bioethical Committee at the Maria Sklodowska-Curie Memorial Cancer Centre and Institute of Oncology, Warsaw, Poland (ethical licenses 25/2006 and 25/2006/2007).
DNA extraction
Genomic DNAs were extracted from either EDTA-blood or intestinal biopsies using the QIAamp DNA Mini Kit (QIAGEN, Hombrechtikon, Switzerland) or the TRIzol reagent (Invitrogen, Basel, Switzerland), respectively, according to the manufacturer’s instructions. The genomic DNAs were quantified with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) and diluted to a final concentration of 10 ng/μl.
Genotyping of NR1H4 single nucleotide polymorphisms
Genotyping of the five NR1H4 SNPs was performed using TaqMan allelic discrimination assays. The cycling was performed on an 7900HT Fast Real-Time PCR system (Applied Biosystems, Rotkreuz, Switzerland) by using the inventoried TaqMan SNP Genotyping Assays C_28000279_10, C_25598395, C_25598386_10, C_2366616_10, C_2800610_10 for the SNPs rs3863377, rs7138843, rs56163822, rs35724, and rs1086060, respectively. Twenty nanograms of each genomic DNA was used per PCR reaction in a volume of 5 μl. The amplification run conditions were: Once 50°C for 2 min, once 95°C for 10 min, 45 times 95°C for 15 sec, and 60°C for 1 min.
Statistical analysis
Statistical analysis for the individual SNP associations was performed using the software package SPSS 18 (SPSS Inc., Chicago, IL). The Chi-square test or Fisher’s exact test were used to determine associations between individual SNPs and subject phenotypes. A P-value of <0.05 was considered as significant in non-corrected statistical tests and of <0.01 after correction for multiple testing for the five SNPs (according to Bonferroni). The software package PSPower (http://biostat.mc.vanderbilt.edu/twiki/bin/view/Main/PowerSampleSize) was used for retrospective power calculations. A retrospective power analysis of the applied statistical tests on genotype distributions revealed a power of 0.525 for the SNP rs3863377 and a power of 0.323 for the SNP rs56163822, when considering a Bonferroni-corrected alpha level of 0.01 and the detected ORs as shown in Table2 for the applied χ2-tests. Linkage disequilibria (LD) were calculated using D’ statistics and the software package Haploview (http://www.haploview.com). Haplotype predictions and frequency estimations were performed using the software tool FAMHAP (http://www.famhap.meb.uni-bonn.de). FAMHAP performs a permutation test on associations between estimated haplotypes and the affection state based on Monte Carlo simulations. The expectation maximization (EM) algorithm was used to obtain maximum-likelihood estimates of the haplotype frequencies of the sample composed of cases and controls. Individuals with several possible haplotype explanations are assigned with a likelihood weight to each possible haplotype and its calculated frequency estimate. A contingency table is constructed summing up all individuals’ weighted haplotype explanations for each haplotype and the chi-square statistics computed. The corresponding P-value is assessed via Monte Carlo simulation, i.e. in each replication of the algorithm a sample composed of a subgroup of case and control samples is randomly drawn and permuted. FAMHAP implements the calculation of the global P-value via Monte Carlo simulations, as the cell counts used in the contingency table are based on haplotype frequency estimates with increased variances, not on real haplotype counts, which, as a result, does not necessarily follow exactly a chi-square distribution [59,60]. A value of P<0.05 was considered to be significant. Bonferroni-corrected P-values (P<0.006, corrected for eight haplotypes that FAMHAP considered to be relevant to test for) were defined as the significance level for single haplotype comparisons in the white sections of Tables 4-6. In addition, haplotypes in best reconstruction (not weighted) were listed for the case and control groups in Tables 4-6 (grey sections) and used for association analysis performing Fisher’s exact tests or, in case of high cell counts (11 or more), Chi-square tests. Bonferroni-corrected significance levels (p<0.003, corrected for 19 haplotypes) were used for significance testing.
