

Abstract
Objectives
This study was designed to evaluate the clinical and prognostic aspects of long QT syndrome-related cardiac events that occur in the first year of life (infancy).
Background
The clinical implications for patients with long QT syndrome who experience cardiac events in infancy have not been studied previously.
Methods
The study population of 3,323 patients with QTc ≥ 450 ms enrolled in the International LQTS Registry involved 20 patients with sudden cardiac death (SCD), 16 patients with aborted cardiac arrest (ACA), 34 patients with syncope, and 3,253 patients who were asymptomatic during the first year of life.
Results
The risk factors for a cardiac event among 212 patients who had an ECG recorded in the first year of life included QTc≥500ms, heart rate ≤100bpm, and female sex. ACA before age 1 year was associated with a hazard ratio of 23.4 (p<0.01) for ACA or SCD during age 1-10 years. During the 10-year follow-up after infancy, beta-blocker therapy was associated with a significant reduction in ACA/SCD only in those with a syncopal episode within 2 years before ACA/SCD, but not for those who survived ACA in infancy.
Conclusions
Patients with LQTS who experience ACA during the first year of life are at very high-risk for subsequent ACA or death during their next 10 years of life, and beta-blockers may not be effective in preventing fatal or near fatal cardiac events in this small but high-risk subset.
Keywords: Long QT Syndrome, Genetics, Infants, Risk Stratification
Long QT Syndrome (LQTS) is a rare genetic disorder caused by mutations involving genes that encode critical channel pore-forming alpha subunits and channel interacting proteins (1). The syndrome presents clinically with delayed ventricular repolarization seen as a prolonged QT interval on the electrocardiogram (ECG). Patients with this disorder are at high risk of experiencing syncope, aborted cardiac arrest (ACA), and sudden cardiac death (SCD) (1-4). Recent studies of LQTS indicate that risk factors for experiencing LQTS-related cardiac events are age and sex related (1). The childhood(5), adolescent(6), and adult(7) periods have been described, but little is known about the clinical course of LQTS patients who experience cardiac events during infancy, i.e., the first year of life.
The management of infants with symptomatic LQTS constitutes a serious clinical problem. It generates major anxiety in the families and significant concerns among the physicians involved. The lack of data leaves considerable uncertainty about the criteria for risk stratification and for the attendant therapeutic strategies that should vary from cautious conservative management to more aggressive and invasive approaches.
The goals of the present study are to assess the clinical risk factors for patients experiencing LQTS-related cardiac events (SCD, ACA, or syncope) in the first year of life and to evaluate the subsequent clinical course of patients with an infantile expression of symptomatic LQTS (ACA or syncope) during their next decade compared to those who reached their first birthday without any LQTS-related cardiac events.
Methods
The study population consisted of 3,323 patients drawn from the International LQTS Registry (8) who met the following criteria: LQTS probands and first and second-degree relatives of probands who had a QTc ≥450ms on a 12-lead ECG; patients from LQTS families who were genotype positive for an LQTS mutation regardless of their QTc interval; and patients from LQTS families who were categorized as having SCD according to established Registry criteria (6) even if they didn't have a recorded ECG. Infancy was defined as the period from birth until age 1 year. Patients with the Jervell and Lange-Nielsen Syndrome, as determined by QTc prolongation and congenital deafness, were excluded from the present analysis because their unusually high risk during their first decade of life has already been described (9,10) . Of the 3,323 patients who made up the study population, there were 3,253 subjects who had no historical evidence of LQTS-related cardiac events in the first year of life, and this group was used, by default, as the no cardiac event comparison group in infancy (Table 1).
