

ABSTRACT
MtrA is a member of the AraC family of transcriptional regulators and has been shown to play an important role in enhancing transcription of the mtrCDE operon, which encodes a tripartite multidrug efflux pump, when gonococci are exposed to a sublethal level of antimicrobials. Heretofore, the DNA-binding properties of MtrA were unknown. In order to understand how MtrA activates mtrCDE expression, we successfully purified MtrA and found that it could bind specifically to the mtrCDE promoter region. The affinity of MtrA for the mtrCDE promoter increased 2-fold in the presence of a known effector and substrate of the MtrCDE pump, the nonionic detergent Triton X-100 (TX-100). When placed in competition with MtrR, the transcriptional repressor of mtrCDE, MtrA was found to bind with apparent lower affinity than MtrR to the same region. However, preincubation of MtrA with TX-100 prior to addition of the promoter-containing DNA probe increased MtrA binding and greatly reduced its dissociation from the promoter upon addition of MtrR. Two independent approaches (DNase I footprinting and a screen for bases important in MtrA binding) defined the MtrA-binding site 20–30 bp upstream of the known MtrR-binding site. Collectively, these results suggest that the MtrA and MtrR-binding sites are sterically close and that addition of an effector increases the affinity of MtrA for the mtrCDE promoter such that MtrR binding is negatively impacted. Our results provide a mechanism for transcriptional activation of mtrCDE by MtrA and highlight the complexity of transcriptional control of drug efflux systems possessed by gonococci.
IMPORTANCE
Antibiotic resistance in Neisseria gonorrhoeae has been increasing in recent years, such that in 2007 the Centers for Disease Control and Prevention listed N. gonorrhoeae as a “superbug.” One of the major contributors to antibiotic resistance in N. gonorrhoeae is the MtrCDE efflux pump. Until now, most work on the regulation of the genes encoding this efflux pump has been done on the transcriptional repressor, MtrR. This study is the first one to purify and define the DNA-binding ability of the transcriptional activator, MtrA. Understanding how levels of the MtrCDE efflux pump are regulated increases our knowledge of gonococcal biology and how the gonococcus can respond to various stresses, including antimicrobials.
Introduction
Neisseria gonorrhoeae is an obligate human pathogen that causes more than 90 million cases of the disease gonorrhea worldwide each year (1). In 2007, the Centers for Disease Control and Prevention added N. gonorrhoeae to the list of “superbugs” due to its increased resistance to a broad spectrum of antibiotics. A common mechanism of resistance to both antibiotics and host-derived immune compounds (e.g., antimicrobial peptides) is their export by the resistance/nodulation/division (RND)-type efflux pump termed MtrCDE. This tripartite efflux pump has been shown to be essential for resistance to β-lactams (penicillin G and nafcillin), macrolides (erythromycin), and host-derived compounds (peptide LL-37 and progesterone) (2, 3) and was essential for growth of gonococci in the lower genital tract of experimentally infected female mice (4).
There are two known transcriptional regulators of the mtrCDE efflux pump operon: a member of the TetR family of repressors, termed MtrR (5, 6), and a member of the AraC family of activators, termed MtrA (7). MtrR has been studied extensively and has been shown to bind to the mtrCDE promoter at a dyad repeat between the −10 and −35 elements (8). The mtrR gene is divergently transcribed and lies 250 bp upstream of the mtrCDE genes. Mutations in the helix-turn-helix DNA-binding domain of MtrR lead to elevated expression of mtrCDE and correspondingly intermediate levels of resistance to known MtrCDE pump substrates (3, 9). Two promoter mutations that lead to even higher levels of both mtrCDE expression and antimicrobial resistance have been characterized. One is a single base pair deletion in an inverted repeat in the overlapping mtrR and mtrCDE promoters that allows higher transcription of mtrCDE than MtrR coding mutations (6). Another is a C-to-T transition located 120 bp upstream of the mtrC start codon that generates a new promoter outside the regulation of MtrR or MtrA (10).
In contrast to the case with MtrR, less is known about how MtrA functions in regulating gene expression in gonococci. MtrA was first characterized as being required for high-level inducible resistance to the detergent Triton X-100 (TX-100) and the spermicide nonoxynol 9 (N-9) (7). MtrA contains a C-terminal DNA-binding domain, with a possible N-terminal dimerization/effector-binding domain typical of most AraC-family transcriptional regulators (11, 12). Loss of MtrA leads to a fitness defect compared to the parent strain FA19 in a competitive female mouse infection model (13). Correspondingly, loss of the repressor, MtrR, leads to a fitness advantage compared to the parent strain FA19 (13), strongly suggesting that regulation of mtrCDE levels is correlated with in vivo fitness.
