

Abstract
Infection with human papilloma virus (HPV) is almost universal and eventually asymptomatic, but pathologic infection with HPV is severe, recurrent, and recalcitrant to therapy. It is also an underappreciated manifestation of primary immunodeficiency. Mutations in EVER1, EVER2, GATA2, CXCR4, and DOCK8 are typically associated with extensive HPV infections, whereas several other primary immune defects have severe HPV much less frequently. We review immunodeficiencies with severe HPV infections and the mechanisms underlying them.
Keywords: Immunodeficiency, human papilloma virus, HPV, warts, squamous, carcinoma, dysplastic
Introduction
Warts are benign virus-induced tumors caused by human papilloma virus (HPV) that are common in the general population at some point in life. There are more than 100 different genotypes of HPV classified according to their tissue tropism (mucosal or cutaneous) and oncogenic potential. Along with host and environmental factors, HPV genotype influences the type and malignant potential of lesions1. The prevalence of cutaneous viral warts in children and adolescents is between 3-5%2, 3 occurring with similar frequency in adults aged 25-34 years4. In 90% of children, warts present by age 11 and clear by 16 years1. Other rates of spontaneous clearance have also been reported: 23% at 2 months5, 6 and 66% by 2 years6. There is no exact definition for recalcitrant cutaneous warts but an accepted rule is failure to respond after five treatments over a period of 6 months7. Mucosal HPV is much more common, with up to 79% of sexually active women acquiring genital HPV during their lifetime, ranging from no phenotype, to koilocytosis, to genital warts, to high grade intraepithelial neoplasms. Spontaneous regression occurs in 30% of women within 4 months8, and median time to clearance with treatment is 6 months9. HPV infection is the most frequent cause of cervical cancer in women but also induces cancer of the vagina, vulva, anus, and penis. HPV has been linked to oral squamous cell carcinomas as well10.
Host defense against HPV relies on intact and functioning cellular immunity including T cell and natural killer (NK) cell cytotoxicity. Therefore, in patients in whom warts are severe or recalcitrant, concern for immune defects is raised. Here we review the frequency, severity, and clinical importance of warts in primary immunodeficiencies. We will discuss separately the syndromes in which HPV is a manifestation in the majority of cases and those syndromes in which HPV is less consistently recognized as a severe manifestation of immunodeficiency.
Epidermodysplasia Verruciformis (EV): EVER1 and EVER2 deficiency
EV is a rare genodermatosis characterized by increased susceptibility to cutaneous HPV. First described by Lewandowsky and Lutz in 192211, EV typically presents in infancy, and has two main phenotypes differentiated by their potential for malignant transformation12. The more benign lesions typically occur on the trunk, neck and extremities as flat, wart-like, hypo- or hyper-pigmented papules that may coalesce as scaly patches or plaques with irregular borders. Lesions with greater malignant potential present as verrucous or seborrheic keratosis-like lesions that occur more frequently on sun exposed surfaces12. The HPV’s that cause disease in EV are beta papillomaviruses, which typically cause asymptomatic infections in the general population. At least 19 different beta HPV genotypes have been found in patients with EV and patients usually are infected with more than 1 genotype. HPV-5 is the most frequent genotype in EV patients followed by HPV-3 and HPV-10 13. HPV-5 is also the most common genotype to undergo malignant conversion. EV specific HPV’s are acquired early in infancy 14 and are prevalent in the normal skin of healthy adults 15. Symptomatic infections and malignant transformation may occur in patients with EV or those with other immunodeficiencies.
Although the immunophenotype of EV may be normal, it may include decreased total T lymphocyte counts16, reduced cell mediated immunity as measured by reduced responsiveness to mitogens and antigens as well as cutaneous anergy to recall antigens16-22, and defective cell mediated immunity toward EV-specific HPV types23. Reduced cell mediated immunity may be related to the length of infection with EV specific HPV’s17, 18 and is independent of oncologic potential18-20, 22. NK cell cytotoxic responses are increased in EV16, 24, 25, primarily in patients with early premalignancies or malignancies, but are reduced in disease induced by EV associated HPV-3 displaying a specific reduction of NK cell mediated cytotoxicity against disease specific target cells25. Hypoproductive polymorphisms of the interleukin (IL)-10 gene promoter have also been linked to EV. Low IL-10 levels allow for higher production of inflammatory cytokines and may contribute to HPV persistence by inhibiting infected Langerhan’s cells from migrating to regional lymph nodes26.
EV is historically inherited in an autosomal recessive fashion, but X-linked27 and autosomal dominant28 inheritance patterns have been reported. Homozygous mutations in EVER1 or EVER2 have been reported in ~75% of patients with EV13. The EVER genes are predicted to encode highly conserved transmembrane proteins29 that are important regulators of zinc homeostasis30. EVER proteins are expressed in T and B lymphocytes, NK cells, endothelial cells, myeloid cells, and dendritic cells29. Products of EVER1 and EVER2 act as dominant restriction factors for HPV. Loss of EVER zinc homeostasis enhances expression of viral genes, specifically the pro-oncogenic E6 and E7, contributing to HPV mediated carcinogenesis30.
EV lesions are refractory to conventional therapies. Non-surgical interventions with topical 5-fluorouracil31, 5% imiquimod32, tacalcitol33, systemic retinoids combined with interferon (IFN)α34, cimetidine35, and topical 5-aminolevulinic acid photodynamic therapy36 yield inconsistent results. Approximately one third of patients go on to develop malignancy with an average of 24 years between development of benign lesions and cancer37. Invasive skin cancers are typically squamous cell carcinomas that often retain features of Bowen’s carcinomas. They develop slowly and are locally destructive38.
Warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) Syndrome: CXCR4 deficiency
This rare autosomal dominant immunodeficiency was described by Zuelzer39 and Krill et al 40 in 1964 in a 10 year old girl with congenital neutropenia and recurrent infections. WHIM is characterized by recurrent bacterial infections in infancy and early childhood, most commonly pulmonary, gastrointestinal, and cutaneous. Kawai and Malech pooled information from 37 published cases41. At first presentation, 78.6% had warts, 89.6% had hypogammaglobulinemia, and 91.7% had neutropenia. Specific clinical features and age at diagnosis were variable. All patients had recurrent bacterial infections in childhood including pneumonias, sinusitis, cellulitis, urinary tract infections, thrombophlebitis, omphalitis, osteomyelitis, deep soft tissue abscesses, and skin infections. Common pathogens include Haemophilus influenzae, Streptococcus pneumoniae, Klebsiella pneumoniae, Staphylococcus pneumoniae, and Proteus mirabilis. Infections typically respond well to antibiotics and run a benign course. However, recurrent pneumonias can result in bronchiectasis and lead to chronic infection with Pseudomonas aeruginosa and Burkholderia cepacia41.
HPV related disease is the predominant feature. Numerous cutaneous warts occur on the feet, hands, and trunk; genital condyloma accuminata with papillomatosis may become dysplastic or neoplastic. There are also reports of severe recurrent oral and genital herpes 42, and herpes zoster 43 as well as two reports of patients developing Epstein Barr virus (EBV) -related lymphoproliferative disorders, indicating susceptibility to herpes family viruses as well44, 45. HPV related carcinomas are a significant cause of morbidity with lesions of mucosal or transitional areas of the skin at the highest risk of dysplasia. Frequent monitoring with biopsies or surgical removal is often required 41.
The primary laboratory abnormality is neutropenia with bone marrow hypercellularity due to a defect in the release of mature neutrophils41, 46. Myeloid hypercellularity with cytoplasmic vacuolation, chromatin hypercondensation, and hypersegmented pyknotic nuclei all contribute to the pattern referred to as myelokathexis 47. Despite severe neutropenia, patients are not at risk for overwhelming sepsis, presumably because functional neutrophils are released from the bone marrow during stress. Other laboratory abnormalities include lymphopenia of CD27+ memory B cells as well as both unswitched immunoglobulin D (IgD+) and switched (IgD-) B lymphocytes 48. Hypogammaglobulinemia primarily involves IgG but can also affect IgM, but responses to immunization demonstrate that the defect in humoral immunity is incomplete. T lymphopenia with normal CD4/CD8 ratio and normal proliferative responses to mitogens occurs as well48.
