

Abstract
D-Ribulose 1,5-bisphosphate carboxylase/oxygenases (RuBisCOs) are promiscuous, catalyzing not only carboxylation and oxygenation of D-ribulose 1,5-bisphosphate but also other promiscuous, presumably nonphysiological, reactions initiated by abstraction of the 3-proton of D-ribulose 1,5-bisphosphate. Also, RuBisCO has homologues that do not catalyze carboxylation; these are designated RuBisCO-like proteins or RLPs. Members of the two families of RLPs catalyze reactions in the recycling of 5′-methylthioadenosine (MTA) generated by polyamine synthesis: 1) the 2,3-diketo-5-methylthiopentane 1-phosphate (DK-MTP 1-P) “enolase” reaction in the well-known “methionine salvage” pathway in species of Bacilli; and 2) the 5-methylthio-D-ribulose 1-phosphate (MTRu 1-P) 1,3-isomerase reaction in the recently discovered “MTA-isoprenoid shunt” that generates 1-deoxy-D-xylulose 5-phosphate (DXP) for nonmevalonate isoprene synthesis in Rhodospirillum rubrum. We first studied the structure and reactivity of DK-MTP 1-P which was reported to decompose rapidly [Ashida, H., Saito, Y., Kojima, C., and Yokota, A. (2008) Biosci Biotechnol Biochem 72, 959–67]. The 2-carbonyl group of DK-MTP 1-P is rapidly hydrated in solution and can undergo enolization both nonenzymatically and enzymatically via the small amount of unhydrated material that is present. We then examined the ability of RuBisCO from R. rubrum to catalyze both of the RLP-catalyzed reactions. Contrary to a previous report [Ashida, H., Saito, Y., Kojima, C., Kobayashi, K., Ogasawara, N., and Yokota, A. (2003) Science 302, 286–290], we were unable to confirm that this RuBisCO catalyzes the DK-MTP 1-P “enolase” reaction either in vitro or in vivo. We also determined that this RuBisCO does not catalyze the MTRu 1-P 1,3-isomerase reaction in vitro. Thus, although RuBisCOs can be functionally promiscuous, RuBisCO from R. rubrum is not promiscuous for either of the known RLP-catalyzed reactions.
INTRODUCTION
D-Ribulose 1,5-bisphosphate carboxylase/oxygenase (RuBisCO1) catalyzes carboxylation of D-ribulose 1,5-bisphosphate with atmospheric CO2 to generate two molecules of 3-phosphoglycerate (3-PGA) in the Calvin-Benson-Bassham cycle as well as nonproductive oxygenation to generate one molecule of 3-PGA and one molecule of phosphoglycolate (2). The reactions are initiated by abstraction of the substrate’s 3-proton by a carboxylated active site Lys to generate an enediolate anion intermediate that is stabilized by coordination to an active site Mg2+ (Figure 1, panel A). In addition, RuBisCOs catalyze other competing reactions in vitro that also are initiated by abstraction of the 3-proton [epimerization to generate D-xylulose 1,5-bisphosphate, 1,2-proton transfer to generate 3-ketoarabinitol 1,5-bisphosphate, and elimination of phosphate to generate 1-deoxy-D-glycero-2,3-pentdiulose 5-phosphate] (3–5).
Figure 1.

Mechanisms of RLP-catalzyed reactions. Panel A, RuBisCO. Panel B, DK-MTP 1-P “enolase”. Panel C, MTRu 1-P isomerase.
The potential for RuBisCO to be promiscuous evidently has been exploited by Nature in the divergent evolution of homologues that do not catalyze the carboxylation of D-ribulose 1,5-bisphosphate, although the pathway(s) for the evolution of different physiological functions is not known. These homologues, designated RuBisCO-like proteins or RLPs, lack one or more functional groups necessary for carboxylation (6, 7). RLPs can be partitioned into several families [a sequence similarity network (8) for the RLPs is shown in Figure 2], some retaining cationic side chains that form the binding site for the 5-phosphate group of D-ribulose 1,5-bisphosphate in RuBisCO and others having hydrophobic substitutions for these residues that would prevent the binding of metabolites with 5-phosphate groups [all retain the binding site for the 1-phosphate group at the ends of the seventh and eighth β-strands in the (β/α)8-barrel domain] (9).
Figure 2.

Sequence similarity network for the RLPs (8). Each symbol (node) is a protein sequence; each line (edge) represents sequence similarity associated with the BLASTP e-value relating the sequences. This network was constructed using 10−80 as the threshold e-value, i.e., the edges are associated with values <10−80. The nodes colored with red are associated with the DK-MTP 1-P “enolase” function; the nodes colored with blue are associated with the MTRu 1-P 1,3-isomerase function. The functions associated with the other colors are currently unknown. The nodes colored light blue, light green, and green retain the residues for binding the ω-phosphate group of a ketose 1-phosphate substrate; the nodes colored magenta and yellow contain substitutions for these residues that suggest that the substrates for these are ketose 1-phosphates.
The members of two RLP families that lack the residues for binding a 5-phosphate group and, therefore, are predicted to utilize ketose 1-phosphates as substrates (instead of ketose 1,5-bisphosphates) participate in distinct pathways for recycling 5′-methylthioadenosine (MTA) generated in polyamine synthesis. The members of the first family (red nodes in Figure 2) catalyze the 2,3-diketo-5-methylthiopentane 1-phosphate (DK-MTP 1-P) “enolase”2 reaction (Figure 1, panel B) in the well-known “methionine salvage” pathway in species of Bacilli in which the 5′-methylthioribose moiety of MTA is converted to methionine (Figure 3, panel A; in other organisms, the “enolase” reaction is catalyzed by an analogous enzyme); the product of the “enolase” reaction is 2-hydroxy-3-keto-5-methylthiopent-1-ene 1-phosphate (HK-MTP 1-P) (10). The members of the second family (blue nodes in Figure 2) catalyze the 5-methylthio-D-ribulose 1-phosphate (MTRu 1-P) 1,3-isomerase reaction (Figure 1, panel C) in a recently discovered nonmevalonate isoprene synthesis pathway (“MTA-isoprenoid shunt”) in Rhodospirillum rubrum (Figure 3, panel B) (11); the 1,3-isomerase converts MTRu 1-P to 1-methylthioxylulose 5-phosphate (MTXu 5-P) that is the immediate precursor to 1-deoxy-D-xylulose 5-phosphate (DXP) in the isoprene pathway and methanethiol that is captured, in part, for methionine synthesis (12).
Figure 3.

The two characterized pathways for MTA recycling. Panel A, the classical methionine salvage pathway in which methionine is quantitatively recovered from MTA (10). Panel B, the recently discovered pathway in which MTA is ribose moiety of MTA is converted to DXP and the 5′-methylthio group is released as methanethiol that can be captured, in part, to form methionine (12).