Prior to this study, DNA binding studies of MtrA had not been done because it had not been purified. AraC-family proteins are difficult to purify because they often precipitate out of solution and are difficult to solubilize without denaturation (14). In this study, we were able to successfully purify soluble MtrA with N-terminal maltose-binding protein (MBP) or hexahistidine fusion tags. This enabled DNA binding studies to be done in order to characterize the binding of MtrA and MtrR to the mtrCDE promoter region. Here we have shown that the binding of MtrA to the DNA sequence upstream of mtrCDE is enhanced in the presence of certain MtrCDE substrates and MtrA effectors (e.g., TX-100 and N-9). The superior DNA-binding ability of MtrA in the presence of effectors was found to reduce the ability of the MtrR repressor to bind within the same region. Our results provide a model for how gonococci (and potentially other bacteria) can counteract transcriptional repression mediated by MtrR so as to enhance their resistance to antimicrobials.
RESULTS
MtrA binds to mtrCDE promoter region.
A previous study showed that MtrA is responsible for an increase in transcription from the mtrCDE promoter in the presence of sublethal levels of Triton X-100 and the structurally related spermicide nonoxynol-9 (7). Negative control of mtrCDE expression by the TetR family repressor, MtrR, has been well characterized. Thus, MtrR binds as a dimer to two pseudorepeats between the −10 and −35 regions of the mtrCDE promoter (8). Mutations in the DNA-binding region of MtrR (9) or loss of MtrR increase transcription of mtrCDE, leading to increased resistance of N. gonorrhoeae to a range of antimicrobial compounds and innate immune effectors (3). In contrast to the case with MtrR, the DNA-binding activity of MtrA was heretofore unknown despite genetic evidence that it can activate mtrCDE transcription in gonococci even in the presence of MtrR (7). Accordingly, we sought to define the DNA-binding action of MtrA in the presence of effectors that induce mtrCDE expression and the negative regulator of MtrR. In order to address these issues, it was necessary to purify MtrA for DNA binding studies, a task that has proved difficult for AraC-like transcriptional activators (14). We successfully purified MtrA (data not presented) with N-terminal MBP (MBP-MtrA) or an N-terminal His tag (MtrA-His); the former was used because the MtrR protein used in competition experiments (see below) was an MBP fusion protein (5), while only the latter proved useful in DNase I protection assays.
Since MtrA was found to be required for enhanced expression of mtrCDE when gonococci were grown in sublethal levels of TX-100 (7), we first used an electrophoretic mobility shift assay (EMSA) to test if MtrA could bind to the mtrCDE intergenic region that also contains the MtrR-binding site. Using 2-fold stepwise increases in the protein concentration, the EMSA demonstrated that 8 μg of MBP-MtrA could bind and completely shift the mtrCDE promoter region (Fig. 1A). This binding was shown to be specific, since it could be competed by addition of unlabeled mtrCDE promoter DNA probe but not a nonspecific probe of similar length (data not shown).
FIG 1 .
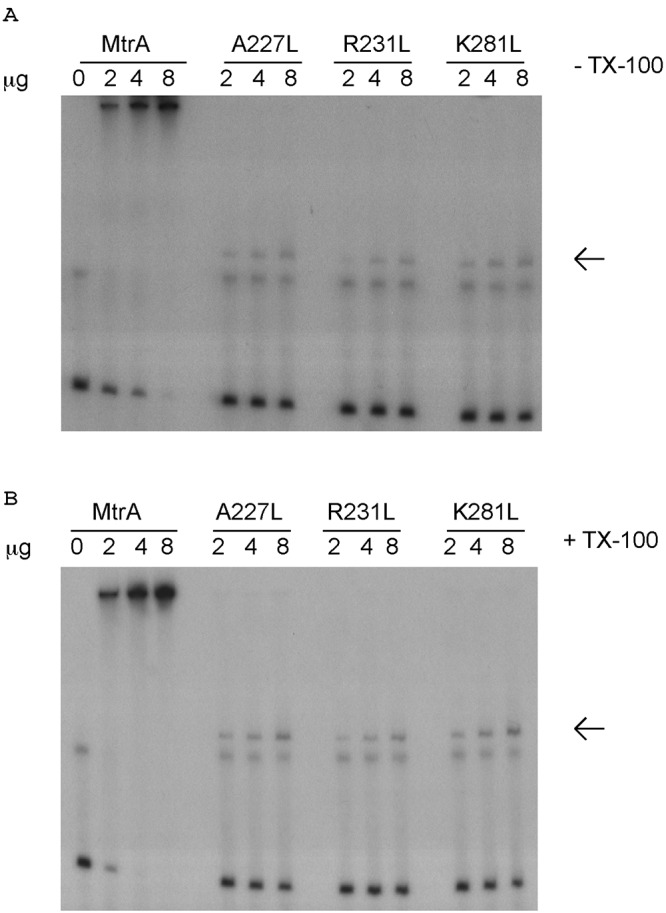
Relative binding to the mtrCDE promoter of wild-type MBP-MtrA and MBP-MtrA containing mutated residues predicted to be involved in DNA binding. The specific mutations are given above the lanes of the gel. The EMSA was done in the absence (A) or presence (B) of Triton X-100. The arrow indicates the protein-DNA complex for the mutated MtrA construct.