WHIM is caused by dominant heterozygous gain-of-function mutations in the chemokine receptor, CXCR4. Four mutations have been described: 1 frameshift and 3 nonsense mutations, which result in truncation of the cytoplasmic C-terminal portion of the receptor49. CXCR4 is a G-protein coupled receptor that selectively binds stromal cell derived factor – 1 (SDF-1, or CXCL12). Mutations in CXCR4 impair SDF-1 mediated signaling due to absence of intracellular phosphorylation sites on the receptor.43 Leukocytes expressing the truncated CXCR4 demonstrate enhanced chemotactic responses to SDF-1 which likely impairs their trafficking 43, 50. The C-terminal portion of the receptor contains canonical phosphorylation sites that are targets of G protein coupled receptor kinases. Phosphorylation results in binding of β -arrestins to CXCR4 and interaction with C-terminal sequences that result in endocytic internalization of the receptor and desensitization to ligand stimulation 50. Two cases of WHIM have no mutation identified so far 43, 51. In the patients lacking specific mutations, a marked decrease in G-protein coupled receptor kinase 3 (GRK3) has been described. Overexpression of GRK3 restored ligand-mediated internalization of CXCR4 in response to SDF-1 to normal levels, showing that CXCR4 hyperactivity is the common biochemical feature in all affected patients. SDF-1 and CXCR4 are expressed in normal Langerhans’ cells and keratinocytes52 and increased levels of SDF-1 are expressed in HPV infected dermis43. These observations suggest that host susceptibility to HPV infection is mediated by upregulation of CXCR4. Defects in number and function of myeloid and, more impressively, plasmacytoid dendritic cells, have also been demonstrated, likely enhancing susceptibility to HPV53.
Early diagnosis and aggressive measures to decrease bacterial infections improves prognosis. Treatment with granulocyte colony stimulating factor (G-CSF), granulocyte macrophage colony stimulating factor (GM-CSF), intravenous immunoglobulin (IVIg), and prophylactic antibiotics provide benefit41, 54, 55. The mechanism of action of G-CSF involves a positive feedback loop that increases neutrophil counts and enhances release of neutrophil elastase. Neutrophil elastase cleaves SDF-1 and CXCR4, reducing its activity and releasing mature neutrophils from the bone marrow into the peripheral blood54, 55. Therapy with plerixafor, a CXCR4 antagonist, has shown promising results, reversing neutropenia in 3 WHIM patients56.
Autosomal Recessive Hyper IgE Syndrome (DOCK8 Deficiency)
Mutations in dedicator of cytokinesis 8 (DOCK8) cause an autosomal recessive combined immunodeficiency with hyper IgE. Some features are in common with STAT3 mutated hyper IgE (Job’s syndrome), such as atopic dermatitis, Staphylococcus aureus skin abscesses and soft tissue infections, pneumonias, elevated serum IgE and eosinophilia. However, a distinguishing feature of DOCK8 deficiency is susceptibility to cutaneous viral infections, most commonly herpes simplex virus (HSV), HPV, molluscum contagiosum virus (MCV), and varicella zoster virus (VZV). These infections are extensive, can be disfiguring, occur concurrently, and are difficult to control. Examples include chronic orolabial or anogenital infections, herpes simplex keratitis, and eczema herpeticum. HPV presents as flat and verrucous warts. In 21 patients followed at the National Institutes of Health (NIH), 62% had histories of significant warts 57. An increased incidence of genital warts has not been reported and it is unclear whether these patients are at increased risk for them. Molluscum lesions are often confluent and can be disfiguring as well57-59.
DOCK8 deficiency is associated with recurrent upper and lower respiratory tract infections, sinusitis, otitis media, mastoiditis, and pneumonia from a wide spectrum of Gram positive and Gram negative bacteria as well as intracellular fungi, such as Histoplasma capsulatum. Mucocutaneous candidiasis and recurrent infections of the gastrointestinal tract are common. DOCK8 deficiency is highly associated with the development of malignancies in childhood or young adulthood, including squamous cell carcinomas, Burkitt’s and other lymphomas, and T cell leukemia 58, 59.
The lymphopenia of DOCK8 deficiency progresses with age, predominantly affecting T cells, especially CD4 T cells, and to a lesser extent NK cells and B cells. CD8 T cells from DOCK8 deficient patients fail to activate, divide, and expand following T cell receptor stimulation and have poor production of IFNγ and tumor necrosis factor (TNF)α 60. Impaired functioning of CD8 T cells likely contributes to the viral susceptibility of DOCK8 deficiency61. Decreased CD4 T helper type 17 cells (Th17) may explain the increased susceptibility of some DOCK8 deficient patients to mucocutaneous candidiasis 62. Besides their elevated IgE, DOCK8 deficient patients have low IgM, high or normal IgG, and low, high, or normal IgA. Antibody responses to previously encountered protein antigens and polysaccharides are variable60.
Warts are extremely difficult to eradicate; imiquimod, cidofovir, and other conventional treatments are usually unsuccessful. Eczema is frequently exacerbated by Staphylococcus aureus. Reduction of colonization of the skin with bleach baths is recommended, as is the use of topical corticosteroids, although the latter may exacerbate cutaneous viral infections. Patients with antibody deficiency may benefit from IVIg 63. Mortality due to malignancy and infections is high. Hematopoietic stem cell transplant (HSCT) has led to immune reconstitution and viral eradication 64. However, DOCK8’s role in non-hematopoietic tissues and the ability of HSCT to prevent malignancies is undefined.
Idiopathic CD4 Lymphopenia
Idiopathic CD4 lymphopenia (ICL) was first defined by the Centers for Disease Control in 1992 as a disease with “a documented absolute CD4 T lymphocyte count less than 300 cells/mm3 or of less than 20% on more than one occasion, no evidence of infection on HIV testing, and the absence of any defined immunodeficiency or therapy65.” ICL is a heterogeneous disorder that is usually detected after an opportunistic infection in a person without a known immunodeficiency. Cryptococcosis, HPV, and nontuberculous mycobacterial infection were the three most common infectious presentations in a cohort of ICL patients followed at the NIH from 1992-200666. ICL typically presents in the fifth decade. Less common infections included histoplasmosis, mucosal candidiasis, VZV, and cytomegalovirus (CMV). Pneumocystis jirovecii pneumonia is uncommon.
Warts in ICL typically present as disseminated verrucae or flat warts on the extremities, face, and genitals. A few reports have found HPV types -2, -3, -6, and -49 in cutaneous lesions67-71. HPV related dysplasia and carcinoma can be the presentation of ICL, as can juvenile laryngeal papillomatosis 72 and recurrent vulvar intraepithelial neoplasia 66. Some patients do well with topical treatments and destructive therapies, whereas others require systemic immunomodulators, such as IFNα; some may require radical surgery. Of 40 ICL patients currently followed at the NIH, 15 have had cutaneous or genital HPV. Five of the nine patients with genital HPV have developed dysplasia or carcinoma. One patient with cutaneous HPV has developed cutaneous squamous cell cancer.
Autoimmune phenomena and autoantibodies occur frequently, with thyroid disorders and anti-nuclear antigens being the most common. Systemic lupus erythematosus, antiphospholipid antibody syndrome, autoimmune hemolytic anemia, ulcerative colitis, psoriasis, and vitiligo also occur66. The temporal relationship of autoimmune disease to ICL and the exact nature of this correlation are not known.
The majority of patients have an inverted CD4/CD8 ratio, but a normal CD4/CD8 ratio is found in some with concomitant depletion of CD8 T lymphocytes. Low absolute B lymphocyte counts with normal immunoglobulin production and response to vaccination and low absolute NK cells counts are common. T cells are more activated (human leukocyte antigen [HLA]-DR+), have increased CD4 T cell turnover as measured by Ki-67, higher percentages but lower absolute number of regulatory T cells as measured by forkhead box p3 (Foxp3) expression, and a lower proportion of naïve CD4 T cells as measured by expression of CD127 (the alpha chain of the IL-7 receptor, IL-7Rα) 66. CD4 autoantibodies have been hypothesized and reported in at least one patient 73.
Despite the high rate of opportunistic infections and HPV, the overall prognosis of ICL is quite good. From the cohort followed from 1992-2006, there were 7 deaths, 4 related to opportunistic infections; there have been no deaths in the cohort followed currently. Three patients had pulmonary Mycobacterium avium complex (MAC), progressive multifocal leukoencephalopathy, and EBV-B cell lymphoma, respectively. One patient with severe genital HPV died of metastatic squamous cell carcinoma 42 months after initial diagnosis. All 4 deaths were in patients who had concurrent CD8 lymphopenia. The three non-opportunistic infection related deaths were due to pneumonia and metastatic prostate cancer (age 84 years). There were no deaths in the group that had no opportunistic infections and were incidentally found to have ICL 66.
