

Abstract
Most cancers use glucose as substrate for aerobic glycolysis in preference to oxidative phosphorylation. However, variable glucose concentrations within the in-vivo tumor microenvironment may necessitate metabolic plasticity. Furthermore, little information exists on a role for estrogen receptors in modulating possible metabolic adaptations in breast cancer cells. Here we find that MCF-7 cells switch between metabolic pathways depending on glucose availability and 17β-estradiol (E2) potentiates adaptation. In high glucose conditions E2 up-regulates glycolysis via enhanced AKT kinase activity and suppresses tricarboxylic acid cycle activity. After a decrease in extracellular glucose, mitochondrial pathways are activated in preference to glycolysis. In this setting, E2 suppresses glycolysis and rescues cell viability by stimulating the tricarboxylic acid cycle via the up-regulation of pyruvate dehydrogenase (PDH) activity. E2 also increases ATP in low glucose-cultured cells, and the novel phosphorylation of PDH by AMP kinase is required for these metabolic compensations. Capitalizing on metabolic vulnerability, knockdown of PDH in the low-glucose state strongly potentiates ionizing radiation-induced apoptosis and reverses the cell survival effects of E2. We propose that lowering glucose substrate and inhibiting PDH may augment adjuvant therapies for estrogen receptor-positive breast cancer.
In cancer, energy production occurs predominantly via aerobic glycolysis unlike less proliferative cells that mostly utilize the mitochondrial tricarboxylic acid cycle (TCA) and oxidative phosphorylation (oxphos) pathways. Such metabolism is referred to as the “Warburg effect” (1, 2). The altered metabolic phenotype is important for cancer cell biology and has been linked to epithelial tumor progression and poor clinical prognosis (3). Breast carcinoma cell lines, similar to other cancer cell types, exhibit glucose dependency and derive the majority of energy as ATP from high throughput glycolysis (3). Additionally, several laboratories have shown that intermediate glucose metabolites are important to tumor biology, unrelated to ATP production. For example, glucose-6-phosphate (G6P) is shunted into the pentose phosphate pathway, thereby enhancing reduced nicotinamide adenine dinucleotide (NAD) phosphate and nucleotide generation, respectively, favoring lipid and nucleic acid synthesis required by highly proliferative tumors (4).
In vivo, uneven blood flow to cancer exists and results in hypoxia and differential glucose concentrations within the microenvironment of the tumor (5, 6). Hypoxia causes a shift toward glycolysis in cancer cells through the up-regulation of HIF-1α that stimulates glycolytic and glucose transporter gene expression (7). Switching cancer cells to an aglycemic environment induces stress and cell death particularly in cells that exhibit increased AKT activity (8). However, adaptation to more physiological variations in glucose is poorly understood. Adaptation likely occurs not only at the level of the tumor but also by the organism, because crossing mouse models of mammary cancer to diabetic mice produces a highly proliferative, accelerated tumor phenotype (9, 10). Carefully controlling the metabolic syndrome in these models significantly diminishes the aggressiveness of the tumors in vivo (10).
17β-Estradiol (E2) and the estrogen receptor (ER) α have been implicated in promoting the proliferation, survival, and migration of breast cancer cells through multiple mechanisms thereby strongly contributing to tumor biology (11, 12). However, it is unknown whether ER promotes metabolic adaptation in breast cancer. Here we investigated the metabolic response of breast cancer cells that were switched from high glucose medium to medium containing a decreased but physiological level of glucose that is characteristic in vivo. We found that rapid signaling by ER enhances tumor cell adaptation to lower glucose substrate, increasing TCA cycle activity and ATP production. These adaptations were necessary for E2-stimulated viability of the cells in low glucose and identified a metabolic vulnerability that we used to promote enhanced cancer cell death by radiation.
Materials and Methods
Reagents
E2, ICI 182,780 (ERα antagonist), 4,4′,4′-(4-propyl-[1H]-pyrazole-1,3,5-triyl) trisphenol [ERα agonist propyl pyrazole triol (PPT)], diarylpropionnitrile [ERβ agonist (DPN)], and an AMP kinase (AMPK)-specific inhibitor Compound C, were obtained from Tocris Bioscience (Ellisville, MO). Antibodies to phospho-specific AMPK (Tyr172), total AMPKα, phospho-specific AKT (Ser473), and total AKT were from Cell Signaling Technologies (Danvers, MA). Antibodies to total PDH kinase (PDK)1–4 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The PDHE1α subunit for the kinase assays was from Abcam (Cambridge, MA). Lactate and citrate assay kits were from Biovision, Inc. (Milpitas, CA). The PDH enzyme activity kit (Mitosciences, Eugene, OR), ATP measurement kit (Invitrogen, Carlsbad, CA) and the cell viability assay 3-(4,5-dimethylthiazol 2yl)-2,5-diphenyttetrazolium bromide (MTT) kits (Promega Corp., Madison, WI) were purchased as noted. Two specific small interfering RNA (siRNA) oligonucleotides to AKT, PDHE1α, PDK 1–4 and AMPK each were from Santa Cruz Biotechnology. siRNAs to ERα and β were obtained from QIAGEN (Chatsworth, CA).
Cell culture
The ER-positive MCF-7 breast carcinoma cell line (American Type Culture Collection, Manassas, VA) was used between passages 18–30. Cultures were maintained under a 5% CO2/O2 (normoxia) environment. The cells were seeded into 100-mm Petri dishes, six-well dishes, or 96-well plates depending on the particular assay. Cells were grown in DMEM-Ham's F-12 nutrient mixture medium (DMEM-F12) (Sigma, St. Louis, MO) without phenol red, containing 10% fetal calf serum, 1% antibiotic-antimycotic (Invitrogen), and the standard tissue culture glucose concentration of 450 mg/dl. This is representative of a diabetic milieu and was therefore referred to as “high glucose” (HG). Cells were cultured in DMEM-F12 with a glucose concentration of 100 mg/dl for 1 d, 1 wk, or 2 months depending on the assay requirement. This glucose concentration is physiological and representative of the in vivo concentration in human serum. In the cell culture setting this is the “low glucose” (LG) condition. ZR-75–1 breast cancer cells (ATCC) were similarly cultured. At 24 h before the experiments cells were switched to fetal calf serum-free media in most studies.
siRNA transfection
The cells were seeded into six-well dishes and grown to 50–60% confluency and were transfected with one of two separate and specific siRNAs to each target. The transfection mix used was 4 μl of Oligofectamine, 100 μl Opti-Mem, and 1 μg of siRNA. The Oligofectamine and siRNA solutions were combined, and 200 μl of this transfection mix were combined with 800 μl Opti-Mem in each dish. The cells were incubated overnight with the complete transfection mix and incubated the following day with E2 (1 nm) or other compounds for the designated times. The knockdowns were validated by quantitative real-time PCR (qRT-PCR). siRNAs for PDHE1α, AMPK, and PDK 1–4 were validated in this study (Supplemental Fig. 1 published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org), and we previously validated the AKT and ER isoform knockdown efficiencies in MCF-7 cells (13–15).