In the functional characterization of the RLP from Bacillus subtilis as the “enolase” in methionine salvage pathway, data were provided that RuBisCO from R. rubrum could catalyze the “enolase” reaction (10). A plasmid encoding RuBisCO from R. rubrum was reported to trans-complement a B. subtilis RLP− strain when MTA was provided as the sole sulfur source, although the growth rate was slower than that of the wild type strain. Purified RuBisCO from R. rubrum also was reported to catalyze the “enolase” reaction, although in contrast to the reaction observed with the authentic “enolase” from B. subtilis, the reaction catalyzed by the RuBisCO showed a curious lag in the production of HK-MTP 1-P product.
Given our interest in the functions and mechanisms of the functionally characterized RLPs (1, 7, 11, 13), we decided to (re)investigate the ability of RuBisCO from R. rubrum to catalyze both the “enolase” and MTRu 1-P 1,3-isomerase reactions. In particular, we were intrigued that the specificity determining residues differ and, also, the proton abstraction reactions are catalyzed by structurally distinct general basic catalysts located on opposite faces of the 2-keto groups of the substrates that are coordinated to active site Mg2+ ions that stabilize the enediolate intermediates [the 3-proton of D-ribulose 1,5-bisphosphate by the carboxylated Lys in RuBisCO and the 1-proS proton of MTRu 1-P by a Lys that is present in all DK-MTP 1-P “enolases” but absent in RuBisCOs; we discuss the likely identities of the general basic/acidic catalysts in the 1,3-isomerase]. In this article, we report that we are unable to reproduce the reported promiscuity of RuBisCO from R. rubrum to catalyze the “enolase” reaction both in vitro and in vivo or detect the MTRu 1-P 1,3-isomerase reaction in vitro. We also comment on the limitations of using in vivo complementation without either detailed in vitro enzymology or a complete knowledge of in vivo metabolic pathways to discover functional promiscuity in the RuBisCO superfamily.
MATERIALS AND METHODS
Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich or Fisher Scientific. PCR amplification reactions were performed with Invitrogen Pfx polymerase according to the manufacturer’s instructions; restriction digests were performed with enzymes obtained from New England Biolabs and used according to the manufacturer’s instructions. 1H NMR spectra were obtained with a Varian Unity INOVA 500NB spectrophotometer; 13C NMR spectra were acquired with a Varian Unity Inova 600 MHz spectrometer. All kinetic assays were performed with a Cary 300 Bio UV/VIS spectrometer.
5-Methylthio-D-ribose (MTR) was synthesized by published procedures (1, 11). RuBisCO from R. rubrum (14), DK-MTP 1-P “enolase” from G. kaustophilus (1), MTRu 1-P 1,3-isomerase from R. rubrum (11), MTR 1-P 1,2-isomerase from B. subtilis (1), and MTRu 1-P dehydratase from B. subtilis (1) were prepared according to procedures reported by our laboratories. H218O (95% enrichment) was purchased from Cambridge Isotope Laboratories, Inc.
[2-13C]MTR
[2-13C]MTR was prepared from [2-13C]-D-ribose (Omicron Biochemicals) according to one of the procedures described for unlabeled MTR (1).
MTR 1-P and [2-13C]MTR 1-P
MTR 1-P and [2-13C]MTR 1-P were prepared enzymatically using MTR kinase (1).
1H NMR Assay for RuBisCO
The reaction mixture (0.8 mL) contained 20 mM K+ phosphate, pH 7.8, 5 mM MgCl2, 15 mM NaHCO3, 2.5 mM D-ribulose 1,5-bisphosphate, and 025 μM RuBisCO. 1H NMR spectra were accumulated as a function of time. The intensity of the resonance associated with the 3-proton decreased linearly with time; the rate of this decrease was used to calculate the value of kcat. The value of ratio of carboxylation to oxygenation in the assay was calculated from the intensities of the resonances associated with the α-protons of 3-phosphoglyerate and 2-phosphoglycolate products.
Spectrophotometric Assay for Conversion of DK-MTP 1-P to HK-MTP 1-P (“Enolase” Reaction)
The initial reaction mixtures (0.2 mL) contained 20 mM Na+ HEPES, pH 7.9, 5 mM MgCl2, 15 mM NaHCO3, and MTR 1-P. MTRu 1-P was generated in situ by addition of MTR 1-P 1,2-isomerase to a final concentration of 1 μM followed by incubation at room temperature for five minutes. MTRu 1-P dehydratase then was added to a final concentration of 10 μM, and the reaction mixture was incubated for 5 min to allow formation of the hydrate of DK-MTP 1-P (vide infra).
To study the nonenzymatic conversion of DK-MTP 1-P to HK-MTP 1-P, the increase in absorbance at 270 nm was measured. To study the enzymatic conversion, either “enolase” or RuBisCO was added, and the increase in absorbance at 270 nm was measured. The rate of formation of HK-MTP 1-P was calculated using an extinction coefficient of 9.1 × 103 M−1 cm−1 at 270 nm for the enone chromophore (15).
For assays measuring the dependence of rate on enzyme concentration, the reactions contained 100 μM DK-MTP 1-P (from MTR 1-P). For assays measuring the dependence rate on HK-MTP 1-P concentration, “enolase” or RuBisCO was added to final concentrations of 1 μM or 10 μM, respectively.
Spectrophotometric Assay for Conversion of MTRu 1-P to MTXu 5-P (1,3-Isomerase Reaction)
The initial reaction mixtures (0.2 mL) contained 100 mM Tris-HCl, pH 7.9, 5 mM MgCl2, 15 mM NaHCO3, 0.4 mM NADPH, 10 mM DTT, 10 μM D-xylulose 5-P reductoisomerase, 1 μM 1-methythio-D-xylulose 5-phosphate methylsulfurylase, and varying concentrations of MTR 1-P (12). MTR 1-P 1,2-isomerase was added to a final concentration of 10 μM; after 5 min at room temperature, 1,3-isomerase or RuBisCO was added to a final concentration of 0.25 μM or 10 μM, respectively, and the reaction was monitored at 360 nm (ε = 4,300 M−1 cm−1).
1H NMR for Conversion of DK-MTP 1-P to HK-MTP 1-P (“Enolase” Reaction)
The initial reaction mixtures (0.8 mL) contained 20 mM K+ phosphate, pH 7.8, 5 mM MgCl2, 15 mM NaHCO3, and 5 mM MTR 1-P. (As specified in the figure legends, some reactions were performed in 100% D2O; others were performed in 90% H2O/10% D2O.) MTR 1-P 1,2-isomerase was added to a final concentration of 5 μM, and the reaction mixture was incubated for 30 minutes to generate MTRu 1-P. For enzyme-catalyzed reactions, either “enolase” or RuBisCO was added to a final concentration of 1 μM before MTRu 1-P dehydratase was added to a final concentration of 1 μM to generate HK-MTP 1-P. For nonenzymatic reactions, only dehydratase was added. The reactions were incubated overnight at room temperature before obtaining the 1H NMR spectra.