GATA2 Deficiency
Deficiency in GATA2, an important transcription factor involved in hematopoiesis and maintenance of the stem cell compartment 74, 75, leads to a syndrome characterized by opportunistic infections and myelodysplasia or leukemia 76-78. GATA2 deficiency has a variety of presentations including MonoMAC (monocytopenia and M. avium complex infection) 78, familial myelodysplastic and leukemia syndrome 77, DCML (dendritic cell, monocyte, B cell, and NK cell lymphoid deficiency) 76, and Emberger syndrome (a primary lymphedema with myelodysplasia or acute myelogenous leukemia) 79. Missense and null mutations in GATA2 have been found with all four phenotypes, suggesting that the mechanism of disease is haploinsufficiency 77, 80-82. This syndrome typically presents in later childhood or adulthood with profound circulating monocytopenia and NK, B cell, and dendritic cell lymphocytopenia 76, 78. T cell counts and function are variable. Patients are susceptible to nontuberculous mycobacteria, HPV, Histoplasma, Cryptococcus and Aspergillus. In the largest series, HPV occurred in more than 75%, followed by herpes family viruses (HSV, VZV, and EBV) 78. Other features include pulmonary alveolar proteinosis, panniculitis, pulmonary hypertension, thromboembolic phenomena, and recurrent miscarriages. Hypotelorism, epicanthal folds, webbed neck, long tapering fingers, and high frequency sensorineural deafness were also noted in patients described as having Emberger syndrome 83-86. Despite specific cytopenias described, macrophages and plasma cells are seen at sites of inflammation, and patients produce normal quantities of immunoglobulin 78.
Generalized warts occur frequently and can be severe leading to dysplasia and neoplasia. In 18 patients from 5 families with GATA2 deficiency followed at the NIH, 14 had severe or disseminated HPV; in most it was the first manifestation of disease. Warts typically presented in adolescence or early adulthood as verruca plana or vulgaris on the face or extremities and genital condylomata. Severe genital HPV commonly led to cervical dysplasia and carcinoma 78.
The high rate of myelodysplasia and leukemias contributes to the high mortality rate in patients with GATA2 deficiency78, 87, 88. Abnormal cytogenetics are common. Allogeneic hematopoietic stem cell transplantation seems to be curative. 87. Graft versus host disease (GVHD) has occurred both early and late after transplantation but was usually manageable.
WILD Syndrome
Warts, immunodeficiency, lymphedema, and dysplasia (WILD) syndrome is described in two cases as a combined immunodeficiency with disseminated warts, lymphedema, and anogenital dysplasias89, 90. Both cases had lower extremity lymphedema presenting at 6 months that progressed to involve the entire lower and upper extremities and groin. Warts began in adolescence with eventual development of anogenital dysplasia or cancer. Both patients had B and T cell lymphopenia and depressed mitogen stimulated lymphocyte proliferation. This may well represent another manifestation of GATA2 deficiency.
Netherton Syndrome (SPINK5 deficiency)
Netherton syndrome is a rare autosomal recessive disorder characterized by congenital icthyosiform erythroderma, tricorrexis invaginata (bamboo hair), and atopy, food allergies and asthma. Chronic inflammation of the skin leads to dehydration, life threatening infections, and sepsis, leading to a high mortality rate in the first year of life 91,92.
Patients with Netherton syndrome have increased susceptibility to viral skin infections, specifically HSV and HPV, although there are few published reports of the latter 92, 95-98. The Netherton syndrome has high IgE, normal to elevated IgG, hypercomplentemia (C3 and C4), and NK cell lymphocytopenia 92. Interestingly, IgE levels are normal at birth, reach their peak by age 4, and then decrease over time. CD4 lymphopenia, impaired skin tests to microbial antigens, and impaired lymphocyte proliferation to mitogens have also been reported 97, 99. Susceptibility to cutaneous and systemic infections may reflect the proteolytic breakdown of antimicrobial peptides, specifically the cathelicidin carboxy fragment, LL-37.
Serine protease inhibitor Kazal-type 5 (SPINK5) encodes the serine protease inhibitor, lympho-epithelial Kazal-type-related inhibitor (LEKTI), which is normally cleaved into 15 active domains that suppress serine protease activity93. SPINK5 mutations truncate LEKTI and permit unopposed serine protease activity. There is a strong genotype-phenotype correlation with the length of he residual LEKTI molecule 94.
Warts in Netherton syndrome typically present in adolescence or adulthood as common genitoanal, plane and verrucous lesions. Depending on the HPV type, they may progress to carcinoma. EV-associated HPV types have been found on biopsy and skin cancers occur in these patients 97. Lesions are typically recalcitrant to therapy but there has been some success with use of retinoids, psoralen plus ultraviolet A (PUVA) therapy, and IFNα 92, 95-98.
MST1 (MST1 or STK4 deficiency)
Homozygous mutations in the serine-threonine kinase 4 (STK4) gene cause a combined immunodeficiency that is primarily characterized by a dramatically reduced amount and survival of circulating naïve T cells. Missense mutations and deletions in STK4 in 4 patients from two consanguineous unrelated families from Turkey100 and in 3 patients from a consanguineous Iranian family101 result in truncation of the STK4 protein, previously named Mammalian sterile 20-like protein (MST1).
Patients with STK4 deficiency suffer from recurrent bacterial skin and respiratory tract infections; recurrent cutaneous viral infections with HSV, VZV, and MCV are common. Persistent EBV viremia is common and can lead to EBV associated lymphoproliferative syndrome and EBV related malignancies. Chronic dermatitis resembling eczema has been described in 3 patients. Autoantibodies and autoimmune hemolytic disease have occurred in 2. All 3 patients in the Iranian family, and 1 in the Turkish cohort had structural heart disease including atrial septal defect; patent foramen ovale; mitral, tricuspid, and pulmonary insufficiency; and right ventricular hypertrophy100, 101.
Cutaneous warts were described in all 3 patients from the Iranian family. Histologic analysis showed orthokeratosis and epithelial hyperplasia secondary to HPV-57 and -84 and HPV -71, -3, and -25 in 2101.
Progressive CD4 T cell lymphopenia with profoundly low naïve CD4 T cell counts is hallmark while CD8 T cells and NK cells are within normal range. T cell proliferation responses to both antigens and mitogens are markedly impaired. B cell counts are mildly low with hypergammaglobulinemia of IgG and variable increases in IgA and IgE. Antibody responses are defective. Persistent neutropenia occurs with normal maturation of neutrophils in the bone marrow 100, 101.
STK4 is involved in several pathways controlling T cell survival and death and its activity can be regulated by caspase-induced cleavage. STK4 is necessary for activation of forkhead box protein O1 (FOXO1), an important transcription factor in T cell homeostasis. Loss of STK4 results in significantly lower expression of FOXO1. Decreased expression of FOXO1 leads to decreased expression of IL-7Rα, CCR7, and CD62 ligand (CD62L) on naïve T cells, possibly impairing T cell homing100, 101. STK4 deficient T cells have enhanced apoptosis mediated through increased Fas expression, decreased B cell lymphoma 2 (BCL-2) expression100, and loss of mitochondrial transmembrane potential101. STK4 deficiency may also impair development or maintenance of T regulatory cells with decreased absolute numbers described in these patients100.
Therapy relies on treatment of the underlying infections. EBV related diseases were intermittently controlled with anti CD20 therapy. HSCT was curative in 1 patient; 2 died within 6 months due to GVHD100.
NEMO (NEMO or IKBKG deficiency)
Hypomorphic mutations in the nuclear factor (NF)κB essential modulator (NEMO or IKBKG) gene cause a combined immunodeficiency with early susceptibility to pyogenic and mycobacterial infections. NEMO is a 419 amino acid regulatory protein encoded by 10 exons on the X chromosome that is critical for NFκB activation. After specific receptor activation, the inhibitor of κB (Iκb) kinase complex (IKK) phosphorylates IκB, leading to its degradation and the release of NFκB, which activates the transcription of various genes 102. Amorphic NEMO mutations are lethal in boys and cause an ectodermal phenotype known as incontinentia pigmenti in girls, characterized by dermal scarring and skin hyperpigmentation. Hypomorphic mutations in boys can result in immunodeficiency with or without ectodermal dysplasia. The ectodermal dysplasia is characterized by dental abnormalities, eccrine sweat gland dysgenesis, and fine sparse hair 103 because of impaired ectodysplasin A signaling via NFκB.