Table 1. Clinical Characteristics of the Study Population in the First Year of Life.
| Clinical Characteristics | Sudden Cardiac Death | Aborted Cardiac Arrest† | Syncope Only | No Cardiac Event |
|---|---|---|---|---|
| #of patients | 20 | 16 | 34 | 3,253 |
| Female, n (%) | 8 (40)* | 11 (69) | 22 (65) | 2,014 (62) |
| Age at ECG, years | 0.2±0.3* | 3.5±5.5* | 6.7±9.1* | 29.4±21.4 |
| (number of ECGs) | (n=8) | (n=16) | (n=33) | (n=3,155) |
| QTc (ms) | 549±55* | 545±65* | 522±72* | 489±47 |
| Family LQT1, n, (%) | 6 (30) | 0 (0) | 7 (21) | 806 (25) |
| Family LQT2, n (%) | 0 (0) | 4 (25) | 7 (21) | 679 (21) |
| Family LQT3, n, (%) | 1 (5) | 1 (6) | 1 (3) | 173 (5) |
| Genotype unknown (%) | (65) | (69) | (55) | (49) |
| Beta-blockers anytime, n (%) | 5(25) | 12 (75)* | 24 (71) | 476 (15) |
| Left cardiac sympathetic denervation, n (%) | 1 (5.) | 3 (19)* | 2 (6)* | 23 (0.7) |
| Pacemaker, n (%) | 1 (5) | 5 (31)* | 1 (3) | 36 (1) |
| Implanted cardiac defibrillator, n (%) | 0 (0) | 3 (19)* | 1 (3) | 29 (0.9) |
P-value <0.05 versus No Cardiac Event
Ten of the 16 infants who had an aborted cardiac arrest had a prior syncopal episode.
The International LQTS Registry study was approved by the University of Rochester Institutional Review Board, and informed consent was obtained from study participants. The Registry included data on patients' demographics, clinical characteristics, clinical history, information on LQTS-related cardiac events, and therapy as described previously (8). Copies of past ECGs were obtained at the time of enrollment and additional ECGs, if available, were obtained at yearly follow-up. The first recorded ECG was used for the current analysis. Bazett's formula was used to correct the QT interval for heart rate (QTc).
Patients were categorized into one of four LQTS groups: 1) infants who had SCD and were from an identified LQTS family; 2) infants who had ACA; 3) infants who had a syncopal event defined as transient unexplained loss of consciousness during awake daily activity with abrupt onset and offset of the episode that was observed by a parent or an adult supervising the infant; and 4) infants who did not have an LQTS-related cardiac event. The primary outcome during age 1-10 years among patients who experienced ACA, syncope, or no cardiac event in the first year of life was a near-fatal or fatal cardiac event (ACA or SCD).
Statistical analysis involved the t-test for continuous variables and chi-square or Fisher's exact tests for categorical variables. Follow-up time after the first year of life was censored at 10 years of age because of the limited number of patients who experienced cardiac events beyond that age. The graphical Kaplan-Meier method and the Cox proportion hazards model were used to evaluate the risk of pre-specified clinical factors of interest to cardiac events during age 1-10 years. Time-dependent syncope and time-dependent β-blocker use were included in the Cox models. Relevant interactions among the risk variables and outcome were explored in the Cox models. All models were stratified by the decade in which the patient was born (before 1970, 1970-1980, 1980-1990, or after 1990) to account for differences in the baseline hazard function for historically different time periods in which different LQTS-related therapies were used. The statistical software used to perform the analyses was SAS version 9.1.3. All statistical tests were 2-sided, and the significance level was set at p<0.05.
Results
LQTS during Infancy (age 0-1 year)
In the study population of 3,323 LQTS subjects, 20 died suddenly, 16 experienced ACA, and 34 experienced syncope in the first year of life; the remaining 3,253 subjects had no evident LQTS-related symptoms before their first birthday. The clinical characteristics of the four groups of patients are presented in Table 1.
Descriptive characteristics of the 20 patients who died suddenly during the first year of life are presented in Table 2. Eight (40%) of the patients were female, and all 8 who had an ECG had a QTc ≥500 ms. Data were available for at least one parent's QTc in 15 patients, and 10 out of 15 patients had a parent with a QTC ≥450 ms. Four of the 20 infants with SCD experienced a prior cardiac event before death, and 4 patients were on beta-blockers at the time of death. One patient who died was treated with a pacemaker, left sympathetic denervation, and β-blockers.
