

Abstract
Familial distal renal tubular acidosis (dRTA) can be caused by mutations in the Cl−/HCO3− exchanger of the renal Type A intercalated cell, kidney AE1/SLC4A1. dRTA-associated AE1 mutations have been reported in families from North America, Europe, Thailand, Malaysia, Papua-New Guinea, Taiwan, and the Philipines, but not India. The dRTA mutation AE1 A858D has been detected only in the context of compound heterozygosity. We report here two unrelated Indian patients with combined hemolytic anemia and dRTA who share homozygous A858D mutations of the AE1/SLC4A1 gene. The mutation creates a novel restriction site that is validated for diagnostic screening.
Keywords: Band 3, SAO, dRTA, anemia, Eosin-5-maleimide
Distal urinary acidification requires coordinated function of several proteins in the Type A intercalated cell of the renal collecting duct: the multi-subunit, vacuolar (v)H+-ATPase of the apical membrane, cytoplasmic carbonic anhydrase II, and the SLC4A1/AE1 Cl−/HCO3− exchanger (kidney Band 3, kAE1). Mutations in any one of these genes can cause dRTA, and AE1 mutations have been linked to both recessive and dominant forms of dRTA (1). AE1 is expressed not only in Type A intercalated cells, but also in late erythroid precursors, reticulocytes, and circulating red cells. AE1 mutations associated with dominant hereditary spherocytic and stomatocytic anemias are generally unassociated with a renal phenotype. Conversely, dRTA mutations associated with dominant and recessive dRTA were long believed to be without associated erythroid phenotype (1). However, compound heterozygotes have been described with combined spherostomatocytic anemia and dRTA (2). More recently, subclinical cation-leak stomatocytosis has been associated with AE1 mutation-associated dRTA (3).
Case reports of non-sporadic dRTA have appeared from India. Among 40 pediatric nephrocalcinosis cases from New Delhi, 50% presented with dRTA between ages 3 and 6 years (4). However, molecular diagnoses of dRTA have only recently begun to appear from India. A 12 year old with osteopetrosis, renal tubular acidosis, and cerebral calcifications was homozygous for the carbonic anhydrase II mutation S29P (5), and siblings with dRTA and sensorineural deafness were homozygous for R31X truncations in the B1 subunit of vacuolar H+-ATPase (6).
We report here the first two cases of dRTA from India associated with mutations in the SLC4A1/AE1 gene. Two unrelated patients from Maharashtra with combined hemolytic anemia and dRTA were found to be homozygous for the AE1 A858D mutation, previously detected only in the heterozygous or compound heterozygous state.
Patient 1, a 15 year old from Maharasthtra of Kunbi caste born of a non-consanguinous marriage, presented at age 6 years with growth retardation, jaundice and a history of fracture on falling. Physical exam revealed a weight of 13 kg (<5th %ile for age) and rickets. Abdominal sonogram revealed splenomegaly, bilateral renal medullary calcification, and two simple left renal cortical cysts (unchanged 3 years later). Additional findings of “metabolic acidosis with alkaline urine pH” (detailed medical records are unavailable) led to the clinical diagnosis of distal renal tubular acidosis. Treatment with oral sodium bicarbonate and potassium citrate reportedly resolved the acidosis.
In May 2009, the patient presented with jaundice and history of a recent blood transfusion. Exam revealed normal stature and weight with persistent splenomegaly. Peripheral blood smear revealed ovalocytes and tear drop cells without stomatocytes (Figure 1A). Hematological indices revealed macrocytic anemia with Hct 13.2%, Hgb 4.9 g/dL, reticulocyte count 7%, mean corpuscular volume 129 fL, mean corpuscular hemoglobin concentration 37.1 g/dL, and relative density width 27.2. E5M mean channel fluorescence (MCF) indicating AE1/Band3 surface abundance was 850 au (normal range 1080-1300 au). Serum Vitamin B12 was low at 110 pg/mL (normal range 211 - 911 pg /mL). Total bilirubin was 4.9 mg/dL with indirect bilirubin 4.7 mg/dL, and serum alkaline phosphatase was elevated at 530 IU/L. Serum K+ was normal at 4.1 mM. Serum bicarbonate was 26.7 mM with blood pH 7.41 while on continued oral supplementation with bicarbonate and citrate. Blood urea nitrogen of 9 mg% and serum creatinine of 0.4 mg% indicated normal renal glomerular function. 24 hr urinary calcium excretion was normal at 2.2 mmol per 5.3 mmol creatinine. The patient’s parents and one older sib had no clinical history of hematological or renal disease. The patient’s mother had normal hematological indices, but E5M MCF was reduced (847 au vs. normal control 1103 au).
