

Abstract
The “fetal origins hypothesis” argued that physiological changes consequent to in utero exposures ultimately contribute to disease susceptibility in later life. The dramatic increase in asthma prevalence is attributed to early exposures acting on preexisting asthma-susceptible genotypes. We showed previously that distinct transcriptome signatures distinguish the developmental respiratory phenotype of atopic (Brown Norway, BN) and normoresponsive (Lewis) rats. We aimed to determine whether maternal allergen exposure would influence asthma pathogenesis by reprogramming primary patterns of developmental lung gene expression. Postnatal offspring of dams sensitized to ovalbumin before mating and challenged during pregnancy were assessed for lung function, inflammatory biomarkers, and respiratory gene expression. Although maternal ovalbumin exposure resulted in characteristic features of an allergic response (bronchoalveolar lavage neutrophils, IgE, methacholine-induced lung resistance) in offspring of both strains, substantial strain-specific differences were observed in respiratory gene expression. Of 799 probes representing the top 5% of transcriptomic variation, only 112 (14%) were affected in both strains. Strain-specific gene signatures also exhibited marked differences in enrichment for gene ontologies, with immune regulation and cell proliferation being prominent in the BN strain, cell cycle and microtubule assembly gene sets in the Lewis strain. Multiple ovalbumin-specific probes in both strains were also differentially expressed in lymphoblastoid cell lines from human asthmatic vs. nonasthmatic sibling pairs. Our data point to the existence of distinct, genetically programmed responses to maternal exposures in developing lung. These different response patterns, if recapitulated in human fetal development, can contribute to long-term pulmonary health including interindividual susceptibility to asthma.
Keywords: animal models, asthma, gene-environment interactions, in utero exposure, lung development
first proposed by david barker, the “fetal origins hypothesis” argued that specific in utero exposures contribute to disease susceptibility in later life (3). Barker's hypothesis was based on the idea that, during critical developmental windows, the organism is more “plastic” to its environment. Although over time this “plasticity” is lost, the early developmental “programming” can generate permanent changes to organs and systems impacting life-long disease risk. A growing body of epidemiological data support this hypothesis. However, very little is known about the mechanisms whereby environmental insults modify long-term disease susceptibility.
Whereas Barker's original hypothesis focused on the consequences of in utero nutrition, there is now evidence that other exposures, including maternal cigarette smoke and allergens, are strong risk factors for development of asthma. More than 500 articles examining the relationship of maternal and postnatal cigarette smoke exposure to asthma and lung function outcomes consistently show a 30–40% increase in the odds of asthma or persistent wheezing in early life (33). Similarly, maternal allergen exposure increases risk of atopy in children (21, 47).
Little is known about how maternal exposure impacts the developing airways. However, there are clear links between airway remodeling early in childhood and increased asthma susceptibility. Wide interindividual variability in normal airway development (16, 43) and responsiveness (34, 43) is evident in the postnatal period, and reduced neonatal lung function has been associated with lower lung function in childhood, increased airway responsiveness, wheezing, and early asthma diagnosis (28, 45). A thickened subepithelial lamina reticularis is apparent in bronchial biopsies of children several years before asthma diagnosis (36, 39). Bronchial epithelium of children with asthma is less differentiated, has a greater rate of proliferation, and dysregulated repair, all of which may contribute to initiation of lung remodeling (10, 20).
The Brown Norway (BN) rat is used as an experimental model of asthma (27). The normoresponsive Lewis rat serves as a control. Although appearing normal at baseline, the defining features of asthma can be reproduced in Lewis rat following allergen exposure (9). The BN rat is naturally atopic (31). In common with human allergic asthma, BN rats have early and late allergic responses and eosinophilic airway inflammation (9). With repeated allergen challenge, they develop airway hyperresponsiveness to methacholine (MCh) (4) and structural changes in the airways (40).
We showed previously that distinct transcriptome signatures distinguish the developmental respiratory phenotypes of BN and Lewis rat (7). We hypothesized that maternal allergen exposure would influence asthma pathogenesis in offspring by reprogramming these primary patterns of lung gene expression. We show that prenatal allergen exposure alters the developmental trajectory of lung gene expression in a strain-specific fashion and influences postnatal respiratory phenotype in both atopic BN and normoresponsive Lewis rat.
MATERIALS AND METHODS
Animals.
Lewis and BN male and female rats (Charles River Laboratories, Saint Constant, QC, Canada) were housed in the Montreal Children's Hospital Animal Facility. Animal procedures were approved by the Canadian Council for Animal Care and the Animal Care Committee of the McGill University Health Centre.
Allergen sensitization and challenge.
Ovalbumin (OVA) sensitization was performed on 8–10-wk-old female rats by simultaneous injections of 1 mg of OVA (Sigma-Aldrich, Oakville, ON) absorbed to 100 mg of aluminum hydroxide in 1 ml of saline (SAL) subcutaneously (s.c.) and 2 × 109 heat-killed Bordetella pertussis in 0.5 ml SAL by intraperitoneal injection (provided by T. Issekutz, Dalhousie University, Halifax, NS, Canada). Control rats received SAL injections. Animals were bred 14 days later. Pregnant dams were challenged for 30 min at embryonic days (E)1, E7, E13, and E19 with 1% aerosolized OVA in SAL or SAL alone, using the InExpose system (SCIREQ, Montreal, QC, Canada). Pups were collected at postnatal days (PN)1, 7, or 14 (Fig. 1).
Fig. 1.
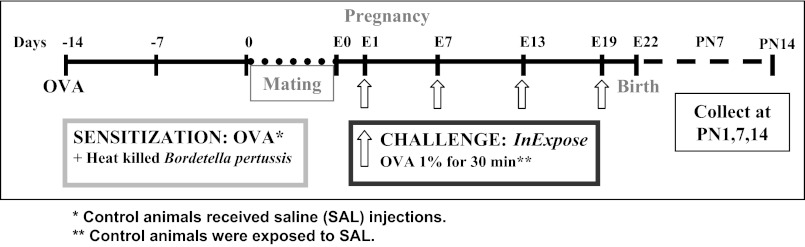
Ovalbumin (OVA) exposure protocol. Adult female rats were sensitized to OVA 14 days before mating. Control rats received saline (SAL). Pregnant rats sensitized to OVA were challenged with OVA for 30 min, 4 times during the pregnancy. Control rats received SAL. Pups were collected at postnatal day 1 (PN1), 7, or 14. E1, embryonic day 1.
Airway responsiveness.
Airway responsiveness was measured in 14-day-old pups using a computer-controlled small animal ventilator (flexiVent, SCIREQ) as we described (7). Pups were deeply anaesthetized by an intraperitoneal (ip) injection of xylazine (8 mg/kg) and pentobarbital (70 mg/kg), tracheotomized, and ventilated quasisinusoidally at a frequency of 150 breaths/min and a tidal volume of 10 ml/kg at positive end-expiratory pressure level of 3 cm of H2O. Subsequently, they were paralyzed by injection of pancuronium bromide (0.8 mg/kg ip). Baseline respiratory resistance was measured at 150 breaths/min. Maximal resistance was recorded before and after increasing doses of aerosolized MCh (6.25, 12.5, 25.0, and 50.0 mg/ml).
Bronchoalveolar lavage.