Table 2. Descriptive Characteristics of the 20 LQTS Infants Who Died in the First Year of Life.
| Gender (year of birth) | Age at Death (days) | QTc (ms) | RR interval (ms) | Parent* Max QT (ms) | Prior Syncope (n) | Prior Aborted Cardiac Arrest (n) | Treatment(s) | |
|---|---|---|---|---|---|---|---|---|
| 1 | F (1985) | 176 | 560 | 420 | -- | 0 | 1 | |
| 2 | M (1987) | 193 | 500 | 460 | 440 | 0 | 3 | BB |
| 3 | M (1991) | 352 | 600 | 440 | 420 | 0 | 2 | BB, PM, LCSD |
| 4 | F (1991) | 79 | 620 | 600 | 470 | 0 | 0 | BB |
| 5 | M (1945) | 5 | -- | -- | 490* | 0 | 0 | |
| 6 | F (1960) | 1 | -- | -- | 480 | 0 | 0 | |
| 7 | F (1978) | 75 | -- | -- | -- | 0 | 0 | |
| 8 | M (1957) | 312 | -- | -- | -- | 0 | 0 | |
| 9 | M (1962) | 122 | -- | -- | 580 | 0 | 0 | |
| 10 | M (1997) | 1 | 500 | 780 | 500* | 0 | 0 | |
| 11 | M (1999) | 3 | -- | -- | 400* | 0 | 0 | |
| 12 | F (1992) | 33 | 610 | 720 | 490 | 0 | 0 | BB |
| 13 | M (1953) | 2 | -- | -- | 450* | 0 | 0 | |
| 14 | F (1955) | 60 | -- | -- | 450* | 0 | 0 | |
| 15 | M (1970) | 93 | -- | -- | 410 | 3 | 0 | |
| 16 | M (1967) | 120 | -- | -- | 430 | 0 | 0 | |
| 17 | M (1993) | 47 | -- | -- | -- | 0 | 0 | |
| 18 | F (1992) | 88 | 500 | 360 | -- | 0 | 0 | |
| 19 | M (1999) | 326 | 500 | 530 | 490 | 0 | 0 | BB (d/c before death) |
| 20 | F (1999) | 60 | -- | -- | 500 | 0 | 0 |
ACA: aborted cardiac arrest; BB: β-blockers; LCSD: left cardiac sympathetic denervation; PM: pacemaker. The 3 infants who had neither an ECG nor a parental ECG were first degree relatives of family members with well-documented LQTS.
Only mother's QTc recorded.
The distribution of risk factors for the different cardiac event groups for the 212 patients who had an ECG during the first year of life are presented in Table 3. Cox proportional hazard regression analysis revealed that the following risk factors were associated with a cardiac event in infancy (n=28; SCD, ACA, or syncope only): QTc≥500ms (hazard ratio (HR)=4.31, p=0.002), RR≥600ms (HR=2.38, p=0.04), and female sex (HR=2.21, p=0.05); age at ECG, β-blocker at time of ECG, and LQT1, 2, and 3 genotypes did not make significant contributions to the risk model.
Table 3. Clinical Characteristics of Patients with an ECG in the First Year of Life.
| Clinical Characteristic | Sudden Cardiac Death | Aborted Cardiac Arrest | Syncope Only | No Cardiac Event |
|---|---|---|---|---|
| # of patients | 8 | 6 | 14 | 184 |
| Female, n (%) | 4 (50) | 4 (67) | 8 (57) | 86 (47) |
| Age at ECG, year | 0.2±0.3 | 0.2±0.3 | 0.2±0.2 | 0.2±0.3 |
| Beta-blocker at time of ECG, n (%) | 0 (0) | 1 (17) | 3 (21) | 20 (11) |
| QTc (ms) | 549±55* | 588±58* | 534±74* | 491±48 |
| QTc ≥500ms, n (%) | 8 (100)* | 5 (83)* | 7 (50) | 65 (35) |
| RR (ms) | 538±150 | 723±151* | 561±195 | 496±111 |
| RR≥600ms, n (%) | 3 (38)* | 5 (83)* | 1 (7) | 16 (9) |
| Family LQT1, n (%) | 2 (25) | 0 (0) | 5(36) | 63 (34) |
| Family LQT2, n (%) | 0 (0) | 1 (17) | 4 (29) | 43 (23) |
| Family LQT3, n (%) | 1 (13) | 1 (17) | 0 (0) | 11 (6) |
P-value <0.05 versus No Cardiac Event in the first year of life.