Metabolic assays
PDH activity was determined by an ELISA-based kit. Citrate, G6P, and lactate production was determined using colorimetric assays. Cell counts were used to normalize the experiments. Cells in six-well dishes were grown to 70% confluency and synchronized by complete serum removal for 24 h. The cells were exposed to E2 (0.1–10 nm) for various time points, and the cells were lysed and centrifuged at 1000 × g for 10 min and the supernatant was removed. The lysis buffer supplied with the kits was supplemented with protease inhibitor cocktail and 100 μm phenylmethylsulfonyl fluoride. For PDH activity, the lysed cell supernatant was incubated in a 96-well dish coated with an anti-PDH antibody. After an immunocapture step, a reaction mix containing NAD+ was added, and the conversion of NAD+ to reduced NAD was measured over 15 min; PDH activity was determined as the slope of the line of absorbance constructed by measuring each sample every minute for 15 min to obtain the line. The reaction utilizes a PDH reporter the concentration of which is measured by absorbance at 450 nm using a 96-well plate reader. Citrate was measured in cell supernatants incubated in a 96-well dish with an enzyme mix that promotes the conversion of citrate to oxaloacetate; the higher oxaloacetate produced in the assay dish indicates increased citrate production. To measure either G6P or lactate levels, a 50-μl aliquot of the lysed cellular supernatant was incubated with specific probes to each metabolic enzyme producing a colored product. For G6P and lactate, each kit utilizes probes that conjugate each glucose metabolite producing an intense color measured at 450 nm.
PDHE1α mutation
The PDHE1α 390-amino acid sequence was analyzed for the presence of the AMPK canonical motif, LRRVxSxxNL, or a homologous sequence by the CLUSTAL W (1.83) program. A putative AMPK substrate motif was found surrounding the serine 19 residue (SRVLSGASQ). Serine 19 was mutated to alanine (Genscript), and both wild-type (WT) and mutated PDHE1α DNAs were cloned into pUC57 vectors. Bacterial cells transformed with WT or mutant PDHE1a plasmids were harvested, lysed, centrifuged at 10,000 × g for 10 min, decanted, and the weight of the pellet determined. The cell pellet was resuspended in Bugbuster reagent (Novagen, Madison, WI), 5 ml per g of wet cell paste. Protease inhibitor cocktail (Sigma) and 25 U of benzonase (Novagen) per ml of Bugbuster reagent were added, and cell suspensions were incubated on a rotating mixer at a slow setting for 10–20 min at room temperature. Insoluble cell debris was then removed by centrifugation at 16000 × g for 20 min at 4 C, and the supernatants were transferred to a fresh tube. The supernatants were used directly for purification to near homogeneity by batch-wise adsorption onto a nickel-charged nitrilotriacetic acid-agarose resin (Ni-NTA-agarose) (Qiagen) according to the manufacturer's protocol. Resin suspension (4 ml) was added into a 15-ml centrifuge tube and centrifuged at 2000 rpm for 2 min to pellet the resin, and the supernatants were removed. The resin was washed with H2O (6 ml × two washes) followed by one wash with binding buffer [20 mm Tris, 0.5 m NaCl, 5 mm imidazole, 10 mm mercaptoethanol (pH 7.9)]. To the packed resin, bacterial supernatant was added and rotated at 4 C for 1 h. The resin was spun down and the supernatant removed. The resin was washed six times with 6 ml of wash buffer [20 mm Tris, 0.5 m NaCl, 60 mm imidazole, 10 mm mercaptoethanol (pH 7.9)]. The resin was eluted with 1 ml elution buffer for 1 h at 4 C on the rotator. The resin was spun down, and the supernatant was dialyzed against one liter of 50 mm Tris, 10% glycerol, 10 mm mercaptoethanol (pH 8.0) overnight. Dialyzed samples were used as a substrate protein in kinase assay. Subsequent studies used the PDH E1a mutant for AMPK studies.
Kinase activities
For PDK and AMPK activity assays, the cells were synchronized for 24 h in serum-free media. The cells were then exposed to E2 (1 nm) for 5 and 15 min, then lysed in a kinase-specific buffer lysis buffer: [50 mm Tris-HCl (pH 7.5), 5 mm EDTA, 100 mm NaCl, 50 mm NaF, 100 μm phenylmethylsulfonyl fluoride and protease inhibitor cocktail]. After centrifugation, the protein from supernatants was immunoprecipitated overnight at 4 C with protein A agarose conjugated to PDK1–4 or AMPK antibodies. Kinase activities were determined by incubating the precipitates with 40 μl reaction mix containing 10 ng PDHE1α peptide, 25 mm HEPES (pH 7.5), 10 mm MgAc, 40 μm ATP, 10 μCi [32P]ATP, and 2 mm dithiothreitol incubated for 30 min at 30 C. The reaction was stopped by adding 2× Laemmli buffer and boiling for 5 min. Samples were separated by 10% SDS-PAGE followed by gel drying and autoradiography.
The activating phosphorylations of AMPK and AKT were determined by Western blot analysis using phospho-specific antibodies. Protein normalization was done using the Bradford assay (Pierce Chemical Co., Rockford, IL). Protein (50 μg) from each experimental condition was dissolved in a 4× Laemmli buffer (containing 2-mercaptoethanol) and boiled for 5 min. The samples were separated on a 10% sodium dodecyl sulfate gel and transferred to polyvinylidene fluoride membrane and then subjected to autoradiography. The phosphorylated (activated) kinases were detected using phospho-specific antibodies against tyrosine 172 and serine 473 of AMPK and AKT, respectively. The membranes were stripped and reprobed for total amounts of the kinase proteins as controls.
Cell viability
Breast cancer cell viability was determined by MTT assay from Promega. Cells (5000) were seeded per well of a 96-well dish. Cells were serum starved for 24 h for synchronization and then treated with E2 (1 nm) for 24 h under either HG or LG conditions. of Culture media (100 μl) (containing 15 μl of MTT dye solution were then added to each well, and the cells were incubated at 37 C for 4 h. The formed formazan crystals in the cells were solubilized with a stop solution provided by the manufacturer, and absorbance was measured at 570 nm. The more intense signal reflects the greater viability of the cells.
ATP measurement
ATP was measured using a bioluminescent assay. Cells were counted and seeded into six-well dishes to 70% confluency, synchronized overnight, and treated with E2 (1 nm, 2–24 h). The cells were lysed and centrifuged at 10,000 rpm for 10 min. Sample aliquots were then incubated with recombinant firefly luciferase and substrate d-luciferin. The light produced was measured at an emission wavelength of 570 nm (Nova-Star 2000 Livonia, MI).
Terminal deoxynucleotide transferase-mediated dUTP nick end labeling (TUNEL) assay
The TUNEL assay (Roche Pharmaceuticals, Indianapolis, IN) uses terminal deoxynucleotidyl transferase dUTP to nick end label fragmented DNA in the nucleus. The cells were also stained with 4′,6-diamidino-2-phenylindole to enable counting of TUNEL-negative cells. For each experimental condition, 200 cells were assessed for apoptosis and were expressed as a percentage of the total cell count. Immunofluorescent images of the cells were obtained using a NIKON Eclipse TE-200 microscope (Nikon, Melville, NY) at ×200 magnification. A diagnostic instruments camera (model 3.2.0) and the Spot Advance software program captured the images for cell counts.