Bronchoalveolar lavage (BAL) was performed by instilling the lungs four times with 0.4, 0.7, and 1.0 ml of cold PBS for PN1, 7, and 14, respectively, through a tracheal cannula. Lavage fluid was centrifuged, and pellets were resuspended in 0.5 ml cold saline. Total cell numbers were counted with a hemacytometer. Cytospin slides (Cytospin4; Shandon, Pittsburgh, PA) prepared for differential cells counts were stained with Diff-Quick (Hema3 stainset; Fisher Scientific, Middletown, VA) and counted (>200 cells/slide) to determine the percentage of each cell type.
Serum IgE.
At PN14 blood was collected from killed pups. Plasma was isolated by centrifuging the blood samples at 12,000 rpm for 10 min. Total IgE levels were measured using the Rat IgE ELISA Quantitation Set according to the manufacturer's procedure (Bethyl Laboratories, Montgomery, TX). Briefly, 96-well plates were coated with sheep anti-rat IgE antibody and blocked for 30 min. Rat IgE added to the wells was detected with horseradish peroxidase-conjugated sheep anti-rat IgE antibody and tetramethyl benzidine peroxidase substrate. After the reaction was stopped, absorbance was measured using an ELISA plate reader set at 450 nm.
Inflammatory cytokines.
The MESO multi-spot immunoassay system (Meso Scale Diagnostics, Gaithersburg, MD) was used for detection of rat IL-1β, KC/GRO (CXCL1), IL-4, IL-5, TNF-α, IFN-γ, and IL-13 according to the supplier.
RNA isolation.
RNA from whole lung tissue was isolated using Trizol (Invitrogen, Burlington, ON, Canada) according to the manufacturer. RNA was resuspended in 1× RNASecure (Ambion, Austin, TX) and treated with the Turbo DNase-free kit (Ambion) to remove traces of DNA.
Quantitative real-time RT-PCR.
Quantitative real-time RT-PCR was performed on the Mx4000 QPCR system (Stratagene, La Jolla, CA) using the QuantiTect SYBR green RT-PCR kit (Qiagen, Mississauga, ON, Canada). Gene-specific primers for SYBR green detection of Edg4, Sftpd, Orm1, Tagln, Lgals, Timp1, Myl9, and Gapdh were predesigned (Qiagen QuantiTect primer assays). cDNA was prepared from an initial 250 ng/μl of RNA. RNA was incubated at 65°C for 5 min with 1 μg/μl of random primers and 10 mM dNTPs. To this, 5× first strand buffer, 1 mM DTT, 1 μl of RNase OUT (Invitrogen), and 1 μl of Superscript II (Invitrogen) was added and incubated at 42°C for 1 h and 70°C for 15 min. RT-PCR was performed in 25-μl reactions for 40 cycles using 1 μl of cDNA. Relative mRNA expression levels were analyzed using the ΔΔ cycle threshold method (n ≥ 4 for all assays).
Statistical analyses.
All results for respiratory phenotypes are presented as means ± SEM. Statistical significance of difference of group averages was determined by one-way or two-way ANOVA. Pair-wise comparisons for respiratory phenotypes were assessed using the two-tailed Student's t-test with significance defined as P ≤ 0.05.
Microarray data analysis.
Global gene expression analysis was carried out at the McGill University and Genome Quebec Innovation Centre on the Illumina RatRef 12 microarray, which contains 22,523 gene probes that interrogate 11,631 unique rat genes. Raw data were quantile normalized using the R Bioconductor package, lumi. (http://www.bioconductor.org). Principal component analysis (PCA) and linear correlation analysis were used to assess patterns of global gene expression in whole lung. Microarray data are available at the National Center for Biotechnology Information Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) GSE32622.
Four biological replicate samples were microarray profiled for each condition (strain-age-exposure). There are 12 (2 rat strains × 3 ages × 2 exposures) unique conditions. PCA was used to investigate the sample variation in 22,523 gene probe expression space and to select two replicate samples per condition for subsequent gene expression profile reproducibility and differential analyses. Firstly for each gene probe, we computed the linear correlation across the 12 unique conditions between the first and second replicate sample profiles to quantify the reproducibility of the gene probe expression (measurement) profile. Depending on the context, a gene probe was considered reproducible if this correlation value was >0.7 or 0.8. Secondly for the differential analysis (SAL vs. OVA), we considered both the PCA contribution of the gene probe and its correlation value between OVA vs. SAL conditions for each rat strain. The earlier PCA returned a graphical representation of the sample variation in principal components 1–3 (PC1–3) space, where each principal component (PC) is a linear combination of 22,253 gene probe features. For example, PC1 = a1*g1 + a2*g2 + ... + aj*gj + ... + a22,253*g22,253, where aj is a real-valued loading coefficient whose magnitude represents the contribution of the gene probe gj to sample variation along PC1 (18, 19). Depending on the context, the top 5% or 10% highest loading magnitude gene probes in any one of PC1–3 were selected for further differential analysis: SAL vs. OVA. For each rat strain and each gene probe, the OVA vs. SAL linear correlation value across three ages × two replicates was used to quantify the change in the developmental time profile of the gene between the two conditions. We considered a gene to be differentially expressed if it is 1) reproducible, i.e., with correlation between first and second replicate profiles >0.7 or 0.8, 2) a top 5% or 10% PC1–3 gene described earlier, and 3) its OVA vs. SAL correlation value was <0.0 or −0.1.
RESULTS
Maternal allergen exposure enhances effects of MCh provoked airway resistance.
We measured airway resistance at PN14 at baseline and after exposure to increasing doses of aerosolized MCh using the Flexivent small animal ventilator (Scireq). Maternal OVA exposure was associated with increased airway resistance in BN pups at higher doses of MCh (Fig. 2). A modest but significant induction of lung resistance was also observed in OVA-exposed Lewis rat at lower MCh dose.
Fig. 2.

Lung resistance is elevated in Brown Norway (BN) and Lewis pups following maternal allergen exposure. Airway resistance is significantly increased after OVA exposure in Lewis pups (top) after 12.5 mg of methacholine (MCh) (*P < 0.05, n = 4 SAL, n = 15 OVA), in BN (bottom) after 50 mg/ml of MCh (*P < 0.05, n = 5 SAL, n = 16 OVA). All measurements were taken at PN14.
Maternal allergen exposure increases inflammatory cells in BAL.
Inflammatory cell infiltrates were assessed in BAL fluid isolated from rat pups at PN1, 7, and 14. Figure 3A illustrates the percent increase in BAL neutrophils and eosinophils in each of the rat strains. Maternal OVA exposure was associated with increase in percentage of neutrophils in both strains at all time points with the exception of BN pups at PN14. OVA-exposed BN pups also had a significantly higher number of neutrophils than OVA-exposed Lewis pups at PN1. OVA-exposed BN pups had elevated eosinophils at PN7 and 14. At all time points, OVA-exposed BN pups had significantly higher eosinophil levels than OVA-exposed Lewis pups.
Fig. 3.

Inflammatory cell infiltrates in bronchoalveolar lavage (BAL) fluid isolated from rat pups at PN1, 7, and 14. The percentage (A) and total number of inflammatory cell infiltrates (B) were assessed in BAL fluid isolated from rat pups at PN1, 7, and 14 (n > 4 all groups, except Lewis SAL and BN OVA PN14, n = 3). A: percentage of neutrophils was elevated in OVA-exposed Lewis pups at PN7 and 14 and BN pups at PN1 and 7. Following maternal OVA exposure, BN pups display higher neutrophil levels than Lewis pups at PN1. Eosinophil percentage was significantly increased in BN pups after maternal OVA exposure at PN7 and 14 and was higher than in Lewis pups at all 3 time points. B: OVA exposure was associated with an increase in the total number of neutrophils from BAL fluid in Lewis pups at PN14 and in BN pups at PN1 and 7. The total number of eosinophils was elevated in OVA-exposed BN pups at PN1 and 7 and was significantly higher than that observed in Lewis pups at PN1 (*P < 0.05 OVA vs. SAL; #P < 0.05 vs. Lewis rat).