In order to confirm the DNA-binding capacity of MtrA, we constructed defined missense mutations (A227L, R231L, and K281L) that would cause radical amino acid replacements in this AraC-like protein. The predicted structure of MtrA was derived using the Swiss-Model structural prediction algorithm on the ExPASy Web server (see Materials and Methods). Using this prediction model, the DNA-binding domain of MtrA exhibited high (>98%) homology to the DNA-binding domain of Rob (PDB no. 1D5Y) (15). Rob is involved in the transcriptional activation of acrAB, which encodes components of the Escherichia coli AcrAB-TolC efflux pump (16, 17). The Rob crystal structure was determined in complex with DNA (15), which allowed the determination of residues that are involved in its DNA-binding activity. Accordingly, we aligned the putative DNA-binding domain of MtrA with Rob in complex with DNA using the software program MacPyMol (see Materials and Methods) to find the corresponding residues on MtrA likely involved in DNA binding. The MtrA residues predicted to be involved in DNA binding were A227, R231, and L281. To confirm that they were important for DNA binding, all three of these residues were mutated to leucine, and the DNA-binding abilities of these mutant proteins were assessed by EMSA using the labeled mtrCDE promoter probe. All three of these mutant proteins displayed markedly less DNA-binding ability than wild-type MtrA (Fig 1A). In addition, the migration of the protein-DNA complex for the missense mutations was much lower in the gel, indicating an altered ratio of protein-DNA complex compared to that for wild-type MtrA. Binding of these missense mutants to DNA is most likely done as a monomer, whereas the larger complex of wild-type MtrA-DNA is likely a dimer or higher-order structure.
To identify bases important for DNA binding, we first used mutagenic PCR to generate a series of mutations in the mtrR-mtrCDE intergenic region and screened for products with weaker MtrA binding in the presence of TX-100 (see Materials and Methods). The result of this screen identified three base changes positioned 20 to 40 bp upstream of the published MtrR-binding site (Fig. 2B). The changes consisted of two substitutions, T to C and A to G, at positions −208 and −209, respectively, and a deletion of a T from position −224 to −226. Taken together, results from the EMSA and mutagenesis experiments confirmed that MtrA can bind specifically to the DNA sequence upstream of the mtrCDE operon near the site recognized by MtrR.
FIG 2 .

(A) DNase I footprint of MtrA on the mtrCDE promoter in the presence of 100 μg TX-100. The black bars represent areas of protection observed. The gel shows protection of the coding strand (5′) of a 94-bp probe. (B) mtrCDE promoter, indicating bases that were revealed by the mutagenesis experiment to potentially be important for MtrA binding. Shown are the predicted −10 and −35 elements of the mtrCDE promoter, the published MtrR-binding site, and the putative MtrA-binding site.
Effectors increase the affinity of MtrA for the mtrCDE promoter region.
The N-terminal domain of AraC/XylS family proteins often binds effector molecules, which can alter their DNA-binding affinity (18, 19). One such example is AraC, which in the presence of arabinose alters its conformation to allow induction of the arabinose operon by binding to different sites in the promoter region (20). Since it was previously shown that TX-100 and N-9 could induce expression of the mtrCDE efflux pump in an MtrA-specific manner (7), we tested if these compounds could alter the DNA-binding ability of MtrA. Accordingly, MtrA was preincubated with TX-100 or N-9 prior to the addition of target DNA, and complexes were resolved by EMSA. The results showed that the affinity of MtrA for the mtrCDE promoter region was increased approximately 2-fold when it was preincubated with TX-100 (Fig. 1A and B) or N-9 (data not shown), as estimated by the amount of protein needed to completely shift 5 ng of labeled probe. Thus, for MBP-MtrA not preincubated with TX-100, 8 μg of protein was required to completely shift the probe (Fig. 1A), whereas only 4 μg of protein was required when it was preincubated with TX-100 (Fig. 1B). Importantly, this effect was consistently observed with repeated EMSAs and was replicated when a second effector (N-9) was employed. In support of the conclusions drawn from these experiments, we also found that MtrA containing the radical amino acid replacements, which are described above, did not afford increased binding to the target DNA when the mutant proteins were preincubated with TX-100 (Fig. 1A and B).