Clinical Course During Ages 1-10 Years
The clinical course of patients who experienced ACA, syncope, or no cardiac event in the first year of life was evaluated during the next 10 years of life. Those who experienced ACA in the first year had a significantly higher probability of experiencing another ACA event or a LQTS-related SCD during ages 1-10 years (Figure 1). ACA in the first year of life was associated with a hazard ratio of 23.4 (p<0.01) for a subsequent ACA/LQTS-related SCD during the 1-10 year period (Table 4). However, syncope only during infancy was not associated with an increased risk for ACA/LQTS-related SCD during follow-up. Recent syncope within the past 2 years, QTc ≥500 ms, and male gender were significant risk factors for ACA/LQTS-related SCD in the follow-up period. Beta-blocker use was not associated with a significant reduction in ACA/LQTS-related SCD risk during 10-year follow-up (Table 4). In interaction analyses, beta-blocker therapy was associated with a significant reduction in these events in those with a syncopal episode within 2 years before ACA/SCD (HR=0.35, p=0.03, CI: 0.14-0.90), but not for those with remote syncope more than 2 years before or ACA in infancy.
Figure 1.
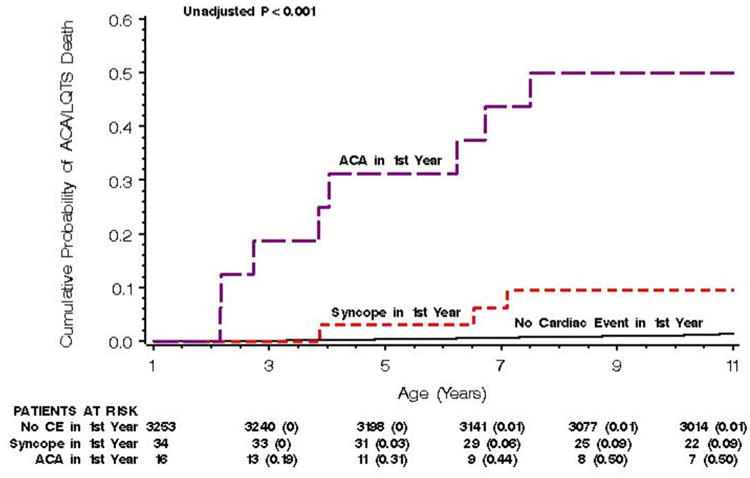
Cumulative Probability of Aborted Cardiac Arrest/LQTS-related Death During Ages 1-10 years Among Those with Aborted Cardiac Arrest, Syncope, or No Cardiac Event in the First Year of Life.
Table 4. Multivariate Analysis: Predictors of Aborted Cardiac Arrest or LQTS-related Sudden Death During Ages 1-10 Years.
| Factor | Number of 1st Cardiac Events [ACA/SCD] in Patients with the Specific Factor | Hazard Ratio | 95% CI | P-value |
|---|---|---|---|---|
| ACA in first year of life (n=16) | 8 [6/2] | 23.4 | 7.6-71.9 | <0.01 |
| Syncope in first year of life (n=44) | 7 [4/3] | 0.7 | 0.2-2.5 | 0.58 |
| ≥1 syncope in the past 2 years | 24[20/4] | 9.7 | 5.2-18.0 | <0.01 |
| ≥1 syncope more than 2 years ago and no syncope within the past 2 years | 4[3/2] | 1.6 | 0.5-5.4 | 0.47 |
| QTc ≥ 500 ms (n=1,062) | 38 [26/12] | 3.4 | 1.9-6.0 | <0.01 |
| Male (n=1,256) | 38 [27/11] | 2.2 | 1.3-3.9 | 0.01 |
Note: This analysis involves only patients who survived the first year of life and includes 99 patients with “Missing ECGs” who had only 2 deaths during the 10-year follow-up. The hazard ratio is the risk of a cardiac event per unit of time with the factor present to the risk for patients with the factor absent. The numbers in parentheses refer to the number of patients with the factor present. During the 10 year follow-up, there were 58 first cardiac events consisting of 42 aborted cardiac arrests (ACA) and 16 LQTS-related sudden cardiac deaths. Time-dependent β-blocker did not make a significant main-effect contribution to this model (hazard ratio=0.71, p=0.30), but was included as a factor in the multivariate analysis; 559 patients were on beta-blockers at one time or another during the 1-10 year follow-up. *Syncope within the past 2 years and remote syncope more than 2 years ago are time-dependent factors with patients moving into and out of each of these risk groups over time during the 1-10 year follow-up. The numbers of patients with these risk factors cannot be provided as simple raw number counts for these time-dependent variables.