A significant increase in the total number of neutrophils following OVA exposure was observed in Lewis pups at PN14 and in BN pups at PN1 and PN7. At PN1 and PN7, total neutrophils in OVA-exposed BN pups were significantly higher than in OVA-exposed Lewis pups. Total BAL eosinophils were elevated in BN pups at PN1 and PN7.
Increased serum OVA-specific IgE and inflammatory cytokines in pups of dams exposed to allergen.
We assessed whether elevated BAL eosinophil levels observed in BN pups would be associated with increased OVA-specific IgE (Fig. 4A). We noted significantly elevated OVA-specific IgE in both BN and Lewis pups at PN14. Levels of IgE in OVA-exposed BN pups were significantly higher than corresponding values in OVA-exposed Lewis pups at all time points. We further assessed BAL levels of a panel of inflammatory serum cytokines including IFN-γ, IL-1β, IL-4, IL-13, KC/GRO (also known as CXCL1, GROα), and TNF-α. Maternal OVA-exposed BN but not Lewis pups had elevated levels of IL-1β, TNF-α, and KC/GRO (Fig. 4B).
Fig. 4.

A: Serum IgE levels are increased in Lewis and BN pups after maternal OVA exposure. OVA exposure is associated with increased serum IgE in BN pups at PN14 (n = 4, *P < 0.05 OVA vs. SAL, #P < 0.05 vs. Lewis). B: BAL levels of IL-1β, TNF-α, and GRO/KC are elevated in OVA-exposed BN pups at PN14. BAL levels of a panel of inflammatory serum cytokines including IFN-γ, IL-1β, IL-4, IL-13, KC/GRO, and TNF-α were assessed. OVA-exposed BN but not Lewis pups had elevated levels of IL-1β, TNF-α, and KC/GRO (n = 4, *P < 0.05).
Maternal allergen exposure reprograms the developmental trajectory of lung gene expression.
Having demonstrated developmental differences in the respiratory phenotype of maternal OVA-exposed pups, we next examined the impact of this exposure on global gene expression. In this study, our primary objective was to examine the intrastrain differences in the transcriptome following maternal exposures. Total RNA isolated from whole lung at PN1, 7, and 14 from OVA- and SAL-exposed pups was profiled using Illumina RatRef12 microarrays. Quadruplicate microarray measurements were made for each of the 12 conditions: two strains × three ages × two exposures.
PCA was used to qualitatively assess the sample variation in the postnatal developing lung transcriptome (18, 19). To visualize lung developmental trajectories for the rat strains and exposures in 22,523-probe transcriptome space, replicate sample points were coclustered (data not shown); therefore, we selected two of four replicate samples to represent each strain-age-exposure for subsequent analyses. Figure 5 illustrates the principal component coordinate centroids of each strain-age-exposure with connecting lines between centroids to mark developmental trajectories. Visually, PC1 appeared to correspond to developmental age, PC2 to strain, and PC3 to a combination of age and strain. Therefore, we considered the top 5% of probes contributing to any one of PC1–3 as potentially important contributors to age and strain variation. The greatest distances between OVA and SAL trajectories were seen at PN1 for BN pups and at PN7 for Lewis pups.
Fig. 5.

Principal component analysis of whole lung samples in 22,523-gene probe expression space. 2 replicate samples were selected per condition: 2 strains × 3 ages × 2 exposures. The principal components 1–3 (PC1–3) coordinate centroids are plotted for each condition. Samples are marked Strain.Age. Connecting lines are colored gray for SAL and black for OVA. PC1 appears to correspond to developmental age, PC2 to strain, and PC3 to a combination of age and strain.
We next investigated genes that were affected by OVA exposures. We identified 3,693 probes with reproducible profiles, i.e., linear correlation >0.8 between replicate profiles across strain-age-exposure. To assess the change in the expression time series between OVA and SAL for each strain and each probe, we computed the linear correlation between the OVA vs. SAL profiles across three ages × two replicates. If the expression time series of a gene is different in OVA vs. SAL, its correlation value will be low. An example for two hypothetical profiles of genes A and B is illustrated in Fig. 6A. The expression time series for gene A is well correlated in SAL vs. OVA and poorly correlated for gene B. The expression of gene B during lung development is potentially affected by OVA exposure. Note that, if OVA exposure causes a uniform basal shift in gene expression from SAL exposure as in gene A, the OVA-SAL correlation is 1, and therefore gene A is not considered to be affected by OVA exposure when correlation is used to measure similarity. Figure 6B shows the histograms of OVA-SAL time series correlations for the 3,693 reproducible probes for each strain. Overall, the Lewis pups have more poorly correlated genes than BN pups.
Fig. 6.

Gene expression trajectories perturbed by maternal OVA exposure. A: 2 hypothetical genes illustrating the use of linear correlation between developmental time series expression profiles to identify developmental expression affected by maternal SAL vs. OVA exposures. The SAL vs. OVA correlation value is inversely proportional to effect of OVA exposure. The expression time series for gene A is well correlated in SAL vs. OVA and poorly correlated for gene B. The expression of gene B during lung development is potentially affected by OVA exposure. If OVA exposure causes a uniform basal shift in gene expression from SAL exposure as in gene A, the OVA-SAL correlation is 1, and therefore gene A is not considered to be affected by OVA exposure when correlation is used to measure similarity. B: 3,693 reproducible probes (correlation between 1st and 2nd replicate profiles > 0.8) and the histogram of their OVA vs. SAL correlation values for each rat strain. The dashed vertical line marks the threshold for OVA effect (OVA vs. SAL correlation < −0.1), and we count the number of probes below/above this threshold. The light gray solid vertical line marks the median of OVA vs. SAL correlation values for the strain. C: of the 3,693 reproducible probe from B, 1,889 probes have loading magnitudes that are in the top 5% in PC1–3, which correspond to age-strain variation, and 799 of these are affected by OVA exposure, OVA vs. SAL correlation < −0.1 for either strain. D: for gene ontology enrichment analyses, we relaxed the criteria in C. For determining whether a probe is affected by OVA exposure, we identified 5,724 reproducible probes correlation >0.7 between 1st and 2nd replicate profiles. Of these, 3,669 probes have loading magnitudes that are in the top 10% in PC1–3, which correspond to age-strain variation, and 1,806 of these are affected by OVA exposure, OVA vs. SAL correlation < 0.0 for either rat strain.
Among the 3,693 reproducible probes, 1,889 were among the top 5% contributors to PC1–3 age and strain variation. For each of these 1,889 probes in each strain, we computed the OVA-SAL correlation and consider probes with correlation <−0.1 as representing genes whose developmental expression profile is affected by OVA exposure, Fig. 6C. There were 799 such OVA-affected probes: 112 were affected in both strains, 176 in BN only, and 511 in Lewis only. Table 1 summarizes a subset of probes whose developmental time series is affected by maternal OVA exposure. Many of these represent genes previously identified to have a role in lung development. Some have been associated with asthma based on a comprehensive literature search. The expression time series of genes identified by microarray analysis are illustrated in Fig. 7 including Edg4, Orm1 (negative OVA-SAL correlation for BN and Lewis pups, see Table 2) Sfpd, Tgln (negative OVA-SAL correlation for BN pups only), Lgals1, Timp1, and My19 (negative OVA-SAL correlation for Lewis pups only). Figure 8 illustrates qRT-PCR validation for a subset of significantly affected genes.