Reactive oxygen species (ROS) quantification
Cultured cells were loaded with 10 μm 2′7′-dichlorodihydrofluorescin diacetate (CM-H2DCFDA; Molecular Probes, Eugene, OR) for 1 h before radiation or addition of estrogen. In some cells, transfection of siRNA to PDH was done 24 h earlier. After oxidation by ROS, CM-H2DCF is converted to green-fluorescing dichlorofluorescein (DCF) and was quantified in a NovaStar spectrofluorometer (excitation wavelengths, 396 and 510 nm; emission measured at 580 nm). Studies were carried out three times.
qRT-PCR
Total RNA was extracted using the Qiagen RNeasy Mini Kit. RNA purity and concentrations were measured by UV-spectrophotometer (A260 and A280). cDNA was synthesized using approximately 500 ng RNA and Oligo dT primers with the Improm-II reverse transcription system (Promega). qRT-PCR was used to examine the relative expression of AMPK, PDH, and PDK 1–4 and was normalized to the housekeeper GAPDH gene expression. Primers were designed using Primer 3 (http://frodo.wi.mit.edu/). Blast analysis was used to check specificity (Primers: AMPK F (5′-ggagccttgatgtggtagga-3′); AMPK R(5′-tttcatccagccttccattc-3′); PDH F (5′-ttaccgttaccacggacaca-3′); PDH R (5′-ctcttccaaaggtggctcag-3′); PDK1 F(5′-cacgctgggtaatgaggatt-3′), PDK1 R (5′-actgcatctgtcccgtaacc-3′); PDK2 F (5′-atggcagtcctcctctctga-3′), PDK2 R (5′-cacccaccctcttcctaaca-3′); PDK3 Variant 1 F (5′-tgggaaagtccaggtggtag-3′); PDK3 variant 1 R (5′-cccagggatgcttcaaaata-3′); PDK3 Variant 2 F (5′-aacagccaagatgctgtgtg-3′); PDK3 Variant 2 R (5′-agcagggtagccctcttttc-3′); PDK4 F (5′-cctttggctggttttggtta-3′); PDK4 R (5′-cctgcttgggatacaccagt-3′); GAPDH F (5′-ccacagtccatgccatca-3′); GAPDH R (5′-ggatgaccttgcccacag-3′). Primers were designed for an annealing temperature of 55 C and to amplify regions of 150–200 bp. PCR amplicon sizes were confirmed by agarose gel electrophoresis before qRT-PCR analysis. For qRT-PCR analysis, cDNA was combined in a 50-μl reaction with 25 μl of SYBR GreenER qPCR Supermix (Invitrogen), 1 μl of 10 μm forward/reverse primer stocks, and nuclease-free water. Thermocycling was carried out using the iCycler (Bio-Rad Laboratories, Inc., Hercules, CA) with a melting curve temperature of 60 C. Relative mRNA levels were calculated using the Ct method.
Statistics
The results are shown as bar graphs based on data from at least three experiments combined. Bars on graphs indicate means ± sems, and significance was determined by ANOVA plus Schefe's post hoc test. Significance of differences was taken at P < 0.05.
Results
Differential glycolytic and oxphos activities in cells cultured in high and LG conditions
MCF-7 breast cancer cells were cultured in standard medium containing 450 mg/dl (HG), and some cells were switched to medium containing physiological glucose, 100 mg/dl (LG) for 1 wk for comparison. The cells were maintained in fetal calf serum under either HG or LG conditions to simulate the in vivo states. Activity of PDH, the rate limiting enzyme for glucose metabolite entry into the TCA cycle (16), was significantly increased in LG-incubated cells compared with HG cells. This suggested an increase in TCA cycle activity in LG conditions (Fig. 1A). Citrate levels were also significantly increased in LG compared with HG grown cells, further supporting increased TCA cycle activity (Fig. 1B). In contrast, lactate levels were significantly higher in cells in the hyperglycemic environment, supporting a preference for metabolism by aerobic glycolysis (Fig. 1C). These results support the fundamental tenets of the Warburg hypothesis during HG availability to cancer cells. However, we report the novel finding that breast cancer cells metabolically compensate for decreased glucose substrate by up-regulating TCA cycle activity.
Fig. 1.
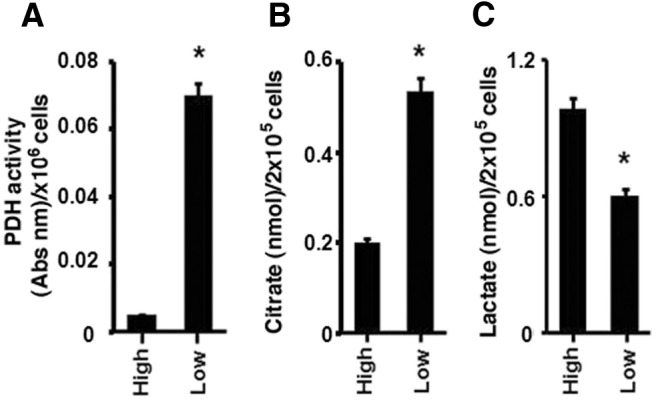
Metabolic adaptation of breast cancer cells to LG. MCF7 cells were switched from a HG media (450 mg/dl) and cultured in a physiological LG media (100 mg/dl) for 1 wk. PDH activity (A) and citrate levels (B) were determined as described in Materials and Methods. C, Lactate concentrations were measured in HG and LG cell culture conditions by kit. Each condition was performed in triplicate per each experiment. Bar graphs represent data from four experiments combined and are mean ± sem. *, P < 0.05 for HG vs. LG. Statistical analysis was obtained by ANOVA plus Schefe's test. Abs nm, Absolute number.
E2 differentially regulates glycolysis and TCA cycle activity as a function of glucose
Estrogen is an important factor driving the pathogenesis and tumor progression of 70% of human breast cancers. We therefore investigated a possible modulation of metabolism by E2. Exposure (15 min) to physiological concentrations of the sex steroid (0.1–10 nm) significantly stimulated lactate production from HG-incubated cells, compared with control (no steroid) (Fig. 2A). This suggests that E2 rapidly promotes glycolysis when glucose substrate is plentiful. In contrast, a similar short exposure to E2 caused a significant decrease in lactate production in cells grown under LG conditions for 1 wk (Fig. 2B).
Fig. 2.

Estrogen differentially modulates glycolysis under HG and LG conditions in breast cancer cells. MCF-7 cells cultured in HG (A) or LG (B), the latter for 1 wk, were then exposed to E2 for 15 min and lactate levels were measured. C, E2 exposure for 15 min stimulates PI3K/AKT activity (phosphorylation of AKT at Ser473) upon culturing cells in HG conditions and (D) inhibits AKT activity in cells exposed to LG (1 wk). *, P < 0.05 for control (untreated) vs. E2 (0.01, 1, or 10 nm). E, E2 increases lactate levels via the activation of AKT during HG exposure. MCF7 cells were transfected with an AKT-specific siRNA, recovered for 24 h, and then exposed to E2 for 10 min. siRNA validation is shown by Western blot. +, P < 0.05 for E2 vs. AKT siRNA and E2. F, Wortmanin (PI3K inhibitor) reduces E2-stimulated lactate production. *, P < 0.05 for control (untreated) vs. E2; +, P < 0.05 for E2 vs. wortmanin and E2. E2 stimulates glucose-6-phosphate concentration at 15 min in HG cultured cells (G) and decreases G6P in LG (H). The bar graphs are from four experiments combined. *, P < 0.05 for control (untreated) vs. E2 (0.01, 1, or 10 nm).
The rapid effects of the sex steroid could be consistent with modulation of metabolic pathways by signaling from membrane-localized ER (13). It has been shown that rapid and prolonged activation of kinase activity originates from this receptor pool both in vitro and in vivo (17–19). The phosphatidylinositol 3-kinase (PI3K)/AKT pathway has been implicated to up-regulate glycolysis through multiple mechanisms (17). We found that E2 (1 nm) stimulated a PI3K-dependent activation of AKT after 5 min exposure of HG cells to the sex steroid (Fig. 2C). Interestingly, under LG conditions, E2 suppressed AKT phosphorylation (Fig. 2D), suggesting that E2 modulates glycolysis in MCF-7 cells by differential rapid signaling through PI3K/AKT as a function of glucose concentration. Inhibiting AKT by using siRNA to this serine/threonine kinase, as we previously validated (13) and also show here, significantly prevented the ability of E2 to stimulate lactate in HG cells (Fig. 2E). We also found that inhibiting PI3K with wortmannin also prevented E2 stimulation of lactate production under HG conditions (Fig. 2F). To further support the regulation of glycolysis by E2 we measured the levels of another glycolytic metabolite, glucose-6-phosphate (G6P). In a dose-responsive manner, E2 increased G6P concentrations in the cells at 15 min exposure in HG conditions, while conversely reducing levels in LG cells (Fig. 2, G and H). Thus, E2 augments or suppresses glycolysis as a function of glucose availability.
