

ABSTRACT
Bacteria and archaea face continual onslaughts of rapidly diversifying viruses and plasmids. Many prokaryotes maintain adaptive immune systems known as clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated genes (Cas). CRISPR-Cas systems are genomic sensors that serially acquire viral and plasmid DNA fragments (spacers) that are utilized to target and cleave matching viral and plasmid DNA in subsequent genomic invasions, offering critical immunological memory. Only 50% of sequenced bacteria possess CRISPR-Cas immunity, in contrast to over 90% of sequenced archaea. To probe why half of bacteria lack CRISPR-Cas immunity, we combined comparative genomics and mathematical modeling. Analysis of hundreds of diverse prokaryotic genomes shows that CRISPR-Cas systems are substantially more prevalent in thermophiles than in mesophiles. With sequenced bacteria disproportionately mesophilic and sequenced archaea mostly thermophilic, the presence of CRISPR-Cas appears to depend more on environmental temperature than on bacterial-archaeal taxonomy. Mutation rates are typically severalfold higher in mesophilic prokaryotes than in thermophilic prokaryotes. To quantitatively test whether accelerated viral mutation leads microbes to lose CRISPR-Cas systems, we developed a stochastic model of virus-CRISPR coevolution. The model competes CRISPR-Cas-positive (CRISPR-Cas+) prokaryotes against CRISPR-Cas-negative (CRISPR-Cas−) prokaryotes, continually weighing the antiviral benefits conferred by CRISPR-Cas immunity against its fitness costs. Tracking this cost-benefit analysis across parameter space reveals viral mutation rate thresholds beyond which CRISPR-Cas cannot provide sufficient immunity and is purged from host populations. These results offer a simple, testable viral diversity hypothesis to explain why mesophilic bacteria disproportionately lack CRISPR-Cas immunity. More generally, fundamental limits on the adaptability of biological sensors (Lamarckian evolution) are predicted.
IMPORTANCE
A remarkable recent discovery in microbiology is that bacteria and archaea possess systems conferring immunological memory and adaptive immunity. Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated genes (CRISPR-Cas) are genomic sensors that allow prokaryotes to acquire DNA fragments from invading viruses and plasmids. Providing immunological memory, these stored fragments destroy matching DNA in future viral and plasmid invasions. CRISPR-Cas systems also provide adaptive immunity, keeping up with mutating viruses and plasmids by continually acquiring new DNA fragments. Surprisingly, less than 50% of mesophilic bacteria, in contrast to almost 90% of thermophilic bacteria and Archaea, maintain CRISPR-Cas immunity. Using mathematical modeling, we probe this dichotomy, showing how increased viral mutation rates can explain the reduced prevalence of CRISPR-Cas systems in mesophiles. Rapidly mutating viruses outrun CRISPR-Cas immune systems, likely decreasing their prevalence in bacterial populations. Thus, viral adaptability may select against, rather than for, immune adaptability in prokaryotes.
Introduction
A fundamental tenet of Darwinian evolution is that random mutations drive adaptation (1–3). Yet, most nucleotide substitutions are deleterious to host fitness (4–8), making undirected mutation wasteful. What if organisms could sense their changing environments and acquire only those mutations that increased fitness?
In multicellular eukaryotes, sensor-based, Lamarckian evolution appears unlikely, with soma-germ line barriers generally inhibiting the inheritance of environmentally acquired mutations (9–11). In contrast, single-celled bacteria and archaea lack a dedicated germ line. With Lamarckian evolution thus apparently possible in prokaryotes, we sought to capture the conditions under which natural selection favors sensor-based adaptation in bacteria and archaea.
As a model system to quantitatively probe the prevalence of sensor-based adaptation, we studied an adaptive immune system found in many, but not all, bacteria and archaea (12–15). This microbial sensor-based immune system is a genomic locus comprised of two adjacent regions. The first region is an array of interspersed repetitive sequences termed clustered regularly interspaced short palindromic repeats (CRISPR). The second region contains critical accessory genes termed CRISPR-associated (Cas) genes. The protein products of the Cas genes serve as the machinery driving CRISPR-based immunity, enabling CRISPR loci to serially target and incorporate 30- to 84-bp DNA fragments from invading viruses and plasmids between CRISPR repeat sequences (16–18). These CRISPR-incorporated fragments are known as “spacers,” whereas the corresponding viral or plasmid sequences are termed “protospacers.”
Spacers make CRISPR-Cas an adaptive immune system, immunizing bacterial and archaeal hosts against subsequent invasions by viruses or plasmids with matching protospacers (16–18). In many ways, analogous to the RNA interference system of eukaryotes (19), spacer-mediated immunity is RNA guided. The CRISPR locus is first transcribed into a single long RNA sequence; this “pre-CRISPR RNA” is then cleaved into individual spacer repeat units by a complex of Cas proteins (20–24). Aided by additional Cas proteins, the single bound spacer senses and degrades cognate protospacers, inactivating invading viruses or plasmids (12, 18, 20, 22, 25–27). Viruses can evade CRISPR-Cas through minimal changes in targeted protospacer regions. In several experiments, single protospacer mutations have rendered CRISPR-Cas ineffectual (16, 28–30). Conversely, hosts have regained antiviral immunity through new spacer additions (28, 29, 31, 32), driving potential coevolutionary arms races between mutating virus and spacer-incorporating host.
Previously, we combined metagenomic time series data with a mathematical model to track the arms race between CRISPR spacer incorporation and viral protospacer mutation across a multiyear period in an acid mine drainage system (33). To focus on spacer/protospacer coevolution, the previous mathematical model assumed that all prokaryotes contained CRISPR-Cas loci. Similarly, the metagenomic reconstructions targeted CRISPR-Cas regions, limiting most of our analysis to CRISPR-Cas-positive (CRISPR-Cas+) hosts. In actuality, however, less than half of all sequenced bacteria contain CRISPR-Cas loci (15).
Here we investigate why only ~45% of sequenced bacterial genomes maintain CRISPR-Cas systems, in contrast to the over 90% of sequenced archaeal genomes that are CRISPR-Cas+. The relative dearth of bacterial CRISPR-Cas systems appears especially surprising given the extensive diversity of lytic bacterial viruses (34, 35) against which CRISPR-Cas would be expected to provide critical adaptive immunity.
One potential driver of the dichotomous prevalence of CRISPR-Cas between bacteria and archaea could be that most sequenced archaea are thermophiles, whereas most sequenced bacteria are mesophiles. Recent biophysical models show that mutations are more likely to be lethal in thermophilic environments, because high temperatures reduce protein stability (36–40). With an increased cost to mutation, thermophilic genomes are predicted to have lower mutation rates than mesophilic genomes (38, 40). The results of several experiments match these predictions, reporting substantially reduced genomic mutation rates in archaeal and bacterial thermophiles (41–43). A further indicator of the increased cost of mutation in thermophiles is that the average ratio of nonsynonomous to synonomous substitutions, i.e., the dN/dS ratio, averaged across thousands of pairs of orthologous genes, drops from 0.14 in mesophiles to 0.09 in thermophiles (44).
These predictions and measurements indicate that viruses infecting thermophiles are afforded fewer viable protospacer mutations to evade CRISPR-Cas targeting. With viable viral mutation rates reduced, each CRISPR-incorporated spacer provides antiviral immunity for a longer period of time. Armed with more beneficial spacers, the entire CRISPR-Cas system would provide greater immunity in mutationally constrained thermophilic environments. We thus hypothesized that decreased viral mutation rates select for the increased presence of CRISPR-Cas in thermophiles, explaining the disproportionate presence of CRISPR-Cas in archaea.
To quantitatively test the hypothesis that decreased viral mutation rates increase the prevalence of CRISPR-Cas, we developed a population genetic model in which hosts with and without CRISPR-Cas compete under pressure from mutating, lytic viruses. CRISPR-Cas+ hosts serially acquire antiviral spacers, but CRISPR-Cas also comes with a parameterized fitness cost. Weighing the fixed fitness cost of CRISPR-Cas against its changing immunological benefit, the model calculates the evolutionary stability of CRISPR-Cas across the parameter space. In agreement with the thermophilicity hypothesis, simulations capture striking phase transitions in which CRISPR-Cas is highly prevalent at reduced viral mutation rates but eradicated once viral mutation rates surpass cost-dependent thresholds. Thus, increasing viral adaptability appears to depress host immune adaptability.
RESULTS
CRISPR-Cas is disproportionately present in bacterial and archaeal thermophiles.