Table 1.
Function and disease association with Brown Norway and Lewis rats
| Accession | Symbol | Corr BN | Corr L | Function and Disease Association |
|---|---|---|---|---|
| Brown Norway Rat | ||||
| XM_001069955.1 | Edg4 (LPA2) | −0.486 | −0.556 | LPA2, potent bioactive phospholipid, induces cell proliferation, migration, and cytokine release. (52) |
| NM_019195.2 | Cd47 | −0.926 | 0.069 | Antiangiogenic signaling: CD47 ligation to TSP1 inhibits Vegfr2 signaling. (17) |
| NM_001017445.1 | Faf2 | −0.836 | 0.278 | Highly expressed in peripheral blood of patients with AD. (1) |
| NM_020308.1 | Adam15 | −0.830 | 0.569 | Involved in cell adhesion and processing of cytokines. (1) |
| XM_001059464.1 | Cdh11 | −0.808 | −0.937 | Expression regulated by mediators of inflammation such as TNF-α. Plays a role in cartilage destruction in arthritis. (46) |
| NM_053963.1 | Mmp12 | −0.803 | 0.947 | Role in ECM remodeling. Degrades collagens I, III and elastin. Role in emphysema. Mmp12 allele associated with positive effect on lung function in children with asthma. (15, 44) |
| NM_031549.1 | Tagln(SM22) | −0.729 | 0.429 | Transgelin. Actin cross-linking/gelling protein found in fibroblasts and smooth muscle. Is a direct target of TGF-β/Smad3-dependent epithelial cell migration in lung fibrosis. Increased expression in asthmatic lung. (24, 50) |
| NM_053288.1 | Orm1 | −0.596 | −0.267 | Mediator of sphingolipids homeostasis. Genetic variation in Ormdl3 identified as risk factor in childhood asthma. (6, 48) |
| NM_080698.1 | Fmod | −0.576 | −0.311 | Enhanced deposition in the airway wall of atopic asthmatics. (14) |
| NM_012878.1 | Sftpd | −0.546 | 0.315 | Surfactant associated protein with role in immune modulation. Sftpd expression markedly increases in response to acute lung injury and inflammation. (11) |
| XM_001078237.1 | Trim16 (EBBP) | −0.531 | 0.257 | Estrogen responsive B box protein (Ebbp). Role in innate immunity by enhancing pathway of interleukin-1β secretion. (30) |
| XM_001066152.1 | Egfl6 | −0.511 | 0.456 | Member of EGF family with key role in developmental processes in the lung. (1) |
| NM_139111.1 | Cklf | −0.510 | 0.938 | Chemokine-like factor. Potent chemoattractant for neutrophils, monocytes, and lymphocytes with role in inflammation. Stimulates antigen-presenting capability of immature dendritic cells. (41) |
| NM_031775.2 | Casp6 | −0.403 | 0.401 | Sequential activation of caspases regulate apoptosis. Neutrophils activate alveolar macrophages via Casp6-mediated cleavage of IL-1 Receptor-associated kinase-M. (22) |
| Lewis Rat | ||||
| XM_001069955.1 | Edg4 (LPA2) | −0.486 | −0.556 | LPA2. Potent bioactive phospholipid, induces cell proliferation, migration, and cytokine release. (52) |
| NM_053288.1 | Orm1 | −0.596 | −0.267 | Mediator of sphingolipid homeostasis. Genetic variation in Ormdl3 identified as risk factor in childhood asthma. (6, 48) |
| XM_001067182.1 | Myl9 | 0.063 | −0.975 | Myosin light chain, regulate muscle contraction by modulating the ATPase activity of myosin heads. (1) |
| NM_019904.1 | Lgals1 | 0.533 | −0.963 | Lectin, galactosidase-binding, soluble 1. Modulates cell-cell, cell-matrix interactions and cell proliferation. Mediates dendritic cell energy. Modulates release of cytokines involved in autoimmunity. (13, 23) |
| XM_001076104.1 | Mgst3 | 0.229 | −0.910 | Microsomal glutathione S-transferase 3. Involved in the production of leukotrienes and prostaglandin E, important mediators of inflammation. (1) |
| XM_342219.3 | Cpa3 | 0.975 | −0.907 | Carboxypeptidase A. Mast cell Cpa3 has a role in regulating innate immune responses. (35) |
| XM_579614.1 | Ifi271 | 0.528 | −0.898 | Interferon, α-inducible protein 27 interacts with steroid hormone and IFN in endometrium at implantation. (26) |
| XM_343604.3 | Serpine2 | 0.712 | −0.891 | Proposed susceptibility gene for COPD and related phenotypes. May be a risk factor for the development of emphysema. (12) |
| NM_053819.1 | Timp1 | 0.505 | −0.836 | Inhibitor of the MMPs involved in extracellular matrix degradation. Regulates proliferation/apoptosis Transcription induced by cytokines and hormones. (1) |
BN, Brown Norway; L, Lewis; LPA2, lysophosphatidic acid receptor; TSP1, thrombospondin 1; AD, atopic dermatitis; ECM, extracellular matrix; MMP, matrix metalloproteinase.
Fig. 7.

Select strain-specific gene probes whose PN1, 7, and 14 lung expression profiles were affected by maternal OVA exposure as reported by Illumina Rat microarray (12). Plots of the means ± SD of quantile normalized logarithm base 2 expression signal of gene probes in 6 conditions (3 ages × 2 exposures, duplicate measurements per condition). Gray and black represents SAL and OVA exposures, respectively.
Table 2.