13C-NMR to Characterize DK-MTP 1-P
For natural abundance 13C NMR spectra, the reactions (0.8 mL) contained 20 mM K+ phosphate, pH 7.8, 5 mM MgCl2, and 10 mM MTR 1-P in 90% H2O/10% D2O. MTRu 1-P was formed in situ by the addition of MTR 1-P 1,2-isomerase to a final concentration of 5 μM. After 1 hr, MTRu 1-P dehydratase was added to a final concentration of 1 μM, and the reaction was immediately cooled to 4°C in the spectrometer to suppress the rate of enolization to HK-MTP 1-P. A total of 22,576 transients were acquired over 18 hours.
13C NMR Spectra of [2-13C]MTR 1-P, [2-13C]MTRu 1-P, and [2-13C]DK-MTP 1-P
For the spectrum of [2-13C]MTR 1-P, the reaction mixture (0.8 ml) contained 20 mM K+ phosphate, pH 7.8, 5 mM MgCl2, and 5 mM [2-13C]MTR 1-P in 50% H218O/40% H216O/10 % D216O. MTR 1-P 1,2-isomerase then was added to a final concentration of 10 μM; after 1 hr the 13C spectrum of [2-13C]MTRu 1-P was recorded. MTRu 1-P dehydratase then was added to a final concentration of 4 μM; after 1 hr the 13C spectrum of [2-13C] DK-MTP 1-P was recorded.
1H-NMR for Conversion of MTRu 1-P to MTXu 5-P/MTRu 5-P (1,3-Isomerase Reaction)
The reaction mixtures (0.8 mL) contained 20 mM K+ phosphate, pH 7.8, 5 mM MgCl2, 15 mM NaHCO3, and 5 mM MTR 1-P in 100% D2O. MTRu 1-P was formed in situ by addition of MTR 1-P 1,2-isomerase to a final concentration of 5 μM. After 30 minutes, either 1,3-isomerase or RuBisCO was added to a final concentration of 10 μM. The 1H NMR spectrum was recorded after overnight incubation at room temperature.
Insertional Disruption of the Gene Encoding DK-MTP 1-P “Enolase” in B. subtilis (MtnW::pMUTIN4)
The gene encoding the “enolase” (mtnW) in B. subtilis was inactivated by insertional disruption using the erythromycin resistance conferring plasmid pMUTIN4 (16) according to the published procedure (10). A ~350 bp segment of the N-terminal section of mtnW was amplified by PCR with primers containing a 5′ BamHI site and a 3′ HindIII site (5′-GGAGGATCCTACGCCAAACTTCGGTCCCGG-3′ and 5′-GGCATGAAGCTTTTATTAGCGACATATCTCCTGACCG-3′). The product was digested and ligated into the disruption vector pMUTIN4.
Wild type B. subtilis 168 was transformed with the pMUTIN4 construct (17) and spread on LB plates containing 1 μg/ml erythromycin, 1 mM IPTG, and 1 mM L-methionine. Colonies were allowed to grow at 37°C for 48 hours before screening. Extracted genomic DNA was screened by PCR amplification of the insert junctions using the following primers: 5′-GGCTGCCTGTGCCTGAAGGCTATACTTCTGAG-3′ and 5′-GGATGTGCTGCAAGGCGATTAAGTTGGGTAACG-3′ for the 5′-junction site; and 5′-GGAGCTAAAGAGGTCCCTAGACTCTAGACCCGG-3′ and 5′-GCCATAATCGGAACTGGGATTTCAGGATCTTCTG-3′ for the 3′-junction site. The strain, designated MtnW::pMUTIN4, was unable to utilize MTA as sole sulfur source in minimal medium.
Complementation with pDR67 MtnXYZ by insertion into amyE
For addressing polar effects due to the insertional disruption of MtnW, the downstream genes encoding the phosphatase (MtnX), dehydratase (MtnY), and dioxygenase (MtnZ) were amplified (18) using the following primers: 5′-GGTCTAGACGCTAGATAAATGGGGAAAGG-3′ that introduces an XbaI cut site and 5′-GACAGCGTGAATCAATAAGCGAGATCTGCG-3′ that introduces a BglII cut site. After double digestion, the insert was ligated into pDR67 and screened for presence of the insert. Prior to transformation, the pDR67 MtnXYZ plasmid was linearized with AatII overnight at 37°C. The transformation of B. subtilis 168 (wild type and MtnW::pMUTIN4) with linearized pDR67 MtnXYZ was performed as described above.
Complementation with pEB112, pEB112 containing B. subtilis RLP, and pEB112 containing R. rubrum RuBisCO
The gene encoding MtnW was PCR amplified using the following primers: 5′-TTTTCGGATCCTCATACGGCTTCAGCCTTTCCCCATTTATCTA-GCG-3′ and 5′-GCGACCCGGGTTAAGGAGGATTTTTTGATATGGATGAAAATGAAA-GG-3′ that introduce BamHI and XmaI restriction sites, respectively. The gene encoding R. rubrum RuBisCO was PCR amplified from gDNA using the following primers: 5′-CGAGGATCCTTAAGGAGGATTTTTTGATATGGACAGTCATCTCGTTACGTCAATC-TGG-3′ and 5′-GACGCGGAGGGGAGGGATCCCGCGTCTTCTGCTTTGACCGGCCGTCC-AACC-3′ that introduce BamHI restriction sites, respectively. Both fragments were digested according to the manufacturer’s specifications. Digested fragments were gel extracted using a Qiaquick Gel extraction kit and then ligated into pre-digested pEB112. Ligation mixtures were used to transform E. coli XL-1 blue competent cells by electroporation. Colonies were screened for insert and correct orientation before using them to transform B. subtilis 168 wild type or B. subtilis 168 MtnW::pMUTIN4. Transformations were plated on LB agar plates supplemented with 50 μg/mL kanamycin.
Media and conditions used for growth curves
The measurement of growth curves used the ED minimal medium described (19) with the addition of 1 mM IPTG. When necessary, the antibiotics were added: 1 μg/mL erythromycin, 1 μg/mL kanamycin, and/or 1 μg/mL chloramphenicol.