Endnotes
aMembers of the Swiss Inflammatory Bowel Disease Cohort Study (SIBDCS) group: Pierluigi Ballabeni, Peter Bauerfeind, Christoph Beglinger, Stefan Begré, José Bengoa, Janek Binek, Daniel Boller, Jan Borovicka, Christian Braegger, Patrick Brun, Patrick Bühr, Bernard Burnand, Rafael Camara, Dominique Criblez, Philippe de Saussure, Lukas Degen, Joakim Delarive, Tobias Ehmann, Matthias Engelmann, Ali El Wafa, Christian Felley, Alain Frei, Remus Frei, Michael Fried, Florian Froehlich, Suzanne Gallot-Lavallée, Tilman Gerlach, Martin Geyer, Marc Girardin, Oliver Goetze, Horst Haack, Serge Hediger, Peter Hengstler, Klaas Heyland, Patrick Janiak, Pascal Juillerat, Vera Kessler Brondolo, Christoph Knoblauch, Gerd A. Kullak-Ublick, Michael Manz, Rémy Meier, Christa Meyenberger, Pierre Michetti, Christian Mottet, Christoph Müller, Beat Müllhaupt, Thierry Nicolet, Andreas Nydegger, Isabelle Pache, Franziska Piccoli, Julia Pilz, Valérie Pittet, Ronald Rentsch, Jean-Pierre Rey, Silvia Rihs, Daniela Rogler, Gerhard Rogler, Markus Sagmeister, Bernhard Sauter, Niklaus Schaub, Susanne Schibli, Alain Schoepfer, Franck Seibold, Johannes Spalinger, Philippe Stadler, Michael Steuerwald, Alex Straumann, Michael Sulz, Michela Schäppi, Joël Thorens, John-Paul Vader, Stephan Vavricka, Jürg Vögtlin, Roland Von Känel, Gert Wachter, Jürg Wermuth, Paul Wiesel.
Abbreviations
IBD: Inflammatory bowel disease; UC: Ulcerative colitis; CD: Crohn’s disease; FXR: Farnesoid X receptor; SNP: Single nucleotide polymorphism; NR1H4: Nuclear receptor subfamily 1, group H, member 4; OR: Odds ratio; CI: Confidence interval; MAF: Minor allele frequency.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RA performed the genotyping, interpreted data, and wrote the first draft of the manuscript. JM performed the statistical analysis of the data. KT, JR, and PG performed the sample collection and management. GK and GR contributed to the supervision and design of the study. JJE conceived and supervised the study, and contributed to the data interpretation and writing of the manuscript. All authors read and approved of the final manuscript.
Contributor Information
Ragam Attinkara, Email: ragam.attinkara@usz.ch.
Jessica Mwinyi, Email: jessica.mwinyi@usz.ch.
Kaspar Truninger, Email: kaspar.truninger@unibas.ch.
Jaroslaw Regula, Email: jregula@coi.waw.pl.
Pawel Gaj, Email: pawelgaj@tlen.pl.
Gerhard Rogler, Email: gerhard.rogler@usz.ch.
Gerd A Kullak-Ublick, Email: gerd.kullak@usz.ch.
Jyrki J Eloranta, Email: jyrki.eloranta@uzh.ch.
Acknowledgements
We thank Christian Hiller for excellent technical assistance, and members of our team for fruitful discussions. This study was financially supported by the Swiss National Science Foundation (grant 320030_120463), the Swiss IBD Cohort Study (SNF grant 33CSC0-108792), Zurich University Research Priority Programme “Integrative Human Physiology” (ZIHP), the Center of Clinical Research at the University Hospital Zurich, and the Novartis Foundation for Biomedical Research.
Author details
1Department of Clinical Pharmacology and Toxicology, University Hospital, Zurich, Switzerland. 2Department of Biomedicine, Institute of Biochemistry and Genetics, University of Basel, Switzerland and Division of Gastroenterology, Regional Hospital, Langenthal, Switzerland. 3Department of Gastroenterology and Hepatology, Medical Center for Postgraduate Education and the Maria Skłodowska Curie Memorial Cancer Center, Institute of Oncology, Warsaw, Poland. 4Division of Gastroenterology and Hepatology, University Hospital, Zurich, Switzerland.