Given the enhanced DNA-binding capacity of MtrA in the presence of TX-100, we used DNase I protection to identify sites for MtrA binding. Using purified MtrA-His preincubated with TX-100 for 15 min prior to addition of the radiolabeled probe, we noted a region of protection upstream of the known MtrR-binding site (8) and the −10 region of the mtrCDE promoter (Fig 2A). The defined region of protection was from position −215 to −199 (GCGGATTATAAAAGAC). Importantly, this region of protection spans the sequence bearing point mutations, which was recovered from the mutagenic experiments that identified nucleotides that reduced MtrA binding.
Transcriptional activation versus repression of mtrCDE as determined by MtrA and MtrR.
Since the bases identified as being important for MtrA binding and the region identified by DNase I as the MtrA-binding site are adjacent to the MtrR-binding site (8), we asked how the binding of one protein to this region was influenced by the presence of the other and, if competition existed, whether the presence of an effector would influence DNA-binding activities. To determine the relative binding abilities of MtrR and MtrA to the mtrCDE promoter region, the proteins were used singularly or together in EMSAs. Both MtrA and MtrR (each containing an N-terminal MBP tag) were able to completely shift the mtrCDE promoter region, giving two distinct bands (Fig. 3, lanes 1 and 2), which enabled us to distinguish between DNA bound by MtrA or MtrR. When used together in the DNA-binding reaction, the two transcription factors were used at their lowest micromolar concentrations that result in a full shift of the target DNA (2.6 × 10−5 μM for MtrA and 7.5 × 10−6 μM for MtrR). At these concentrations, the two transcription factors were added either at the same time or 15 min apart (Fig. 3, lanes 3 to 5). Adding both regulators at the same time or MtrR first and then MtrA 15 min later resulted in DNA being almost completely bound by MtrR (Fig 3, lanes 3 and 5). Thus, under noninducing conditions, MtrR seemed to have a higher affinity for the mtrCDE promoter region than MtrA. In contrast, in the presence of an effector (TX-100), very different results were obtained. When both proteins were added at the same time, there was a slight increase in MtrA binding to the probe, consistent with a 2-fold increase in affinity for the probe (Fig 3, lane 8). When MtrA was added first, however, there was only a band shift for MtrA (Fig 3, lane 9), indicating that the binding sites for MtrA and MtrR were sterically close, such that when MtrA binds near the promoter in the presence of TX-100, it prevents the binding of the MtrR. Addition of TX-100 had no impact on MtrR binding ability. Thus, in the presence of TX-100, MtrA binding to the mtrCDE promoter region gave evidence of reducing subsequent binding by MtrR, which was not observed in the absence of TX-100.
FIG 3 .

Competition between MtrR and MtrA for binding to the mtrCDE promoter region. Lanes 1 to 5 were done in the absence Triton X-100, while lanes 6 to 10 were done in the presence of Triton X-100. The amounts of MtrR and MtrA used were the minimal amounts that would completely shift the mtrCDE promoter region. The “+” sign indicates the transcription factor incubated with the radiolabeled DNA for 15 min prior to the addition of the second transcription factor. The arrow indicates unbound probe.
DISCUSSION
This study is the first characterization of the DNA-binding properties of MtrA for the mtrCDE promoter region, which is important for understanding how the efflux pump operon is regulated at the level of transcription. We have shown that MtrA binds to the mtrCDE promoter region and that its binding site is sterically close to that of MtrR, the known repressor of mtrCDE transcription. The binding characteristics of these two known regulators of mtrCDE were tested in the presence or absence of known effectors, TX-100 or N-9. In the absence of an effector, MtrR seems to bind to the promoter region with higher affinity than MtrA. However, in the presence of these effectors, MtrA seems to have increased affinity for the mtrCDE promoter region. Taken together with our earlier work on MtrR binding to this region, the binding sites for MtrA and MtrR are sterically close. Thus, the positions of the binding sites for these regulators and the increased binding of MtrA in the presence of effectors likely affect its ability to reduce binding of MtrR (Fig. 3). We propose that this competition would allow for transient up-regulation of mtrCDE expression until the concentration of the inducer is reduced intracellularly, possibly by export via the MtrCDE pump, allowing the repressor, MtrR, to bind to the promoter region.
In a previous study, we showed that the loss of MtrA reduces fitness of gonococci approximately 500-fold in a female mouse model of lower genital tract infection (13). This might suggest that MtrA could serve as a “druggable” target in the same fashion that drug efflux pumps have served as targets for inhibitors, given that clinically relevant levels of antibiotic resistance can be caused by increased expression of efflux pumps (21). However, the impact on fitness due to loss of MtrA in the female mouse model could be counteracted by second-site mutations in mtrR that enhanced efflux pump gene expression (13). These findings have two important implications. First, they strongly suggest that higher levels of the pump, either through induction by MtrA or derepression through loss of MtrR, are important for survival in vivo. Second, they also suggest that targeting transcriptional regulators of drug efflux pump genes might not prove useful, since there exist many ways for bacteria to increase expression of drug efflux pumps.