RESULTS AND DISCUSSION
Structure and Chemical Reactivity of DK-MTP 1-P
Knowledge of the solution structure and chemical properties of DK-MTP 1-P, the substrate for “enolase”, is essential for understanding both its nonenzymatic and, also, enzymatic reactivity as investigated in this article. DK-MTP 1-P is generated in situ from MTRu 1-P by the action of MTRu 1-P dehydratase (Figure 3, panel A). In the published characterization of dehydratase (19), the DK-MTP 1-P product was reported to be labile, “decomposing” with a rate constant of 0.048 sec−1 (t1/2 ~ 15 sec). The decomposition of DK-MTP 1-P was quantitated by the loss of absorbance at 270 nm (the α,β-diketo chromophore in DK-MTP 1-P) after its rapid in situ generation from MTRu 1-P. When dehydratase and “enolase” were present together in a reaction containing MTRu 1-P, the rate of formation of enolized HK-MTP 1-P product of “enolase” was significant; however, when “enolase” was added several minutes after dehydratase [~6 minutes or 24 half-times for decomposition of DK-MTP 1-P; Figure 4 in (19)], the rate of formation of HK-MTP 1-P was significantly decreased. When additional MTRu 1-P was added to the reaction mixture, the rate of formation of HK-MTP 1-P again was significant (as expected because the dehydratase and “enolase” were both present).
We used 1H and 13C NMR spectroscopies to characterize the structure of the “decomposed” DK-MTP 1-P product of dehydratase. The 1H NMR spectrum (in 90% H2O/10% D2O) of a solution containing DK-MTP 1-P obtained within two minutes after generation with dehydratase is shown in Figure 4, panel A (8 half-times for decomposition, therefore “decomposed”). The spectrum appears that expected for the structure of DK-MTP 1-P: a doublet associated with the protons on carbon-1 (3.8 pm; coupled to the 31P of the 1-phosphate group), two triplets associated with the protons on carbons-4 and -5 (3.05 and 2.65 ppm, respectively), and a singlet associated with the thiomethyl group (2.0 ppm).
Figure 4.
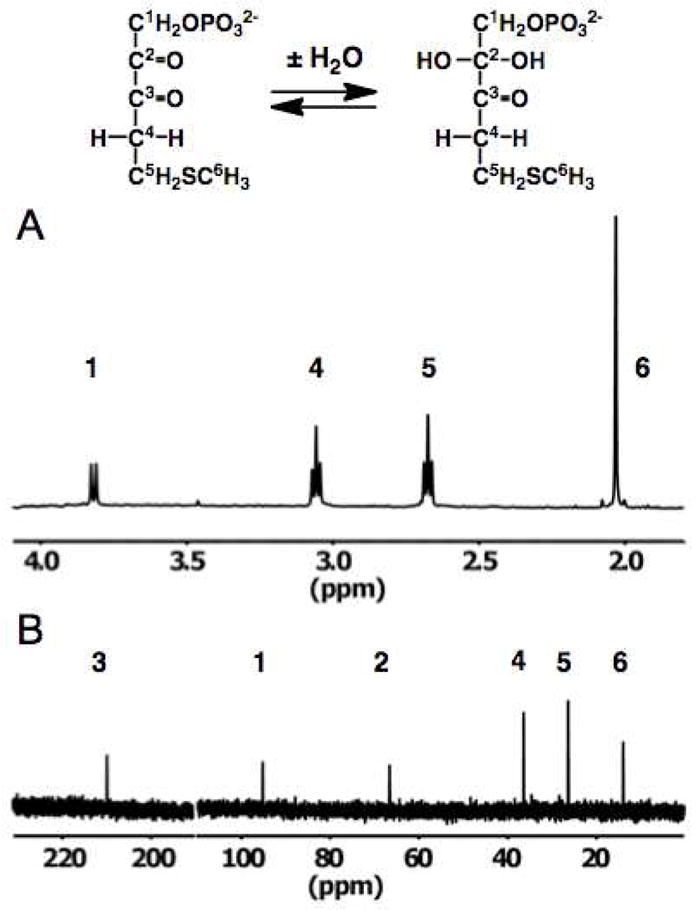
Solution characterization of DK-MTP 1-P. Panel A, 1H NMR spectrum immediately after in situ generation from MTRu 1-P with MTRu 1-P dehydratase. Panel B, 1H decoupled natural abundance 13C NMR spectrum.
The natural abundance 13C NMR spectrum (Figure 4, panel B) contained six resonances. However, only a single resonance was observed in the downfield region for carbonyl carbons (210 ppm); also, one resonance was observed in the region expected for a carbon with two singly-bonded oxygens (95 ppm) and only a single resonance for a carbon with one singly-bonded oxygen (68 ppm). But, as expected, the spectrum contained a resonance for a carbon adjacent to a carbonyl carbon (38 ppm) and two for carbons adjacent to sulfur (26 and 15 ppm). From this spectrum we concluded that one of the α,β-diketo carbonyl carbons is hydrated (gem-diol) in “decomposed” DK-MTP 1-P. A straightforward interpretation of the lack of enzymatic reactivity would be that the 2-carbonyl group is hydrated, thereby decreasing the acidity of the 1-proton that would be abstracted in the “enolase” reaction. Alternatively, hydration of the 3-carbonyl group might prevent its binding to the Mg2+ that stabilizes the enolate anion intermediate in the active site of “enolase”.
Verification that the 2-carbonyl group is, in fact, hydrated in the “decomposed” DK-MTP 1-P was established using [2-13C]MTR 1-P as the precursor and performing successive in situ generation of [2-13C]MTRu 1-P with MTR 1-P 1,2-isomerase and [2-13C]DK-MTP 1-P with dehydratase in 50% H218O/40% H216O/10% D216O. The 1H decoupled spectrum of [2-13C]MTR 1-P is a doublet because of coupling of the 2-13C to the 31P of the 1-phosphate group (Figure 5, panel A). After isomerization, the 1H decoupled spectrum of [2-13C]MTRu 1-P appears as a doublet of doublets (Figure 5, panel B): 1) the 2-13C is coupled to the 31P of the 1-phosphate group; and 2) the carbonyl oxygen is in equilibrium with the 1:1 16O/18O isotopic composition of the solvent (with the 18O causing an upfield shift in the 13C resonance). After dehydration, the 1H decoupled spectrum appears as a doublet of triplets (Figure 5, panel C), establishing that the 2-carbonyl group is hydrated (present as a gem-diol): 1) the 2-13C is coupled to the 31P of the 1-phosphate group; and 2) both oxygens of the hydrate are equilibrated with the solvent, with the “triplet” reflecting the expected 1:2:1 isotopic distribution of the [16O2]-, [16O18O]-, and [18O2]-labeled hydrates.
Figure 5.

13C NMR spectra of [2-13C] labeled materials in 50% H218O/40% H216O/10% D216O (i.e., the isotopic composition of the solvent is 50% 16O, 50% 18O). Panel A, [2-13C]MTR 1-P. Panel B, [2-13C]MTRu 1-P. Panel C, [2-13C]DK-MTP 1-P.