The basic premise of our thermophilicity hypothesis is that, by reducing viral mutation rates, increased environmental temperatures increase the prevalence of CRISPR-Cas. Thus, a high frequency of CRISPR-Cas is predicted for the minority of bacteria that are thermophiles. To test this prediction, we sampled a representative set of 383 bacterial and archaeal genomes from the collection of all fully sequenced prokaryotes (Materials and Methods). Only one sequence per genus was generally sampled, increasing statistical independence. We then analyzed the sampled genomes for the presence of putatively functional CRISPR-Cas loci, using established bioinformatics methods (45, 46).
In agreement with the prediction of the thermophilicity hypothesis, approximately 90% of bacterial thermophiles possess CRISPR-Cas systems, whereas only 46% of bacterial mesophiles are CRISPR-Cas+ (Fig. 1, top). Archaeal thermophiles are also more likely to contain CRISPR-Cas than are archaeal mesophiles. Across all prokaryotes, thermophilicity and the presence of CRISPR-Cas are highly correlated (P < 10−11 by Fisher’s exact test). Multivariate logistic regression verifies that the strong correlation between thermophilicity and the presence of CRISPR-Cas exists independent of whether the thermophiles are archaea or bacteria (P < 10−6). In addition to the strong environmental correlation, there is a weak correlation between archaeal-bacterial taxonomic affiliation and CRISPR-Cas presence (P = 0.02). To test whether the presence of CRISPR-Cas hinges more strongly on thermophilic environment or on archaeal taxonomic affiliation, we used the Akaike information criterion (AIC), a model selection method (47). The AIC computes relative goodness of fit for statistical models, with a lower AIC indicating a better fit. Computing the AIC values shows that the presence of CRISPR-Cas is better predicted by thermophilic environment alone (AIC = 479) than by archaeal taxonomy alone (AIC = 509).
FIG 1.
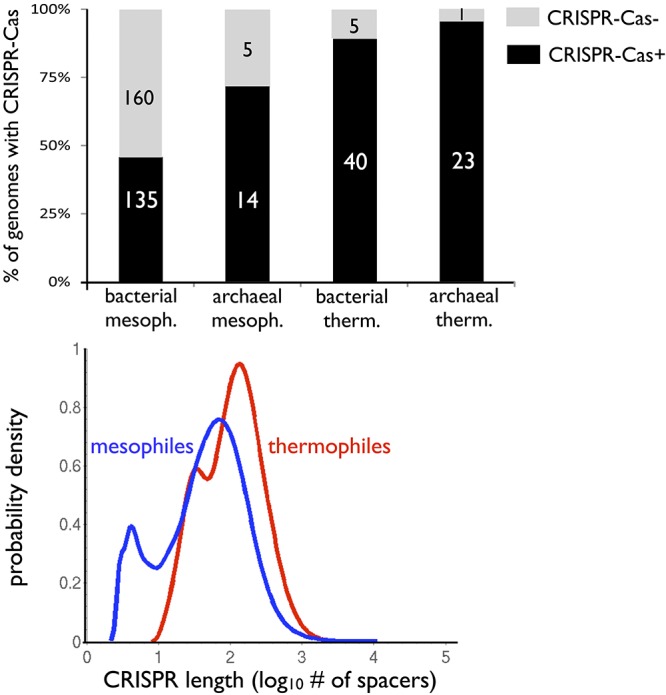
CRISPR-Cas is disproportionately prevalent in thermophiles. (Top) Bar graph showing the percentage of CRISPR-Cas+ and CRISPR-Cas− prokaryotes in mesophilic (mesoph.) and thermophilic (therm.) environments. The numbers in white or black shown on the bars are the numbers of species. Across 383 diversified bacterial and archaeal genomes (Materials and Methods), CRISPR-Cas is disproportionately sampled in thermophiles (P < 10−6). (Bottom) CRISPR locus length distributions. The distributions of locus lengths for CRISPR-Cas+ thermophiles and mesophiles fit to log10 of the total number of spacers per genome (Fig. S1 shows histograms in logarithmic and nonlogarithmic scales). On average, thermophilic loci possess more spacers (P < 10−7). However, CRISPR locus lengths show greater variance in mesophiles (P = 5 × 10−3).
To further analyze the environmental dependence of CRISPR-Cas, we estimated the distribution of the number of spacers per CRISPR-Cas+ host in mesophilic and thermophilic environments (Fig. 1, bottom; see Fig. S1 in the supplemental material). On average, thermophiles possess a greater number of CRISPR spacers per genome than do mesophiles (P < 10−7 by Welch’s t test). However, the variance in the per-genome number of spacers is greater in mesophiles than in thermophiles (P = 5 × 10−3 by the F test), with the greatest number of spacers found in a mesophile.
A mutation-selection-drift model for the evolution of CRISPR-Cas.
To quantitatively probe the high prevalence of CRISPR-Cas in thermophiles, we developed a population genetic model. Similar to previous mathematical models of CRISPR-virus coevolution (33, 48–50), the model implements basic events known to occur during viral infections such as unidirectional host spacer addition and viral protospacer mutation. However, in contrast to earlier models, here we include horizontal gene transfer (HGT) events that occur independent of viral infection. During HGT events, hosts can acquire or delete entire CRISPR-Cas loci. This allows us to compete the resulting CRISPR-Cas-positive (CRISPR-Cas+) and CRISPR-Cas-negative (CRISPR-Cas−) subpopulations across wide swaths of parameter space, yielding thresholds for the maintenance of CRISPR-Cas systems.
All model events occur during discrete, nonoverlapping iterations, with model parameters determining event probabilities (see Table S1 in the supplemental material). The full model algorithm is detailed in the supplemental material; below we describe the key steps involved in each iteration (Fig. 2).
FIG 2.

Model of virus-host coevolution. Schematic of a representative model iteration. Host and viral populations are divided into strains labeled B1 and B2 (B stands for bacteria) and V1 and V2 (V stands for virus), respectively. Host strains are shown as ovals and viral strains as hexagons; bars within the strains reflect host spacers and viral protospacers. Identical spacer and protospacer sequences have the same colored bar. Host strains lacking CRISPR-Cas are shown as empty ovals. In the first step of a model iteration, host and viral strains encounter one another. In the next step, mutations occur (single and double quotation marks). Because viruses mutate only in productive encounters and hosts incorporate spacers only if they survive infection, mutations are concentrated in immune hosts and infective viruses. In the next step, more mutations can occur if CRISPR-Cas+ hosts delete spacers or lose entire CRISPR-Cas loci. Similarly, CRISPR-Cas− hosts can acquire CRISPR-Cas loci. In the fourth step, selection modifies strain frequencies according to equations 1 and 2 in the text. Finally, each iteration ends with the model randomly sampling approximately fixed numbers of hosts and viruses.
(i) Step 1. Virus-host encounters.
In each iteration, a fixed and parameterized number of virus-host encounters occur. These encounters are divided among the host and viral strains according to the products of host and viral strain frequencies (mass action). Each virus-host encounter is then classified as either “immune” or “productive” based on the outcome: either the immune host clears the virus, or the productive virus successfully infects (i.e., lyses) the host.
Immune encounters arise in one of two ways: (i) CRISPR-Cas+ hosts can survive viral infection by possessing spacers matching viral protospacers, or (ii) CRISPR-Cas+ and CRISPR-Cas− hosts can survive through non-CRISPR-based resistance mechanisms such as restriction modification (35, 51). When both CRISPR and innate resistance mechanisms fail, the virus kills the host in a lytic encounter. Offering CRISPR-Cas a strong selective advantage in the model and in agreement with the results of viral challenge assays performed in two distinct model systems (28, 30), we parameterize CRISPR-Cas to be 105-fold more protective than non-CRISPR-based resistance mechanisms (see Table S1 in the supplemental material). Importantly, CRISPR-Cas is protective only when it contains a spacer matching a viral protospacer. Innate resistance mechanisms are thus vital to host survival when the host lacks spacers matching an invading virus.
(ii) Step 2. Immune hosts can add spacers, and infective viruses can mutate protospacers.
In a parameterized fraction of immune (but not productive) virus-host encounters, a CRISPR-Cas+ host strain can unidirectionally incorporate a new spacer. Analogous to host spacer addition, in a parameterized fraction of productive virus-host encounters, a viral strain can mutate a random protospacer. Given the >30-bp size of each protospacer, a previously unseen protospacer is placed in the slot of the mutated protospacer (“infinite allele” assumption). Virus and host mutants are initialized with an abundance of 1.
(iii) Step 3. CRISPR-Cas+ hosts can lose CRISPR-Cas or delete spacers, and CRISPR-Cas− hosts can gain CRISPR-Cas.