We also examined TCA cycle activity. In HG cells, E2 induced a significant and rapid decrease in PDH activity (Fig. 3A). Under LG conditions, E2 stimulated PDH activity compared with LG alone or in comparison with E2 action in HG cells (Fig. 3B). The sex steroid also induced a significant increase in citrate levels in LG cells but decreased citrate levels under HG conditions (Fig. 3, C and D). These results support the concept that E2 rapidly stimulates TCA cycle activity in MCF-7 cells during reduced glucose availability while down-regulating this pathway under glucose-replete conditions. Also, the cancer cells show inherent metabolic plasticity of both cytosolic (glycolysis) and mitochondrial (TCA) pathways in response to glucose availability in the absence of E2.
Fig. 3.

Estrogen modulates TCA activity depending upon glucose conditions. A, E2 inhibits PDH activity (15 min) in a dose-dependent manner in MCF7 cells exposed to HG and (B) stimulates PDH activity under LG conditions (after 1 wk LG exposure). C, E2 decreases the citrate concentration (15 min) in cells exposed to HG but (D) increases citrate production from cells exposed to LG. E, E2 increases citrate via the activation of PDH during LG exposure. MCF7 cells were transfected with a PDH E1α subunit-specific siRNA and recovered for 24 h; the cells were exposed to E2 and citrate production was determined by kit. The bar graphs are from four experiments combined. *, P < 0.05 for control (untreated) vs. E2 treatment (0.01, 1, or 10 nm); +, P < 0.05 for E2 vs. PDH siRNA and E2.
To support the idea that citrate comes from TCA cycle and not cytosolic citrate production, we measured the citrate increase to E2 in the presence of PDH-specific siRNA (see Supplemental Fig. 1 for siRNA validation). PDH knockdown blocked the rapid induction of citrate by E2, indicating that this metabolite was produced by the mitochondrial TCA cycle (Fig. 3E).
To further support metabolic compensation in breast cancer, we used an additional ER-positive breast cancer cell line (ZR-75-1) and demonstrated comparable changes in the basal responses to HG and LG conditions. We also found similar E2 modulation of PDH activity and lactate concentration changes in these cell conditions (Supplemental Fig. 2).
E2 rapidly increases ATP production in LG setting via oxphos
An important function of the TCA cycle is to generate reduced NAD+ that serves as an electron donor for the oxphos complexes that produce ATP. We therefore determined whether the enhanced TCA response to LG promoted increased ATP production and whether E2 facilitated this. In cells incubated in LG, exposure to 1 nm E2 induced an increase in ATP production at 2 h, persisting for the 24 h of experimentation (Fig. 4A). siRNA knockdown of PDH protein blocked the E2-stimulated increase in ATP production, linking the effects of E2 on the TCA cycle to activating oxphos (Fig. 4B). Conversely, MCF-7 cells exposed to HG and E2 showed no effect on ATP production at 2 h (Supplemental Fig. 3). These latter results suggest that the stimulation of glycolysis by E2 in the HG setting is not primarily to generate energy in a relatively short period.
Fig. 4.
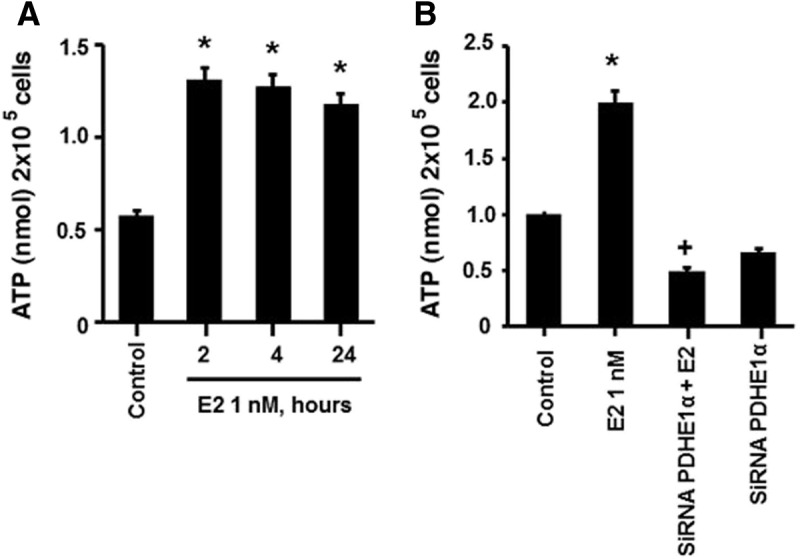
Effects of estrogen on ATP production. A, E2 exposure induces rapid ATP production (2 h) in LG cells. *, P < 0.05 for control (untreated) vs. E2 (0.01, 1, or 10 nm). B, Induction of ATP by E2 is dependent on PDH activity. MCF7 cells were transfected with a PDH-specific siRNA, recovered for 24 h, and then exposed to E2 for 2 h. *, P < 0.05 for control vs. E2. +, P < 0.05 for E2 vs. PDH siRNA and E2. *, P < 0.05 for control vs. E2 (0.01, 1. or 10 nm). Data are from four experiments combined.
ERα/β modulates oxphos, ATP production, and cell viability under LG conditions
We then investigated which ER isoform is involved in the regulation of PDH activity and ATP production in the LG setting. E2 stimulation of PDH activity (15 min) was blocked by the general ER inhibitor ICI 182,780 (Fulvestrant, 1 μm) (Fig. 5A). Using agonists of ERα, PPT (1 nm), or ERβ, DPN (1 nm), the induction of PDH activity was similar to stimulation by equimolar E2 indicating the involvement of both receptor subtypes (Fig. 5A). E2-induced PDH activity was also significantly inhibited by specific siRNAs to either ERα or ERβ, which were previously validated (14) (Fig. 5B).
Fig. 5.

Estrogen modulates metabolism via ERα and ERβ. A, E2 (1 nm), PPT (ERα agonist), or DPN (ERβ agonist) comparably stimulate PDH activity at 15 min. The E2 induction of PDH activity is blocked by the ER agonist ICI 182,780 (1 μm). *, P < 0.05 for control (untreated) vs. E2, PPT, or DPN; +, P < 0.05 for E2 vs. ICI 182,780 and E2. B, An ER siRNA approach further validates the regulation of PDH by both ERα and ERβ. MCF7 were transfected with ERα- and ERβ-specific siRNAs, recovered for 24 h, and then exposed to E2 (1 nm). *, P < 0.05 for control (untreated) vs. E2. +, P < 0.05 for E2 vs. E2 combined with an ERα siRNA or ERβ siRNA. C, E2 (1 nm) induces ATP production at 2 h via ERα and ERβ. *, P < 0.05 for control (untreated) vs. E2, PPT, or DPN. +, P < 0.05 for E2 vs. ICI 182,780 (1 μm) and E2. D, E2 rescues the viability of cells exposed to LG conditions via ERα and ERβ. MCF7 cells were switched from HG to LG media for 24 h. The decrease in cell viability (after LG exposure) was reversed by E2 (1 nm) and both the ERα and ERβ agonists (PPT and DPN, 1 nm). *, P < 0.05 for HG vs. LG, HG vs. LG, ICI 182,780, and E2. +, P < 0.05 for LG vs. LG and E2; LG vs. LG and PPT; LG vs. LG and DPN. E, E2 rescues cell viability after acute exposure to LG via induction of the citric acid cycle. MCF7 cells were first transfected with a PDH-specific siRNA. Estrogen induced rescue of cell viability in LG was blocked by PDH knockdown. *, P < 0.05 for HG vs. LG, HG vs. LG, PDHE1α siRNA, and E2. +, P < 0.05 for LG vs. LG and E2. The bar graphs represent four experiments combined, and significance was determined by ANOVA plus Schefes's test.