Boys with defects in NEMO are susceptible to bacterial, viral, pneumocystis and mycobacterial infections starting within the first year of life. They can present with pneumonia, bacteremia, skin and soft tissue abscesses, enteritis or colitis, encephalitis or meningitis, sinusitis, or osteomyelitis. Approximately 20% of patients develop HSV, CMV or MCV. However, patients are also susceptible to severe disseminated HPV, typically flat warts104. Inflammatory colitis is also common 105.
The immunodeficiency of NEMO can include hypogammaglobulinemia with elevated IgM and IgA due to impaired CD40-mediated B cell class switch recombination. Toll-like receptor (TLR) signaling and NK cell cytotoxicity are diminished as well. T cell numbers and proliferation to mitogens tend to be preserved. In the patients with HPV infection, CD4 lymphopenia was common with decreased NK cell cytotoxicity in one patient 105.
Treatment of patients with defects in NEMO relies on prevention and aggressive treatment of infections. Replacement IVIg and antimicrobial prophylaxis are recommended.
Severe Combined Immunodeficiency
Severe Combined Immunodeficiency (SCID), characterized by absence of T cells and profoundly impaired adaptive immunity, has an incidence of 1 in 40,000-75,000 live births. Glanzmann and Riniker first described SCID in 1950 in Swiss infants who were profoundly lymphopenic and died of infection within the first two years of life 106. Adenosine deaminase (ADA) deficiency was recognized in 1972 as the first molecular cause of SCID 107. Now, mutations in more than 30 different genes have been described as causes of SCID108.
SCID classically presents in infancy with recurrent severe infections, chronic diarrhea, and failure to thrive. Persistent mucocutaneous candidiasis, adenovirus, CMV, EBV, rotavirus, respiratory syncitial virus, VZV, HSV, measles, influenza, and parainfluenza 3, Pneumocystis jirovecii, and vaccine related infections with polio, rotavirus, varicella and Bacillus Calmette-Guerin are common and can be fatal. Transplacental passage of alloreactive maternal T cells or transfusion of blood products containing viable lymphocytes can cause GVHD109.
Typical laboratory abnormalities include severe T cell lymphopenia, abnormal lymphocyte subpopulations, absent or very low T cell proliferative responses, and hypogammaglobulinemia. However, absolute lymphocyte counts may be normal in the setting of maternal T cell engraftment and hypogammaglobulinemia may be masked by transferred maternal IgG in infants less than 6 months.
Widespread seborrhea-like dermatitis, morbilliform eruptions, and extensive eczema are common in SCID. However, there are a few cases of warts as the presenting cutaneous sign of SCID. Three adults with lifelong histories of frequent sinopulmonary infections and mucosal candidiasis developed warts as adults and were found to have ADA deficiency 110-112. Presumably, the defect in cellular immunity caused their HPV susceptibility. However, ADA is present in somatic cells in addition to the myeloid compartment and ADA itself may play a role in prevention of HPV. Widespread cutaneous and plantar warts have also been described in patients with IL-2 receptor common gamma chain (γc) and Janus kinase 3 (JAK3) deficiencies following hematopoietic stem cell transplantation 113, 114. Laffort et al found warts a mean of 8 years after HSCT, primarily on the hands and feet, but no genital or pre-malignant lesions were seen. HPV genotypes associated with EV (HPV-5, -14, -36), common warts (HPV-2 and -57) and flat warts (HPV-3) were identified in the lesions of 6 patients. Types HPV-5 and -14 are potentially oncogenic and were noted only in patients with EV-like lesions. Chimerism patterns after transplantation were similar in patients with mutations in γc-deficient and JAK3- deficient SCID, regardless of HPV status. T and NK cells were of donor origin in all patients tested and B cells were of donor origin in 2/7 patients tested with HPV disease and 1/8 without HPV. There was no difference in NK cell number between the groups with and without HPV. One patient had complete resolution with a combination of laser therapy, acitretin, and topical imiquimod. All other patients’ lesions were recalcitrant to therapy. The etiology for increased susceptibility to warts after HSCT in patients with IL2Rγc/JAK3 mutations is unknown, but IL2Rγc-dependent cytokines may induce production or amplification of immune responsive molecules in keratinocytes. 113
CVID
Common variable immunodeficiency (CVID) is the most prevalent immunodeficiency, affecting 1 in 25,000 – 1 in 50,000 white patients, but a single unifying genetic etiology is still elusive. Most patients are diagnosed at 20-40 years but both children and adults have been described. CVID is heterogeneous with a broad range of clinical manifestations including chronic infections, inflammatory and autoimmune disease, and increased cancer and lymphoma. CVID is characterized by low IgG, IgA, and/or IgM with poor or absent antibody production115.
Pneumonias due to Streptococcus pneumoniae, Haempohilus influenzae or Mycoplasma species are common before diagnosis and treatment are initiated. Warts are uncommon in CVID, but severe disseminated warts are reported. Three cases with ages of wart onset of 9, 13, and 55 were associated with T-lymphopenia and impaired T cell response to mitogens116, 117. One had intestinal lymphangiectasia as well 118. An 18-year-old man with CVID had normal T cell numbers but decreased T cell proliferation to antigen and multiple warts on his arms, hands, and feet. His lesions completely resolved after treatment with subcutaneous immunoglobulin replacement 119. T cell deficiency likely contributes to susceptibility to warts in CVID. These cases may represent other recognized genetic defects masquerading as CVID.
LAD-1 (CD18 deficiency)
Leukocyte adhesion deficiency type-1 (LAD-1) was described in the 1970s 120 as an autosomal recessive disease, eventually recognized as caused by mutations in integrin B2 (ITGB2), encoding the common beta chain of the beta 2 integrin family (CD18) 120-122. Mutations resulting in dramatically reduced or nonfunctional forms of CD18 also prevent surface expression of alpha subunits of the three heterodimers A1/B2 (lymphocyte function-associated 1 [LFA-1], CD11a), A2/B2 (macrophage-1 antigen [Mac-1], CD11b), and A3/B2 (p150,95, CD11c) as well as impair binding to intracellular adhesion molecules, involved in firm adhesion of leukocytes to vascular endothelium. This impairment leads to defective polymorphonuclear chemotaxis, margination, adherence, phagocytosis of complement coated particles, bacterial killing, and diminished NK cell and cytotoxic T cell activity 122, 123. Clinical characteristics include frequent systemic, skin, and soft tissue infections, impaired wound healing, gingivitis and periodontitis, and inflammatory bowel disease, all without pus formation 122-124. Disease phenotypes correlate with complete or partial absence of CD18125. Somatic reversion mutations have also been reported 126.
Uzel et al., recently reported severe warts in 5 patients with milder forms of LAD-1 127. Two siblings with deletions of exons 12 and 13 developed extensive warts over their upper extremities in childhood. The younger sibling had leukocytoclastic vasculitis, myositis, and Graves’ disease, as well. A 30-year-old woman with extensive colitis, perianal fistula and subtotal colectomy developed extensive vulvovaginal warts at age 28. A 30-year-old man developed extensive perianal warts requiring resection and treatment with pegylated IFNα. A 37-year-old man with colitis developed warts over his hands. The exact susceptibility to HPV in LAD-1 is unknown; decreased T and NK cell cytotoxicity and immunodysregulation in patients with residual CD18 levels and inflammatory bowel disease may be related. It may be that viral susceptibility is also high in severe forms of LAD-1, but they are transplanted or die earlier in life. Allogeneic HSCT succeeds in LAD-1. Gene therapy in a canine LAD-1 model is promising 128.
Wiskott-Aldrich Syndrome (WASP deficiency)
Wiskott Aldrich Syndrome (WAS) is an X-linked immunodeficiency affecting approximately 1 in 100,000 live births first described by Wiskott in 1937 after noticing the triad of thrombocytopenia, eczema, and frequent pyogenic infections129, 130. This syndrome was re-discovered by Aldrich in 1954 who noted its X-linked inheritance pattern131. Subsequently, other clinical features of WAS have been described including specific immunodeficiency and high rates of autoimmunity and malignancy129, 132, 133.