The binding of MtrA to the mtrCDE promoter appears to be as a dimer or higher-order structure, given the very slow electrophoretic mobility of the protein-DNA complex. Most AraC family proteins have a C-terminal DNA-binding domain and an N-terminal dimerization and/or effector-binding domain (19). Based on the results of this study, we propose that the N terminus of MtrA is a dimerization and effector-binding domain, while the C-terminal domain is a DNA-binding domain, which has high similarity to Rob of E. coli; direct evidence for dimerization or higher-order complexes of MtrA is presently lacking but under investigation. However, we have found that the DNA-MtrA complex formed in the presence of SDS showed resolution of an MtrA monomer to the target DNA (data not shown). Also, the three targeted MtrA mutants made in this study could shift DNA only as a much smaller complex, most likely as a monomer. Therefore, it is likely that these mutations in the DNA-binding region of MtrA affect dimerization. There is also evidence that these mutations affect the ability of MtrA to increase affinity for target DNA in the presence of an effector (Fig. 1A and B). Further structural studies are under way to clarify the DNA- and effector-binding ability of MtrA.
Expression of RND efflux pumps is often under control of a variety of regulatory proteins. It is common for RND pump gene expression to be under control of a repressor (17), and increased expression of the efflux pump is often achieved through mutations of the repressor (22). In some cases, there are also activators of RND pump operons, which can allow conditional activation of gene expression in the presence of various effectors. It is much less common for the pump to be under the sole control of an activator, although the MexEF-OprN system in Pseudomonas aeruginosa is one such system, under control of the activator MexT (23). A similar mode of regulation seen with the mtrCDE efflux pump operon (one under control of both an activator and a repressor) can be found with the acrAB locus in E. coli, but this system is controlled by three AraC-like proteins: Rob (16), MarA (24), and SoxS (25), along with the TetR family repressor AcrR (26). In fact, given that the DNA-binding domain of MtrA is predicted to have a high homology to Rob (>98%), it is likely that common regulatory activities modulate expression of both systems. Indeed, increased expression of the acrAB operon by the various activators is achieved by two different mechanisms. One is through increased expression of the activators MarA and SoxS. These members of the AraC family of regulators do not have an N-terminal effector-binding domain and as a consequence do not respond directly to stimuli. Instead, increased expression of marA and soxS are mediated by their specific regulators MarR and SoxR, respectively, which respond to their own stimuli within the cell (17, 22).
Rob, unlike most AraC family proteins, has an N-terminal DNA-binding domain and a C-terminal effector-binding domain (15). It has been shown to respond to bile salts and fatty acids (27), and increased expression leads to organic solvent tolerance increased antibiotic resistance (28) through induction of acrAB expression. While the effector-binding domains of MtrA and Rob are not homologous, they do share the ability to respond to stimuli, and the results of this study suggest that MtrA increases its affinity for the mtrCDE promoter in the presence of hydrophobic compounds. Although AcrR (the repressor of the acrAB operon), Rob, SoxS, and MarA all compete for binding to the acrAB operon, to date no detailed study on their interactions has been reported. In this respect, we have shown that MtrR binds to the mtrCDE promoter region with higher affinity than MtrA. However, in the presence of an effector, the binding affinity of MtrA is increased for the promoter, which can explain the increased expression of mtrCDE under inducing conditions (7). The dueling regulatory properties of these two transcription factors in gonococci are of likely importance in determining bacterial fitness in vivo and may be common to other bacterial efflux systems.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All N. gonorrhoeae strains are derived from strain FA19, which possesses wild-type mtrR and mtrA genes. All cloning steps were done with E. coli strain DH5a, which was grown in Luria-Bertani broth or on Luria-Bertani agar plates supplemented with the appropriate antibiotic at 37°C. N. gonorrhoeae strains were grown on gonococcal medium base (GCB) agar (Difco Laboratories, Detroit, MI) containing glucose and iron supplements at 37°C under 5% (vol/vol) CO2 and supplemented with the appropriate antibiotic. The concentrations of antibiotics used in this study were as follows: ampicillin, 100 mg/ml; kanamycin, 50 mg/ml; and chloramphenicol, 1 mg/ml.
Construction of MBP-MtrA and MtrA-His.
Primers were designed to amplify mtrA from FA19 genomic DNA with an EcoRI site on the forward primer (AAGAGGAATTCGACATTCTGGACAAACTG) and a PstI site on the reverse primer (TTCACTGCAGATTTGCGTTTGAAGCC). The PCR product was digested with EcoRI and PstI, ligated into the pMal-c2x vector (NEB, Ipswich, MA), digested with the same enzymes, and transformed into E. coli DH5a. The mtrA gene from the putative clones was sequenced (Beckman Coulter Genomics) to confirm that a wild-type mtrA gene was cloned into pMal-c2x. This created an MBP-MtrA fusion.