Figure 1.

Wright-Giemsa blood smears of Patient 1 before (A) and (B) after supplementation with folate and vitamin B12.
After transfusion, the patient was treated with intravenous vitamin B12 and with chronic oral folate supplementation. 13 months later, splenomegaly persisted, but hematological indices showed improvement: Hgb 12.0 g/dL, Hct 31.2 %, MCV 81.3 fL, MCHC 38.5 mg/dL, reticulocyte count 2%. Total bilirubin was 3.7 mg/dL (direct 0.5 mg/dL), and alkaline phosphatase 238 U/L. The smear at that time revealed that resolution of macrocytosis was accompanied by more prominent elliptocytosis and stomatocytosis and acanthocytosis (Figure 1B). Both parents have been asymptomatic, and hematologic indices of the mother were unremarkable.
The clinical diagnosis was treated dRTA with ovalocytic, hemolytic anemia and ineffective erythropoiesis complicated by folate/B12 deficiency responsive to supplementation. The combined anemia and dRTA suggested evaluation of erythroid band 3/AE1. Reduced E5M MCF further supported moderate erythroid Band 3 deficiency.
Patient 2, a 6 year old male from Maharashtra, of Kunbi caste born to first cousin parents (Figure 2A), presented at age 16 months with sudden onset hypotonia without diarrhea or vomiting. Physical examination revealed hepatosplenomegaly. Hb was 4.4 g/dL with reticulocytosis, and peripheral blood smear showed spherocytes, ovalocytes, acanthocytes, and schizocytes (not shown). Sonography detected hepatosplenomegaly and nephrocalcinosis. Metabolic acidosis was noted concomitant with spot urine pH of 8 and absence of glucosuria. The patient was diagnosed with hemolytic anemia and dRTA (detailed medical records not available). He was transfused and treated with oral sodium bicarbonate and potassium citrate. From 3 years of age, the tranfusion requirement gradually increased to a current frequency of every 30-45 days. At age 4 ½ years the patient developed multi-drug resistant pulmonary tuberculosis, initially treated with kanamycin, ethionamide, pyrazinamide, capreomycin, clofazimine, and ofloxacin, and more recently switched to a regimen of ethionamide, cycloserine, sodium aminosalicylate, moxifloxacin, and clofazermine.
Figure 2.
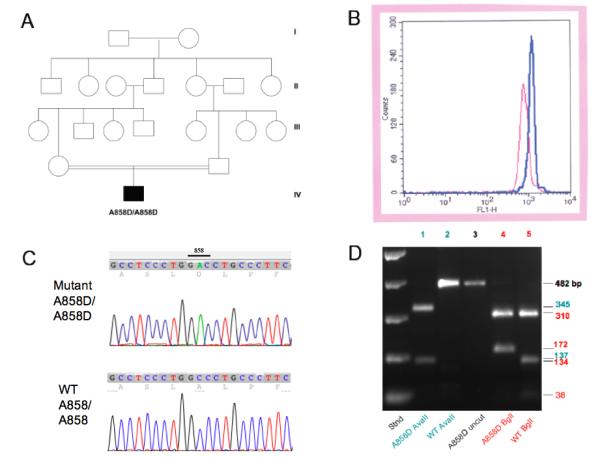
A. Consanguinous pedigree of patient 2. B. Eosin-5′-maleimide surface staining profiles of Patient 2 (red) and of control red cells (red). C. Sequence traces from the AE1 gene exon 19 of Patient 1 (upper) showing the homozygous GCC-GAC mutation encoding the missense substitution A858D, and from a control individual (lower) with the homozygous WT sequence. D. Diagnostic restriction digest of an AE1 gene amplicon from Patient 1 showing acquisition of a novel AvaII site and loss of a BglI site in the mutant allele. The same pattern was noted with genomic DNA from Patient 2 (not shown).