Gene ontology enrichment analysis highlights distinct biological processes affected by in utero OVA exposure in developing lungs of atopic Brown Norway and normoresponsive Lewis rat
| Gene Ontology Groups Highlighted In In Utero OVA vs. SAL-exposed Rat | No. of Genes in Group | Human Genes | Fold Enrichment | |
|---|---|---|---|---|
| Brown Norway Rat | ||||
| Lipid metabolism | α-1-Acid glycoprotein | 2 | ORM1, ORM2 | 43.50 |
| Phospholipid dephosphorylation | 3 | PPAP2A, PTEN, PP3R1 | 20.75 | |
| Oxidative stress | Age-dependent oxidative stress | 2 | CLN8, SOD2 | 41.50 |
| Intramolecular oxidoreductase | 3 | PDIA4, PTGIS, GREM1 | 11.61 | |
| Nitric oxide mediated signal transduction | 3 | RASD1, DDAH1, APOE | 8.89 | |
| Response to hydrogen peroxide | 7 | HMOX1, TXNIP, GLRX2, SOD2, PRDX3, SDC1, CASP6 | 5.18 | |
| Immune regulation and function | Macrophage chemotaxis | 3 | EDNRB, SFTPD, CKLF | 13.83 |
| Regulation of leukocyte chemotaxis | 3 | CXCL12, IL6R, GREM1 | 11.32 | |
| Positive regulation of phagocytosis | 3 | CALR, SFTPD, MFGE8 | 10.37 | |
| Regulation of phagocytosis | 4 | CALR, PTEN, SFTPD, MFGE8 | 10.37 | |
| Metalloproteinase | 4 | ACE, ADAM15, MMP12, MME | 7.74 | |
| Acute phase | 4 | IL6R, ORM1, ORM2, CFB | 6.41 | |
| Acute inflammatory response | 10 | EPHX2, IL6R, C5, ORM1, C1QC, PTGER3, ORM2, CASP6, KLKB1, CFB | 4.34 | |
| Cell death | Caspases | 3 | CASP7, CFLAR, CASP6 | 10.18 |
| hsa04210:Apoptosis | 8 | MYD88, CYCS, CASP7, CFLAR, BIRC2, CASP6, PPP3CA, PPP3R1 | 3.00 | |
| Negative regulation of apoptosis | 24 | EDNRB, PTEN, BCL2A1, CLN8, SOD2, SOX4, TAX1BP1, ILK, CD74 | 2.98 | |
| Aging | 7 | CALR, SOCS2, CLN8, PTEN, SOD2, ILK, TIMP3 | 2.71 | |
| Regulation of cell death | 44 | CLN8, GCH1, RTEL1, BLOC1S2, PCSK6, IL6R, ACTN4, JAK2, CASP6 | 2.37 | |
| Cell death | 37 | CLN8, GLRX2, CASP6, CASP7, ECE1, RTEL1, ITPR1, PTGER3, HMOX1, DBC1 | 2.24 | |
| Cell proliferation | Regulation of cell growth | 10 | TAF9B, SOCS2, CYR61, CTH, SPHK1, ILK, GREM1, HTRA3, SEMA4F | 2.30 |
| Regulation of cell proliferation | 37 | EDNRB, SFTPD, SOX4, VCAM1, ADAMTS1, MMP12, CD47, COMT, GAL | 2.07 | |
| Positive regulation of cell proliferation | 17 | CCND1, CALR, IL6R, AGGF1, NBN, SOX4, MMP12, CD47, ILK, FIGF, | 1.78 | |
| Negative regulation of cell proliferation | 18 | TOB1, FRK, PTEN, SOD2, BTG2, SFTPD, ADAMTS1, ILK, COMT, GPC3 | 2.12 | |
| Lewis Rat | ||||
| Lipid metabolism | 3-Hydroxyacyl-CoA dehydrogenase | 3 | CRYL1, HADH, HADHA | 18.84 |
| Inositol polyphosphate kinase | 4 | ITPKA, IPMK, IP6K1, ITPKC | 14.36 | |
| Fatty acid elongation in mitochondria | 4 | HADH, MECR, HADHA, PPT2 | 9.74 | |
| Inositol metabolic process | 5 | ITPKA, PTEN, IPMK, SLC5A3 | 5.99 | |
| Angiogenesis/angiotensin | Regulation of angiotensin metabolic process | 3 | ECE1, ACE, ATP6AP2 | 13.48 |
| Cell cycle | Positive regulation of angiogenesis | 6 | RUNX1, BTG1, NOS3, GATA2, ANGPTL4, SPHK1 | 5.18 |
| Microtubules | Cyclins_&_p27_cell cycle | 3 | CDK4, CDKN1B, CDC2 | 11.93 |
| cell cycle control | 6 | CDC25B, CCNA2, MKI67, RBL2, CDK4, CDC2 | 5.16 | |
| DNA replication | 13 | ORC6L, MCM3, GINS4, POLD1, POLD2, RRM2, RFC3, RFC2, DSCC1, LIG1, NASP, RPA2, CHAF1B | 3.94 | |
| Mitosis | 26 | CETN3, CDC2, FAM33A, SETD8, CEP55, PLK1, CDCA3, BUB1B, ACTB, HAUS4, CDCA8, CCNA2, KIFC1, RUVBL1, CENPE, RAN, NCAPD2, SPC25, CDC25B, ARHGEF2, CCNB2, KIF2C, NUF2, TIMELESS, NDE1, CDC20 | 3.79 | |
| Positive regulation of cell cycle | 8 | CALR, TGFA, ID2, SPHK1, EGF, CENPE, PEBP1, ESPL1 | 3.15 | |
| β-Tubulin, autoregulation binding site | 4 | TUBB2C, KIFC2, TUBB6, TUBB | 5.58 | |
| Kinetochore | 11 | SPC25, FAM33A, CENPT, KIF2C, NUF2, NDE1, PLK1, BUB1B, CENPE, NUP107, NUP85 | 4.66 | |
| Microtubule binding | 10 | FAM33A, UXT, ARHGEF2, NDE1, CDK5RAP2, GABARAPL2, KLC3, SBDS, RAE1, TRIM54 | 3.29 | |
| Spindle | 20 | CDCA8, KIF15, SNCG, TUBG1, KIFC1, KIF22, KIF4A, CENPE, SBDS, NUP85, AURKA, CDC2, FAM33A, ARHGEF2, NDE1, PLK1, CDC20, BUB1B, HAUS4, ROCK2 | 3.17 | |
| Microtubule organizing center | 28 | CETN3, ESPL1, AURKA, CEP55, TACC1, PLK1, BUB1B, HAUS4, ROCK2, RAB3IP, MCM3, TSGA14, UXT, KIF15, SNCG, TUBG1, CDK5RAP2, KIFC1, NCRNA00153, RUVBL1, FAM125A, EZR, CDC25B, NME1, NME2, NDE1, NDRG2, ARL2, NDN | 2.58 | |
Not all genes in larger ontology groups are listed. Fold enrichment represents odds ratio that the difference in developmental pattern of expression for genes in the ontology group are a consequence of in utero ovalbumin (OVA) exposure. SAL, saline.
Fig. 8.
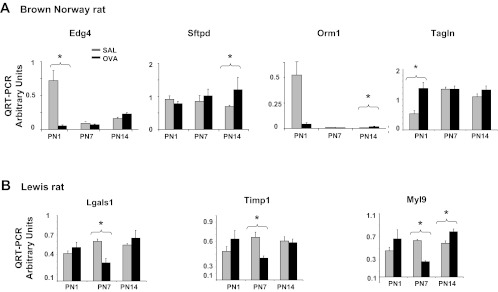
Validation of strain-specific perturbation of trajectories of respiratory gene expression in OVA-exposed pups by qRT-PCR. mRNA expression in whole lung tissue isolated from OVA or SAL exposed BN (A) and Lewis (B) pups was assessed by quantitative real-time PCR at PN1, 7, and 14. (n = 4, *P < 0.05, all genes BN and Lewis).
Multiple OVA-specific probes are differentially expressed in lymphoblastoid cell lines from human asthmatic vs. nonasthmatic sibling pairs.
We compared genes affected by OVA exposure in the developing lungs of our rat strains to those differentially expressed in lymphoblastoid cells from asthmatic vs. nonasthmatic human sibling pairs (Fig. 9, Table 1) (29). Of the 148 human homologs corresponding to the 288 OVA-affected probes in BN rat, 36 (24.3%) were differentially expressed in asthma. Of the 326 human homologs corresponding to the 623 OVA-affected probes in Lewis rats, 61 (18.7%) were differentially expressed in asthma. Both gene sets were enriched for asthmatic vs. nonasthmatic differential expression, 2.1-fold for BN, 1.5-fold for Lewis-Fisher exact test P < 0.0033. There was greater overlap between the OVA-affected probes in the atopic BN than in the Lewis rat.