The MtrA-His construct was made in the pET15b vector. Primers were designed with an NdeI site on the forward primer (TCGATCCATATGGACATTCTGGACAA) and a BamHI site on the reverse primer (GATCGGATCCTTATTTTTGCCCGCCTTC). The resulting PCR product and pET15b vectors were digested with NdeI and BamHI and then ligated and transformed in E. coli DH5a.
Construction of residue changes in MBP-MtrA.
Splice overlap PCR was employed to alter MtrA residues predicted to be involved in DNA binding to leucine. Internal complementary primers were designed to change A227, R231, and L281 of MtrA to leucine and were used with the flanking forward and reverse primers to generate a PCR product that contains the desired change. These were cloned into pMal-c2x and purified as described below.
Structural comparison of MtrA.
Structural prediction of MtrA was determined using the Swiss-Model server (http://swissmodel.expasy.org). Comparison of MtrA prediction to Rob (PDB 1D5Y) was done using MacPyMol.
Purification of MBP-MtrA.
All growth of pMal-c2-mtrA culture was done at 28°C and in rich medium (LB broth plus 2% glucose). A 10-ml MBP-MtrA culture was grown overnight and added to 600 ml of rich medium. The culture was grown until mid-log phase (optical density [OD] of ~0.5) and then induced with 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h. Cells were pelleted by centrifugation and left overnight at −20°C. Cells were resuspended in column buffer (20 mM Tris-HCl [pH 7.5], 200 mM NaCl, and 1 mM EDTA) containing 1 tablet of EDTA-free protease inhibitor cocktail catalog no. 11873580001; Roche) and lysed by use of a French press by passing through 3 cycles at 1,000 mPa. The cell lysate was centrifuged at 16,442 × g, and the supernatant was passed by gravity flow over a 2-ml amylose column and washed with 20 ml column buffer. MBP-MtrA was eluted in 2-ml fractions with column buffer with 10 mM maltose. The fractions containing the protein were dialyzed 3× in 2 liters of storage buffer (50 mM Tris-HCl [pH 7.5], 100 mM KCl, and 1 mM EDTA) at 4°C. The protein was then concentrated through a 30-kDa Centricon filter (Millipore), and glycerol and dithiothreitol (DTT) were added to concentrations of 10% (vol/vol) and 1 mM, respectively. MtrR-MBP was purified as described previously (5). Purity was assessed by SDS-PAGE (29), and protein was visualized by staining with Coomassie brilliant blue.
Purification of MtrA-His.
All purification of MtrA-His was done in E. coli BL21(DE3). The pET15b-mtrA construct was coexpressed with the pG-KJE8 plasmid (catalog no. 3340; TaKaRa Bio Inc.), which inducibly expressed the GroEL/S and DnaK/J-GrpE chaperones. Briefly, 10-ml cultures of BL21(DE3) harboring both pET15b-mtrA and pG-KJE8 were grown overnight at 37°C and added to 1.2 liters of LB broth the next morning. Expression of DnaK/J-GrpE and GroEL/S was induced with 0.5 mg/ml l-arabinose and 5 ng/ml tetracycline, respectively. The culture was grown at 37°C until mid-log phase and then induced with 0.3 mM IPTG and grown at room temperature overnight. Cells were harvested and resuspended in 20 ml buffer (10 mM Tris [pH 8] and 200 mM NaCl) and lysed as for MtrA-MBP (see above). MtrA-His was purified over a 3-ml nickel-nitrilotriacetic acid (Ni-NTA) column. The column was washed with 50 mM imidazole, and MtrA-His was eluted in fractions containing 100 and 250 mM imidazole. The fractions containing MtrA-His were concentrated and buffer exchanged into storage buffer (50 mM Tris-HCl [pH 7.5], 100 mM KCl, and 1 mM EDTA). Purity was assessed by using the SDS-PAGE gel described above.
EMSA.
The ability of MBP-MtrA to bind DNA was assayed by electrophoretic mobility shift assay (EMSA) as described elsewhere (30). Briefly, mtrCDE promoter DNA was amplified from FA19 chromosomal DNA using the primers KH9_2 (CGTTTCGGGTCGGTTTGACG) and mtrC_R (CATCGCCTTAGAAGCATAAAAAGCC). From the resulting 266-bp PCR product, 500 ng was end labeled with [γ-32P]dATP with T4 polynucleotide kinase (NEB, Beverly, MA). In the EMSA reaction, MBP-MtrA was incubated with 5 ng of end-labeled PCR product in reaction buffer [20 mM Tris, pH 8, 200 mM NaCl, 1 mM DTT, and 1 μg/μl poly(dI/dC)] and incubated for 15 min at room temperature. If an effector was used (TX-100 [100 μg] or nonoxynol-9 [0.3% {vol/vol}]), it was added 15 min prior to addition of the radiolabeled DNA. The reactions were stopped by adding loading buffer (33% [wt/vol] sucrose, 10 mM Tris-HCl [pH 7.5], and 1 mM EDTA), loaded on a 6% polyacrylamide gel, and run at 4°C at 250 V. The gel was dried and exposed to autoradiography.