Recent evaluation of patient 2 revealed continued growth retardation, hepatosplenomegaly, and nephrocalcinosis. Peripheral blood smear was remarkable for spherostomatocytosis, anisopoikilocytosis, acanthocytosis, and elliptocytes. Hematological indices revealed Hgb 4.9 g/dL, Hct 15.9%, MCV 80.7 fL, MCHC 30.8 g/dL, reticulocyte count 5%. Alkaline phosphatase was elevated at 273 IU/L. Serum K+ was 3.4 mM. On oral bicarbonate/citrate supplementation, blood pH was 7.38 and serum bicarbonate was 23.4 mM, Blood urea nitrogen was 8 mg/dL and serum creatinine was 0.3 mg/dL Spot urine revealed pH 8.0, trace proteinuria, and no glucosuria. Analysis of red cell AE1/Band 3 surface abundance by E5M flow-cytometry revealed MCF of 710 a.u. (normal 1070-1300; Fig. 2B). Erythroid osmotic fragility was slightly increased (0.52% vs control 0.42 - 0.48%). Both parents were asymptomatic. The patient’s father had normal hematological indices, but moderate reduction in red cell E5M MCF of 896 a.u. The patient’s mother had mild microcytic anemia (Hb 10.7, Hct 32.2, MCV 73). No further family history or laboratory workup was available.
The presence of band 3-deficient ovalocytic hemolytic anemia together with evidence of distal renal tubular acidosis suggested possible mutation of the SLC4A1 gene encoding erythroid and kidney isoforms of Band 3/AE1/SLC4A1. The entire coding region and all splice junctions and flanking sequences of the AE1 gene were sequenced. Complete DNA sequences were also obtained for introns 2, 5, 7, 8, 10-12, 14, 15, 18, and 19 from both patients. In addition, introns 3 and 6 were sequenced completely from patient 1, and partially from patient 2.
As shown in Fig. 2C, nominally nonconsanguinous patient 1 was homozygous for the SLC4A1/AE1/Band 3 missense mutation A858D, encoded by a C-to-A transversion changing codon GCC to GAC in exon 19. The same homozygous mutation was found in consanguinous patient 2 genomic DNA (not shown). The A858D mutation has been reported previously only in heterozygous or compound heterozygous states. Neither patient exhibited the Southeast Asian ovalocytosis deletion mutation, Δ400-408, or the tightly linked Band 3 Memphis I polymorphism, E56K.
The C-to-A nucleotide substitution encoding the AE1 A858D mutation introduces a novel AvaII restriction site not present in the wildtype AE1 gene. The presence of the AvaII site can be used diagnostically to confirm the presence of the A858D mutation in appropriate PCR amplification products, as shown in Fig. 2D. The PCR product of 482 bp (lane 3) remains uncut in wildtype genomic DNA (lane 2), whereas the PCR product from A858D/A858D genomic DNA is cleaved at a single site, giving rise to two AvaII fragments of 345 and 137 bp (lane 1). Fig. 2D also shows that the C-to-A nucleotide substitution of the A858D mutation also abolishes a BglI site in the same PCR product, such that wildtype BglI fragments of 134 and 38 bp (lane 5) are contained in a single 172 bp fragment in A858D/A858D genomic DNA (lane 4).
Several intronic sequence variants were detected in both patients. Patients 1 and 2 each were homozygous for intron 3 substitutions c.11079G>A (SNP rs999716) and the apparently novel c.11200C>A, both within the putative kAE1 promoter region. Both patients were also homozygous for intron 17 substitutions c.20332T>G (SNP rs2857078) and c.20381G>A (SNP rs1476512). Genomic DNA was unavailable from either set of nominally obligate heterozygote parents or from the sib of patient 1. The two dRTA patients with hemolytic anemia described in this paper are the first dRTA patients from India with defined mutations in the SLC4A1/AE1 gene encoding kAE1 of the Type A intercalated cell basolateral membrane and eAE1 or the erythocyte. These patients are also the first reported with with homozygous AE1 A858D mutations. This substitution mutation introduces a novel AvaII restriction site easily diagnosed by restriction digest of a genomic PCR product.