When “decomposed”, actually hydrated, DK-MTP 1-P is incubated in the absence of “enolase”, the absorbance at 270 nm (also the enone chromophore in HK-MTP 1-P) slowly increases (Figure 6, panel A) and, also, the 1H NMR spectrum reveals the formation of the enolized HK-MTP 1-P as judged by the characteristic vinyl resonance [a doublet because of coupling to the 31P of the 1-phosphate group (Figure 6, panel B)]. The rate constant describing the initial formation of HK-MTP 1-P, 0.0004 sec−1 (t1/2 ~ 30 min) is independent of the substrate concentration, i.e., the enolization is first-order in DK-MTP 1-P. As shown in Figure 6, panel A, the enolization is biphasic, “quickly” approaching ~60% completion in the time predicted by the measured rate constant; this phase is followed by a slower increase in absorbance until the reaction reaches equilibrium (~4:1 HK-MTP 1-P/DK-MTP 1-P). No change is observed in the chemical shifts of the resonances in 1H NMR spectrum during the second phase nor does a second set of resonances that can be associated with a different structure for HK-MTP 1-P appear. One explanation is that HK-MTP 1-P exists as a mixture of geometric isomers, i.e., cis- and trans-enediols, with one of these favored kinetically. Irrespective of the explanation, the hydrate of DK-MTP 1-P is not “decomposed” irreversibly but is capable of nonenzymatic enolization via the small amount of unhydrated DK-MTP 1-P present in equilibrium with the hydrate.
Figure 6.

Nonenzymatic enolization of DK-MTP 1-P to HK-MTP 1-P. Panel A, time course of the reaction at several concentration of DK-MTP monitored at 270 nm. Panel B, time course of the reaction (5 mM DK-MTP 1-P) monitored by 1H NMR spectroscopy. The assignments of the resonances are provided in Figure 4, panel A.
In vitro Reactivity of DK-MTP 1-P with “Enolase” and RuBisCO (“Enolase” Reaction)
Having characterized the structure of DK-MTP 1-P and the rate of its nonenzymatic “enolization”, we studied the ability of both “enolase” and RuBisCO to catalyze the DK-MTP 1-P “enolase” reaction. The dependence of the reaction rate on enzyme concentration in shown in Figure 7, panel A; the dependence of reaction rate on substrate concentration is shown in Figure 7, panel B. [The kcat for RuBisCO under these conditions measured by 1H NMR spectroscopy is 0.85 sec−1; the ratio of carboxylation to oxygenation is ~ 1.]
Figure 7.

Reactivity of DK-MTP with DK-MTP 1-P “enolase” and RuBisCO. Panel A, dependence of reaction velocity on enzyme concentration using 100 μM DK-MTP 1-P. Circles: “enolase”; Squares, RuBisCO. Panel B, dependence of reaction velocity on substrate concentration. Circles: 1 μM “enolase”; Squares, 10 μM RuBisCO.
“Enolase” catalyzes the slow, but easily measurable, formation, of HK-MTP 1-P (kcat/Km = 1,300 M−1 sec−1); the low value of kcat/Km is explained by our demonstration (previous section) that the reactive, unhydrated DK-MTP 1-P constitutes only a minor fraction of the total “substrate” pool (even with [2-13C]DK-MTP 1-P,3 no resonance associated with the 2-keto group can be detected). The value of kcat/Km for “enolase” is 1.3 × 107 M−1 sec−1 when the reaction is initiated by the rapid formation of DK-MTP 1-P by addition of dehydratase to an assay mixture containing MTRu 1-P, i.e., the initial rate is measured before a significant amount of the hydrate is formed (20). The ratio of the value of kcat/Km we measure after the hydrate is formed and the value before the hydrate is formed measures the fraction of DK-MTP 1-P that exists as the reactive diketo form, i.e., 0.01%.
We attribute the “saturation” in rate as the enzyme concentration is increased to rate-limiting dehydration of the hydrate to the reactive unhydrated molecule. In contrast, even at high enzyme or substrate concentrations, no reaction is observed with RuBisCO and the in situ produced DK-MTP 1-P [kcat/Km = −1.9 (± 0.3) M−1 sec−1]. Thus, we conclude that in vitro RuBisCO is not promiscuous for the “enolase” reaction.
We note that the genome of R. rubrum does not encode the classical methionine salvage pathway that includes the DK-MTP 1-P “enolase” (Figure 3, panel A); instead, it converts MTA to DXP and methanethiol via the MTA-isoprenoid shunt, a newly discovered pathway, that includes the MTXu 5-P 1,3-isomerase reaction (Figure 3, panel B) that also is catalyzed by an RLP (11). Thus, even if observed, no functional significance would have been easily attributed to a promiscuous DK-MTP 1-P “enolase” reaction catalyzed by RuBisCO. Finally, the lack of promiscuity of RuBisCO for the “enolase” reaction is expected because the single general basic catalysts in RuBisCO and the “enolase” active sites are located on opposite faces of their substituted D-ribulose substrates (vide infra).
In vivo Reactivity of DK-MTP 1-P with RuBisCO
As noted previously, RuBisCO was reported to catalyze the “enolase” reaction in a B. subtilis strain in which the gene encoding the “enolase” (MtnW) had been insertionally disrupted (RLP− strain) as judged by its ability to restore growth on MTA as sole sulfur source (10). This strain was constructed to have the downstream genes in the same operon encoding the dehydratase, phosphatase, and dioxygenase (Figure 3, panel A; MtnXYZ) under the control of a separate promoter to eliminate polar effects. Because we observed that the RuBisCO was not able to catalyze the “enolase” reaction in vitro (vide supra), we repeated these complementation experiments.
In our experiments, the gene encoding the “enolase” was insertionally disrupted using pMUTIN4, i.e., we generated an independent RLP− strain; as expected, this strain (MtnW::pMUTIN4) was unable to utilize MTA as sole sulfur source. Irrespective of whether it was transformed with a plasmid (pEB112) containing no insert (“empty” vector), the gene encoding “enolase”, or the gene encoding RuBisCO, no complementation was observed, i.e., no growth on MTA as sole sulfur source (data not shown). We attributed the lack of complementation by “enolase” to polar effects: as noted previously, the gene encoding “enolase” is upstream of those encoding dehydratase, phosphate, and dioxygenase so its disruption likely decreases the efficiency of transcription of the downstream genes.
To circumvent polar effects, we inserted the genes encoding dehydratase, phosphate, and dioxygenase (MtnXYZ) in the amyE locus of our RLP− strain (MtnW::pMUTIN4) using to generate the strain designated as MtnW::pMUTIN4 amyE::pDR67. With this strain, pEB112 containing the gene for “enolase” complemented the disruption but pEB112 without an insert or containing the gene for RuBisCO did not restore the ability to use MTA as sole sulfur source (Figure 8). [The RLP− strain transformed with the “empty” vector is able to grow slowly with MTA as sole sulfur source, likely because the DK-MTP 1-P produced the residual dehydratase nonenzymatically “enolizes” slowly in the absence of the “enolase” (insertionally disrupted so it is not present) and can be used as substrate by the low levels of phosphatase and dioxygenase that also are present. We cannot provide an explanation for why the RLP− strain transformed with the vector containing the RuBisCO gene grew more slowly than the strain transformed with the empty vector.]