Fig. 9.

Genes perturbed by OVA exposure are also differentially expressed in lymphoblastoid cell lines from asthmatic patients. Maternal OVA exposure affects genes in BN (n = 36) and Lewis (n = 61) rat strains that are also differentially expressed in lymphoblastoid cell lines of asthmatic vs. nonasthmatic human sibling pairs in Moffatt et al. 2007 (21). The contingency tables show the number of overlapping genes between the rat and human data. The significance of overlap was assessed using the Fisher exact test. The odds ratio represents the fold enrichment or strength of the overlap.
Gene ontology enrichment analysis highlights strain-specific biological processes affected by maternal OVA exposure.
We performed gene ontology (GO) enrichment analysis on the human gene homologs of rat genes affected by OVA exposure in each strain. Here we relaxed the above criteria for determining whether a gene is affected by OVA exposure to enlarge the set of genes for GO analysis (Fig. 6D). The reproducibility criterion was relaxed to correlation >0.7 (compared to >0.8 above) between replicate sample expression profiles across all 12 strain-age-exposure conditions resulting in 5,724 reproducible probes. Next, we restricted to 3,669 probes in the top 10% highest contributors (compared to top 5% above) to transcriptomic variance along PC1–3. Finally, 771 and 1,344 probes were found to be affected by OVA exposure with correlation <0.0 (compared to <−0.1 above) between OVA vs. SAL profiles across age in BN and Lewis strains, respectively. Strain-specific biological processes enriched in these probe sets include lipid metabolism, oxidative stress, immune regulation and function, cell death and cell proliferation in BN pups, lipid metabolism, angiogenesis, cell cycle, and microtubules in Lewis pups (Table 2).
DISCUSSION
Alveolar development begins at 28 wk of gestation, and alveoli increase in number, size, and complexity until 3–4 yr (8). In rodents, alveogenesis is exclusively postnatal. Respiratory function tracks from the first year of life into adulthood, and poor lung development is a risk factor for asthma (2). Multiple studies on the association between exposures and subsequent asthma risk (reviewed in Ref. 32) have confirmed that timing of exposure is a critical variable in determining asthma risk. The maternal environment is the first exposure that contributes to this risk. Although epidemiological studies have revealed many environmental risk factors for asthma, how individual genotypes modulate responses to these factors is poorly understood.
We reported on the impact of asthma susceptibility traits on respiratory development (7). A rat strain that modeled the trait of atopy (BN) had a distinct developmental respiratory phenotype associated with a unique transcriptome signature. We hypothesized that these transcriptomic differences would be exaggerated in pups exposed to allergen in utero. However, we did not predict the extent to which maternal OVA exposure would impact developmental pulmonary gene expression and respiratory phenotype in the normoresponsive rat; nor did we expect that the gene sets that are most profoundly influenced by allergen exposure would be almost entirely different from those that confer innate genetic susceptibility in the naturally atopic rat. The effect of OVA on the BN and Lewis transcriptomes was distinct with only 14% overlap in allergen-induced alterations in the developmental trajectories of gene expression in the alveolarizing lung. Of note, human homologs of OVA-specific probes in rat models were enriched in human lymphoblastoid cell lines prepared from asthmatic patients. Moreover the atopic BN rat showed a greater percentage of overlap and greater fold enrichment than the nonatopic Lewis rat.
The BN rat has long been considered the most suitable animal model for the study of allergic inflammation (38). Differences in the allergic response to antigen between adult Lewis and BN rats are well documented. In BN rats, injections of OVA induce a high IgE-specific antibody response and high total serum IgE levels accompanied by T cell-mediated airway eosinophilia and expression of Th2 cytokines. Lewis rats produce a significantly lower response (1). The BN dam may be considered analogous to allergic mothers who are at higher risk of having an asthmatic child. Our findings support specific roles for environmental exposures in influencing fetal respiratory development in offspring of atopic vs. nonatopic mothers. These distinct effects of maternal exposure on lung development in the two strains may reflect both direct effects of OVA on in utero development and indirect effects of maternal inflammation in the atopic vs. nonatopic dams. To our knowledge, this is the first report of the effects of maternal gene-environment interactions on in utero lung development with relevance to asthma risk. Our findings in the Lewis rat also underscore the profound impact of maternal exposure on lung development and allergic reaction even in the absence of preexisting genetic predisposition. These findings also have implications for human asthma studies and emphasize the need to identify children born to mothers in settings of dramatically increased exposures.
Maternal inflammatory mediators are also present in mother's milk, raising the question of whether effects on lung development may be accounted for by postnatal nutrition. However, our findings of profound neutrophilia and eosinophilia and of the most pronounced effects on the lung transcriptome at PN1 in the atopic BN rat underscore the effects of prenatal exposure to OVA in this model. Moreover, we show that a significant percentage of genes affected in Lewis pups at PN7 are the very same as those affected in BN pups at PN1, suggesting that there is a common effect that is temporally distinct in the two strains and that this effect occurs before postnatal nutrition.
MCh-induced lung resistance, BAL neutrophilia, and elevated serum OVA-specific IgE were observed in pups of both strains. BN pups also exhibited BAL eosinophilia with elevated levels of TNF-α, IL-1β, and KC/GRO, cytokines more often associated with inflammatory lung injury at birth, bronchopulmonary dysplasia (BPD) and chronic obstructive pulmonary disease (COPD). Increased risk for asthma and COPD (2) in BPD survivors has been attributed to developmental impairment of lung function. The distinct phenotypes exhibited in the postnatal period were associated with unique trajectories of global gene expression. Many of the strain-specific genes whose trajectories of expression were altered following OVA exposure have a documented role in lung development. Some have also been implicated in respiratory disease including asthma. Although individual genes affected were unique to each of the strains, their potential impact on critical biological processes in respiratory development may be similar. We briefly discuss how these processes may be affected by perturbation of expression of several of these genes.
Cellular phospholipids serve multiple roles in respiratory development and immune defense. Maternal exposures that perturb expression of genes essential to normal phospholipid functions may have subtle effects on the lung that compromise its ability to respond to future environmental insults. The trajectory of expression of Edg4 (Lpa2), a lysophosphatidic acid (LPA) receptor and Orm1 (Human ORMDL3), was affected by OVA in both strains. LPA acts via Edg4 to transactivate factors with roles in developmental remodeling and in remodeling in asthma (49, 42). Orm proteins regulate sphingolipid homeostasis. Human ORMDL3 variation confers increased asthma risk in children estimated at up to 20% (29). Our observed effects on Orm1 in developing lungs provide a unique link between allergen exposure, sphingolipid homeostasis, and lung development.
Surfactant deficiency in severe prematurity causes respiratory distress syndrome and may lead to BPD. Surfactant protein D (SP-D), encoded by Sftpd, produced by alveolar cells in late gestation, functions in innate immune defense. SP-D is induced in acute asthma and is believed to be involved in allergen-induced airway changes. The interaction between SP-D and allergen exposure in the developing lung may have implications for long-term immune defense.
Myoblastic differentiation of lung mesenchymal cells depends on the timely stimulation of contractile proteins including smooth muscle protein 22 (SM22, encoded by Tagln) (37). SM22 also has a key role in pulmonary repair (50, 51). Elevated Tgln expression observed in BN pups is also a characteristic feature of endobronchial biopsies from asthmatic patients (25). The newborn lung is likely to be particularly sensitive to fluctuations in Tagln expression.