Syncope within the past 2 years and remote syncope more than 2 years ago are time-dependent factors with patients moving into and out of each of these risk groups over time during the 1-10 year follow-up. The numbers of patients with these risk factors cannot be provided as simple raw number counts for these time-dependent variables.
Because of the increased long-term risk associated with ACA that occurred in the first year of life (Table 4), we compared the risk of ACA in the first year of life to the risk of a first ACA after age 1 year for second ACA or LQTS-related SCD during ages 1-10 years. Infants who had an ACA in the first year of life had a 2.3-fold greater risk (p=0.03) for a second ACA or SCD during ages 1-10 years than patients who experienced their first ACA after age 1 (Table 5).
Table 5. Multivariate Analysis: Predictors for 2nd Aborted Cardiac Arrest or LQTS-related Death During Age 1-10 Years by 1st Aborted Cardiac Arrest in the First Year of Life vs. 1st Aborted Cardiac Arrest Age 1-10 Years.
| Factor | Hazard Ratio | 95% CI | P-value |
|---|---|---|---|
| 1st ACA in the first year of life | 2.3 | 1.1-4.8 | 0.03 |
| QTc ≥ 500 ms | 2.3 | 0.9-5.5 | 0.07 |
| Male | 0.9 | 0.4-2.0 | 0.7 |
Note: QTc ≥500ms and Male were included in this model in view of their contribution to cardiac events in Table 4.
ACA=aborted cardiac arrest.
Discussion
This International LQTS Registry study with focus on cardiac events occurring in the first year of life and on the prognostic significance of these events to age 10 has provided four major findings. Infants with QTc prolongation, relative slow heart rate, and female sex are at increased risk for cardiac events during the first year of life. LQTS infants who experience ACA in the first year of life were at very high risk for near fatal or fatal cardiac events during the next 10 years of life. Syncope in infancy was not a major risk factor for subsequent ACA/SCD during age 1-10 years, but this negative finding may relate to the difficulty parents have in accurately recalling or identifying a syncopal episode with transient loss of consciousness in their infant child. For infants who survive the first year of life, we found no statistical evidence of an association between the use β-blockers and reduction in ACA or LQTS-related SCD during the next decade of life. These results carry significant clinical implications and provide previously unavailable data to help physicians make rational choices for the management of LQTS infants with syncope and aborted cardiac arrest in the first year of life.
Goldenberg et al showed that time-dependent β-blocker therapy was associated with a significant 53% reduction in the risk of ACA or LQTS-related SCD during ages 1-12 years (5). Goldenberg's study did not include infants who experienced ACA in the first year of life. In the current study, β-blockers were associated with a 65% reduction in 1-10 year cardiac events in those who had recent syncope, i.e., within 2 years of the end point, but not for patients who experienced ACA in infancy.
In a study of LQTS neonates and children, Villain et al reported an overall favorable outcome for children treated with β-blocker therapy (11). Direct comparison with our study is difficult because their definition of neonates extended only to the first six months. However, there were two deaths in patients on β-blocker therapy, and they involved two infants both with complete atrioventricular block who died at 3 and 9 days of age following a pacemaker implant and related complications. In agreement with our findings, 16 children required additional therapies because of insufficient protection from β-blocker therapy for cardiac events, and 10 of these 16 children had symptoms in the first year of life.