To further support the involvement of both receptor types, ATP production at 2 h of steroid exposure was measured; E2 increased the levels of ATP blocked by Fulvestrant, and stimulation was comparable from equimolar PPT or DPN exposure (Fig. 5C).
We then assessed whether these changes in metabolism promoted by E2 in breast cancer cells are a mechanism for rescuing the cells from the effects of reduced glucose substrate. Removing all glucose from cancer cells that are routinely cultured in HG medium causes cell death (4). However, complete lack of glucose has not been demonstrated for in vivo tumors without the complete loss of blood flow to the tumor. We therefore determined the viability response to acutely switching cells to a physiological but LG level, a model that more likely simulates the changing glucose availability to in vivo tumors (5, 6). Cells were acutely switched from HG to LG culture media for 24 h and compared with cells that continued in HG culture. Cells switched to LG showed a significant loss of cell viability but this was significantly prevented by E2 (Fig. 5D). The response to the steroid was blocked by ICI 182,780, and the effect was replicated with equimolar PPT and DPN (Fig. 5D).
We hypothesized the rescue effect of E2 on cell viability in LG conditions required activation of the TCA/oxphos pathway. Upon PDH knockdown, the E2-induced increase in cell viability was significantly reversed (Fig. 5E). These results suggest that the sex steroid stimulates compensatory metabolic pathways to enhance cell viability during metabolic stress, a novel mechanism.
Increased PDH activity in response to E2 occurs from AMPK activation
Because PDH is the key enzyme for the entry of the glucose metabolite pyruvate into the TCA cycle, we investigated the mechanism by which E2 stimulates the enzyme activity in LG conditions. PDH activity is negatively regulated by PDKs and positively regulated by PDH phosphatases, comprising the PDH complex (16). To date, no other signal transduction proteins have been implicated in the direct regulation of the PDH complex. AMPK serves as a metabolic sensor that is activated in cells cultured in LG (20) and therefore, this kinase could play a role in E2 action. E2 rapidly induced AMPK activation (5 min) as measured by phosphorylation at tyrosine 172 in the LG environment (Fig. 6A) but not in HG cells (Supplemental Fig. 4). AMPK activation significantly contributed to the E2 up-regulation of PDH activity because AMPK siRNA blocked PDH induction (Fig. 6B) (validation of siRNA in Supplemental Fig. 1). We also found that the an AMPK inhibitor, Compound C, blocked the ability of E2 to stimulate PDH activity (Fig. 6B).
Fig. 6.

Estrogen-activated AMPK directly stimulates PDH activity A, E2 (1 nm) rapidly induces AMPK activity (phospho or p-AMPK) in LG conditions (1 wk exposure to LG). *, P < 0.05 for control (untreated) vs. E2 (5 and 10 min). B, E2 induces PDH activation via AMPK induction. Left, MCF7 were transfected with an AMPK-specific siRNA, recovered for 24 h, and then exposed to E2. *, P < 0.05 for control (untreated) vs. E2; +, P < 0.05 for E2 vs. AMPK siRNA and E2. Right, compound C (CC), an AMPK inhibitor, prevents E2 activation of PDH activity. *, P < 0.05 for control (untreated) vs. E2; +, P < 0.05 for E2 vs. CC and E2. C, Estrogen-induced AMPK in LG conditions phosphorylates a PDHE1α subunit peptide in vitro and (D) reduces the PDH phosphorylation in HG conditions. MCF-7 cells were incubated with E2 (1 nm, 5–15 min), and AMPK was immunoprecipitated from the lysed cells and combined with PDHE1α peptide in a tube for an in vitro kinase activity assay. Phosphorylation of the subunit by AMPK was determined by [32P]ATP incorporation. *, P < 0.05 for control (untreated) vs. E2 (5 min). E, AMPK directly phosphorylates the PDHE1α subunit at the serine 19 residue in LG conditions. Estrogen-treated cells (5 min, 1 nm) were lysed, and total AMPK was immunoprecipitated. Isolated AMPK was incubated with WT or mutated (S19A) PDHE1α peptides as substrate, and kinase activity was determined. *, P < 0.05 for control (WT PDH) vs. WT PDH and E2. +, P < 0.05 for WT PDH and E2 vs. mut PDH and E2; WT PDH and E2 vs. WT PDH, CC (AMPK inhibitor), and E2. F, AMPK directly modulates PDH activity in vivo by activation of the PDHE1α subunit at serine 19. MCF-7 cells were transfected with plasmids containing WT or mutant (S19A) PDHE1α and recovered after 24 h. *, P < 0.05 for control (no transfection) vs. E2 (5 min, 1 nm); control vs. WT PDH and E2. +, P < 0.05 for E2 vs. AMPK inhibitor (CC) and E2; E2 vs. Mut PDH and E2; WT PDH and E2 vs. Mut PDH and E2. The bar graphs represent four experiments combined. mut, Mutant.
We then determined whether the regulation of PDH activity by AMPK occurred by direct phosphorylation of the PDHE1α-regulatory subunit. We used an in vitro kinase assay with PDHE1α as peptide substrate and AMPK immunoprecipitated from each experimental condition. In cells cultured in LG, E2 rapidly stimulated the AMPK-dependent phosphorylation of the PDH subunit (Fig. 6C). However, under HG conditions, E2 treatment suppressed basal AMPK-mediated phosphorylation of PDHE1α (Fig. 6D), consistent with steroid temporal suppression of the TCA cycle under HG conditions (Fig. 3A).
We therefore determined what residue in the PDHE1α structure might be a direct substrate for AMPK. We first examined the amino acid sequence of the catalytic PDHE1α subunit and found a sequence including serine 19 (SRVLSGASQ) that could serve as substrate for AMPK. A serine 19 to alanine point mutation (S19A) was introduced into the WT PDHE1α subunit and both mutated and WT PDH proteins were expressed and purified. The LG MCF-7 cells were incubated with or without E2 (1 nm, 5 min), and AMPK was immunoprecipitated from the cells for the in vitro kinase assay with either WT or mutated (S19A) PDH peptides serving as substrate. E2 stimulated AMPK phosphorylation of the WT PDHE1α, and this effect was prevented by coincubation of the cells with the AMPK inhibitor, Compound C (Fig. 6E). However, AMPK precipitated from E2-treated cells did not phosphorylate the mutant PDH subunit, indicating Ser19 is an AMPK phosphorylation site.
It has been demonstrated in a previous study that expression of a different PDH mutant construct served as a dominant negative for endogenous PDH (21). We transfected plasmids containing WT or mutated PDH in the MCF7 cells cultured under LG conditions. E2 stimulated an AMPK-dependent increase in PDH activity in WT enzyme-expressing cells because the soluble inhibitor of AMPK (Compound C) blocked this effect of the sex steroid (Fig. 6F). In contrast, mutant PDH expression prevented E2 from stimulating PDH activity (Fig. 6F). This suggests that the mutant construct served as a dominant negative for endogenous PDH activity. Our results indicate that the phosphorylation by AMPK of the PDHE1α subunit at Ser19 is required for E2-induced PDH activity. This is the first activating phosphorylation of PDH to be identified and supports a new mechanism of regulation of this important metabolic enzyme.