Figure 8.

Growth curves of wild type B. subtilis and B. subtilis with the “enolase” insertional deletion (MtnW::pMUTIN4). The data are for 1) wild type B. subtilis with the genes encoding the dehydratase, phosphatase and dioxygenase inserted in amyE (amyE::pDR67 MtnXYZ) and transformed with empty pEB112 (green circles); 2) wild type amyE::pDR67 transformed with pEB112 containing the “enolase” (purple diamonds); 3); wild type amyE::pDR67 MtnXYZ transformed with pEB112 containing the RuBisCO form R. rubrum (blue triangles); 4) B. subtilis with the insertional disruption of the “enolase” (MtnW::pMUTIN4) and amyE::pDR67 MtnXYZ transformed with empty pEB112 (red-orange squares); 5) MtnW::pMUTIN4 and amyE::pDR67 MtnXYZ transformed with pEB112 containing the “enolase” (brown triangles); and 6) MtnW::pMUTIN4 and amyE::pDR67 MtnXYZ transformed with pEB112 containing the RuBisCO form R. rubrum (orange inverted triangles).
Thus, we conclude from both our in vitro and in vivo experiments that RuBisCO is unable to catalyze the “enolase” reaction. Although we cannot provide an explanation for why the results of our in vivo experiments differed from those reported previously (10), we are confident that in vitro RuBisCO cannot catalyze the “enolase” reaction based on our thorough characterization of both the solution structure of DK-MTP 1-P as well as its reactivity for formation of HK-MTP 1-P both in the absence and presence of “enolase”.
Reactivity of MTRu 1-P with RuBisCO (1,3-Isomerase Reaction)
Finally, we investigated whether RuBisCO is promiscuous for the reaction catalyzed by the RLP encoded by its genome, i.e. the 1,3-isomerase reaction (Figure 3, panel B). We first used 1H NMR spectroscopy to determine whether RuBisCO catalyzes the 1,3-isomerase reaction. After incubation of MTRu 1-P (Figure 9, panel A) with 1,3-isomerase in D2O, the previously documented 1:3 mixture of MTXu 5-P and MTRu 5-P was formed [Figure 9, panel B; (13)]. [Note that the isomerase-catalyzed reaction is accompanied by incorporation of solvent hydrogen at carbons-1, -4, and -5.] However, no reaction was observed in the presence of RuBisCO (Figure 9, panel C).
Figure 9.

Reactivity of MTRu 1-P. Panel A, 1H NMR spectrum of MTRu 1-P. Panel B, 1H NMR spectrum of MTRu 1-P after overnight incubation with 1 μM MTRu 1-P 1,3-isomerase. Panel C, 1H NMR spectrum of MTRu 1-P after overnight incubation with 10 μM RuBisCO.
We also used a coupled-enzyme spectrophotometric assay to quantitate the activity of RuBisCO for the 1,3-isomerase reaction (12); the formation of MTXu 5-P from MTRu 1-P is detected by its further conversion to DXP by DXP reductoisomerase (12). With this assay, virtually no activity could be detected: kcat/Km = 0.1 (± 0.9) M−1 sec−1 for RuBisCO; kcat = 1.3 sec−1, Km = 50 μM, kcat/Km = 2.7 × 104 M−1 sec−1 for the 1,3-isomerase reaction. Thus, we conclude that in vitro RuBisCO is not promiscuous for the 1,3-isomerase reaction.
We previously demonstrated that a mutant strain of R. rubrum in which the genes encoding both RuBisCO and 1,3-isomerase had been disrupted could not utilize MTA aerobically. Furthermore, although the gene encoding 1,3-isomerase could complement the aerobic growth phenotype, the gene encoding RuBisCO could not (21). Thus, in vivo RuBisCO is not promiscuous for the 1,3-isomerase reaction.
An unliganded structure (PDB 3QFW) is available for a presumed orthologous 1,3-isomerase from Rhodopseudomonas palustris. The active sites of activated (carboxylated) RuBisCO from R. rubrum complexed with D-ribulose 1,5-bisphosphate (D-Ru 1,5-bisP; highlighted in cyan; Panel A; PDB 9RUB), activated (carboxylated) “enolase” from G. kaustophilus complexed with 2-hydroxy-3-keto-5-hydroxypentane 1-phosphate (DK-H 1-P; highlighted in magenta; PDB 2OEM), and 1,3-isomerase are compared in Figure 10. In RuBisCO, the carboxylated Lys 191 (CX-Lys 191) is the general base for abstraction of the 3-proton as well as a ligand for the essential Mg2+ [Lys 166 and 168 are required for enolization; His 321 catalyzes hydration (2)]. In “enolase”, Lys 98* (from the symmetry-related polypeptide in the obligate dimer) is the general base for abstraction of the 1-proton, the carboxylated Lys 173 (CX-Lys 173) is a ligand for the essential Mg2+ (1), and Lys 147 is conserved in all RLPs The 1,3-isomerase is not activated, i.e., Lys 164 is not carboxylated nor does the structure contain Mg2+; Asp 166 and His 264 are likely ligands for Mg2+, although no biochemical or structural data are available to support this proposal. Glu 35* (from the symmetry-related polypeptide in the obligate dimer) is positioned to be a general acid/base catalyst as discussed in the next paragraph.
Figure 10.

Comparison of the active sites of RuBisCO from R. rubrum (Panel A), “enolase” from G. kaustophilus (Panel B), and the presumed MTRu 1-P 1,3-isomerase from R. palustris (Panel C). Details are provided in the text.
Although no structure-function studies have been performed with the 1,3-isomerase from R. palustris or R. rubrum (13), a Glu residue (Glu 35 in R. palustris; Glu 32 in R. rubrum) conserved in all members of the 1,3-isomerase family, but not in either RuBisCOs or “enolases”, is located on the opposite face of the active site from the carboxylated Lys. The curious, but unexplained, promiscuity of 1,3-isomerase to generate a 3:1 mixture (13) of MTXu 5-P that is the precursor of DXP in the MTA-isoprenoid shunt (Figure 3, panel B) and MTRu 5-P that is a “dead-end” metabolite (11) may reflect the absence of a general acid to protonate carbon-2 of the Mg2+-stabilized enediolate anion in the initial 1,2-proton transfer reaction initiated by the carboxylated Lys (13). As a result, a mixture of diastereomeric (“racemic”) 5-methylthio-3-ulose 1-phosphates is produced by release of the enediolate anion to solvent (only the “productive” diastereomer is shown in Figure 1, panel C). Both diastereomers are then substrates for the second 1,2-proton transfer reaction in which the observed 3:1 mixture of MTXu 5-P and MTRu 5-P products is generated; in this 1,2-proton transfer reaction, the conserved Glu is appropriately positioned to function as the general base that abstracts the 4-proton from both diastereomers of the 3-ulose intermediate and, also, the general acid that protonates carbon-3 of the mixture of enediolate anion intermediates. Thus, even in the absence of experimental studies, we conclude based on stereochemical and structural considerations that the mechanism of the reaction catalyzed by 1,3-isomerase requires general acid/base catalysts on both faces of the active site. Consequently, as for the “enolase” reaction, we do not expect RuBisCO to possess the general acid/base catalysts necessary for the 1,3-isomerase reaction. Therefore, the lack of promiscuity observed both in vitro and in vivo is expected.