Beta-blockers are currently first-line therapy for patients with symptomatic LQTS (12,13), but they are not always effective in the young (14). Nevertheless, β-blockers should be initiated in all infants with LQTS unless a specific contraindication exists such as asthma or an allergy to this class of drugs. Infants who survive an ACA are potential candidates for an implanted defibrillator (15), but this therapy is rarely used in very young patients because of the potential complications associated with device implantation in small subjects. Pacemakers (16) and left cardiac sympathetic denervation (17-19) have been used in young, high-risk LQTS patients, but these therapies as well as implanted defibrillators were used infrequently in the study population, and thus there was insufficient statistical power to properly evaluate their efficacy in this age group. Nevertheless, the reported high success rate of left cardiac sympathetic denervation in LQTS in older patients (19) and its feasibility in infants (20,21) provides a rationale for considering this intervention together with β-blocker therapy as a logical first step for therapy in high-risk infants who have experienced an ACA event. This therapeutic approach does not preclude the possibility of an implanted defibrillator later on, if necessary, when the child is older and is of sufficient size for defibrillator implantation.
LQTS in the first year of life is of special interest because of its association with Sudden Infant Death Syndrome (SIDS), and newborn ECG screening has been used to identify these at-risk patients (22,23). The risks and benefits of such a diagnostic approach has been discussed in a recent editorial by Berul and Perry (24). In a large prospective study looking at the mechanism of SIDS, Arnestad et al genotyped SIDS victims for LQTS mutations and estimated that LQTS was the cause of at least 9.5% of SIDS cases (25). Currently, it is cost prohibitive to routinely genotype every newborn for LQTS mutations, but it is advisable to genotype newborns in LQTS families, and newborns found to have a clear QT prolongation during an ECG screening performed for whatever reason.
Limitations in the present study are the absence of ECGs in 60% of those who died in infancy, the limited numbers of infants who received therapy of any kind prior to SCD in the first year of life, and the incomplete genotyping of the study population. The goal of the present study was to evaluate all LQTS probands and affected first- and second-degree relatives of LQTS probands during the first year of life in the International LQTS Registry. Restriction of the study population only to those with ECGs before cardiac events in infancy, those with therapy, and patients with genotype data would have provided an incomplete picture of the severity of the disease process in infancy. We cannot draw any conclusions about the impact of preventive therapy on SCD in infants with LQTS. In view of the low event counts, the findings from this study need to be interpreted with caution since over-fitting may be an issue.
LQTS patients who experience ACA during the first year of life comprise a small (<2%) but very high-risk group for subsequent near-fatal and fatal cardiac events during the first decade of life. These high-risk patients require aggressive age-related treatment and careful follow-up to reduce their LQTS-related morbidity and mortality.
Acknowledgments
This work was supported in part by research grants HL-33843 (Moss), HL-51618 (Moss), HL-68880 (Schwartz), and HD-42569 (Ackerman) from the National Institutes of Health, Bethesda, Maryland, USA; by an unrestricted research grant from BioReference Labs, Inc., Elmwood Park, New Jersey (Moss); and by grant GGP07016 from Telethon-Italy (Schwartz).