Both patients are also homozygous for identical, linked, intronic polymorphisms. Though nominally unrelated, both patients are of the same Maratha ethnic group from the Mumbai region. The allele frequency of the A858D mutation or other dRTA-associated mutations among Marathas or other Indians is presently unknown. The A858D mutation was not found among 206 healthy individuals from Southern Thailand (7). Neither patient exhibited the Southeast Asian Ovalocytosis (SAO) mutation AE1 Δ400-408 or its tightly linked Memphis I polymorphism E56K.
Patient 1 presented with paradoxical macrocytosis which turned out to be coincident vitamin B12 deficiency, possibly compounded with folate deficiency. The macrocytosis resolved after repletion of cobalamin and folate, but the smear remained atypical. The course of patient 2 was complicated by multidrug-resistant tuberculosis, requiring treatment with nephrotoxic antibiotics, including aminoglycosides and some drugs previously associated with inflammatory intersitial nephritis. Both patients responded to oral base supplementation with resolution of acidosis, but without reversal of nephrocalcinosis. Patient 1 remains moderately anemic, requiring only occasional tranfusion. However, patient 2 is severely anemic with dependence on frequent transfusion, and is at risk for progressive nephrotoxicity from renal hemosiderosis (8) and chronic scarring (9) in the setting of nephrocalcinosis.
The AE1 A858D mutation has been described in compound heterozygosity with AE1 SAO in a Malay family, with AE1 ΔV850 in a family from Papua New Guinea (2), and with AE1 G701D in two Thais (10). All of these compound heterozygous patients exhibited complete dRTA. Both compound heterozygous and homozygous A858D dRTA patients responded to oral base supplementation with normalization of acidosis and resumption of normal growth.
The homozygous A858D patients presented with severe anemia. Anemia was also moderate-to-severe for A858D/SAO and A858D/ΔV850 patients, but A858D/G701D patients and A858D/+ heterozygotes had minimal if any anemia, with slight reticulocytosis. Most A858D/SAO red cells were small atypical elliptocytes. The more frequently elliptocytic A858D/ΔV850 red cells included microcytes, poikilocytes, and schizocytes (2), while A858D/G701D red cells included xerocytes, pincer cells, echinocytes, and acanthocytes (11). The smears of homozygous A858D patients resembled those of compound heterozygotes with Δ850 and G701D (Fig. 1), but A858D/+ red cells were normal or acanthocytic (2). The mechanism by which particular AE1 mutations give rise to specific morphologies of circulating erythrocytes remains unclear.
A858D/+ red cells were of normal rigidity, but A858D/SAO red cells exhibited greater rigidity than SAO/+ red cells. In the A858D/SAO red cell membrane, AE1 A858D polypeptide was less abundant than AE1 SAO polypeptide. DIDS-sensitive sulfate influx, was 80% of normal in intact A858D/+ red cells, but only 30% of normal in A858D/ΔV850 cells (2).
Recombinant kAE1 A858D expressed in Xenopus oocytes exhibited 28% of wildtype activity, a value increased to 64% by coexpression of human glycophorin A (GPA) through increased surface expression. AE1 SAO polypeptide coexpression exerted a dominant negative effect on AE1 A858D even in the presence of glycophorin A (2). AE1 A858D coexpressed with GPA also conferred on Xenopus oocytes low level cation leak activity, a property shared by other AE1 mutants associated with dRTA (3), and with apparently isolated stomatocytosis (12, 13).