Estrogen regulates PDK impacting PDH activity
As mentioned, the PDK family negatively regulates PDH activity by causing an inhibitory phosphorylation of the PDHE1α subunit at one of three serine residues (depending on the PDK isoform involved (16). We determined whether PDK plays a role in the regulation of PDH activity by E2 in LG and HG cells. There are four isoforms of PDK, and all are expressed in MCF-7 cells (Supplemental Fig. 5). We found that only the siRNA knockdown of PDK4 significantly increased PDH activity in LG cells, indicating this isoform is likely to be a regulator of PDH under our LG conditions (Fig. 7A) (siRNA validation is shown in Supplemental Fig. 1A).
Fig. 7.

Estrogen modulates PDK4 activity to regulate PDH. A, LG MCF7 cells were transfected with PDK1–4 (separately)-specific siRNAs, recovered for 24 h, and cultured in the presence of serum for 24 h. *, P < 0.05 for control (no siRNA) vs. PDK4 siRNA. B, E2 decreases the level of activated PDK4 in LG cells as measured by the amount of phosphorylated PDHE1α substrate and (C) increases PDK4 activation in HG cells. MCF-7 cells were treated with E2 (1 nm), and PDK4 was immunoprecipitated and added to the PDHE1α subunit. Phosphorylation of the PDH subunit by PDK4 was measured by [32P]ATP labeling. *, P < 0.05 for control (untreated) vs. E2. D, E2 activated PDK4 is not AMPK dependent. LG cultured cells were pretreated with compound C (AMPK inhibitor) before E2 (1 nm) addition. *, P < 0.05 control vs. E2; control vs. CC and E2. The bar graphs represent four experiments combined. CC, Compound C.
Using an in vitro kinase approach, PDK isoforms 1–4 were each immunoprecipitated from control or E2-treated cells and added to tubes containing WT PDHE1α peptide to assess PDK activity. The level of phosphorylated PDHE1α was decreased compared with control in E2-treated LG cells, but this was only seen from using the PDK4 precipitates (Fig. 7B) (PDK1–3 isoforms shown in Supplemental Fig. 6). Conversely, under HG, immunoprecipitated PDK4 from E2-exposed cells stimulated PDHE1α phosphorylation (Fig. 7C). Therefore, E2 enhances PDK4 phosphorylation of PDHE1α, thus contributing to the inhibition of the TCA cycle in HG conditions. In summary E2 differentially regulates PDK4 activity, contributing to the down-regulation of PDH activity in HG conditions and the activation of PDH in LG conditions.
We then determined the possible role of AMPK to inactivate PDK activity. LG cells were exposed to E2 without or with the AMPK inhibitor, Compound C. No change in phosphorylated WT PDHE1α was noted using PDK4 immunoprecipitated from E2 alone vs. E2 plus AMPK antagonist conditions (Fig. 7D). The results suggest that AMPK and PDK are involved at divergent parts of E2 regulation of PDH activity.
Breast cancer cells exhibit long-term preference for glycolysis
Switching the cells to LG media decreased glycolysis, increasing the activity of the TCA cycle and resulting in additional metabolic pathways as potentiated by estrogen. We hypothesized that this switch to oxphos might be a temporary mechanism adopted by the cells and driven by estrogen to prevent the loss of viability caused by reduced glucose substrate. To test this idea, MCF7 cells were cultured for 8 wk in LG, and the cells were assayed for glycolytic and TCA/oxphos components. Similar to the previous observations, at 1 wk of LG exposure the basal PDH activity was increased (Fig. 8A). Interestingly, after 8 wk of LG incubation, the cells down-regulated PDH activity to levels seen originally in HG conditions. E2 further decreased PDH activity accompanied by increased lactate levels, demonstrating potentiation of the Warburg effect (Fig. 8, B and C). The results demonstrate that the cancer cells reset glucose utilization at the lower level of substrate, favoring glycolysis compared with shunting glucose metabolites through the mitochondrial pathways.
Fig. 8.

Breast cancer cells revert to glycolysis after long-term exposure to LG conditions. A, MCF-7 cells were switched to LG media and cultured for 1 or 8 wk. *, P < 0.05 for HG culture vs. switching to LG culture for 1 wk and 8 wk. B, After 8 wk in LG media, E2 (1 nm, 15 min) decreases PDH activity and (C) stimulates lactate levels. *, P < 0.05 for control vs. E2 .The bar graphs represent four experiments combined.
Inhibiting metabolic compensation potentiates adjuvant therapy
γ-Ionizing radiation is commonly used as an adjuvant to limited surgical removal of breast cancer in women and is a cytotoxic treatment that causes the apoptotic death of many types of cancer cells. We speculated that we might utilize the metabolic vulnerability of cells during LG to enhance the apoptotic effects of ionizing radiation (IR). Cells in both HG and acute LG culture conditions were exposed to a single dose of 5 Gray IR, a dose that is similar to the therapeutic amount administered to breast cancer patients. Some cells were also pretreated with either E2 alone (1 nm, 24 h) or with E2 preceded by siRNA to PDHE1α. In the absence of radiation, switching cells to LG from HG increased the number of cells undergoing apoptosis that was prevented by E2 (Fig. 9A). In LG cells treated with IR, apoptosis was significantly increased compared with LG alone, and these effects were again significantly prevented by E2 (Fig. 9A). IR also increased cell death in HG cells, but more modestly. However, after knockdown of the PDHE1α subunit, estrogen was unable to rescue LG cells from the effects of IR. In addition, the number of cells undergoing cell death from LG exposure and IR in the absence of E2 substantially increased from the loss of PDH activity. These results suggest that targeting the TCA cycle during metabolic compensation to LG may be a strategy to sensitize the cells to IR therapy in breast cancer.
Fig. 9.

Estrogen rescues breast cancer cells from γ-radiation treatment via up-regulation of PDH activity and subsquent TCA induction. A, Cells were maintained in HG or some cells switched to LG for 24 h, and apoptosis was measured in the presence or absence of additional parameters (E2, IR, E2+ IR, PDH siRNA + E2 or IR or both). The cells were exposed to 5 gray (Gy) IR. Exposure of cells to IR followed 24 h in LG conditions and at the same time, HG cells. The number of cells undergoing apoptosis was measured by TUNEL assay. *, P < 0.05 for each control vs. IR + LG or HG; +, P < 0.05 for E2 vs. IR in LG; ++, P < 0.05 for IR vs. E2 + IR in LG; IR vs. IR + siRNA PDH1; E2 + IR vs. same + siRNA PDH, all in LG conditions. The bar graphs represent counts from at least 200 cells in three experiments each. The table shows percent apoptotic cells. B, ROS formation by DHF fluorescence (see Materials and Methods) was determined under similar conditions. Significant differences among conditions were seen as for apoptotic cell comparisons; studies were repeated three times.
Including radiation, many stimuli for apoptosis require oxidative stress to activate cell death pathways. We therefore investigated whether E2 down-regulated IR and LG-induced ROS formation and whether this was dependent on PDH activity. Cells transitioned to LG in the absence or presence of IR showed higher ROS levels than HG cells (Fig. 9B). E2 especially prevented LG-related higher ROS formation, but this was reversed by PDH knockdown. Similarly, IR-induced ROS formation in LG conditions was inhibited by PDH knockdown, in the absence of E2. ROS amounts correlated to the effects of LG, IR, and PDH diminution from siRNA for apoptosis (Fig. 9A). Although HG cells showed similar trends, the magnitude of ROS formation was much smaller, correlating to reduced apoptosis under these conditions (Fig. 9).