Independent of the virus-host encounters and mutations in steps 1 and 2, all host strains can undergo homologous recombination or plasmid-driven HGT. This leads to spacer deletions and acquisitions and losses of entire CRISPR-Cas systems. When a CRISPR-Cas+ host is chosen to delete spacers, two random spacers in its CRISPR locus are sampled. The new mutant deletes all spacers between the two chosen spacers. When a CRISPR-Cas+ host loses CRISPR-Cas, the entire CRISPR-Cas locus is deleted, with all attendant spacers. Finally, when a CRISPR-Cas− host acquires CRISPR-Cas by HGT, the CRISPR-transferring donor strain is chosen by randomly sampling all host strains. If the chosen donor strain lacks CRISPR-Cas, the mutant receives a functional CRISPR-Cas locus but no spacers. Thus, the model continually reintroduces CRISPR-Cas into CRISPR-Cas− populations, testing the stability of CRISPR-Cas− populations to mutational invasion by CRISPR-Cas+ strains. Accordingly, at each point in the parameter space, we can average the prevalence of CRISPR-Cas across large numbers of iterations, instead of averaging the results of many independent simulations (the continual reintroduction of CRISPR-Cas fits an ergodic assumption). As in step 2, all host mutants are generated independently at an initial abundance of 1.
(iv) Step 4. Selection for immune hosts and infective viruses.
Selection modulates the frequencies of host and viral strains according to the fitness functions defined below. This frequency adjustment is performed only for the “parent” strains that underwent the virus-host encounters in step 1: mutants from steps 2 and 3 remain at frequencies of 1/N, where N is the respective host or viral population size.
To increase in fitness, viruses need to productively infect hosts, whereas hosts need to immunize themselves without paying too high a fitness cost. One potential cost of the CRISPR-Cas system could be autoimmunity, stemming from the documented acquisition of self-spacers matching host DNA in ~18% of known CRISPR-Cas loci (52–54). An additional cost is that CRISPR-Cas might hamper all forms of HGT, including the uptake of beneficial genetic material such as antibiotic resistance genes (54, 55). With these sources of fitness costs in mind, the model includes a fixed, parameterized cost for the CRISPR-Cas locus. Importantly, hosts were not penalized for each ~100-bp spacer repeat unit added.
The fixed CRISPR-Cas cost, C, lowers the relative growth rate (r) of CRISPR-Cas+ strains by the factor r = 1/(1 + C). If a host strain lacks CRISPR-Cas, r is defined to equal 1. Weighing this relative fitness cost against the relative immunity of a host strain, selection resets the frequencies of each host strain to the following:
| 1 |
where fBj stands for the frequency of host (Bacterial) strain j, Vi stands for virus, Bj stands for host (Bacterial) strain j, and Immunei,j stands for number of immune encounters between virus strain i and bacterial strain j.
Thus, the new frequency of a host strain is its fraction of all host immune encounters, with the caveat that CRISPR-Cas+ strains pay a relative growth cost.
To determine viral strain frequencies, we consider only the ability of a viral strain to productively infect the host strains. The new frequency of a viral strain is then its fraction of all productive encounters undergone by the viral population:
| 2 |
where fVi stands for the frequency of viral strain i and Productivei,j stands for the number of productive encounters between viral strain i and host strain j.
After selection, the model calculates the number of mutants, m, created by each host and viral parent strain in steps 2 and 3. The cumulative frequency of these mutants, m/NB (for host mutants) where NB stands for the total host bacterial population and m/NV (for viral mutants) where NV stands for the total virus population, is then deducted from the frequency of the parent strain. When the frequency of a parent strain falls below 0, it is cleared from the model.
(v) Step 5. Sampling.
Collecting all parent and mutant strains from the previous steps, we multiply the host and viral strain frequencies by the total population sizes, NB and NV, of the host and viral populations. This yields an abundance for each strain (with an abundance of 1 for each mutant). We then sample (with replacement) an average of NB hosts and NV viruses. Sampled hosts and viruses remain in the model, whereas unsampled strains are removed.
Sampling mimics genetic drift by challenging new mutants that arise at the low abundance of 1 with stochastic extinction, regardless of their fitness. Further, sampling randomly removes old strains which selection has reduced in abundance. Thus, mutation creates diversity, whereas selection and sampling limit diversity, allowing the model to implement mutation-selection-drift balance.
Finally, at the end of an iteration, host and viral strain frequencies are renormalized to ensure that both sum to 1. The model then returns to step 1 in all but the final iteration.
Cost-benefit threshold for CRISPR-Cas systems.
To analytically derive the dependence of CRISPR-Cas prevalence on experimentally measurable parameters, we calculated when CRISPR-Cas is under positive selection in a given model iteration. According to equation 1, the selection equations set the frequency of a host strain to be its fraction of all immune encounters, and CRISPR-Cas+ strains have their immune encounters reduced by a cost. Thus, the postselection prevalence of CRISPR-Cas is the sum of the cost-reduced fractions of immune encounters for all CRISPR-Cas+ host strains. When the postselection prevalence of CRISPR-Cas is greater than its preselection prevalence, CRISPR-Cas is under positive selection in the model. As derived in the supplemental material, CRISPR-Cas is under positive selective when:
| 3 |
Here C is the fitness cost of maintaining CRISPR-Cas, whereas fB∩V is the probability that a randomly chosen CRISPR-Cas+ host strain shares at least one spacer with a randomly chosen viral strain. Given the extremely small failure rate measured for CRISPR-Cas systems (16, 30), this is effectively the probability of CRISPR-Cas providing immunity in a model iteration. Conversely, P0 is the fraction of viral encounters that a host strain survives when CRISPR-Cas is absent or fails.
Because fB∩V is a nonlinear function dependent on all model parameters, equation 3 is not in itself predictive. However, fB∩V can be divided into measurable components, yielding a predictive threshold for CRISPR-Cas in a simplified setting. For simplicity, we assume that the CRISPR-Cas+ strains initially lack spacers against the current viruses and that the number of protospacers per virus is one.
Lacking protective spacers in advance, fB∩V is the combined probability that a CRISPR-Cas+ host survives viral infection due to innate resistance, adds a spacer, and then encounters a virus with a protospacer matching the acquired spacer. With the three events independent, fB∩V = (P0)(Ps_add)(), where P0 is the probability of innate resistance and Ps_add is the probability of adding a spacer in an immune encounter (P0). Ps_add is thus the probability that a spacer addition occurs. The probability of the acquired protospacer being protospacer i is si, where si is the fraction of viruses containing protospacer i. Because the next virus encountered will also contain protospacer i with probability si, once a spacer addition occurs, is the probability that the next virus encountered has the same protospacer. Critically, the si values (i.e., the protospacer frequencies) are directly measurable in both laboratory and metagenomic samplings.
In addition to being measurable, has an immediate connection to a well-studied measure of population diversity known as the Simpson diversity index D (56) that is defined to equal 1 − , i.e., the probability that two randomly sampled protospacers are distinct. Combining equation 3 with the decomposition of fB∩V, in the simplified model setting, selection promotes CRISPR-Cas+ strains when:
| 4 |
Two key predictions arise from this inequality. First, equation 4 is a cost-benefit threshold that quantifies when the cost of CRISPR-Cas is less than the immunological benefit conferred by CRISPR-Cas. The immunological benefit of CRISPR-Cas reflects its ability to acquire protospacers shared by many viruses together with the likelihood that competing, innate resistance mechanisms are nonprotective. Second, this inequality predicts that, if viral diversity is sufficiently high, CRISPR-Cas cannot rise in frequency. As protospacer diversity gets large, 1 − D approaches 0, whereas Ps_add and the 1 − P0 term cannot surpass 1. Thus, for any nonnegligible cost, high viral diversities are predicted to purge CRISPR-Cas from a population, irrespective of the spacer addition rate.
CRISPR-Cas emerges only at intermediate levels of innate resistance.
Inequalities 3 and 4 appear to imply that increasing the probability of innate resistance (P0) decreases the selective advantage of CRISPR-Cas (i.e., reduces the maximal cost at which CRISPR-Cas can be maintained). However, this is not always the correct interpretation. Increasing P0 also decreases viral diversity (D) by decreasing the frequency of productive encounters in which the viruses can mutate. Thus, when increasing P0 increases the 1 − D term more than it decreases the 1 − P0 term, increasing P0 actually promotes CRISPR-Cas+ strains. Increasing innate immunity offers a second advantage to CRISPR-Cas by providing more immune encounters to prime the CRISPR locus with spacers against new viruses.