In polarized MDCK cell monolayers, kAE1 A858D polypeptide was only slightly impaired in exit from endoplasmic reticulum, and trafficked normally to the basolateral membrane, where it exhibited some Cl−/HCO3− exchange activity, despite altered SITS-agarose binding of the detergent-solubilized mutant protein (14). kAE1 A858D coexpression with kAE1 SAO (15) or with kAE1 G701D polypeptides (16) promoted intracellular retention of both mutants with decreased surface expression. The Cys 858 residue is located near the exofacial surface of AE1 (17).
The two patients described in the current report are the first patients from India in whom dRTA has been linked to mutations in the SLC4A1/AE1 gene. They also represent the first reported cases of homozygous AE1 A858D mutations. The associated combined syndrome of hemolytic anema and distal renal tubular acidosis provides another example of an AE1 mutation in which hematologic and renal phenotypes are not segregated.
Methods
Blood samples were obtained during normal clinical care at Mumbai’s B. J. Wadia Children’s Hospital. Non-routine diagnostic studies were performed according to protocols approved by the Institutional Review Boards of the National Institute of Immunohematology (Mumbai) and Beth Israel Deaconess Medical Center (Boston). Hematological indices were measured in Mumbai with Sysmex automated cell counters (KX 21 or XS 800i, Kobe). Eosin-5′-maleimide (E5M) flow cytometry measured red cell surface abundance of Band 3/AE1/SLC4A1 polypeptide (18).
Genomic DNA was isolated from whole blood samples (Qiagen Blood and Tissue Dneasy Mini Kit) . The SLC4A1/AE1/Band 3 gene was PCR-amplified in 5 fragments encompassing all 20 exons with their splice junctions and flanking regions, and the putative kAE1 promoter in intron 3. Reaction mixes containing 100-500 ng of human genomic DNA and HotStart DNA Polymerase (Qiagen), made up to 50 μL reaction volume with the suppliers’ recommended buffer, were incubated at 95°C for 15 min, then subjected to 36 cycles of PCR (45 sec denaturation at 94°C, 2 min annealing at 60°C, 3-5 min elongation at 72°C). A 7 min final extension at 72°C was terminated by rapid cooling to room temperature. PCR products were separated in 1% agarose gels, visualized by ethidium bromide staining, excised, purified (QIAquick Gel Extraction Kit), and sequenced. Table 1 lists the oligonucleotides used to amplify and sequence AE1 genomic DNA.
TABLE 1.
Oligonucleotides for PCR amplification and sequencing of the human SLC4A1/AE1 gene.
| Oligo | SEQUENCE | Direction | Location | Use | Oligo location (nt per NG_007498) |
Amplicon Length |
|---|---|---|---|---|---|---|
| hAE1.I1F | 5′-GAGACCCTGGGATGCCCTGTG-3′ | Forward | Intron 1 | PCR-1, Seq | 10151-10161 | 1490 bp |
| hAE1.I4R | 5′-CCCTTGCTCCTCTCTTCCCTG-3′ | Reverse | Intron 4 | PCR-1, Seq | 11640-11620 | |
| hAE1.I4F | 5′-AAGCCTCACAAGCACAAGCCTC-3′ | Forward | Intron 4 | PCR-2, Seq | 12224-12245 | 1837 bp |
| hAE1.I9R | 5′-AGTAACCCAGGCCAGGTTGGAG-3′ | Reverse | Intron 9 | PCR-2, Seq | 14060-14039 | |
| hAE1.I6F | 5′-TGTCTACCCTTGATGTCTACC-3′ | Forward | Intron 6 | Seq (PCR-2) | 13090-13110 | -- |
| hAE1.I9F | 5′-TGTGGACTCCTGGGTCCTTTCC-3′ | Forward | Intron 9 | PCR-3, Seq | 14447-14467 | 1,440 bp |
| hAE1.I13R | 5′-CTTATACACAACCTCCCGTGTG-3′ | Reverse | Inron 13 | PCR-3, Seq | 15886-15865 | |