Competition between MBP-MtrR and MBP-MtrA.
The smallest amounts of MBP-MtrR and MBP-MtrA that resulted in a complete shift of the mtrCDE promoter region (1 μg and 4 μg, respectively) were used in this competition experiment. The reactions were carried out in the presence and absence of 100 μg TX-100, respectively. For direct competition, both MBP-MtrR and MBP-MtrA were incubated with radiolabeled DNA at either the same time or 15 min apart. The reactions were completed as described as above.
Detection of bases important for MtrA-MBP binding.
The mtrCDE promoter region was amplified using a low-fidelity Taq polymerase (catalog no. 200550; Stratagene) as per the manufacturer’s instructions that would give errors of approximately 1 per 250 bp. This was used as the template in a DNA-binding reaction using reaction buffer (as above) in the presence of 100 μg TX-100 at room temperature. The same reaction mixture containing wild-type (error-free) DNA was used in parallel. After incubating with MBP-MtrA for 15 min, the reaction mixture was added to a 30-kDa Microcon device (catalog no. UFC503096; Millipore) and spun at 6,200 × g at room temperature for 10 min. The flowthrough (containing unbound DNA) was used as the template for a PCR and amplified with proofreading Taq polymerase (F-530L; Finnzymes), A-tailed, and then cloned into the pCR2.1-TOPO vector (Invitrogen, K4510). These steps were done only if the PCR using the flowthrough of the wild-type reaction had no product. The cloned PCR products were transformed into chemically competent E. coli DH5a, and the plasmids were purified and sequenced (Beckman Coulter Genomics) to identify base changes.
DNase I protection assays.
Probes were generated by PCR using one primer labeled with [γ-32P]dATP and a cold primer. The PCR product was purified and used in an EMSA reaction (see above), and then 3 μl DNase I (catalog no. 5025; Sigma) was added and incubated at 37°C for 1 min. Reactions were stopped with addition of stop solution (3 M sodium acetate and 70% ethanol), precipitated, resuspended in elution buffer (10 mM Tris pH 8.0, 1 mM EDTA), and run on a 6% polyacrylamide gel. The probe used for the protection assay was generated with KH9_2 and mtrC_10 (GCAGTCTCAATTTTATGGGTT).
ACKNOWLEDGMENTS
We thank Elizabeth Ohneck and Lane Pucko for their reading and editing of the manuscript.
This work was supported by NIH grants AI021150-28 to W.M.S. and U19 AI031496 (P. F. Sparling, University of North Carolina-Chapel Hill) and a Merit Award from the Medical Research Service of the Department of Veterans Affairs (to W.M.S.). NHMRC Biomedical Training Fellowship 569913 supported Y.M.Z. W.M.S. is the recipient of a Senior Research Career Scholarship Award from the VA Medical Research Service.
Footnotes
Citation Zalucki YM, Dhulipala V, Shafer WM. 2012. Dueling regulatory properties of a transcriptional activator (MtrA) and repressor (MtrR) that control efflux pump gene expression in Neisseria gonorrhoeae. mBio 3(6):e00446-12. doi:10.1128/mBio.00446-12.