While initial increases in innate immunity promote CRISPR-Cas+ strains, inequalities 3 and 4 show that CRISPR-Cas+ strains will be selected against when P0 increases to become close to 1. Inequality 3 shows that even perfect CRISPR-Cas systems, with immunity against 100% of the viruses, cannot evolve when P0 > 2/3 (at the parameterized CRISPR-Cas cost of 0.5). This is because there is no benefit to maintaining a costly CRISPR-Cas system when the cost-free alternative (innate immunity) provides almost complete protection.
With simple analytics implying that innate immunity has competing effects on the evolution of CRISPR-Cas, we designed a model simulation to directly probe the prevalence of CRISPR-Cas as a function of the level of innate immunity (see Fig. S2 in the supplemental material). The model simulations confirm that innate immunity must be increased above a basal, “priming” threshold to maintain CRISPR-Cas. Similarly, CRISPR-Cas loci are lost from populations at extremely high levels of innate resistance. Only at intermediate levels of innate immunity does CRISPR-Cas dominate populations.
High viral mutation rates overwhelm CRISPR-Cas systems.
While the prevalence of CRISPR-Cas nonmonotonically depends on the probability of innate immunity, inequalities 3 and 4 predict a simpler dependence of the prevalence of CRISPR-Cas on the level of viral mutation. Increasing the viral mutation rate lowers the probability that a host’s spacers match future viral protospacers, directly decreasing the benefit of CRISPR-Cas. To test whether this decreased immunological benefit purges CRISPR-Cas from host populations, we simulated the model across thousands of iterations at gradually increasing viral mutation rates.
At low viral mutation rates, all hosts maintain CRISPR-Cas immunity (Fig. 3a; see Fig. S3 in the supplemental material). Because viral diversity is depressed at low viral mutation rates (Fig. S3 and Fig. S4), few spacers are required to provide immunity against the entire viral population. With spacer deletion outpacing spacer addition in the model (see Table S1 in the supplemental material), CRISPR loci delete all but the few antiviral spacers that selection maintains. The CRISPR loci are thus kept small at low viral mutation rates. As the viral mutation rate increases, CRISPR loci gradually increase in length, requiring more and more spacers to maintain immunity against an increasingly diverse viral population. The model predicts that CRISPR loci contain hundreds of spacers per locus at intermediate viral mutation rates, matching the largest experimentally observed CRISPR-Cas systems (Fig. 1, bottom). Further increases in the rate of viral mutation cannot be matched with further increases in CRISPR lengths. Beyond a viral mutation rate threshold, average locus lengths plunge to zero and CRISPR-Cas is purged from populations. Thus, viral mutation overwhelms the CRISPR-Cas system.
FIG 3.
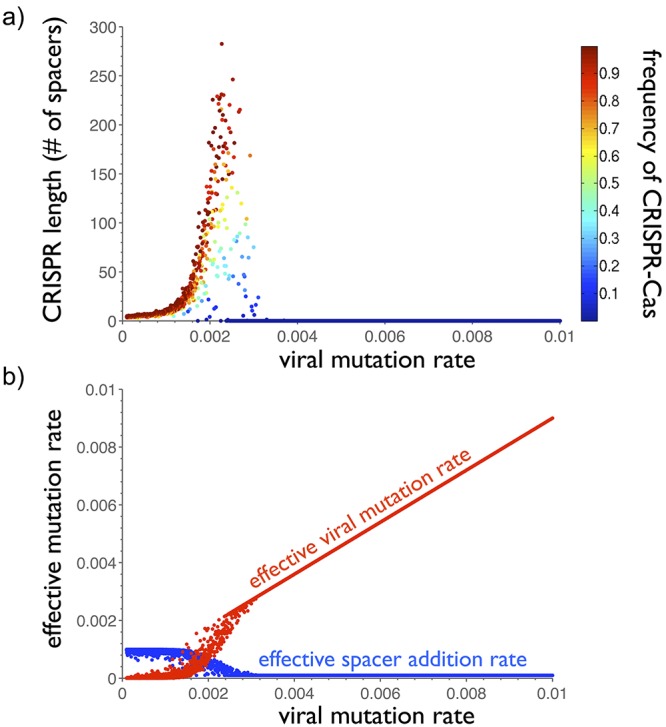
Rapid viral mutation overwhelms CRISPR-Cas systems. (a) Average CRISPR locus length (y axis) and prevalence (heatmap) as functions of the viral mutation rate. Model averages are “time averages” taken across 100,000 (10N) iterations. Viral mutation rates range from 10−4 to 10−2 per genome per productive infection. Since spacer deletion rates outpace spacer addition rates (see Table S1 in the supplemental material), evolution prunes CRISPR loci to the smallest sizes at which they provide antiviral immunity. At low viral mutation rates, few spacers are required against the highly similar viruses. As the viral mutation rate increases, locus lengths increase, with more spacers required against the diversifying viruses (see Fig. S3 in the supplemental material). Once viral mutation rates increase above a threshold, however, CRISPR-Cas systems can no longer keep pace. Overwhelmed by viral diversity, locus lengths crash to 0, and the system is purged from host populations. (b) Effective host and viral mutation rates as functions of the viral mutation rate. Since viruses mutate protospacers only during productive virus-host encounters, the effective viral mutation rate in a simulation is the product of the fraction of productive encounters and the (changing) viral mutation rate. Similarly, since hosts add spacers only during immune virus-host encounters, the effective host mutation rate in a simulation is the product of the fraction of immune encounters and the (fixed) spacer addition rate. Notably, the longest CRISPR loci emerge at reduced effective spacer addition rates. In fact, these long loci emerge in the small window in which the effective mutation rates of both host and virus are nonnegligible. Locus lengths thus reflect the strength of virus-host coevolution, rather than the rate of host spacer addition.
Before the viral mutation rate Pv_mut is increased to the point that it purges CRISPR-Cas from host populations, an intermediate viral mutation regime emerges (0.001 < Pv_mut < 0.003) in which the average prevalence of CRISPR-Cas is often strictly between 0 and 1 (Fig. 3a). Two explanations for this intermediate CRISPR-Cas prevalence are conceivable: either mixed CRISPR-Cas+ and CRISPR-Cas− populations coexist in individual iterations, or the model oscillates between entirely CRISPR-Cas+ and entirely CRISPR-Cas− iterations, yielding an intermediate time-averaged prevalence. To discriminate between these cases, we analyzed all individual iterations of the 600 simulations in which 0.001 < Pv_mut < 0.003. Only 0.2% of the individual model iterations contained mixed CRISPR-Cas+ and CRISPR-Cas− populations (see Fig. S5 in the supplemental material). Thus, at intermediate viral mutation rates (i.e., the separatrix points), hosts occasionally undergo rapid phase transitions between 100% CRISPR-Cas− and 100% CRISPR-Cas+ states (Fig. S4, middle panel). Across thousands of model iterations, the average prevalence of CRISPR-Cas can thus fall anywhere between 0 and 1, depending on the residence times at the CRISPR-Cas+ and CRISPR-Cas− quasi-steady states.
The viral mutation parameter probed above is not the sole determinant of viral diversity. Because viral mutants emerge only in productive virus-host encounters, the probability of a viral mutation is the product of the parameterized viral mutation rate and the probability that virus-host encounters are productive. We denote this product the “effective” viral mutation rate. Similarly, the effective host spacer addition rate is the product of the host spacer addition rate and the probability that a random virus-host encounter is immune (nonproductive). Plotting the effective viral mutation rate and the effective spacer addition rate as functions of the parameterized viral mutation rate reveals an initial inverse symmetry: as the viral mutation parameter increases, slow increases in the effective viral mutation rate match slow decreases in the effective host spacer addition rate (Fig. 3b). These changes in the effective adaptation rates are initially buffered because a still-functioning CRISPR-Cas system keeps most encounters immune (see Fig. S3 in the supplemental material). However, when the viral mutation parameter increases to an intermediate level, a narrow regime emerges in which both the effective spacer addition rate and effective viral mutation rate are nonnegligible. This is the regime of most intensive coevolution, in which hosts frequently add spacers and viruses frequently mutate protospacers. Moreover, selection maintains the host spacer addition in the face of the rapid spacer deletion because the level of viral diversity necessitates extra spacers. Thus, the maximal locus lengths in Fig. 3a reflect maximal virus-host coevolution. Beyond this intermediate regime of maximal coevolution and maximal locus lengths, the effective host spacer addition rate plunges to 0, whereas the effective viral mutation rate increases linearly. The linear increase at high viral mutation rates shows that all but a constant (innate immune) fraction of encounters are productive absent CRISPR-Cas.
High costs and rapid viral mutation eradicate CRISPR-Cas.