List of Abbreviations
- ACA
aborted cardiac arrest
- ECG
electrocardiogram
- LQTS
Long QT Syndrome
- HR
hazard ratio
- QTc
QT interval corrected for heart rate
- SCD
sudden cardiac death
Footnotes
Dr. Michael J. Ackerman is a consultant to and has a license agreement/royalty arrangement with PGxHealth (New Haven, CT), a commercial genetic testing company that is division of Clinical Data, Inc. Dr. Kaufman has received research support from CardioDx and St. Jude Medical (no conflict of interest with current topic).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. 2008;51:2291–300. doi: 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- 2.Moss AJ. Long QT Syndrome. JAMA. 2003;289:2041–4. doi: 10.1001/jama.289.16.2041. [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–74. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 5.Goldenberg I, Moss AJ, Peterson DR, et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation. 2008;117:2184–91. doi: 10.1161/CIRCULATIONAHA.107.701243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hobbs JB, Peterson DR, Moss AJ, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. Jama. 2006;296:1249–54. doi: 10.1001/jama.296.10.1249. [DOI] [PubMed] [Google Scholar]
- 7.Sauer AJ, Moss AJ, McNitt S, et al. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329–37. doi: 10.1016/j.jacc.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 8.Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation. 1991;84:1136–44. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- 9.Goldenberg I, Moss AJ, Zareba W, et al. Clinical course and risk stratification of patients affected with the Jervell and Lange-Nielsen syndrome. J Cardiovasc Electrophysiol. 2006;17:1161–8. doi: 10.1111/j.1540-8167.2006.00587.x. [DOI] [PubMed] [Google Scholar]
- 10.Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783–90. doi: 10.1161/CIRCULATIONAHA.105.592899. [DOI] [PubMed] [Google Scholar]
- 11.Villain E, Denjoy I, Lupoglazoff JM, et al. Low incidence of cardiac events with beta-blocking therapy in children with long QT syndrome. Eur Heart J. 2004;25:1405–11. doi: 10.1016/j.ehj.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 12.Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–23. doi: 10.1161/01.cir.101.6.616. [DOI] [PubMed] [Google Scholar]
- 13.Vincent GM, Schwartz PJ, Denjoy I, et al. High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215–21. doi: 10.1161/CIRCULATIONAHA.108.772533. [DOI] [PubMed] [Google Scholar]
- 14.Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. Jama. 2004;292:1341–4. doi: 10.1001/jama.292.11.1341. [DOI] [PubMed] [Google Scholar]
- 15.Zareba W, Moss AJ, Daubert JP, Hall WJ, Robinson JL, Andrews M. Implantable cardioverter defibrillator in high-risk long QT syndrome patients. J Cardiovasc Electrophysiol. 2003;14:337–41. doi: 10.1046/j.1540-8167.2003.02545.x. [DOI] [PubMed] [Google Scholar]
- 16.Moss AJ, Liu JE, Gottlieb S, Locati EH, Schwartz PJ, Robinson JL. Efficacy of permanent pacing in the management of high-risk patients with long QT syndrome. Circulation. 1991;84:1524–9. doi: 10.1161/01.cir.84.4.1524. [DOI] [PubMed] [Google Scholar]
- 17.Moss AJ, McDonald J. Unilateral cervicothoracic sympathetic ganglionectomy for the treatment of long QT interval syndrome. N Engl J Med. 1971;285:903–4. doi: 10.1056/NEJM197110142851607. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz PJ, Locati EH, Moss AJ, Crampton RS, Trazzi R, Ruberti U. Left cardiac sympathetic denervation in the therapy of congenital long QT syndrome. A worldwide report [see comments] Circulation. 1991;84:503–11. doi: 10.1161/01.cir.84.2.503. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation. 2004;109:1826–33. doi: 10.1161/01.CIR.0000125523.14403.1E. [DOI] [PubMed] [Google Scholar]
- 20.Collura CA, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 2009 doi: 10.1016/j.hrthm.2009.03.024. in press. [DOI] [PubMed] [Google Scholar]
- 21.Ouriel K, Moss AJ. Long QT syndrome: an indication for cervicothoracic sympathectomy. Cardiovasc Surg. 1995;3:475–8. doi: 10.1016/0967-2109(95)94444-2. [DOI] [PubMed] [Google Scholar]
- 22.Quaglini S, Rognoni C, Spazzolini C, Priori SG, Mannarino S, Schwartz PJ. Cost-effectiveness of neonatal ECG screening for the long QT syndrome. Eur Heart J. 2006;27:1824–32. doi: 10.1093/eurheartj/ehl115. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz PJ. Pro: Newborn ECG screening to prevent sudden cardiac death. Heart Rhythm. 2006;3:1353–5. doi: 10.1016/j.hrthm.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 24.Berul CI, Perry JC. Contribution of long-QT syndrome genes to sudden infant death syndrome: is it time to consider newborn electrocardiographic screening? Circulation. 2007;115:294–6. doi: 10.1161/CIRCULATIONAHA.106.675470. [DOI] [PubMed] [Google Scholar]
- 25.Arnestad M, Crotti L, Rognum TO, et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–7. doi: 10.1161/CIRCULATIONAHA.106.658021. [DOI] [PubMed] [Google Scholar]