References
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jess T, Riis L, Jespersgaard C, Hougs L, Andersen PS, Orholm MK, Binder V, Munkholm P. Disease concordance, zygosity, and NOD2/CARD15 status: follow-up of a population-based cohort of Danish twins with inflammatory bowel disease. Am J Gastroenterol. 2005;100:2486–2492. doi: 10.1111/j.1572-0241.2005.00224.x. [DOI] [PubMed] [Google Scholar]
- Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P. et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimers, and heterodimers. Endocr Rev. 1994;15:391–407. doi: 10.1210/edrv-15-3-391. [DOI] [PubMed] [Google Scholar]
- Eloranta JJ, Kullak-Ublick GA. Coordinate transcriptional regulation of bile acid homeostasis and drug metabolism. Arch Biochem Biophys. 2005;433:397–412. doi: 10.1016/j.abb.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD. et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- Huber RM, Murphy K, Miao B, Link JR, Cunningham MR, Rupar MJ, Gunyuzlu PL, Haws TF, Kassam A, Powell F. et al. Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene. 2002;290:35–43. doi: 10.1016/S0378-1119(02)00557-7. [DOI] [PubMed] [Google Scholar]
- Anisfeld AM, Kast-Woelbern HR, Meyer ME, Jones SA, Zhang Y, Williams KJ, Willson T, Edwards PA. Syndecan-1 expression is regulated in an isoform-specific manner by the farnesoid-X receptor. J Biol Chem. 2003;278:20420–20428. doi: 10.1074/jbc.M302505200. [DOI] [PubMed] [Google Scholar]
- Laffitte BA, Kast HR, Nguyen CM, Zavacki AM, Moore DD, Edwards PA. Identification of the DNA binding specificity and potential target genes for the farnesoid X-activated receptor. J Biol Chem. 2000;275:10638–10647. doi: 10.1074/jbc.275.14.10638. [DOI] [PubMed] [Google Scholar]
- Eloranta JJ, Kullak-Ublick GA. The role of FXR in disorders of bile acid homeostasis. Physiology (Bethesda) 2008;23:286–295. doi: 10.1152/physiol.00020.2008. [DOI] [PubMed] [Google Scholar]
- Lee H, Zhang Y, Lee FY, Nelson SF, Gonzalez FJ, Edwards PA. FXR regulates organic solute transporters alpha and beta in the adrenal gland, kidney, and intestine. J Lipid Res. 2006;47:201–214. doi: 10.1194/jlr.M500417-JLR200. [DOI] [PubMed] [Google Scholar]
- Landrier JF, Eloranta JJ, Vavricka SR, Kullak-Ublick GA. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and -beta genes. Am J Physiol Gastrointest Liver Physiol. 2006;290:G476–G485. doi: 10.1152/ajpgi.00430.2005. [DOI] [PubMed] [Google Scholar]
- Plass JR, Mol O, Heegsma J, Geuken M, Faber KN, Jansen PL, Muller M. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology. 2002;35:589–596. doi: 10.1053/jhep.2002.31724. [DOI] [PubMed] [Google Scholar]
- Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276:28857–28865. doi: 10.1074/jbc.M011610200. [DOI] [PubMed] [Google Scholar]
- Schuetz EG, Strom S, Yasuda K, Lecureur V, Assem M, Brimer C, Lamba J, Kim RB, Ramachandran V, Komoroski BJ. et al. Disrupted bile acid homeostasis reveals an unexpected interaction among nuclear hormone receptors, transporters, and cytochrome P450. J Biol Chem. 2001;276:39411–39418. doi: 10.1074/jbc.M106340200. [DOI] [PubMed] [Google Scholar]
- Neimark E, Chen F, Li X, Shneider BL. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology. 2004;40:149–156. doi: 10.1002/hep.20295. [DOI] [PubMed] [Google Scholar]
- Zollner G, Wagner M, Fickert P, Geier A, Fuchsbichler A, Silbert D, Gumhold J, Zatloukal K, Kaser A, Tilg H. et al. Role of nuclear receptors and hepatocyte-enriched transcription factors for Ntcp repression in biliary obstruction in mouse liver. Am J Physiol Gastrointest Liver Physiol. 2005;289:G798–G805. doi: 10.1152/ajpgi.00319.2004. [DOI] [PubMed] [Google Scholar]
- Eloranta JJ, Jung D, Kullak-Ublick GA. The human Na + −taurocholate cotransporting polypeptide gene is activated by glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma coactivator-1alpha, and suppressed by bile acids via a small heterodimer partner-dependent mechanism. Mol Endocrinol. 2006;20:65–79. doi: 10.1210/me.2005-0159. [DOI] [PubMed] [Google Scholar]
- Chiang JY, Kimmel R, Weinberger C, Stroup D. Farnesoid X receptor responds to bile acids and represses cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription. J Biol Chem. 2000;275:10918–10924. doi: 10.1074/jbc.275.15.10918. [DOI] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME. et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/S1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Wildenberg ME, van den Brink GR. FXR activation inhibits inflammation and preserves the intestinal barrier in IBD. Gut. 2011;60:432–433. doi: 10.1136/gut.2010.233304. [DOI] [PubMed] [Google Scholar]
- Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol. 2009;183:6251–6261. doi: 10.4049/jimmunol.0803978. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ. et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S, Klomp LW, Siersema PD, Schipper ME, Danese S. et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–472. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- Gadaleta RM, Oldenburg B, Willemsen ECL, Spit M, Murzilli S, Salvatore L, Klomp LWJ, Siersema PD, van Erpecum KJ, van Mil SWC. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-[kappa]B signaling in the intestine. Biochimica et Biophysica Acta (BBA) Molecular Basis of Disease. 2011;1812:851–858. doi: 10.1016/j.bbadis.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Lian F, Xing X, Yuan G, Schafer C, Rauser S, Walch A, Rocken C, Ebeling M, Wright MB, Schmid RM. et al. Farnesoid X receptor protects human and murine gastric epithelial cells against inflammation-induced damage. Biochem J. 2011;438:315–323. doi: 10.1042/BJ20102096. [DOI] [PubMed] [Google Scholar]
- Kovacs P, Kress R, Rocha J, Kurtz U, Miquel JF, Nervi F, Méndez-Sánchez N, Uribe M, Bock HH, Schirin-Sokhan R. et al. Variation of the gene encoding the nuclear bile salt receptor FXR and gallstone susceptibility in mice and humans. J Hepatol. 2008;48:116–124. doi: 10.1016/j.jhep.2007.07.027. [DOI] [PubMed] [Google Scholar]
- Wadsworth CA, Dixon PH, Wong JH, Chapman MH, McKay SC, Sharif A, Spalding DR, Pereira SP, Thomas HC, Taylor-Robinson SD. et al. Genetic factors in the pathogenesis of cholangiocarcinoma. Dig Dis. 2011;29:93–97. doi: 10.1159/000324688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Mil SW, Milona A, Dixon PH, Mullenbach R, Geenes VL, Chambers J, Shevchuk V, Moore GE, Lammert F, Glantz AG. et al. Functional variants of the central bile acid sensor FXR identified in intrahepatic cholestasis of pregnancy. Gastroenterology. 2007;133:507–516. doi: 10.1053/j.gastro.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Chen X-Q, Wang L-L, Shan Q-W, Tang Q, Deng Y-N, Lian S-J, Yun X. A novel heterozygous NRIH4 termination codon mutation in idiopathic infantile cholestasis. World Journal of Pediatrics. 2012;8:67–71. doi: 10.1007/s12519-011-0299-z. [DOI] [PubMed] [Google Scholar]
- Zhang H, Massey D, Tremelling M, Parkes M. Genetics of inflammatory bowel disease: clues to pathogenesis. Br Med Bull. 2008;87:17–30. doi: 10.1093/bmb/ldn031. [DOI] [PubMed] [Google Scholar]
- Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R. et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugot J-P, Chamaillard M, Zouali H, Lesage S, Cezard J-P, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH. et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, Ogura Y, Nunez G. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–27831. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- Inohara N, Ogura Y, Chen FF, Muto A, Nunez G. Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J Biol Chem. 2001;276:2551–2554. doi: 10.1074/jbc.M009728200. [DOI] [PubMed] [Google Scholar]
- Sashio H, Tamura K, Ito R, Yamamoto Y, Bamba H, Kosaka T, Fukui S, Sawada K, Fukuda Y, Satomi M. et al. Polymorphisms of the TNF gene and the TNF receptor superfamily member 1B gene are associated with susceptibility to ulcerative colitis and Crohn’s disease, respectively. Immunogenetics. 2002;53:1020–1027. doi: 10.1007/s00251-001-0423-7. [DOI] [PubMed] [Google Scholar]
- Fisher SA, Tremelling M, Anderson CA, Gwilliam R, Bumpstead S, Prescott NJ, Nimmo ER, Massey D, Berzuini C, Johnson C. et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat Genet. 2008;40:710–712. doi: 10.1038/ng.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostenbrug LE, Drenth JP, de Jong DJ, Nolte IM, Oosterom E, van Dullemen HM, van der Linde K, te Meerman GJ, van der Steege G, Kleibeuker JH. et al. Association between Toll-like receptor 4 and inflammatory bowel disease. Inflamm Bowel Dis. 2005;11:567–575. doi: 10.1097/01.mib.0000161305.81198.0f. [DOI] [PubMed] [Google Scholar]
- Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, Quertinmont E, Abramowicz M, Van Gossum A, Deviere J. et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut. 2004;53:987–992. doi: 10.1136/gut.2003.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, Balschun T, Karlsen TH, Hedderich J, May S, Lu T, Schuldt D, Nikolaus S, Rosenstiel P, Krawczak M. et al. Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008;40:713–715. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- Sugawara K, Olson TS, Moskaluk CA, Stevens BK, Hoang S, Kozaiwa K, Cominelli F, Ley KF, McDuffie M. Linkage to peroxisome proliferator-activated receptor-[gamma] in SAMP1/YitFc mice and in human Crohn’s disease. Gastroenterology. 2005;128:351–360. doi: 10.1053/j.gastro.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Waller S, Tremelling M, Bredin F, Godfrey L, Howson J, Parkes M. Evidence for association of OCTN genes and IBD5 with ulcerative colitis. Gut. 2006;55:809–814. doi: 10.1136/gut.2005.084574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Schaeffeler E, Marx C, Fromm MF, Kaskas B, Metzler J, Stange E, Herfarth H, Schoelmerich J, Gregor M. et al. Association between the C3435T MDR1 gene polymorphism and susceptibility for ulcerative colitis. Gastroenterology. 2003;124:26–33. doi: 10.1053/gast.2003.50010. [DOI] [PubMed] [Google Scholar]
- Brant SR, Panhuysen CIM, Nicolae D, Reddy DM, Bonen DK, Karaliukas R, Zhang L, Swanson E, Datta LW, Moran T. et al. MDR1 Ala893 Polymorphism Is Associated with Inflammatory Bowel Disease. Am J Hum Genet. 2003;73:1282–1292. doi: 10.1086/379927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, Newman B, Van Oene M, Cescon D, Greenberg G. et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–475. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- Kovacs P, Kress R, Rocha J, Kurtz U, Miquel JF, Nervi F, Mendez-Sanchez N, Uribe M, Bock HH, Schirin-Sokhan R. et al. Variation of the gene encoding the nuclear bile salt receptor FXR and gallstone susceptibility in mice and humans. J Hepatol. 2008;48:116–124. doi: 10.1016/j.jhep.2007.07.027. [DOI] [PubMed] [Google Scholar]
- van den Berg SW, Dolle ME, Imholz S, van der AD, van t Slot R, Wijmenga C, Verschuren WM, Strien C, Siezen CL, Hoebee B. et al. Genetic variations in regulatory pathways of fatty acid and glucose metabolism are associated with obesity phenotypes: a population-based cohort study. Int J Obes (Lond) 2009;33:1143–1152. doi: 10.1038/ijo.2009.152. [DOI] [PubMed] [Google Scholar]
- Christophi C, Hughes ER. Hepatobiliary disorders in inflammatory bowel disease. Surg Gynecol Obstet. 1985;160:187–193. [PubMed] [Google Scholar]
- Navaneethan U, Shen B. Hepatopancreatobiliary manifestations and complications associated with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1598–1619. doi: 10.1002/ibd.21219. [DOI] [PubMed] [Google Scholar]
- Marzolini C, Tirona RG, Gervasini G, Poonkuzhali B, Assem M, Lee W, Leake BF, Schuetz JD, Schuetz EG, Kim RB. A Common Polymorphism in the Bile Acid Receptor Farnesoid X Receptor Is Associated with Decreased Hepatic Target Gene Expression. Mol Endocrinol. 2007;21:1769–1780. doi: 10.1210/me.2007-0025. [DOI] [PubMed] [Google Scholar]
- Nijmeijer RM, Gadaleta RM, van Mil SW, van Bodegraven AA, Crusius JB, Dijkstra G, Hommes DW, de Jong DJ, Stokkers PC, Verspaget HW. et al. Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PLoS One. 2011;6:e23745. doi: 10.1371/journal.pone.0023745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner M, Halilbasic E. Nuclear Receptors as New Perspective for the Management of Liver Diseases. Gastroenterology. 2011;140:1120–1125. doi: 10.1053/j.gastro.2011.02.044. e1112. [DOI] [PubMed] [Google Scholar]
- Pittet V, Juillerat P, Mottet C, Felley C, Ballabeni P, Burnand B, Michetti P, Vader JP. Cohort profile: the Swiss Inflammatory Bowel Disease Cohort Study (SIBDCS) Int J Epidemiol. 2009;38:922–931. doi: 10.1093/ije/dyn180. [DOI] [PubMed] [Google Scholar]
- Becker T, Knapp M. A powerful strategy to account for multiple testing in the context of haplotype analysis. Am J Hum Genet. 2004;75:561–570. doi: 10.1086/424390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Schumacher J, Cichon S, Baur MP, Knapp M. Haplotype interaction analysis of unlinked regions. Genet Epidemiol. 2005;29:313–322. doi: 10.1002/gepi.20096. [DOI] [PubMed] [Google Scholar]