Although Fig. 3 shows that CRISPR is lost at high viral mutation rates due to the loss of antiviral immunity (i.e., benefit), inequalities 3 and 4 predict that the prevalence of CRISPR-Cas is a function of both immunity and cost. We thus ran new simulations to track the average prevalence of CRISPR-Cas as a function of both the cost and the viral mutation rate (Fig. 4).
FIG 4.

Cost-benefit analysis of CRISPR-Cas prevalence. Heatmaps of model statistics—averaged over 100,000 iterations—as functions of the cost of CRISPR-Cas and the viral mutation rate. (a) CRISPR-Cas prevalence. Both high costs and high viral mutation rates shift host populations from 100% CRISPR-Cas+ to 0% CRISPR-Cas+. Importantly, the intermediate CRISPR-Cas frequencies found in the midregion separating these two extremes (i.e., the separatrix) do not reflect coexisting CRISPR-Cas+ and CRISPR-Cas− populations. Instead, the separatrix frequencies reflect an average over time as the model alternates between quasi-steady states of entirely CRISPR-Cas+ and entirely CRISPR-Cas− populations (see Fig. S4 and Fig. S5 in the supplemental material). (b) Probability that CRISPR-Cas provides immunity. Given the low failure rate of CRISPR-Cas (Table S1), the probability of CRISPR-Cas providing immunity is effectively the probability that a host spacer matches a viral protospacer. High viral mutation rates thus reduce CRISPR-Cas immunity. In fact, at high viral mutation rates, immunity is absent even when CRISPR-Cas remains 100% prevalent due to low costs (top left of figure 4b). (c) Average (Shannon) diversity of viral protospacers. Increasing the viral mutation rate increases the average diversity across time by increasing genetic diversity at each time point. More surprisingly, increasing the cost of CRISPR-Cas above a threshold rapidly increases viral diversity, because it purges CRISPR-Cas from populations, allowing viruses to freely mutate. (d) Average CRISPR-Cas locus lengths. As in Fig. 3a, which reflects a vertical cross section of Fig. 5d (i.e., locus lengths at a single cost), the largest CRISPR loci emerge at intermediate viral mutation rates.
As shown in Fig. 4a, when the cost of CRISPR-Cas is sufficiently high (C > ~8), CRISPR-Cas cannot persist for any viral mutation rate. Matching these simulations, inequality 3 shows that the maximal cost at which even 100% immunogenic CRISPR-Cas systems can evolve is C = 9 (with P0 parameterized to equal 0.1). Conversely, at very low costs, CRISPR-Cas will be maintained in populations, even for the high viral mutation rates at which CRISPR-Cas provides almost no immunity (Fig. 4b). Thus, sufficiently increasing either the cost or the viral mutation rate takes host populations from entirely CRISPR-Cas+ to entirely CRISPR-Cas−.
To better understand how the loss of CRISPR-Cas both drives and is driven by increased viral diversity, we also tracked how viral diversity varies with the viral mutation rate and CRISPR-Cas cost (Fig. 4c). To quantify viral diversity, the Shannon diversity index (56) of the viral protospacers was calculated during each model iteration. Similar to Simpson’s diversity index, the Shannon index reflects the unpredictability of a randomly chosen viral protospacer. Mathematically, the Shannon index is defined to equal , where si denotes the fraction of viruses containing protospacer i. Providing a reliable metric of viral diversity, the Shannon index sums to 0 when the viruses are all identical (e.g., in the absence of viral mutation). The Shannon index then increases as increasing viral mutation diversifies the viral protospacer population (Fig. 4c). Importantly, the Shannon index can also increase when the viral mutation rate is kept constant. This occurs when the cost of CRISPR-Cas is increased to the point that CRISPR-Cas is purged from host populations, offering the viruses new productive encounters in which to mutate (Fig. 4c).
Rapid spacer addition cannot preserve CRISPR-Cas at high viral mutation rates.
One might expect that CRISPR-Cas systems can maintain immunity against rapid viral mutation by simply incorporating spacers at a higher rate. To test whether accelerated spacer addition can preserve CRISPR-Cas loci at high viral mutation rates, we systematically tracked CRISPR-Cas prevalence as a function of both the viral mutation rate and the host spacer addition rate. While viral mutation rates are kept low, CRISPR-Cas-increased spacer addition maintains CRISPR-Cas immunity against increased viral mutation. However, once the rate of viral mutation surpasses a (cost-dependent) threshold, CRISPR-Cas is purged from host populations even when the rate of spacer addition far outpaces the rate of viral mutation (see Fig. S6 in the supplemental material). With hosts unlikely to encounter the same viral protospacers twice, increasing the rate of spacer addition is of little benefit.
DISCUSSION
Despite the ubiquity of lytic prokaryotic viruses, less than 50% of bacteria maintain CRISPR-Cas adaptive immune systems. Here we formulate a testable hypothesis to explain the relative dearth of adaptive immunity in bacteria. Using comparative genomics, we first report that the absence of CRISPR-Cas in bacteria is highly temperature dependent. While the majority of bacteria are mesophilic and contain CRISPR-Cas at the relatively low prevalence of 45%, bacterial thermophiles are 88% CRISPR-Cas+. Both theory and experimental results indicate that mesophilic genomes possess higher mutation rates than thermophilic genomes (38, 41–44). We wondered whether the increased viral mutation rates of mesophiles were sufficient to explain the low prevalence of CRISPR-Cas in mesophilic environments. To test this hypothesis, we developed an evolutionary model to analyze how the prevalence of CRISPR-Cas varies as the viral mutation rate and other basic parameters are varied. Model analytics and simulations support the viral mutation hypothesis, capturing how CRISPR-Cas is purged from host populations as viral mutation rates increase above cost-dependent thresholds. By mutating rapidly, viruses undermine the key benefit of CRISPR-Cas, immunological memory. In other words, hosts gain little fitness advantage from CRISPR-Cas storing viral sequences never again encountered.
Although our theoretical model shows that increased viral mutation rates are sufficient to explain the reduced prevalence of CRISPR-Cas in mesophilic bacteria, other hypotheses are plausible. For example, CRISPR-Cas might be more beneficial in thermophilic environments because high temperature settings might be closed off from their surroundings with limited inflow of new, diverse viruses. This viral immigration hypothesis is essentially equivalent to the viral mutation hypothesis: increasing immigration rates will have the same qualitative effect as increasing mutation rates. We focus on mutation rather than immigration because mutation rates are readily measurable in the laboratory and because previous data have already measured reduced thermophilic mutation. Another counterhypothesis might argue that unique genetic barriers specifically inhibit acquisition of CRISPR-Cas by bacteria. However, CRISPR-Cas is commonly found on mobile plasmids and widely distributed in diverse bacteria and archaea (15), undermining this argument. Finally, increased CRISPR-Cas costs, rather than decreased immunological benefits, might be implicated in the reduced frequency of CRISPR-Cas in mesophiles. These cost-driven hypotheses are compatible with the results of our model. Figure 4 shows that both high costs and high viral mutation rates purge CRISPR-Cas from populations.
One recent study suggesting an increased cost for CRISPR-Cas in mesophiles reports that bacterial CRISPR-Cas loci have a disproportionate number of self-targeting spacers in comparison to archaeal CRISPR-Cas loci (57). Thus, increased autoimmune costs might limit CRISPR-Cas in mesophilic bacteria. However, unlike viral mutation rates, one wonders why the frequency of self-targeting spacers would be temperature dependent. An alternative explanation for the high prevalence of self-targeting spacers in mesophiles is that they represent the effects, not the causes, of CRISPR-Cas failure at moderate temperatures. Self-targeting spacers might indicate bacteria abandoning the immune function of CRISPR-Cas, arguably because it fails to provide robust antiviral immunity in mesophiles, instead coopting CRISPR-Cas for RNA interference (RNAi)-like gene regulation. Two studies have already addressed this possibility, with differing conclusions (53, 58), making further investigations required.
A similar cost-driven hypothesis assumes that mesophiles more frequently require DNA uptake via HGT, which CRISPR-Cas can block. Thus, the HGT hypothesis argues that mesophiles disproportionately lack CRISPR-Cas to disproportionately acquire HGT. However, there is little evidence for reduced HGT in thermophilic communities. Genomic screens have captured frequent genetic transfer among thermophiles, even between archaea and bacteria (59). Further, a basic assumption of the HGT hypothesis is that CRISPR-Cas actually blocks significant amounts of beneficial HGT in nature. Although a previous study has captured a dearth of CRISPR-Cas within Enterococcus faecalis strains with horizontally acquired drug resistance modules (55), no inverse CRISPR-HGT correlation has been shown at the interspecies scale in which demographic biases are better accounted for. Among the 383 species studied in this work, we found no significant difference in the presence of plasmids between the CRISPR-Cas+ and CRISPR-Cas− genomes (P = 0.27 by Fisher’s exact test). In fact, a recent study reports a positive CRISPR-HGT correlation, finding an increased prevalence of CRISPR-Cas systems in competent bacteria than in noncompetent bacteria (58). Future studies will need to disentangle what correlation, if any, exists between CRISPR-Cas and HGT.