Discussion
The original Warburg hypothesis suggested that the mitochondria of neoplastic cells are dysfunctional, rendering the cell incapable of carrying out the process of oxphos (1, 2). However, more recently, numerous studies have demonstrated that the mitochondria of various neoplastic cells are highly functional (4, 22–24), also confirmed in our breast cancer cell models. In vivo, blood flow to tumors is often uneven, resulting in areas of tumor hypoxia and differential glucose concentrations (5, 6). Hypoxia induces the HIF1α gene that enhances the transcription of angiogenesis and glycolytic genes (7). As a response, tumor cells show increased glucose uptake and utilization through aerobic glycolysis (7).
We considered that cells may acutely adapt to lower but physiological glucose availability by altering the use of glucose metabolites such as pyruvate for mitochondrial metabolism. We find that lowering glucose availability to breast cancer cells induces metabolic pathway plasticity to compensate, in part, for a loss of viability. In this setting, lactate production decreases and the mitochondrial TCA/oxphos pathways are increased, the latter leading to enhanced ATP concentrations.
E2/ER promotes the development of breast cancer and stimulates established breast cancer epithelial cell proliferation and survival (11, 12). Because glucose metabolism greatly impacts these essential processes, we hypothesized that E2 might facilitate metabolic compensation for lower glucose availability in ER-positive breast cancer cells. In HG-incubated cells, glycolysis is the preferred route for glucose metabolism, and E2 signaling through both ERα and ERβ enhances this pathway. Enhanced glycolysis in response to E2 occurs through PI3K/AKT activation, signals that are known to stimulate glucose uptake (22) and the expression of glycolytic pathway genes (8). Interestingly, E2 concomitantly inhibits the TCA cycle (reflected as decreased PDH activity and citrate concentrations). PI3K/AKT and other kinase activation by E2/ER occurs mainly through ER pools localized at the plasma membrane, both in vivo (18) and in vitro (19). We suggest that rapid signaling by E2 occurs through extranuclear ER that we have demonstrated in MCF-7 cells (13), impacting tumor metabolism as a novel function in the current studies.
In contrast, in the LG setting, E2 stimulates the TCA cycle and subsequent outputs (citrate and ATP production) through up-regulating PDH activity while suppressing glycolysis. Previous studies of in vitro cancer cell models used the approach of complete glucose withdrawal, resulting in severe ATP depletion and cell apoptosis, particularly in tumor cells that are highly dependent on AKT signaling (8, 22). It has not been shown that most in vivo tumors are exposed to a complete lack of glucose. We therefore modeled the lower availability of glucose substrate that more closely reflects the inconsistent blood flow to in vivo tumors and the overall milieu of normoglycemia in nondiabetic humans. We find that sex steroid-enhanced PDH activity is required to prevent the loss of viability that rapidly results from decreased glucose availability. Thus, E2/ER enhances the metabolic plasticity and compensation of breast cancer cells to altered glucose substrate, serving to preserve cell functions (Fig. 10).
Fig. 10.

Cartoon of estrogen regulation of breast cancer cell metabolism. In HG conditions, E2 activates AKT to enhance glycolysis while suppressing the mitochondrial metabolic pathways (TCA cycle and oxphos). Under LG conditions, E2 stimulates PDH activity via AMPK phosphorylation of serine 19 and inhibition of the activity of PDK 4 that inactivates PDH. This involves both ERα and ERβ. As a result, pyruvate is metabolized to acetyl coenzyme A, leading to the activation of the TCA cycle and oxphos. In this way, estrogen rescues the loss of viability of the breast cancer cells that results from decreased glycolysis due to low glucose substrate.
Regulation of the PDH enzyme via AMPK activation
By what mechanism does E2/ER induce compensatory mitochondrial pathway activity? The key enzyme for the utilization of the glucose metabolite pyruvate by the TCA cycle is PDH. This enzyme converts pyruvate to acetyl-coenzyme A in mitochondria, which is further converted to citrate for processing in the TCA cycle or transport to the cytoplasm for fatty acid, triglyceride, and other lipid synthesis. Negative regulation of PDH activity is attributed to inhibitory phosphorylations by a family of PDH kinases (PDK) (16). In addition, two PDH phosphatases remove the inhibitory phosphorylations from PDK(s) action at serines 264, 271, and 203 on the PDHE1α catalytic subunit (25, 26), thus disinhibiting PDH activity. In contrast, no recognized activating phosphorylations of PDH have been described.
We found that the metabolic sensor kinase AMPK was stimulated by E2 under reduced glucose conditions, and this was required for increasing PDH activity, enhanced citrate production, and the rapid induction of ATP. The latter was necessary for E2 to rescue the cells from loss of viability. A direct relationship between AMPK and modulation of the TCA cycle has not been established previously. Interestingly, E2 was unable to activate this kinase in the HG state, accounting for the differential regulation of the TCA cycle as a function of glucose. AKT activation in vascular smooth muscle cells has recently been shown to suppress AMPK activity (27). Because we found that E2 stimulates AKT in HG conditions, this might provide a reason as to why E2/ER does not stimulate AMPK in this setting.
We identified a novel AMPK substrate site within the PDHE1α regulatory subunit and showed that mutation of the serine 19 residue to alanine (S19A) prevented both the phosphorylation and enhanced activity of WT PDH by E2-stimulated AMPK. E2 stimulation of WT PDH activity was also prevented by siRNA to AMPK and by a soluble inhibitor of AMPK. In addition, E2 inhibited PDK4 phosphorylation (Fig. 7B) in the LG setting. AMPK was not involved in the latter, suggesting that two independent pathways are required for PDH up-regulation by E2. We also linked the regulation of PDH through AMPK to further mitochondrial pathway plasticity. Mitochondrial adaptation in response to AMPK may be more pervasive because this kinase has been implicated in PGC1α gene up-regulation in muscle (28) and PGC1α is a master regulator of mitochondrial biosynthesis and function (28, 29). Furthermore, AMPK activation promotes cell survival in response to various metabolic and genotoxic stresses (30). Thus, there may be additional pathways mediated through E2 and AMPK in the setting of LG that impact cell fate.
Inhibiting metabolic compensation for LG enhances adjuvant therapy
Switching breast cancer cells to LG causes a loss of viability, and shifts metabolism toward the TCA/oxphos pathways and away from glycolysis, all promoted by estrogen. Interestingly, the adaptive response to LG is relatively short lived, because the cell ultimately reverts to glycolysis after 8 wk incubation in the LG conditions. Thus, glycolysis is obviously an important driving force in tumor cells, and estrogen reestablishes its promotion of glycolysis and inhibition of PDH activity at that time. Nevertheless, the period of diminished cell viability might be used in therapeutic approaches.
In this regard, we found that breast cancer cells exposed to both LG and PDH knockdown show markedly enhanced cell death when subjected to a therapeutically relevant dose of γ-radiation. Furthermore, the strong cell survival effect of E2, blocking IR-induced apoptosis, was markedly reversed by PDH knockdown in LG conditions. For all treatments, cells under HG conditions showed much less apoptosis. In addition, we found that cells cultured in LG and exposed to IR generated significant amounts of ROS, blocked by E2 but not in the setting of PDH protein loss from siRNA. In vivo, it might be possible to enhance the response of tumors to adjuvant therapies such as radiation by reducing tumor glucose utilization, using 2-deoxygluxose that is avidly taken up by tumors but is not metabolized. Recently it was shown that cancer stem cells that are resistant to chemotherapy can be strongly resensitized by preventing the stem cell synthesis of several fatty acids (31). We propose that understanding the metabolic plasticity and adaptation by cancer cells to altered energy substrate could provide additional therapeutic approaches to facilitate cell death and overcome the strong survival effects of the sex steroid in ER-positive breast cancer.