Requirements for Discovering Promiscuity for Enzymes in MTA Recycling
Our experiments demonstrate that the presumed nonphysiological functional promiscuity documented for RuBisCOs does not extend to the “enolase” and 1,3-isomerase functions that are catalyzed by the functionally assigned RLPs, at least for RuBisCO from R. rubrum. As we have discussed, we are not surprised, given the different stereochemical requirements for proton abstraction in the carboxylation reaction catalyzed by RuBisCO and the proton transfer reactions catalyzed by “enolase” and 1,3-isomerase, i.e., the abstraction of spatially remote protons requires structurally distinct general basic catalysts, not just the carbamate oxygen of the carboxylated Lys residue that is conserved in both RuBisCOs and RLPs (1).
In the case of the functionally diverse enolase superfamily, promiscuity is observed; however, promiscuity is the result of relaxed substrate specificities that present diverse substrates to a conserved set of active site acid/base catalysts (22). Indeed, in vitro experiments have provided support for the hypothesis that natural divergent evolution of function can occur by successive point substitutions that provide enhanced selective advantage by incremental changes in substrate specificity (23–25). However, for the reactions catalyzed by RuBisCO, DK-MTP “enolase”, and MTRu 1-P 1,3-isomerase, the active site acid/base catalysts are not conserved, so it is unlikely that any of these enzymes can be promiscuous for the other reactions.
Our recent discovery that MTA recycling can occur by (at least) two different pathways, one involving genuine “methionine salvage” from MTA (Figure 3, panel A) (10) and the second liberating the 5′-methylthio group of MTA as methanethiol in the MTA-isoprenoid shunt that can be captured by O-acetylhomoserine sulfhydrylase to generate methionine (Figure 3, panel B) (11), complicates the interpretations of studies of the ability of RuBisCOs to complement mutants deficient in MTA recycling (10, 21, 26). Further difficulties are suggested by the observation that under anaerobic conditions a 1,3-isomerase mutant, but not a RuBisCO mutant, of R. rubrum could utilize MTA as sole sulfur source. As expected, the double mutant could not utilize MTA; however, when the double mutant was transformed with a plasmid encoding the RuBisCO, the MTA utilization phenotype was restored (21). One explanation is that R. rubrum has another pathway for MTA utilization, i.e., neither of those in Figure 3, that recovers at least the methylthio group of MTA for sulfur utilization and that the RuBisCO is involved in this pathway.
Conclusions
Our experiments do not provide evidence that RuBisCO from R. rubrum can catalyze either the DK-MTP 1-P “enolase” or the MTRu 1-P 1,3-isomerase reaction, the two functions now established for RLPs. Perhaps other RuBisCOs are promiscuous for these reactions and/or for reactions catalyzed by RLPs that have not yet been functionally assigned. But, until such promiscuity is discovered, few insights are available into Nature’s strategies for divergent evolution of function in the RuBisCO superfamily.
Acknowledgments
We thank Dr. Shoshana Brown and Professor Patricia C. Babbitt for providing the data for the sequence similarity network shown in Figure 2. We also thank Drs. Tobias J. Erb and Kou-San Ju for valuable discussions. We acknowledge Drs. Travis Muff, Hanna Rao, and George Glekas, and Professor George Ordal for providing B. subtilis 168, pDR67, pMUTIN4, and pEB112 as well as the protocols for constructing mutant strains.
Footnotes
This research was supported by NIH R01GM095742 (to F. R. T.) and R01GM065155 (to J.A.G.).
Abbreviations: RuBisCO, D-ribulose 1,5-bisphosphate carboxylase/oxygenase; 3-PGA, 3-phosphoglycerate; RLP, RuBisCO-like protein; MTA, 52-methylthioadenosine; MTR, 5-methylthio-D-ribose; MTR 1-P, 5-methylthio-D-ribose 1-phosphate; MTRu 1-P, 5-methylthio-D-ribulose 1-phosphate; DK-MTP 1-P, 2,3-diketo-5-methylthiopentane 1-phosphate; HK-MTP 1-P, 2-hydroxy-3-keto-5-methylthiopent-1-ene 1-phosphate; DHK-MTP, 1,2-dihydroxy-3-keto-5-methylthiopent-1-ene; KMTB, 2-keto-4-methylthiobutyrate; MTRu 5-P, 5-methylthio-D-ribulose 5-phosphate; MTXu 1-P, 5-methylthio-D-xylulose 5-phosphate; DXP, 1-deoxy-D-xylulose 5-phosphate.
Although the enzyme activity that accomplishes the keto-enol tautomerization reaction in the methionine salvage pathway has long been termed an “enolase”, the usual name for this reaction is “tautomerase”. Given our interest in the members of the mechanistically diverse enolase superfamily whose members catalyze reactions that are initiated by abstraction of a proton from a carbon adjacent to a carboxylate group, including the authentic enolase in glycolysis, we would prefer not to use “enolase” to describe the keto-enol tautomerization reaction in the methionine salvage pathway. However, given the history, we compromise by using “enolase” (in quotation marks) to refer to the enzyme in the methionine salvage pathway that catalyzes the keto-enol tautomerization reaction. [Reproduced from (1).]