Whether a CRISPR-HGT anticorrelation exists, an important evolutionary issue must be resolved. Unlike spacers that protect against deadly viruses, there seems to be no selective benefit to acquiring spacers that block beneficial plasmids. Thus, the experimental studies demonstrating that CRISPR-Cas blocks beneficial HGT are often forced to artificially engineer CRISPR-Cas loci with the deleterious spacers blocking critical plasmids and DNA. It is worth asking whether these deleterious spacers would naturally rise to high frequencies and thus present real costs to CRISPR-Cas+ hosts in nature. Either way, these experimental studies find little selection against CRISPR-Cas+, spacer controls, implying that beneficial HGT may select against spacers but not the CRISPR-Cas system.
The present hypothesis assumes that thermophilic viruses have reduced mutation rates, although previous experiments noting reduced thermophilic mutation have tracked only the mutation rates of thermophilic hosts (41–44). Our claim is premised on the fact that both thermophilic host and virus share the same environmentally driven mutational constraints. Supporting this assumption, in mesophilic environments, host and virus have been measured to have virtually identical per-genome mutation rates (60). With thermophilic hosts measured to have mutation rates an order of magnitude lower than those measured for both mesophilic host and virus (42), we infer that thermophilic viruses also possess reduced mutation rates. Further, a data-driven biophysical study directly predicts that thermophilic viruses have less mutational plasticity than mesophilic viruses do (38).
Thus, the results of initial experimental and genomic assays are compatible with many of the assumptions of our model. However, to show that in nature viral diversity is limited at high temperatures, better metagenomic resolution is required. Fortunately, next-generation deep-sequencing methods should enable more-detailed gauges of viral nucleotide diversity as a function of temperature. Moreover, the prediction of our model that viral mutability overwhelms CRISPR-Cas is directly testable in the laboratory through challenge experiments with increasingly mutagenized viruses.
Beyond offering a testable hypothesis for the absence of CRISPR-Cas in many bacteria, this work has a more general evolutionary implication. At an abstract level, CRISPR-Cas is a genomic sensor that seeks to directly acquire beneficial mutations in response to a stochastically changing environment (i.e., the virome). Studying the prevalence of CRISPR-Cas can thus provide insight into the conditions under which Lamarckian, directed adaptation is favored in evolution. Seminal analytic work by Kussell and Leibler (61) provides the required mathematical framework by analytically deriving a sensor cost threshold above which genomic sensors are deleterious to their hosts. Surprisingly, Kussell and Leibler’s threshold predicts that the sensor cost threshold increases as the Shannon diversity index (i.e., entropy) of the environmental states increases. In other words, the more a cell requires a sensor because of environmental unpredictability, the more a cell can pay for the sensor. Thus, the results of Kussell and Leibler are opposite to the conclusions that we derive from inequalities 3 and 4 and obtain in the simulations.
To explain the dichotomy between the predictions of our model and those of Kussell and Leibler, we note that the assumptions of Kussell and Leibler are unlikely to apply to adaptive immune systems such as CRISPR-Cas. For analytic tractability, Kussell and Leibler required a model in which the environment remains constant while the population adapts to it. Rapid viral mutation is likely to render this separation of time scales inapplicable in the context of virus-host coevolution. More importantly, Kussell and Leibler’s model assumes that sensors always perfectly adapt to the environment, whatever the environmental entropy. In our model, the efficacy of the sensor is directly reduced by increased environmental entropy (Fig. 4b). Thus, when sensor performance hinges on the difficulty of the sensing task at hand (i.e., environmental entropy), we infer inversion of the predictions of Kussell and Leibler. Future work will aim to capture how this phase transition arises as the assumptions of immediate and perfect sensor performance are relaxed.
A final question can be posed. If CRISPR-Cas sensors are unable to confer antiviral immunity against high levels of viral diversity, why have bacteria and archaea been unable to evolve fitter alternatives over billions of years? In contrast, in just about 500 million years, vertebrates have evolved an adaptive immune system that prefabricates immunity against virtually any viral variant. In principle, an analogous preemptive system could have evolved in prokaryotes, with CRISPR-Cas systems generating unlimited repertoires of random spacers, while keeping in place the necessary Cas and genetic machinery to target and cleave matching foreign sequences. However, no prokaryote is known to possess a genome larger than 13 Mb (62). With more than 50 bp contained in each spacer repeat unit, the vertebrate mode of preemptive adaptive immunity is unlikely to be feasible in compact single-celled microbes. With no way to fit billions of randomly generated spacer sequences in a single prokaryotic cell, perhaps the best microbes can do is to adaptively chase the diversifying viral population, trying to stay apace.
MATERIALS AND METHODS
Comparative genomics of CRISPR-Cas.
Bacterial and archaeal genome sequences were downloaded from the NCBI FTP site ftp://ftp.ncbi.nih.gov/genomes/Bacteria/ in March 2010. At that time, 978 bacterial and 77 archaeal genomes were available. A representative set of 383 genomes (45) used in this work includes the largest genome from each genus (as defined by the NCBI taxonomy database) with greater than 500 annotated protein-coding genes. Exceptions were made for the genus Shigella that was considered to be identical to Escherichia and the genera Escherichia and Bacillus that also included the model genomes Escherichia coli strain K-12 substrain MG1655 and Bacillus subtilis strain 168. Ecological information (environment and growth temperature) was obtained from the NCBI Complete Microbial Genomes Web page (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi). cas genetic loci were identified using the PSI-BLAST profiles (45), while the number of CRISPR repeats was determined using the PILER-CR program (46). Statistical analyses of the data were performed in R version 2.14.
Mathematical model.
The mathematical model (see the supplemental material for the full algorithm) was programmed in MatLab. To probe multidimensional parameter space, thousands of simulations were run in parallel on NIH’s Helix compute cluster and Harvard Medical School’s Orchestra cluster.
SUPPLEMENTAL MATERIAL
Histograms of CRISPR locus lengths in logarithmic and nonlogarithmic scales. Number of spacers per genome in CRISPR-Cas+ thermophiles (top panels) and CRISPR-Cas+ mesophiles (bottom panels), respectively. Separate histograms are plotted in log10 and linear scales, with the log10 histograms more normally distributed. Since the F test for variance assumes normality, the statistical comparisons of length distributions in the text are computed for log10 of the total number of spacers per CRISPR-Cas+ genome. Download Figure S1, PDF file, 0.1 MB.
CRISPR-Cas evolves only at intermediate innate immunities. Average CRISPR-Cas prevalence (y axis) and average CRISPR-Cas immunity (heatmap) as functions of the probability of innate immunity. Innate immunity is the probability that a host survives viral infection when CRISPR-Cas fails, lacks matching spacers, or is absent from a host. At low levels of innate immunity, a new CRISPR-Cas locus has few immune encounters in which to acquire initial spacers. With a parameterized cost and no immunological benefit, CRISPRCas+ hosts are thus purged from populations. At high levels of innate immunity, there is no need for an extra, costly CRISPR-Cas system, and the system is similarly lost. Only at intermediate innate immunities can CRISPR-Cas evolve. At these intermediate innate immunities, there are sufficient innate immune encounters to acquire spacers, but insufficient innate immune encounters to render CRISPR-Cas unnecessary. Matching these simulations, equation 3 in the text shows that given the CRISPR-Cas cost of 0.5 (see Table S1 in the supplemental material), when the probability of innate immunity exceeds 2/3, even 100% immunogenic CRISPR-Cas systems cannot persist. Download Figure S2, PDF file, 0.1 MB.
The ability of CRISPR-Cas to provide immunity crashes at viral mutation threshold. Average CRISPR-Cas immunity (y axis) and average viral diversity (heatmap) as functions of the viral mutation rate. Increasing the rate of viral mutation decreases the immunological benefit of a CRISPR-Cas system, explaining the loss of CRISPR-Cas at high viral mutation rates in Fig. 3. Of course, increasing viral mutation also increases viral diversity. This increase in viral diversity is especially rapid once CRISPR-Cas is purged from populations. Download Figure S3, PDF file, 0.1 MB.