REFERENCES
- 1. Workowski KA, Berman SM, Douglas JM., Jr. 2008. Emerging antimicrobial resistance in Neisseria gonorrhoeae: urgent need to strengthen prevention strategies. Ann. Intern. Med. 148:606–613 [DOI] [PubMed] [Google Scholar]
- 2. Shafer WM, Qu X, Waring AJ, Lehrer RI. 1998. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. U. S. A. 95:1829–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hagman KE, et al. 1995. Resistance of Neisseria gonorrhoeae to antimicrobial hydrophobic agents is modulated by the mtrRCDE efflux system. Microbiology 141(Part 3):611–622 [DOI] [PubMed] [Google Scholar]
- 4. Jerse AE, et al. 2003. A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infect. Immun. 71:5576–5582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lucas CE, Balthazar JT, Hagman KE, Shafer WM. 1997. The MtrR repressor binds the DNA sequence between the mtrR and mtrC genes of Neisseria gonorrhoeae. J. Bacteriol. 179:4123–4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hagman KE, Shafer WM. 1995. Transcriptional control of the mtr efflux system of Neisseria gonorrhoeae. J. Bacteriol. 177:4162–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rouquette C, Harmon JB, Shafer WM. 1999. Induction of the mtrCDE-encoded efflux pump system of Neisseria gonorrhoeae requires MtrA, an AraC-like protein. Mol. Microbiol. 33:651–658 [DOI] [PubMed] [Google Scholar]
- 8. Hoffmann KM, Williams D, Shafer WM, Brennan RG. 2005. Characterization of the multiple transferable resistance repressor, MtrR, from Neisseria gonorrhoeae. J. Bacteriol. 187:5008–5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Veal WL, Nicholas RA, Shafer WM. 2002. Overexpression of the MtrC-MtrD-MtrE efflux pump due to an mtrR mutation is required for chromosomally mediated penicillin resistance in Neisseria gonorrhoeae. J. Bacteriol. 184:5619–5624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ohneck EA, et al. 2011. A novel mechanism of high-level, broad-spectrum antibiotic resistance caused by a single base pair change in Neisseria gonorrhoeae. mBio 2(5):e00187-11 http://dx.doi.org/10.1128/mBio.00187-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL. 1997. Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61:393–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Egan SM. 2002. Growing repertoire of AraC/XylS activators. J. Bacteriol. 184:5529–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Warner DM, Folster JP, Shafer WM, Jerse AE. 2007. Regulation of the MtrC-MtrD-MtrE efflux-pump system modulates the in vivo fitness of Neisseria gonorrhoeae. J. Infect. Dis. 196:1804–1812 [DOI] [PubMed] [Google Scholar]
- 14. Schleif R. 2003. AraC protein: a love-hate relationship. Bioessays 25:274–282 [DOI] [PubMed] [Google Scholar]
- 15. Kwon HJ, Bennik MH, Demple B, Ellenberger T. 2000. Crystal structure of the Escherichia coli Rob transcription factor in complex with DNA. Nat. Struct. Biol. 7:424–430 [DOI] [PubMed] [Google Scholar]
- 16. Jair KW, et al. 1996. Transcriptional activation of promoters of the superoxide and multiple antibiotic resistance regulons by Rob, a binding protein of the Escherichia coli origin of chromosomal replication. J. Bacteriol. 178:2507–2513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grkovic S, Brown MH, Skurray RA. 2002. Regulation of bacterial drug export systems. Microbiol. Mol. Biol. Rev. 66:671–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Plano GV. 2004. Modulation of AraC family member activity by protein ligands. Mol. Microbiol. 54:287–290 [DOI] [PubMed] [Google Scholar]
- 19. Martin RG, Rosner JL. 2001. The AraC transcriptional activators. Curr. Opin. Microbiol. 4:132–137 [DOI] [PubMed] [Google Scholar]
- 20. Schleif R. 2010. AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol. Rev. 34:779–796 [DOI] [PubMed] [Google Scholar]
- 21. Piddock LJ. 2006. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev. 19:382–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nikaido H. 2009. Multidrug resistance in bacteria. Annu. Rev. Biochem. 78:119–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Köhler T, Epp SF, Curty LK, Pechère JC. 1999. Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 181:6300–6305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin RG, Jair KW, Wolf RE, Jr, Rosner JL. 1996. Autoactivation of the marRAB multiple antibiotic resistance operon by the MarA transcriptional activator in Escherichia coli. J. Bacteriol. 178:2216–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jair KW, Fawcett WP, Fujita N, Ishihama A, Wolf RE., Jr. 1996. Ambidextrous transcriptional activation by SoxS: requirement for the C-terminal domain of the RNA polymerase alpha subunit in a subset of Escherichia coli superoxide-inducible genes. Mol. Microbiol. 19:307–317 [DOI] [PubMed] [Google Scholar]
- 26. Ma D, Alberti M, Lynch C, Nikaido H, Hearst JE. 1996. The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol. Microbiol. 19:101–112 [DOI] [PubMed] [Google Scholar]
- 27. Rosenberg EY, Bertenthal D, Nilles ML, Bertrand KP, Nikaido H. 2003. Bile salts and fatty acids induce the expression of Escherichia coli AcrAB multidrug efflux pump through their interaction with Rob regulatory protein. Mol. Microbiol. 48:1609–1619 [DOI] [PubMed] [Google Scholar]
- 28. Nakajima H, Kobayashi K, Kobayashi M, Asako H, Aono R. 1995. Overexpression of the robA gene increases organic solvent tolerance and multiple antibiotic and heavy metal ion resistance in Escherichia coli. Appl. Environ. Microbiol. 61:2302–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 30. Folster JP, Dhulipala V, Nicholas RA, Shafer WM. 2007. Differential regulation of ponA and pilMNOPQ expression by the MtrR transcriptional regulatory protein in Neisseria gonorrhoeae. J. Bacteriol. 189:4569–4577 [DOI] [PMC free article] [PubMed] [Google Scholar]