References
- 1.Imker HJ, Fedorov AA, Fedorov EV, Almo SC, Gerlt JA. Mechanistic diversity in the RuBisCO superfamily: the “enolase” in the methionine salvage pathway in Geobacillus kaustophilus. Biochemistry. 2007;46:4077–4089. doi: 10.1021/bi7000483. [DOI] [PubMed] [Google Scholar]
- 2.Cleland WW, Andrews TJ, Gutteridge S, Hartman FC, Lorimer GH. Mechanism of Rubisco: The Carbamate as General Base. Chem Rev. 1998;98:549–562. doi: 10.1021/cr970010r. [DOI] [PubMed] [Google Scholar]
- 3.Larimer FW, Harpel MR, Hartman FC. Beta-elimination of phosphate from reaction intermediates by site-directed mutants of ribulose-bisphosphate carboxylase/oxygenase. J Biol Chem. 1994;269:11114–11120. [PubMed] [Google Scholar]
- 4.Morell MK, Wilkin JM, Kane HJ, Andrews TJ. Side reactions catalyzed by ribulose-bisphosphate carboxylase in the presence and absence of small subunits. J Biol Chem. 1997;272:5445–5451. doi: 10.1074/jbc.272.9.5445. [DOI] [PubMed] [Google Scholar]
- 5.Lee EH, Harpel MR, Chen YR, Hartman FC. Perturbation of reaction-intermediate partitioning by a site-directed mutant of ribulose-bisphosphate carboxylase/oxygenase. J Biol Chem. 1993;268:26583–26591. [PubMed] [Google Scholar]
- 6.Hanson TE, Tabita FR. A ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO)-like protein from Chlorobium tepidum that is involved with sulfur metabolism and the response to oxidative stress. Proc Natl Acad Sci U S A. 2001;98:4397–4402. doi: 10.1073/pnas.081610398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tabita FR, Hanson TE, Li H, Satagopan S, Singh J, Chan S. Function, structure, and evolution of the RubisCO-like proteins and their RubisCO homologs. Microbiol Mol Biol Rev. 2007;71:576–599. doi: 10.1128/MMBR.00015-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown SD, Babbitt PC. Inference of functional properties from large-scale analysis of enzyme superfamilies. J Biol Chem. 2012;287:35–42. doi: 10.1074/jbc.R111.283408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Sawaya MR, Tabita FR, Eisenberg D. Crystal structure of a RuBisCO-like protein from the green sulfur bacterium Chlorobium tepidum. Structure. 2005;13:779–789. doi: 10.1016/j.str.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 10.Ashida H, Saito Y, Kojima C, Kobayashi K, Ogasawara N, Yokota A. A functional link between RuBisCO-like protein of Bacillus and photosynthetic RuBisCO. Science. 2003;302:286–290. doi: 10.1126/science.1086997. [DOI] [PubMed] [Google Scholar]
- 11.Erb TJ, Evans BS, Cho K, Warlick BP, Sriram J, Wood BM, Imker HJ, Sweedler JV, Tabita FR, Gerlt JA. A RubisCO-like protein links SAM metabolism with isoprenoid biosynthesis. Nature Chem Biol. 2012;8:926–932. doi: 10.1038/nchembio.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warlick BP, Evans BS, Erb TJ, Ramagopal UA, Sriram J, Imker HJ, Sauder MJ, Bonanno JB, Burley SK, Tabita FR, Almo SC, Sweedler JV, Gerlt JA. 1-Methylthio-D-Xylulose 5-Phosphate Methylsulfurylase: A Novel Route to 1-Deoxy-D-Xylulose 5-Phosphate in Rhodospirillum rubrum. Biochemistry. 2012;51:8324–8326. doi: 10.1021/bi301215g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imker HJ, Singh J, Warlick BP, Tabita FR, Gerlt JA. Mechanistic diversity in the RuBisCO superfamily: a novel isomerization reaction catalyzed by the RuBisCO-like protein from Rhodospirillum rubrum. Biochemistry. 2008;47:11171–11173. doi: 10.1021/bi801685f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imker HJ. PhD Thesis. Department of Biochemistry, University of Illinois; Urbana-Champaign, Urbana, IL: 2008. [Google Scholar]
- 15.Ashida H, Saito Y, Kojima C, Yokota A. Enzymatic characterization of 5-methylthioribulose-1-phosphate dehydratase of the methionine salvage pathway in Bacillus subtilis. Biosci Biotechnol Biochem. 2008;72:959–967. doi: 10.1271/bbb.70651. [DOI] [PubMed] [Google Scholar]
- 16.Leonhardt H, Alonso JC. Construction of a shuttle vector for inducible gene expression in Escherichia coli and Bacillus subtilis. J Gen Microbiol. 1988;134:605–609. doi: 10.1099/00221287-134-3-605. [DOI] [PubMed] [Google Scholar]
- 17.Spizizen J. Transformation of Biochemically Deficient Strains of Bacillus Subtilis by Deoxyribonucleate. Proc Natl Acad Sci U S A. 1958;44:1072–1078. doi: 10.1073/pnas.44.10.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carre-Mlouka A, Mejean A, Quillardet P, Ashida H, Saito Y, Yokota A, Callebaut I, Sekowska A, Dittmann E, Bouchier C, de Marsac NT. A new rubisco-like protein coexists with a photosynthetic rubisco in the planktonic cyanobacteria Microcystis. J Biol Chem. 2006;281:24462–24471. doi: 10.1074/jbc.M602973200. [DOI] [PubMed] [Google Scholar]
- 19.Sekowska A, Danchin A. The methionine salvage pathway in Bacillus subtilis. BMC Microbiol. 2002;2:8. doi: 10.1186/1471-2180-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saito Y, Ashida H, Sakiyama T, de Marsac NT, Danchin A, Sekowska A, Yokota A. Structural and functional similarities between a ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO)-like protein from Bacillus subtilis and photosynthetic RuBisCO. J Biol Chem. 2009;284:13256–13264. doi: 10.1074/jbc.M807095200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh J, Tabita FR. Roles of RubisCO and the RubisCO-like protein in 5-methylthioadenosine metabolism in the Nonsulfur purple bacterium Rhodospirillum rubrum. J Bacteriol. 2010;192:1324–1331. doi: 10.1128/JB.01442-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerlt JA, Babbitt PC, Jacobson MP, Almo SC. Divergent evolution in enolase superfamily: strategies for assigning functions. J Biol Chem. 2012;287:29–34. doi: 10.1074/jbc.R111.240945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt DM, Mundorff EC, Dojka M, Bermudez E, Ness JE, Govindarajan S, Babbitt PC, Minshull J, Gerlt JA. Evolutionary potential of (beta/alpha)8-barrels: functional promiscuity produced by single substitutions in the enolase superfamily. Biochemistry. 2003;42:8387–8393. doi: 10.1021/bi034769a. [DOI] [PubMed] [Google Scholar]
- 24.Vick JE, Schmidt DM, Gerlt JA. Evolutionary potential of (beta/alpha)8-barrels: in vitro enhancement of a “new” reaction in the enolase superfamily. Biochemistry. 2005;44:11722–1179. doi: 10.1021/bi050963g. [DOI] [PubMed] [Google Scholar]
- 25.Vick JE, Gerlt JA. Evolutionary potential of (beta/alpha)8-barrels: stepwise evolution of a “new” reaction in the enolase superfamily. Biochemistry. 2007;46:14589–14597. doi: 10.1021/bi7019063. [DOI] [PubMed] [Google Scholar]
- 26.Ashida H, Saito Y, Nakano T, Tandeau de Marsac N, Sekowska A, Danchin A, Yokota A. RuBisCO-like proteins as the enolase enzyme in the methionine salvage pathway: functional and evolutionary relationships between RuBisCO-like proteins and photosynthetic RuBisCO. J Exp Bot. 2008;59:1543–1554. doi: 10.1093/jxb/ern104. [DOI] [PubMed] [Google Scholar]