Time series dynamics of CRISPRCas evolution in three viral mutation regimes. CRISPR-Cas prevalence (y axis) and viral diversity (heatmap) plotted across individual simulations. To better understand the dynamics driving the averaged results of Fig. 3 and Fig. S3 in the supplemental material, full-length simulations are shown for the three basic viral mutation regimes: (i) the low viral mutation regime (Pv_mut = 5 × 10−4), (ii) the intermediate viral mutation regime (Pv_mut = 2 × 10−3), and (iii) the high viral mutation regime (Pv_mut = 10−2). As predicted in Fig. 3 and Fig. S4, at low viral mutation rates, CRISPR-Cas provides complete immunity across time with viral diversity almost entirely depressed. At high viral mutation rates, CRISPR-Cas is similarly absent across time, with viral diversity high. Finally, at an intermediate viral mutation rate, the host population undergoes a rapid phase transition during the simulation, changing from 0% CRISPR-Cas+ to 100% CRISPR-Cas+. After this sweep by CRISPR-Cas+ hosts occurs, viral diversity is rapidly depressed due to CRISPRCas preventing productive virus-host encounters. Download Figure S4, PDF file, 1 MB.
Coexisting populations of CRISPR-Cas+ and CRISPR-Cas− cells are rare. Histogram showing the frequency of CRISPR-Cas in individual model iterations. The prevalence of CRISPR-Cas is tracked across the 60 million individual iterations of the intermediate viral mutation regime in Fig. 3 and Fig S3 in the supplemental material (i.e., all 100,000 iterations of the 600 simulations between Pmut = 0.001 and Pmut = 0.003). Despite CRISPR-Cas’ intermediate prevalence across 100,000 iterations at these viral mutation rates (Fig. 3 and 4), only 0.2% of the individual iterations show a CRISPR-Cas prevalence between 1% and 99%. As suggested in the legend to Fig. S4, intermediate CRISPR-Cas prevalences are thus driven by switching between the quasi-steady states of 0% CRISPR-Cas+ and 100% CRISPR-Cas+, rather than coexisting populations of CRISPR-Cas+ and CRISPR-Cas− cells. Download Figure S5, PDF file, 0.1 MB.
Rapid spacer addition fails to maintain CRISPR-Cas against rapid viral mutation. Average frequency of hosts possessing CRISPR-Cas loci as a function of both the spacer addition rate and the viral mutation rate. Increasing the spacer addition rate fails to maintain CRISPR-Cas at rapid viral mutation rates: hosts are merely adding more and more spacers that are never again encountered as viral protospacers. Download Figure S6, PDF file, 0.1 MB.
Table of parameters used in model.
Supplemental materials and methods. Download Text S1, DOC file, 0.1 MB.
ACKNOWLEDGMENTS
We are grateful to David Kristensen, Sergey Kryazhimskiy, Mateusz Pluciński, Eugene Shakhnovich, and Leor Weinberger for modeling and statistical insights, to Azat Badretdin for help identifying CRISPR-Cas loci in the sequenced genomes and to Chanan Reitblat and Adriana Schulz for expert technical assistance.
We are grateful for the financial support from NIH grant AI072360 (to M.S.G.) and an NIH F32 Postdoctoral Fellowship (to A.D.W.). A.E.L., Y.I.W., and E.V.K. were supported by intramural funding from the U.S. Department of Health and Human Services (National Library of Medicine, NIH). We thank the Kavli Institute of Theoretical Physics (KITP) for hosting a cross-disciplinary workshop at which this collaboration began.
We declare that we have no conflicts of interest.
Footnotes
Citation Weinberger AD, Wolf YI, Lobkovsky AE, Gilmore MS, Koonin EV. 2012. Viral diversity threshold for adaptive immunity in prokaryotes. mBio 3(6):e00456-12. doi:10.1128/mBio.00456-12
REFERENCES
- 1. Darwin C. 1859. On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. John; Murray, London, UK: [PMC free article] [PubMed] [Google Scholar]
- 2. Futuyma D. 2005. Evolution. Sinauer Associates, Sunderland, MA [Google Scholar]
- 3. Lynch M. 2007. The origins of genome architecture. Sinauer Associates, Sunderland, MA [Google Scholar]
- 4. Eyre-Walker A, Keightley PD. 2007. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8:610–618 [DOI] [PubMed] [Google Scholar]
- 5. Carrasco P, de la Iglesia F, Elena SF. 2007. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in tobacco etch virus. J. Virol. 81:12979–12984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peris JB, Davis P, Cuevas JM, Nebot MR, Sanjuán R. 2010. Distribution of fitness effects caused by single-nucleotide substitutions in bacteriophage f1. Genetics 185:603–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Estes S, Phillips PC, Denver DR, Thomas WK, Lynch M. 2004. Mutation accumulation in populations of varying size: the distribution of mutational effects for fitness correlates in Caenorhabditis elegans. Genetics 166:1269–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanjuán R, Moya A, Elena SF. 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. U. S. A. 101:8396–8401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koonin EV, Wolf YI. 2009. Is evolution Darwinian or/and Lamarckian? Biol. Direct 4:42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koonin EV. 2011. The logic of chance: the nature and origin of biological evolution. FT Press, Upper Saddle River, NJ [Google Scholar]
- 11. Weissman A. 1893. The germ-plasm. A theory of heredity. Charles; Scribner: ’s Sons, London, United Kingdom [Google Scholar]
- 12. Wiedenheft B, Sternberg SH, Doudna JA. 2012. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338 [DOI] [PubMed] [Google Scholar]
- 13. Horvath P, Barrangou R. 2010. CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170 [DOI] [PubMed] [Google Scholar]
- 14. Marraffini LA, Sontheimer EJ. 2010. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 11:181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Makarova KS, et al. 2011. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9:467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barrangou R, et al. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712 [DOI] [PubMed] [Google Scholar]
- 17. Marraffini LA, Sontheimer EJ. 2008. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322:1843–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garneau JE, et al. 2010. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67–71 [DOI] [PubMed] [Google Scholar]
- 19. Karginov FV, Hannon GJ. 2010. The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol. Cell 37:7–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brouns SJ, et al. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jore MM, et al. 2011. Structural basis for CRISPR RNA-guided DNA recognition by cascade. Nat. Struct. Mol. Biol. 18:529–536 [DOI] [PubMed] [Google Scholar]
- 22. Wiedenheft B, et al. 2011. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 477:486–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deltcheva E, et al. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. 2010. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329:1355–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Al-Attar S, Westra ER, van der Oost J, Brouns SJ. 2011. Clustered regularly interspaced short palindromic repeats (CRISPRs): the hallmark of an ingenious antiviral defense mechanism in prokaryotes. Biol. Chem. 392:277–289 [DOI] [PubMed] [Google Scholar]
- 26. Westra ER, et al. 2012. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by cascade and Cas3. Mol. Cell 46:595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sashital DG, Wiedenheft B, Doudna JA. 2012. Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol. Cell 46:606–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deveau H, et al. 2008. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 190:1390–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andersson AF, Banfield JF. 2008. Virus population dynamics and acquired virus resistance in natural microbial communities. Science 320:1047–1050 [DOI] [PubMed] [Google Scholar]
- 30. Semenova E, et al. 2011. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U. S. A. 108:10098–10103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heidelberg JF, Nelson WC, Schoenfeld T, Bhaya D. 2009. Germ warfare in a microbial mat community: CRISPRs provide insights into the co-evolution of host and viral genomes. PLoS One 4:e4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Horvath P, et al. 2008. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J. Bacteriol. 190:1401–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weinberger AD, et al. 2012. Persisting viral sequences shape microbial CRISPR-based immunity. PLoS Comput. Biol. 8:e1002475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Edwards RA, Rohwer F. 2005. Viral metagenomics. Nat. Rev. Microbiol. 3:504–510 [DOI] [PubMed] [Google Scholar]
- 35. Labrie SJ, Samson JE, Moineau S. 2010. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8:317–327 [DOI] [PubMed] [Google Scholar]