| hAE1.I13F | 5′-GCTGGGTAGAGTTAGCTGGTGG-3′ | Forward | Intron 13 | PCR-4, Seq | 17208-17229 | 1511 bp |
| hAE1.I16R | 5′-TCTTGCCTGCCTGGGAGAATGC-3′ | Reverse | Intron 16 | PCR-4, Seq | 18718-18697 | |
| hAE1.I16F | 5′-GCTACAAGGACACCAAGTATGG-3′ | Forward | Intron 16 | PCR-5, Seq | 19675-19696 | 3057 bp |
| hAE1.3R1 | 5′-TGGAAGGTGGGGATGTGGAATG-3′ | Reverse | Exon 20(a) | PCR-5, Seq | 22731-22710 | |
| hAE1.I17F | 5′-GTGCCTGTGTTTTATTCCCAGC-3′ | Forward | Intron 17 | Seq (PCR-5) | 21449-21470 | -- |
3′-UTR of the hAE1 mRNA
For diagnostic restriction digestion, genomic DNA of patients and normal controls was subjected to PCR-amplification (conditions as above, except for 2 min elongation time) with this oligonucleotide pair: hAE1.I18F2: 5′-AGGGTGCTGGGGTGTGATAGG-3′ and hAE1.I19R2: 5′-CTCACTCACAGCACACCCTGTC-3′. The amplicon was purified (QIAquick PCR purification Kit, Qiagen), digested with AvaII or BglI (New England Biolabs, Beverley, MA), then analyzed by agarose gel electrophroesis. (Informative restriction digestion did not require amplicon purification from the PCR reaction.)
Acknowledgements
This work was supported by NIH grants DK43495, HL077765 (SLA), DK34854 (The Harvard Digestive Diseases Center), and a grant from the Indian Council for Medical Research (RC and KG).
References
- 1.Kurschat CEAS. Renal Tubular Acidosis. In: Mount DBPM, editor. Molecular and Genetic Basis of Renal Disease. Saunders; Philadelphia: 2008. pp. 269–294. [Google Scholar]
- 2.Bruce LJ, Wrong O, Toye AM, Young MT, Ogle G, Ismail Z, Sinha AK, McMaster P, Hwaihwanje I, Nash GB, Hart S, Lavu E, Palmer R, Othman A, Unwin RJ, Tanner MJ. Band 3 mutations, renal tubular acidosis and South-East Asian ovalocytosis in Malaysia and Papua New Guinea: loss of up to 95% band 3 transport in red cells. The Biochemical journal. 2000;350(Pt 1):41–51. [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh S, Borgese F, Gabillat N, Guizouarn H. Southeast Asian AE1 associated renal tubular acidosis: cation leak is a class effect. Biochemical and biophysical research communications. 2009;382:668–672. doi: 10.1016/j.bbrc.2009.03.062. [DOI] [PubMed] [Google Scholar]
- 4.Mantan M, Bagga A, Virdi VS, Menon S, Hari P. Etiology of nephrocalcinosis in northern Indian children. Pediatric nephrology (Berlin, Germany) 2007;22:829–833. doi: 10.1007/s00467-006-0425-7. [DOI] [PubMed] [Google Scholar]
- 5.Nampoothiri S, Anikster Y. Carbonic anhydrase II deficiency a novel mutation. Indian pediatrics. 2009;46:532–534. [PubMed] [Google Scholar]
- 6.Sethi SK, Singh N, Gil H, Bagga A. Genetic studies in a family with distal renal tubular acidosis and sensorineural deafness. Indian pediatrics. 2009;46:425–427. [PubMed] [Google Scholar]
- 7.Yenchitsomanus PT. Human anion exchanger1 mutations and distal renal tubular acidosis. The Southeast Asian journal of tropical medicine and public health. 2003;34:651–658. [PubMed] [Google Scholar]
- 8.Stehberger PA, Shmukler BE, Stuart-Tilley AK, Peters LL, Alper SL, Wagner CA. Distal renal tubular acidosis in mice lacking the AE1 (band3) Cl−/HCO3− exchanger (slc4a1) J Am Soc Nephrol. 2007;18:1408–1418. doi: 10.1681/ASN.2006101072. [DOI] [PubMed] [Google Scholar]