Our combined data suggest that inflammatory, immune, structural, and physiological changes in the lung can be identified by analyzing phenotypic susceptibilities of inbred strains in conjunction with investigation of gene-environment interactions in the developing animal. The very early presence of these abnormalities suggests that environmental processes, operating in utero and in early postnatal life, may forecast the extent and severity of chronic lung disease later in life. The characterization of such risk in early life has practical, clinical, and prognostic implications and could set the basis for preventative strategies.
Our findings underscore the need to describe a set of “allergen”-sensitive genes that may constitute a unique class of genes as yet not identified by whole genome studies. Our results also emphasize the need for studies in animal models, where only retrospective analyses are feasible in humans. The ultimate goal is to determine how maternal and early life exposures interact with preexisting respiratory genotypes to initiate asthma pathogenesis to enable the development of early preventive strategies that effectively and safely reverse immune dysfunction.
GRANTS
This work was supported by operating grants from the Canadian Institutes of Health Research (F. Kaplan, MOP 74683) and from the NIH (NHLBI HL091124 to A. Kho, NIH R01 HL97144 to K. Tantisira and S. Weiss, and RHL086601 to B. Raby) and by a graduate scholarship from the Montreal Children's Hospital Research Institute (N. Carpe).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: N.C., J.G.M., S.T.W., and F.K. conception and design of research; N.C. and I.M. performed experiments; N.C., A.T.K., W.Q., and F.K. analyzed data; N.C., A.T.K., W.Q., K.G.T., B.A.R., and F.K. interpreted results of experiments; N.C., I.M., and A.T.K. prepared figures; N.C. and F.K. drafted manuscript; N.C., I.M., A.T.K., W.Q., J.G.M., K.G.T., B.A.R., S.T.W., and F.K. approved final version of manuscript; I.M., A.T.K., J.G.M., K.G.T., B.A.R., S.T.W., and F.K. edited and revised manuscript.
REFERENCES
- 1. Abadie A, Prouvost-Danon A. Specific and total IgE responses to antigenic stimuli in Brown Norway, Lewis, and Sprague Dawley rats. Immunology 39: 561– 569, 1980 [PMC free article] [PubMed] [Google Scholar]
- 2. Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med 357: 1946– 1955, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Barker D. Fetal and Neonatal Origins of Adult Disease. London: BMJ, 1992 [Google Scholar]
- 4. Bellofiore S, Martin JG. Antigen challenge of sensitized rats increases airway responsiveness to methacholine. J Appl Physiol 65: 1642– 1646, 1988 [DOI] [PubMed] [Google Scholar]
- 5. Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, Saatian Y. Orm family proteins mediate sphingolipid homeostasis. Nature 463: 1048– 1053, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carpe N, Mandeville I, Ribeiro L, Ponton A, Martin J, Kho A, Chu J, Tantisira K, Weiss S, Raby B, Kaplan F. Genetic influences on asthma susceptibility in developing lung. Am J Respir Cell Mol Biol 43: 1– 11, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Luca G, Olivieri F, Melotti G, Aiello G, Lubrano L, Boner AL. Fetal and early postnatal life roots of asthma. J Matern Fetal Neonatal Med 23: 80– 83, 2010 [DOI] [PubMed] [Google Scholar]
- 9. Eidelman D, Bellofiore S, Martin J. Late airway responses to antigen challenge in sensitized inbred rats. Am Rev Respir Dis 137: 1033– 1037, 1988 [DOI] [PubMed] [Google Scholar]
- 10. Fedorov IA, Wilson SJ, Davies DE, Holgate ST. Epithelial stress and structural remodelling in childhood asthma. Thorax 60: 389– 394, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Forbes LR, Haczku A. SP-D and regulation of the pulmonary innate immune system in allergic airway changes. Clin Exper All 40: 547– 562, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fujimoto K, Ikeda S, Arai T, Tanaka N, Kumasaka T, Ishii T, Kida K, Muramatsu M, Sawabe M. Polymorphism of SERPINE2 gene is associated with pulmonary emphysema in consecutive autopsy cases. BMC Med Genet 11: 159, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gieseke F, B+Âhringer J, Bussolari R, Dominici M, Handgretinger R, Miller I. Human multipotent mesenchymal stromal cells use galectin-1 to inhibit immune effector cells. Blood 116: 3770– 3779, 2010 [DOI] [PubMed] [Google Scholar]
- 14. Huang J, Olivenstein R, Taha R, Hamid Q, Ludwig M. Enhanced proteoglycan deposition in the airway wall of atopic asthmatics. Am J Respir Crit Care Med 160: 725– 729, 1999 [DOI] [PubMed] [Google Scholar]
- 15. Hunninghake GM, Cho MH, Tesfaigzi Y, Soto-Quiros ME, Avila L, Lasky-Su J, Stidley C, Melen E, Soderhall C, Hallberg J, Kull I, Kere J, Svartengren M, Pershagen G, Wickman M, Lange C, DeMeo DL, Hersh CP, Klanderman BJ, Raby BA, Sparrow D, Shapiro SD, Silverman EK, Litonjua AA, Weiss ST, Celedon JC. MMP12, lung function, and COPD in high-risk populations. N Engl J Med 361: 2599– 2608, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jeffrey PK. The development of large and small airways. Am J Respir Crit Care Med 157: S174– S180, 1998 [DOI] [PubMed] [Google Scholar]
- 17. Kaur S, Martin-Manso G, Pendrak ML, Garfield SH, Isenberg JS, Roberts DD. Thrombospondin-1 Inhibits VEGF Receptor-2 Signaling by Disrupting Its Association with CD47. J Biol Chem 285: 38923– 38932, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kho AT, Bhattacharya S, Mecham BH, Hong J, Kohane IS, Mariani TJ. Expression profiles of the mouse lung identify a molecular signature of time-to-birth. Am J Respir Cell Mol Biol 40: 47– 57, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kho AT, Bhattacharya S, Tantisira KG, Carey VJ, Gaedigk R, Leeder JS, Kohane IS, Weiss ST, Mariani TJ. Transcriptomic analysis of human lung development. Am J Respir Crit Care Med 181: 54– 63, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kicic A, Sutanto E, Stevens P, Knight D, Stick S. Intrinsic biochemical and functional differences in bronchial epithelial cells of children with asthma. Am J Respir Crit Care Med 174: 1110– 1118, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Kihlstrom A, Lilja G, Pershagen G, Hedlin G. Exposure to high doses of birch pollen during pregnancy, and risk of sensitization and atopic disease in the child. Allergy 58: 871– 877, 2003 [DOI] [PubMed] [Google Scholar]
- 22. Kobayashi H, Nolan A, Naveed B, Hoshino Y, Segal LN, Fujita Y, Rom WN, Weiden MD. Neutrophils activate alveolar macrophages by producing caspase-6-mediated cleavage of IL-1 receptor-associated kinase-M. J Immunol 186: 403– 410, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuo PL, Hung JY, Huang SK, Chou SH, Cheng DE, Jong YJ, Hung CH, Yang CJ, Tsai YM, Hsu YL, Huang MS. Lung cancer-derived galectin-1 mediates dendritic cell anergy through inhibitor of DNA binding 3/IL-10 signaling pathway. J Immunol 186: 1521– 1530, 2011 [DOI] [PubMed] [Google Scholar]