WAS gene encodes its cognate protein, WASP, which is present in all hematopoietic cells, where it facilitates cellular migration through cytoskeletal reorganization.134 The severity of eczema varies considerably but usually resembles classic atopic dermatitis and occurs more frequently in the first year of life. There are at least 4 distinct WAS phenotypes that correlate with levels of protein expression:135, 136, 137.
Immunodeficiency in WAS affects both humoral and cellular immunity and depends largely on the amount of WASP expression. In classic WAS, the lymphopenia often progresses over time; IgG and IgM are typically low or normal and IgA and IgE are often elevated133. Defective antibody responses to polysaccharide vaccines, diminished lymphocyte proliferation to mitogens, normal to increased numbers of NK cells but reduced cytotoxicity, and impaired chemotaxis of phagocytes are common133, 138, 139. Patients are particularly susceptible to Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis. Opportunistic infections include Pneumocystis jirovecii, MCV, systemic VZV and CMV. Fungal infections consist primarily of Candida132, 133. Autoimmunity occurs in 40- 70% of patients, including hemolytic anemia, vasculitis, inflammatory bowel disease, and renal diseases133, 140. Malignancies usually occur in adolescence and young adulthood with B cell lymphomas and leukemias being the most common133.
Cutaneous HPV is uncommon but has been reported in WAS. Sullivan et al. reported 6/154 patients with WAS had recurrent warts133. Case reports of warts in WAS include a twelve year old 141; a two year old with disseminated warts treated successfully with “transfer factor” who had complete resolution of warts within 2 weeks of treatment142. Most recently, a 25 year old man with 2 years of eczema and recalcitrant warts on his hands and feet 143 had complete resolution after receiving acitretin for 13 weeks. However, once it was tapered, the lesions returned. The exact etiology of HPV infection in WAS is not understood, but defects in NK cellular immunity are likely to be etiologic.
Currently, allogeneic HSCT is the only curative treatment available for WAS with the best results in recipients of matched related donors less than 5 years of age144-146. Gene therapy trials with a lentiviral vector147, 148, are currently underway 149.
Ataxia Telangiectasia (ATM Deficiency)
Ataxia – Telangiectasia (A-T) is an autosomal recessive multisystem disorder characterized by progressive neurodegeneration, oculocutaneous telangiectases, radiosensitivity and combined immunodeficiency150, 151. In 1958 Boder and Sedgewick described 8 patients in 6 families with progressive cerebellar ataxia, oculocutaneous telangiectasia, and frequent sinopulmonary infections 152.
Sinopulmonary infections are common in A-T; chronic lung disease and bacterial pneumonia have been major causes of death 153, 154. In 100 A-T patients recurrent sinopulmonary infections occurred in more than one third. However, infections with P. jirovecii or following live viral vaccination do not occur. Infections with Candida were rare and only in the setting of severe viral infection or chemotherapy. Viral infections include VZV, HSV, MCV, and EBV. HPV occurred in 17 patients and were severe and refractory to treatment in 7 155.
Immunodeficiency in A-T is variable. The most common humoral abnormalities are low B lymphocytes 155; decreased IgG, IgA, IgE, IgG4 and IgG2 156, 157,155; impaired antibody responses 158; and occasional hypergammaglobulinemia 159. CD4 T lymphopenia 155, 160, decreased CD4/CD45RA lymphocytes 160, 161; decreased NK cells 155; and impaired lymphoproliferation to mitogens and antigens are reported162.
A-T results from mutations in the ataxia-telangiectasia mutated (ATM) gene on chromosome 11, a phosphatidylinositol 3-kinase family member involved in mitogenic signal transduction, intracellular protein transport, and cell cycle control 151. In the absence of ATM, repair of DNA breaks such as in V(D)J recombination and breaks caused by ionizing radiation does not occur.
Mortality typically occurs from lymphoreticular malignancies and infections of the respiratory system, further complicated by their neuromuscular deterioration. Median survival in two large cohorts was 25 and 19 years, respectively. However, life expectancy did not correlate well with neurologic impairment 163.
CD40L deficiency (X-linked Hyper IgM Syndrome, XHIGM1)
First described in the 1960’s164, the syndrome was most commonly observed in males as an X-linked recessive trait. HIGM1 is due to mutations in CD154 (CD40L, tumor necrosis factor surface family 5 [TNFSF5]), which encodes CD40L, expressed transiently on activated T cells. These mutations result in failure of T cell signaling to B cells and monocyte/macrophages, resulting in impaired B cell stimulation for class switch recombination, as well as other cell mediated defects165, 166.
Affected males are susceptible to recurrent bacterial and opportunistic infections starting early in life, including Pneumocystis jirovecii pneumonia, occurring in 20-40% of cases. Chronic watery diarrhea due to infection with Cryptosporidium species is common. Liver and biliary tract disease with sclerosing cholangitis due to Cryptosporidium parvum, and infections with hepatitis B and C viruses as well as CMV can result in liver and biliary tract tumors. Neutropenia occurs in about 50% of patients, causing recurrent oral ulcers and proctitis165. Autoimmunity is also common in CD40L deficiency with immune thrombocytopenia, hemolytic anemia, and immune mediated nephritis
CD40L deficient patients have normal numbers of circulating B lymphocytes expressing IgM or IgD. However, cell surface expression of IgG, IgA, and IgE may be profoundly reduced. IgM is not always elevated as its name implies; 50% have normal levels at diagnosis. However, the majority of patients develop hyper IgM at some point. Patients also have low affinity antibody production to T dependent antigens and lack an amnestic response, reflecting their lack of memory B cells. T cell subsets and proliferative responses to mitogens are normal, although in vitro proliferation to T dependent antigens is often reduced. Lymph nodes contain primary follicles but characteristically lack germinal centers165, 167.
Susceptibility to viral infections is not a hallmark of CD40L deficiency. A 3 year old boy with cutaneous histoplasmosis also had common warts on the hands 168. He had elevated IgM, low IgG, and normal IgA as well as CD4 T cell lymphopenia, but no neutropenia. A 28 year old man with recurrent herpes labialis had 7 years of recalcitrant common warts on the elbows and dorsal surfaces of the hands and knees169. Allogeneic HSCT is currently the treatment of choice for CD40L deficiency170. Clinical management is with administration of IVIg and prophylactic antibiotics.
Discussion
Immunity leading to wart regression is essentially universal, but poorly understood. Cellular and cytotoxic immunity provided by T cells and NK cells are necessary for control of HPV infections, but the exact mechanisms are unknown.
Several lines of evidence show the importance of cellular immunity in wart regression and long-lived immunity to HPV. In vivo, delayed type hypersensitivity (DTH) reactions to purified formalin inactivated HPV antigens were highest in patients with prior warts (73%) but lower (51%) in those with active warts. The duration of warts strongly influenced DTH 171, with response highest at 6 months (65%) and 2 years (69%) and then decreasing to 20% in those with warts for more than 10 years172. The importance of CD4+ T cells in control of HPV became more obvious with the advent of human immunodeficiency virus (HIV) infection, in which multiple recurrences of cervical HPV infection173 and genital warts174 and prolonged persistence of high risk HPV occur175 despite treatment with antiretroviral therapy 176. Both CD4 and CD8 T cells are seen histologically in regressing genital warts along with upregulation of adhesion molecules required for lymphocyte trafficking 177. These infiltrating lymphocytes produce pro-inflammatory cytokines including IL-12, TNFα, and IFNγ, characteristic of a Th1 phenotype178. Increasing evidence has shown that CD4 T cell responses to E2 and E6 specific HPV proteins are important in control of HPV-16. Strong Th1 responses to E2 and E6 occur in normal individuals without clinical signs of HPV-16 infection179. These are only occasionally impaired in patients with high-grade cervical intraepithelial neoplasia (CIN) but more impaired in patients with cervical cancer180.
Antigen specific cytotoxic T cells (CTL) and NK cells are the most important effectors for viral infections, including HPV. CTL responses of both CD4 and CD8 T cells, measured by specific lysis after stimulation with HPV specific proteins are reduced in patients with previous and ongoing HPV infection181-183. Reduced NK cell activity against HPV-16 infected keratinocytes has been reported in patients with active HPV-induced neoplasia184.