- 36. Berezovsky IN, Shakhnovich EI. 2005. Physics and evolution of thermophilic adaptation. Proc. Natl. Acad. Sci. U. S. A. 102:12742–12747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zeldovich KB, Berezovsky IN, Shakhnovich EI. 2007. Protein and DNA sequence determinants of thermophilic adaptation. PLoS Comput. Biol. 3:e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zeldovich KB, Chen P, Shakhnovich EI. 2007. Protein stability imposes limits on organism complexity and speed of molecular evolution. Proc. Natl. Acad. Sci. U. S. A. 104:16152–16157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cherry JL. 2010. Highly expressed and slowly evolving proteins share compositional properties with thermophilic proteins. Mol. Biol. Evol. 27:735–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wylie CS, Shakhnovich EI. 2011. A biophysical protein folding model accounts for most mutational fitness effects in viruses. Proc. Natl. Acad. Sci. U. S. A. 108:9916–9921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grogan DW, Carver GT, Drake JW. 2001. Genetic fidelity under harsh conditions: analysis of spontaneous mutation in the thermoacidophilic archaeon Sulfolobus acidocaldarius. Proc. Natl. Acad. Sci. U. S. A. 98:7928–7933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Drake JW. 2009. Avoiding dangerous missense: thermophiles display especially low mutation rates. PLoS Genet. 5:e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mackwan RR, Carver GT, Kissling GE, Drake JW, Grogan DW. 2008. The rate and character of spontaneous mutation in Thermus thermophilus. Genetics 180:17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Friedman R, Drake JW, Hughes AL. 2004. Genome-wide patterns of nucleotide substitution reveal stringent functional constraints on the protein sequences of thermophiles. Genetics 167:1507–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Makarova KS, Wolf YI, Snir S, Koonin EV. 2011. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 193:6039–6056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Edgar RC. 2007. PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8:18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Akaike H. 1974. A new look at the statistical model identification. IEEE Trans. Autom. Contr. 19:716–723 [Google Scholar]
- 48. He J, Deem MW. 2010. Heterogeneous diversity of spacers within CRISPR (clustered regularly interspaced short palindromic repeats). Phys. Rev. Lett. 105:128102 [DOI] [PubMed] [Google Scholar]
- 49. Haerter JO, Trusina A, Sneppen K. 2011. Targeted bacterial immunity buffers phage diversity. J. Virol. 85:10554–10560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Childs LM, Held NL, Young MJ, Whitaker RJ, Weitz JS. 2012. Multiscale model of CRISPR-induced coevolutionary dynamics: diversification at the interface of Lamrack and Darwin. Evolution 66:2015–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilson GG, Murray NE. 1991. Restriction and modification systems. Annu. Rev. Genet. 25:585–627 [DOI] [PubMed] [Google Scholar]
- 52. Marraffini LA, Sontheimer EJ. 2010. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463:568–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stern A, Keren L, Wurtzel O, Amitai G, Sorek R. 2010. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 26:335–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Palmer KL, Kos VN, Gilmore MS. 2010. Horizontal gene transfer and the genomics of enterococcal antibiotic resistance. Curr. Opin. Microbiol. 13:632–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Palmer KL, Gilmore MS. 2010. Multidrug-resistant enterococci lack CRISPR-cas. mBio 1(4):e00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hill TC, Walsh KA, Harris JA, Moffett BF. 2003. Using ecological diversity measures with bacterial communities. FEMS Microbiol. Ecol. 43:1–11 [DOI] [PubMed] [Google Scholar]
- 57. Amitai G, Sorek R. Roles of CRISPR in regulation of physiological processes. In Barrangou R, van der Oost J, CRISPR-Cas systems: RNA-mediated adaptive immunity in Bacteria and Archaea, in press Springer, Berlin, Germany [Google Scholar]
- 58. Jorth P, Whiteley M. 2012. An evolutionary link between natural transformation and CRISPR adaptive immunity. mBio 3(5):e00309-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Aravind L, Tatusov RL, Wolf YI, Walker DR, Koonin EV. 1998. Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet. 14:442–444 [DOI] [PubMed] [Google Scholar]
- 60. Drake JW, Charlesworth B, Charlesworth D, Crow JF. 1998. Rates of spontaneous mutation. Genetics 148:1667–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kussell E, Leibler S. 2005. Phenotypic diversity, population growth, and information in fluctuating environments. Science 309:2075–2078 [DOI] [PubMed] [Google Scholar]
- 62. Kuo CH, Ochman H. 2009. Deletional bias across the three domains of life. Genome Biol. Evol. 1:145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histograms of CRISPR locus lengths in logarithmic and nonlogarithmic scales. Number of spacers per genome in CRISPR-Cas+ thermophiles (top panels) and CRISPR-Cas+ mesophiles (bottom panels), respectively. Separate histograms are plotted in log10 and linear scales, with the log10 histograms more normally distributed. Since the F test for variance assumes normality, the statistical comparisons of length distributions in the text are computed for log10 of the total number of spacers per CRISPR-Cas+ genome. Download Figure S1, PDF file, 0.1 MB.
CRISPR-Cas evolves only at intermediate innate immunities. Average CRISPR-Cas prevalence (y axis) and average CRISPR-Cas immunity (heatmap) as functions of the probability of innate immunity. Innate immunity is the probability that a host survives viral infection when CRISPR-Cas fails, lacks matching spacers, or is absent from a host. At low levels of innate immunity, a new CRISPR-Cas locus has few immune encounters in which to acquire initial spacers. With a parameterized cost and no immunological benefit, CRISPRCas+ hosts are thus purged from populations. At high levels of innate immunity, there is no need for an extra, costly CRISPR-Cas system, and the system is similarly lost. Only at intermediate innate immunities can CRISPR-Cas evolve. At these intermediate innate immunities, there are sufficient innate immune encounters to acquire spacers, but insufficient innate immune encounters to render CRISPR-Cas unnecessary. Matching these simulations, equation 3 in the text shows that given the CRISPR-Cas cost of 0.5 (see Table S1 in the supplemental material), when the probability of innate immunity exceeds 2/3, even 100% immunogenic CRISPR-Cas systems cannot persist. Download Figure S2, PDF file, 0.1 MB.
The ability of CRISPR-Cas to provide immunity crashes at viral mutation threshold. Average CRISPR-Cas immunity (y axis) and average viral diversity (heatmap) as functions of the viral mutation rate. Increasing the rate of viral mutation decreases the immunological benefit of a CRISPR-Cas system, explaining the loss of CRISPR-Cas at high viral mutation rates in Fig. 3. Of course, increasing viral mutation also increases viral diversity. This increase in viral diversity is especially rapid once CRISPR-Cas is purged from populations. Download Figure S3, PDF file, 0.1 MB.
Time series dynamics of CRISPRCas evolution in three viral mutation regimes. CRISPR-Cas prevalence (y axis) and viral diversity (heatmap) plotted across individual simulations. To better understand the dynamics driving the averaged results of Fig. 3 and Fig. S3 in the supplemental material, full-length simulations are shown for the three basic viral mutation regimes: (i) the low viral mutation regime (Pv_mut = 5 × 10−4), (ii) the intermediate viral mutation regime (Pv_mut = 2 × 10−3), and (iii) the high viral mutation regime (Pv_mut = 10−2). As predicted in Fig. 3 and Fig. S4, at low viral mutation rates, CRISPR-Cas provides complete immunity across time with viral diversity almost entirely depressed. At high viral mutation rates, CRISPR-Cas is similarly absent across time, with viral diversity high. Finally, at an intermediate viral mutation rate, the host population undergoes a rapid phase transition during the simulation, changing from 0% CRISPR-Cas+ to 100% CRISPR-Cas+. After this sweep by CRISPR-Cas+ hosts occurs, viral diversity is rapidly depressed due to CRISPRCas preventing productive virus-host encounters. Download Figure S4, PDF file, 1 MB.
Coexisting populations of CRISPR-Cas+ and CRISPR-Cas− cells are rare. Histogram showing the frequency of CRISPR-Cas in individual model iterations. The prevalence of CRISPR-Cas is tracked across the 60 million individual iterations of the intermediate viral mutation regime in Fig. 3 and Fig S3 in the supplemental material (i.e., all 100,000 iterations of the 600 simulations between Pmut = 0.001 and Pmut = 0.003). Despite CRISPR-Cas’ intermediate prevalence across 100,000 iterations at these viral mutation rates (Fig. 3 and 4), only 0.2% of the individual iterations show a CRISPR-Cas prevalence between 1% and 99%. As suggested in the legend to Fig. S4, intermediate CRISPR-Cas prevalences are thus driven by switching between the quasi-steady states of 0% CRISPR-Cas+ and 100% CRISPR-Cas+, rather than coexisting populations of CRISPR-Cas+ and CRISPR-Cas− cells. Download Figure S5, PDF file, 0.1 MB.
Rapid spacer addition fails to maintain CRISPR-Cas against rapid viral mutation. Average frequency of hosts possessing CRISPR-Cas loci as a function of both the spacer addition rate and the viral mutation rate. Increasing the spacer addition rate fails to maintain CRISPR-Cas at rapid viral mutation rates: hosts are merely adding more and more spacers that are never again encountered as viral protospacers. Download Figure S6, PDF file, 0.1 MB.
Table of parameters used in model.
Supplemental materials and methods. Download Text S1, DOC file, 0.1 MB.