- 24. Leguillette R, Laviolette M, Bergeron C, Zitouni N, Kogut P, Solway J, Kachmar L, Hamid Q, Lauzon AM. Myosin, transgelin, and myosin light chain kinase: expression and function in asthma. Am J Respir Crit Care Med 179: 194– 204, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leguillette R, Laviolette M, Lauzon AM. Quantitative smooth muscle RNA analysis of asthmatic human bronchial biopsies. Proc Amer Thorac Soc 2: A338, 2005 [Google Scholar]
- 26. Li Q, Zhang M, Kumar S, Zhu LJ, Chen D, Bagchi MK, Bagchi IC. Identification and implantation stage-specific expression of an interferon-regulated gene in human and rat endometrium. Endocrinology 142: 2390– 2400, 2001 [DOI] [PubMed] [Google Scholar]
- 27. Martin J, Tamaoka M. Rat models of asthma and chronic obstructive lung disease. Pulm Pharmacol Ther 19: 377– 385, 2006 [DOI] [PubMed] [Google Scholar]
- 28. Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, Morgan WJ. Asthma and wheezing in the first six years of life. N Engl J Med 332: 133– 138, 1995 [DOI] [PubMed] [Google Scholar]
- 29. Moffatt MF. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448: 470– 473, 2007 [DOI] [PubMed] [Google Scholar]
- 30. Munding C, Keller M, Niklaus G, Papin S, Tschopp J, Werner S, Beer HD. The estrogen-responsive B box protein: a novel enhancer of interleukin secretion. Cell Death Differ 13: 1938– 1949, 2006 [DOI] [PubMed] [Google Scholar]
- 31. Murphey S, White C, Waters T, Fireman P. Reagin synthesi in inbred rats. J Allergy Clin Immunol 58: 381– 386, 1976 [DOI] [PubMed] [Google Scholar]
- 32. Ober C, Vercelli D. Gene environment interactions in human disease: nuisance or opportunity? Trends Genet 27: 107– 115, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Office on Smoking, and Health, Department of Health and Human Services The health consequences of involuntary exposure to tobacco smoke: a report of the surgeon general. In: Respiratory Effects in Children of Exposure to Second hand Smoke. Atlanta, GA: US Department of Health and Human Services. Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, 2006 [Google Scholar]
- 34. Palmer LJ, Rye PJ, Gibson NA, Burton PR, Landau LI, Lesouef PN. Airway responsiveness in early infancy predicts asthma, lung function, and respiratory symptoms by school age. Am J Respir Crit Care Med 163: 37– 42, 2001 [DOI] [PubMed] [Google Scholar]
- 35. Pejler G, Knight SD, Henningsson F, Wernersson S. Novel insights into the biological function of mast cell carboxypeptidase A. Trends Immunol 30: 401– 408, 2009 [DOI] [PubMed] [Google Scholar]
- 36. Pohunek P, Warner JO, TurzÃ-kovà J, Kudrmann J, Roche WR. Markers of eosinophilic inflammation and tissue re-modelling in children before clinically diagnosed bronchial asthma. Pediatr Allergy Immunol 16: 43– 51, 2005 [DOI] [PubMed] [Google Scholar]
- 37. Popova AP, Bozyk PD, Goldsmith AM, Linn MJ, Lei J, Bentley JK, Hershenson MB. Autocrine production of TGF-β1 promotes myofibroblastic differentiation of neonatal lung mesenchymal stem cells. Am J Physiol Lung Cell Mol Physiol 298: L735– L743, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ramos-Barbon D, Ludwig MS, Martin JG. Airway remodeling: lessons from animal models. Clin Rev Allergy Immunol 27: 3– 21, 2004 [DOI] [PubMed] [Google Scholar]
- 39. Saglani S, Payne DN, Zhu J, Wang Z, Nicholson AG, Bush A, Jeffery PK. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am J Respir Crit Care Med 176: 858– 864, 2007 [DOI] [PubMed] [Google Scholar]
- 40. Sapienza S, Du T, Eidelman DH, Wang NS, Martin JG. Structural changes in the airways of sensitized brown Norway rats after antigen challenge. Am Rev Respir Dis 144: 423– 427, 1991 [DOI] [PubMed] [Google Scholar]
- 41. Shao L, Li T, Mo X, Majdic O, Zhang Y, Seyerl M, Schrauf C, Ma D, Stackl J, Han W. Expressional and functional studies of CKLF1 during dendritic cell maturation. Cell Immunol 263: 188– 195, 2010 [DOI] [PubMed] [Google Scholar]
- 42. Shiomi T, Boudreault F, Padem N, Higashiyama S, Drazen JM, Tschumperlin DJ. Lysophosphatidic acid stimulates epidermal growth factor-family ectodomain shedding and paracrine signaling from human lung fibroblasts. Wound Repair Regen 19: 229– 240, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stick S, Stick S. Pediatric origins of adult lung disease. 1. The contribution of airway development to paediatric and adult lung disease. Thorax 55: 587– 594, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taddese S, Jung MC, Ihling C, Heinz A, Neubert RH, Schmelzer CE. MMP-12 catalytic domain recognizes and cleaves at multiple sites in human skin collagen type I and type III. Biochim Biophys Acta 1804: 731– 739, 2010 [DOI] [PubMed] [Google Scholar]
- 45. Turner SW, Palmer LJ, Rye PJ, Gibson NA, Judge PK, Young S, Landau LI, Le Souef PN. Infants with flow limitation at 4 weeks: outcome at 6 and 11 years. Am J Respir Crit Care Med 165: 1294– 1298, 2002 [DOI] [PubMed] [Google Scholar]
- 46. Vandooren B, Cantaert T, Borg MtT, Noordenbos T, Kuhlman R, Gerlag Dl Bongartz T, Reedquist K, Tak PP, Baeten D. Tumor necrosis factor drives cadherin 11 expression in rheumatoid inflammation. Arthritis Rheum 58: 3051– 3062, 2008 [DOI] [PubMed] [Google Scholar]
- 47. Warner JA, Jones C, Jones A, Warner J. Prenatal origins of allergic disease. J Allergy Clin Immunol 105: S493– S496, 2000 [DOI] [PubMed] [Google Scholar]
- 48. Wu H. Genetic variation in ORM1-like 3 (ORMDL3) and gasdermin-like (GSDML) and childhood asthma. Allergy 64: 629– 635, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yogi A, Callera GE, Aranha AB, Antunes TT, Graham D, McBride M, Dominiczak A, Touyz RM. Sphingosine-1-phosphate-induced inflammation involves receptor tyrosine kinase transactivation in vascular cells: upregulation in hypertension. Hypertension 57: 809– 818, 2011 [DOI] [PubMed] [Google Scholar]
- 50. Yu H, Konigshoff M, Jayachandran A, Handley D, Seeger W, Kaminski N, Eickelberg O. Transgelin is a direct target of TGF-β/Smad3-dependent epithelial cell migration in lung fibrosis. FASEB J 22: 1778– 1789, 2008 [DOI] [PubMed] [Google Scholar]
- 51. Zhang JC, Kim S, Helmke BP, Yu WW, Du KL, Lu MM, Strobeck M, Yu Q, Parmacek MS. Analysis of SM22alpha-deficient mice reveals unanticipated insights into smooth muscle cell differentiation and function. Mol Cell Biol 21: 1336– 1344, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhao Y, Natarajan V. Lysophosphatidic acid signaling in airway epithelium: role in airway inflammation and remodeling. Cell Signal 21: 367– 377, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]