The importance of effective T and NK cell cytotoxic responses is further supported in host defense against recurrent respiratory papillomatosis (RRP). RRP is a rare disease of the larynx caused by HPV-6 and -11. A shift in immunologic tolerance with polarization of T cells to a Th2 and T regulatory cell phenotype as well as defective NK cell cytotoxicity of HPV infected keratinocytes have been described in patients with chronic RRP. Patients with RRP have no other infection susceptibilities and have otherwise normal immunity185.
There is also some role of humoral immunity in the control of HPV. Eventual seroconversion and evolution of type specific antibody to L1, the major HPV coat protein, occurs 6-18 months after infection. However, 20-50% of women with detectable HPV DNA do not have type specific HPV antibodies. When present, these antibodies are long lived, persisting for more than 10 years in 20-25% of women186. Whether these low level antibodies are protective against future infection with the same or different HPV strains is not known.
HPV evades host defense and induces poor immune responses in normal individuals. It has an exclusively intra-epithelial life cycle, infecting basal keratinocytes via micro-abrasions of the epithelium, leaving the basal lamina intact. The subsequent life cycle, including encapsidation, viral assembly, and maturation occur in the most superficial cells of the squamous epithelium. Virus capsid entry typically leads to activating signals for dendritic cells, such as Langerhans’ cells but these cells are not activated after uptake of HPV antigens 187. HPV’s are not lytic, but live in terminally differentiated cells at a high level of viral replication. Because of the infection of “dying” cells, there is little inflammatory response with release of pro-inflammatory mediators, and thus little danger signal to induce innate immune responses. Although HPV’s can enter antigen presenting cells, their replication is confined to keratinocytes, thus decreasing the antigen load within the antigen presenting cell. Lastly, there is no viremic state, leaving the draining lymph nodes where adaptive responses are initiated with poor access to the HPV antigens188, 189. Despite the absence of viral induced cell death, keratinocytes should be able to be activated to have type 1 interferon responses. However, HPV, like many other DNA viruses, can inhibit interferon synthesis and signaling190-193. HPV inhibits downstream signaling from the type 1 interferon receptor preventing translocation, transactivation, and transcription of critical elements in the type 1 interferon response (reviewed in 190). Furthermore, HPV has evolved mechanisms to evade the effects of type 1 interferons. Several human cervical epithelial cell lines immortalized by recombinant HPV-16, -18, and -33 prevent IFNα inhibition of transcription of E6 and E7 190, 194.
Treatment and cure of HPV in the immunocompromised is both imperative and challenging. Multiple modalities are often required including destructive treatments, topical therapies, systemic anti-virals, and immunomodulators. Two HPV vaccines, bivalent and quadrivalent, are commercially available and routine vaccination with the quadrivalent vaccine for prevention of HPV is recommended by the Advisory Committee on Immunization Practices (ACIP) for females and males ages 11-26 years 195, 196. Successful primary treatment of HPV-associated intraepithelial neoplasia with the quadrivalent vaccine has been recently reported 197-200. The role for HPV vaccination in the setting of immunodeficiency is undefined but is likely warranted.
When warts are numerous, recurrent, and recalcitrant, clinicians should suspect underlying immune defects, especially if occurring with other infections, atopy, autoimmunity, or malignancy. The types and locations of other infections (viral vs. fungal vs. opportunistic) can greatly help in formulating a guided differential diagnosis (see table 3). A thorough history and physical examination and focused laboratory testing should help guide an informed and prudent search for the specific immune defect (figure 1).
Table III.
Other diagnostic features of Primary Immune Deficiencies associated with warts
| Warts with other infections | Warts with other features |
|---|---|
|
| |
| Pneumocystis jiroveci pneumonia | Malignancy |
| NEMO | DOCK8 |
| XHIGM1 | GATA2 |
| WAS | STK4 |
| SCID | CVID |
| AT | |
| WAS | |
| Skin – EV, Netherton syndrome | |
|
| |
| Other viral infections (MCV, HSV, VZV) | Atopy |
| DOCK8 | WAS |
| Netherton syndrome | DOCK8 |
| NEMO | Netherton syndrome |
| WAS | |
| ICL | |
| GATA2 | |
| STK4 | |
| SCID | |
|
| |
| Encapsulated bacterial infections | Autoimmunity |
| CVID | ICL |
| LAD-1 | CVID |
| WHIM | LAD-1 |
| AT | WAS |
| WAS | STK4 |
| NEMO | |
| XHIGM1 | |
| SCID | |
| STK4 | |
|
| |
| Nontuberculous mycobacteria | |
| GATA2 | |
| NEMO | |
|
| |
| Histoplasma capsulatum | |
| GATA2 | |
| ICL | |
| XHIGM1 | |
|
| |
| No HPV associated infections | |
| EV | |
| ICL | |
NEMO, Nuclear Factor κ B essential modulator deficiency; XHIGM1, X-linked Hyper IgM syndrome type 1 or CD40 ligand deficiency; WAS, Wiskott-Aldrich Syndrome; SCID, Severe combined Immunodeficiency; DOCK8, dedicator of cytokinesis 8; ICL, Idiopathic CD4 lymphopenia; STK4, serine-threonine protein kinase 4; CVID, Combined variable immunodeficiency; LAD-1, Leukocyte adhesion deficiency type-1; WHIM, Warts, Hypogammaglobulinemia, Infections, Myelokathexis; AT, Ataxia-Telangiectasia; EV, Epidermodysplasia verruciformis
Figure I.
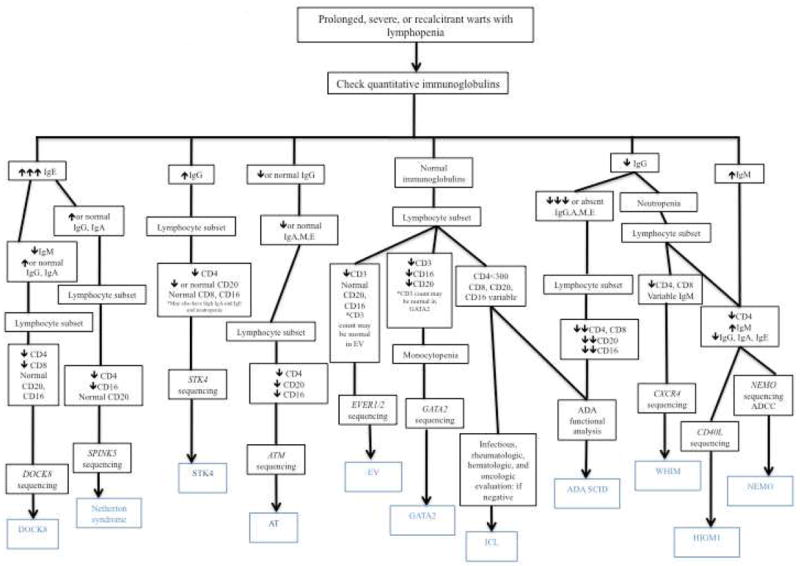
Algorithm for diagnosis of PID with warts and lymphopenia.
DOCK8, dedicator of Cytokinesis 8; EV, Epidermodysplasia verruciformis; ICL, Idiopathic CD4 Lymphopenia; ADA SCID, Adenosine demaminase severe combined immunodeficiency; AT, Ataxia-Telangiectasia; XHIGM1, X-linked Hyper IgM Syndrome type 1/CD40L deficiency; NEMO, Nuclear Factor κB essential modulator deficiency
Host defense against HPV is multifaceted. Investigation of patients with severe recalcitrant HPV has increased our understanding of host defense and cutaneous and systemic immunity. Further study of patients with severe and recalcitrant warts will continue to yield the immune mechanisms that control one of our long-associated fellow travelers, HPV.
Figure II.