- 9.Shayakul C, Jarolim P, Zachlederova M, Prabakaran D, Cortez-Campeao D, Kalabova D, Stuart-Tilley AK, Ideguchi H, Haller C, Alper SL. Characterization of a highly polymorphic marker adjacent to the SLC4A1 gene and of kidney immunostaining in a family with distal renal tubular acidosis. Nephrol Dial Transplant. 2004;19:371–379. doi: 10.1093/ndt/gfg557. [DOI] [PubMed] [Google Scholar]
- 10.Khositseth S, Sirikanerat A, Wongbenjarat K, Opastirakul S, Khoprasert S, Peuksungnern R, Wattanasirichaigoon D, Thongnoppakhun W, Viprakasit V, Yenchitsomanus PT. Distal renal tubular acidosis associated with anion exchanger 1 mutations in children in Thailand. Am J Kidney Dis. 2007;49:841–850. e841. doi: 10.1053/j.ajkd.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Khositseth S, Sirikanaerat A, Khoprasert S, Opastirakul S, Kingwatanakul P, Thongnoppakhun W, Yenchitsomanus PT. Hematological abnormalities in patients with distal renal tubular acidosis and hemoglobinopathies. American journal of hematology. 2008;83:465–471. doi: 10.1002/ajh.21151. [DOI] [PubMed] [Google Scholar]
- 12.Stewart AK, Vandorpe DH, Heneghan JF, Chebib F, Stolpe K, Akhavein A, Edelman EJ, Maksimova Y, Gallagher PG, Alper SL. The GPA-dependent, spherostomatocytosis mutant AE1 E758K induces GPA-independent, endogenous cation transport in amphibian oocytes. American journal of physiology. 2010;298:C283–297. doi: 10.1152/ajpcell.00444.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruce LJ, Robinson HC, Guizouarn H, Borgese F, Harrison P, King MJ, Goede JS, Coles SE, Gore DM, Lutz HU, Ficarella R, Layton DM, Iolascon A, Ellory JC, Stewart GW. Monovalent cation leaks in human red cells caused by single amino-acid substitutions in the transport domain of the band 3 chloride-bicarbonate exchanger, AE1. Nature genetics. 2005;37:1258–1263. doi: 10.1038/ng1656. [DOI] [PubMed] [Google Scholar]
- 14.Cordat E, Kittanakom S, Yenchitsomanus PT, Li J, Du K, Lukacs GL, Reithmeier RA. Dominant and recessive distal renal tubular acidosis mutations of kidney anion exchanger 1 induce distinct trafficking defects in MDCK cells. Traffic (Copenhagen, Denmark) 2006;7:117–128. doi: 10.1111/j.1600-0854.2005.00366.x. [DOI] [PubMed] [Google Scholar]
- 15.Kittanakom S, Cordat E, Reithmeier RA. Dominant-negative effect of Southeast Asian ovalocytosis anion exchanger 1 in compound heterozygous distal renal tubular acidosis. The Biochemical journal. 2008;410:271–281. doi: 10.1042/BJ20070615. [DOI] [PubMed] [Google Scholar]
- 16.Ungsupravate D, Sawasdee N, Khositseth S, Udomchaiprasertkul W, Khoprasert S, Li J, Reithmeier RA, Yenchitsomanus PT. Impaired trafficking and intracellular retention of mutant kidney anion exchanger 1 proteins (G701D and A858D) associated with distal renal tubular acidosis. Molecular membrane biology. 2010;27:92–103. doi: 10.3109/09687681003588020. [DOI] [PubMed] [Google Scholar]
- 17.Fujinaga J, Tang XB, Casey JR. Topology of the membrane domain of human erythrocyte anion exchange protein, AE1. The Journal of biological chemistry. 1999;274:6626–6633. doi: 10.1074/jbc.274.10.6626. [DOI] [PubMed] [Google Scholar]
- 18.Kedar PS, Colah RB, Kulkarni S, Ghosh K, Mohanty D. Experience with eosin-5′-maleimide as a diagnostic tool for red cell membrane cytoskeleton disorders. Clinical and laboratory haematology. 2003;25:373–376. doi: 10.1046/j.0141-9854.2003.00557.x. [DOI] [PubMed] [Google Scholar]