Supplementary Material
Acknowledgments
This work was supported by grants from National Institutes of Health (CA-100366) and the Department of Veterans Affairs Merit Review Program (to E.R.L.). Fiona O'Mahony was cofunded by Marie Curie Actions, the Irish Higher Education Authority Program for Third Level Institutions Cycle 4, and the Italian National Research Council.
Disclosure Summary: The authors have nothing to disclose and no conflicts of interest.
NURSA Molecule Pages:
Ligands: 17β-estradiol.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- AMPK
- AMP kinase
- DCF
- dichlorofluorescein
- DPN
- diarylpropionnitrile
- E2
- 17β-estradiol
- ER
- estrogen receptor
- G6P
- glucose-6-phosphate
- HG
- high glucose
- IR
- ionizing radiation
- LG
- low glucose
- MTT
- 3-(4,5-dimethylthiazol 2yl)-2,5-diphenyttetrazolium bromide
- NAD
- nicotinamide adenine dinucleotide
- oxphos
- oxidative phosphorylation
- PDH
- pyruvate dehydrogenase
- PDK
- PDH kinase
- PI3K
- phosphatidylinositol 3-kinase
- PPT
- propyl pyrazole triol
- qRT-PCR
- quantitative real-time PCR
- ROS
- reactive oxygen species
- siRNA
- small interfering RNA
- TCA
- tricarboxylic acid
- TUNEL
- terminal deoxynucleotide transferase-mediated dUTP nick end labeling
- WT
- wild type.
References
- 1. Warburg O, Wind F, Negelein E. 1927. The metabolism of tumors in the body. J Gen Physiol 8:519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Warburg O, Posener K, Negelein E. 1924. Ueber den Stoffwechsel der Tumoren; Biochemische Zeitschrift, 152; 319–344. Reprinted in English in the book on metabolism of tumors by O. Warburg. London: Constable, London, 1930 [Google Scholar]
- 3. Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. 2007. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104:19345–19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vaupel P, Hockel M. 2000. Blood supply, oxygenation status and metabolic micromilieu of breast cancers: characterization and therapeutic relevance. Int J Oncol 17:869–879 [DOI] [PubMed] [Google Scholar]
- 6. Sutherland R, Freyer J, Mueller-Klieser W, Wilson R, Heacock C, Sciandra J, Sordat B. 1986. Cellular growth and metabolic adaptations to nutrient stress environments in tumor microregions. Int J Radiat Oncol Biol Phys 12:611–615 [DOI] [PubMed] [Google Scholar]
- 7. Semenza GL. 2003. Targeting Hif-1α for cancer therapy. Nat Rev Cancer 3:721–732 [DOI] [PubMed] [Google Scholar]
- 8. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. 2004. AKT stimulates aerobic glycolysis in cancer cells. Cancer Res 64:3892–3899 [DOI] [PubMed] [Google Scholar]
- 9. Novosyadlyy R, Lann DE, Vijayakumar A, Rowzee A, Lazzarino DA, Fierz Y, Carboni JM, Gottardis MM, Pennisi PA, Molinolo AA, Kurshan N, Mejia W, Santopietro S, Yakar S, Wood TL, LeRoith D. 2010. Insulin-mediated acceleration of breast cancer development and progression in a non-obese model of type 2 diabetes. Cancer Res 70:741–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fierz Y, Novosyadlyy R, Vijayakumar A, Yakar S, LeRoith D. 2010. Insulin-sensitizing therapy attenuates type 2 diabetes-mediated mammary tumor progression. Diabetes 59:686–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McGuire WL, Chamness GC, Fuqua SA. 1991. Estrogen receptor variants in clinical breast cancer. Mol Endocrinol 5:1571–1577 [DOI] [PubMed] [Google Scholar]
- 12. Levin ER, Pietras RJ. 2008. Estrogen receptors outside the nucleus in breast cancer. Breast Cancer Res Treat 108:351–361 [DOI] [PubMed] [Google Scholar]
- 13. Pedram A, Razandi M, Levin ER. 2006. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol 20:1996–2009 [DOI] [PubMed] [Google Scholar]
- 14. Pedram A, Razandi M, Wallace D, Levin ER. 2006. Functional estrogen receptors in the mitochondria of breast cancer cells. Mol Biol Cell 17:2125–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pedram A, Razandi M, O'Mahony F, Lubahn D, Levin ER. 2010. Estrogen receptor β inhibits cardiac fibrosis. Mol Endocrinol 24:2152–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sugden MC, Holness MJ. 2006. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem 112:139–149 [DOI] [PubMed] [Google Scholar]
- 17. Razandi M, Pedram A, Greene GL, Levin ER. 1999. Cell membrane and nuclear estrogen receptors derive from a single transcript: studies of ERα and ERβ expressed in CHO cells. Mol Endocrinol 13:307–319 [DOI] [PubMed] [Google Scholar]
- 18. Pedram A, Razandi M, Kim JK, O'Mahony F, Lee EY, Luderer U, Levin ER. 2009. Developmental phenotype of a membrane only estrogen receptor α. J Biol Chem 284:3488–3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pedram A, Razandi M, Evinger AJ, Lee E, Levin ER. 2009. Estrogen inhibits ATR signaling to cell cycle checkpoints and DNA repair. Mol Biol Cell 20:3374–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kahn BB, Alquier T, Carling D, Hardie DG. 2005. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1:15–25 [DOI] [PubMed] [Google Scholar]
- 21. Brown RM, Head RA, Boubriak II, Leonard JV, Brown GK. 2003. A pathogenic glutamate to aspartate substitution (D296E) in the pyruvate dehydrogenase E1 subunit gene PDHA1. Hum Mutat 22:496–497 [DOI] [PubMed] [Google Scholar]
- 22. Buzzai M, Bauer DE, Jones RG, DeBerardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. 2005. The glucose dependence of AKT-transformed cells can be reversed by pharmacologic activation of fatty acid β oxidation. Oncogene 24:4165–4173 [DOI] [PubMed] [Google Scholar]
- 23. Koppenol WH, Bounds PL, Dang CV. 2011. Otto Warburgs contributions to current concepts of cancer metabolism. Nat Rev Cancer 11:325–337 [DOI] [PubMed] [Google Scholar]
- 24. Plas DR, Thompson CB. 2005. AKT-dependent transformation: there is more to growth than just surviving. Oncogene 24:7435–7442 [DOI] [PubMed] [Google Scholar]
- 25. Patel MS, Korotchkina LG. 2006. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans 34:217–222 [DOI] [PubMed] [Google Scholar]
- 26. Sugden MC, Holness MJ. 2003. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by the PDKs. Am J Physiol Endocrinol Metab 284:E855–E862 [DOI] [PubMed] [Google Scholar]
- 27. Ning J, Xi G, Clemmons DR. 2011. Suppression of AMPK activation via S485 phosphorylation by IGF-1 during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology 152:3143–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fernandez-Marcos PJ, Auwerx J. 2011. Regulation of PGC1-α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93:884S–890S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Puigserver P. 2005. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-α. Int J Obes (Lond) 29:(Suppl 1):S5–S9 [DOI] [PubMed] [Google Scholar]
- 30. Wu SB, Wei YH. 2012. AMPK-mediated increase of glycolysis as an adaptive response to oxidative stress in human cells: implication of the cell survival in mitochondrial diseases. Biochim Biophys Acta 1822:233–247 [DOI] [PubMed] [Google Scholar]
- 31. Roodhart JM, Daenen LG, Stigter EC, Prins HJ, Gerrits J, Houthuijzen JM, Gerristen MG, Schipper HS, Backer MJ, van Amersfoot M, Vermaat JS, Moerer P, Ishihara K, Kalkhoven E, Beijnen JH, Derksen PW, Medema RH, Martens AC, Brenkman AB, Voest EE. 2011. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell 20:370–383 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.