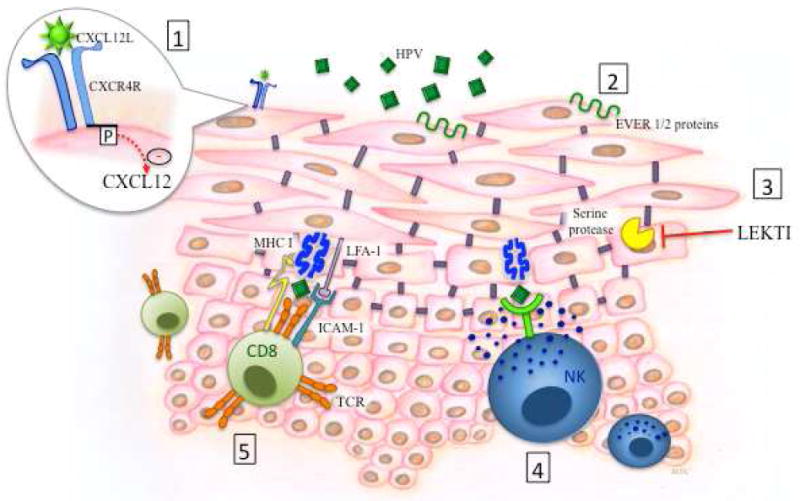
Schematic depiction of the critical elements of host defense against Human Papilloma viruses (HPV) in the skin as determined from primary immune defects.
1. CXCR4: Gain of function mutations inhibit CXCL12 mediated signaling and leukocyte trafficking.
2. EVER 1 and 2: transmembrane proteins that act as restriction factors for HPV.
3. LEKTI inhibits serine protease activity, preventing breakdown of intercellular adhesions. Mutations in SPINK5 lead to decreased LEKTI production and therefore impaired skin integrity.
4, 5. HPV antigen specific activation of natural killer (NK) cells and cytotoxic CD8 T cells results in degranulation of cytotoxic granules.
Figure III.
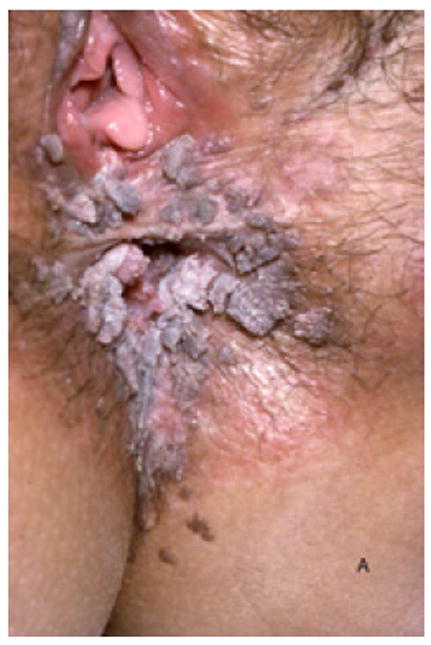
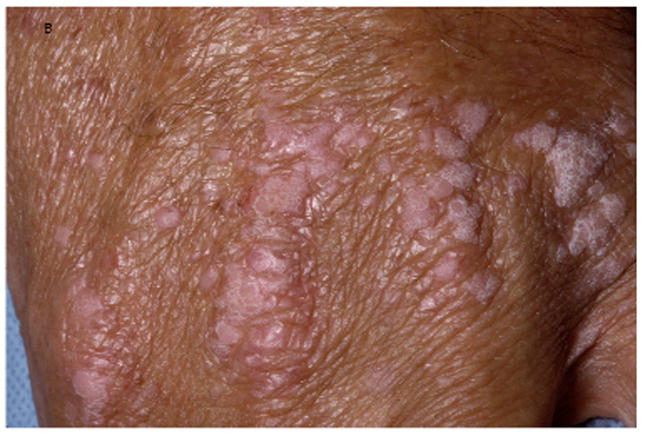
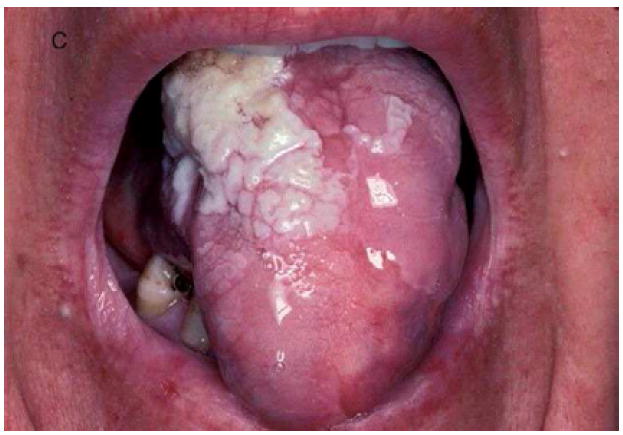
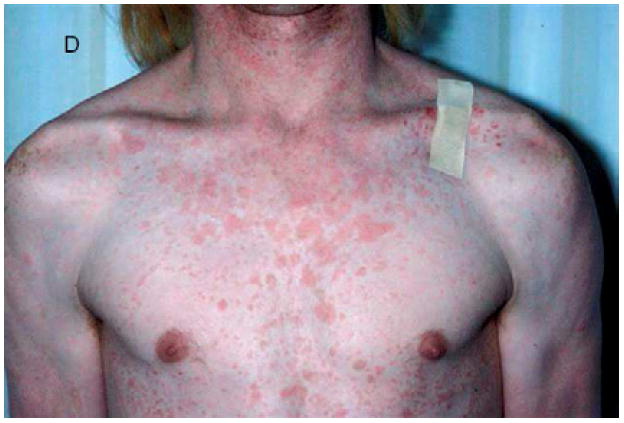
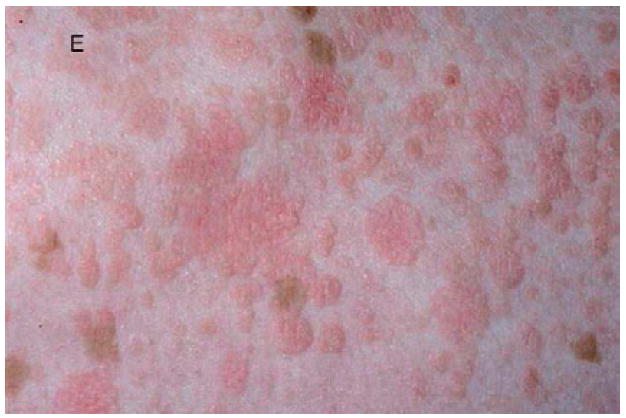
A, Verrucous genital HPV in a patient with GATA2 deficiency. B, Flat warts in a patient with NFκB essential modulator deficiency. C, Warts on the tongue of a patient with epidermodysplasia verruciformis. D and E, Warts on the chest of a patient with epidermodysplasia verruciformis that appear as hyperpigmented lesions similar to tinea versicolor (pictures C, D, and E courtesy of Dr. Maria Turner, NCI, NIH).
Table I.
Primary Immune Deficiencies with warts as a major feature
| PID | Gene / protein mutated | Inheritance | Age at presentation of warts | Types / Location of warts | Other Infections | Other clinical associations | Immune phenotype |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Epidermodysplasia Verruciformis | EVER1 and 2 / epidermodysplasia verruciformis 1 and 2 | AR Reports of XL and AD |
Infancy | Flat – trunk, neck, extremities Verrucous – sun exposed areas Hypo /hyper pigmented lesions resembling pityriasis versicolor | 30-70% lesions undergo malignant transformation |
|
|
|
| |||||||
| WHIM | CXCR4 / chemokine receptor 4 | AD | Variable | Flat, verrucous – extremities, trunk Verrucous - genital | Pneumonia, Cellulitis, Sinusitis, UTI, Thrombophlebitis, Omphalitis, Osteomyelitis, Soft tissue abscesses, HSV, VZV |
|
|
|
| |||||||
| DOCK8 | DOCK8 / DOCK8 | AR | Variable | Flat, verrucous – trunk, face, extremities | S. aureus skin abscesses, Pneumonia, Sinusitis, Soft tissue infections, HSV, MCV, VZV | Eczema, Food allergies, Asthma, Lymphoreticular and squamous cell malignancies |
|
|
| |||||||
| ICL | Unknown | ? | Late adolescence, adult | Flat, verrucous – face, trunk, extremities Verrucous -genital | Opportunistic infections – MAC, Cryptococcus, histoplasmosis, Mucocutaneous Candida, HSV, P. Jirovecii rare | Autoimmune disease |
|
| Autoantibodies | |||||||
| Mucocutaneous | |||||||
| Squamous cell cancer | |||||||
|
| |||||||
| GATA2 deficiency | GATA2/GATA2 binding protein | AD | Late adolescence / adult | Flat, verrucous – face, trunk, extremities, disseminated, genital | NTM | Myelodysplasia, leukemias, Malignancy Panniculitis Thromboembolic phenomena Pulmonary alveolar proteinosis Lymphedema |
|
| MonoMAC | Histoplasma | ||||||
| DCML | Aspergillus | ||||||
| Emberger syndrome | Cryptococcus | ||||||
| VZV | |||||||
| HSV | |||||||
|
| |||||||
| Netherton Syndrome | SPINK5 / LEKTI | AR | Adolescence, adulthood | Common, plane – extremities, trunk Verrucous -genital | Cellulitis | Bamboo hair |
|
| Bacteremia | Icthyosis | ||||||
| Cutaneous HSV | Eczema | ||||||
| Food allergies | |||||||
| Asthma | |||||||
|
| |||||||
| STK4 Deficiency | STK4 / STK4 or MST1 | AR | Unknown | Cutaneous | Cellulitis | Autoimmunity |
|
| Skin Abscesses | EBV related malignancy | ||||||
| Pneumonia | Structural heart disease | ||||||
| Sinusitis | |||||||
| HSV | |||||||
| VZV | |||||||
| MCV | |||||||
| EBV | |||||||
AR, autosomal recessive; AD, autosomal dominant; XL, X-linked; LPM, lymphocyte proliferation to mitogen; WHIM, warts, hypogammaglobulinemia, infections, myelokathexis; UTI, urinary tract infection; HSV, herpes simplex virus; VZV, varicella zoster virus; Ig –G, -A, -M, -E, -D, immunoglobulin –G, -A, -M, -E, -D; LPA, lymphocyte proliferation to antigen; DOCK8, dedicator of cytokinesis 8; MCV, molluscum cantagiosum virus; ICL, Idiopathic CD4 lymphopenia; MAC, Mycobacterium avium complex; MonoMAC, monocytopenia, M. avium complex infection; DCML, dendritic cell, monocyte, B cell and NK cell lymphopenia; NTM, nontuberculous mycobacteria; SPINK5, serine protease inhibitor Kazal-type 5; LEKTI, lympho-epithelial Kazal-type related inhibitor; STK4, serine-threonine protein kinase 4; MST1, mammalian sterile 20-like protein; EBV, Epstein-Barr virus.
Table II.
Primary Immune Deficiencies with reports of warts
| PID | Gene / protein mutated | Inheritance | Age at presentation of warts | Types / Location of warts | Other Infections | Other clinical associations | Immune phenotype | Reference |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| NEMO | IKBKG / NFκB essential modulator | XL | variable | Cutaneous |
|
|
|
Hanson et al. 105 |
| Tobin et al.104 | ||||||||
|
| ||||||||
| Ataxia-telangiectasia | ATM / ATM | AR | Not noted | Not noted |
|
|
|
Nowak et al. 155 |
|
| ||||||||
| SCID | ADA,γc,JAK3 | XL, AR | ADA – adults; γc/JAK3 mean of 8 years following HSCT | ADA – widespread 111, 112; hands and feet 110 γc/JAK3 – flat, EV like, no genital |
|
|
|
Antony et al. 110, Fairbanks et al. 111, Shovlin et al. 112, Laffort et al.113 |
|
| ||||||||
| CVID | ICOS / ICOS | Sporadic | Childhood, Adolescence, Adulthood | 116,119Hands and feet |
|
|
Reid et al. 116, Uluhan et al 117., Lynn et al. 118, Lin et al 119. | |
| CD19 / CD19 | 117Disseminated | |||||||
| CD20 / CD20 | 118palmoplantar | |||||||
| CD81/ CD81 | ||||||||
| BAFFR / BAFFR | ||||||||
| TNFRSF13B / TACI | ||||||||
|
| ||||||||
| LAD-1 | ITGB2/CD18 | AR | Adolescence, adult | Disseminated, genital |
|
|
|
Uzel et al.127 |
|
| ||||||||
| WAS | WASP / WASP | XL | Child, adolescence, adult | Extremities, disseminated |
|
|
|
Sullivan et al. 133, Ormerod et al. 141, Stevens et al. 142, Kim et al. 143 |
|
| ||||||||
| XHIGM1 | CD40L gene / CD40L | XL | Childhood | Common - Extremities |
|
|
|
Yilmaz et al. 168, Chang et al169. |
NEMO, nuclear factor κ B essential modulator; XL, X-linked; NTM, nontuberculous mycobacteria; HSV, herpes simplex virus; CMV, cytomegalovirus; MCV, molluscum cantagiosum virus; Ig –G, -A, -M, -E, immunoglobulin –G, -A, -M, -E; ATM, ataxia-telangiectasia mutated; AR, autosomal recessive; VZV, varicella zoster virus; EBV, Epstein-Barr virus; LPA, lymphocyte proliferation to antigen; LPM, lymphocyte proliferation to mitogen; SCID, Severe combined immunodeficiency; γc, common gamma chain of the interleukin 2 receptor; JAK3, Janus kinase 3; HSCT, hematopoietic stem cell transplantation; GVHD, graft versus host disease; CVID, Common variable immunodeficiency; ICOS, inducible costimulator; BAFFR, B cell activating factor receptor; TNFRSF13B, tumor necrosis factor receptor superfamily 13B; TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor; LAD-1, Leukocyte adhesion deficiency type type-1; ITGB2, integrin beta chain β2; IBD, inflammatory bowel disease; WAS, Wiskott Aldrich Syndrome; WASP, Wiskott Aldrich Syndrome protein; XHIGM1, X-linked Hyper IgM syndrome type 1; HBV, hepatitis B virus; HCV, hepatitis C virus
Acknowledgments
Financial Support: This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health
Abbreviations/Acronyms
- ACIP
Advisory Committee on Immunization Practices
- AD
autosomal dominant
- ADA
adenosine deaminase
- AR
autosomal recessive
- A-T
Ataxia – Telangiectasia
- ATM
ataxia-telangiectasia mutated gene
- BAFFR
B cell activating factor receptor
- BCL-2
B cell lymphoma 2
- CD40L
CD40 ligand
- CIN
cervical intraepithelial neoplasia
- CMV
cytomegalovirus
- CTL
cytotoxic T cells
- CVID
Common variable immunodeficiency
- DCML
Dendritic cell, monocyte, B cell, and natural killer cell deficiency
- DOCK8
dedicator of cytokinesis 8
- DTH
delayed type hypersensitivity
- EBV
Epstein Barr virus
- EV
Epidermodysplasia verruciformis
- FOXO1
forkhead box protein O1
- FOXp3
forkhead box protein 3
- γc
common gamma chain of the interleukin 2 receptor
- G-CSF
granulocyte colony stimulating factor
- GM-CSF
granulocyte macrophage colony stimulating factor
- GRK3
G-protein coupled receptor kinase 3
- GVHD
graft versus host disease
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- HLA
human leukocyte antigen
- HPV
human Papilloma Virus
- HSCT
hematopoietic stem cell transplant
- HSV
herpes simplex virus
- IBD
inflammatory bowel disease
- ICOS
inducible costimulator
- ICL
Idiopathic CD4 lymphopenia
- IFN
interferon
- IgG, -A, -M, -E, -D
immunoglobulin –G, -A, -M, -E, -D
- IκB
inhibitor of κB
- IKK
IκB kinase complex
- IL-7Rα
interleukin 7 receptor alpha
- IL
interleukin
- ITGB2
integrin beta chain β2
- IVIg
intravenous immunoglobulin
- JAK3
Janus kinase 3
- LAD-1
Leukocyte adhesion deficiency type-1
- LEKTI
lympho-epithelial Kazal-type-related inhibitor
- LFA-1
lymphocyte function-associated 1
- LPA
lymphocyte proliferation to antigen
- LPM
lymphocyte proliferation to mitogen
- MAC
Mycobacterium avium complex
- Mac-1
macrophage-1 antigen
- MCV
molluscum contagiosum virus
- MonoMAC
monocytopenia, M. avium complex infection
- MST1
mammalian sterile 20-like protein
- NEMO
nuclear factor κB essential modulator
- NFκB
nuclear factor κB
- NIH
National Institutes of Health
- NTM
nontuberculous mycobacteria
- NK cells
natural killer cells
- PUVA
psoralen plus ultraviolet A
- RRP
recurrent respiratory papillomatosis
- SCID
Severe combined immunodeficiency
- SDF-1
stromal cell derived factor -1
- STK4
serine-threonine kinase 4
- SPINK5
serine protease inhibitor Kazal-type 5
- TACI
transmembrane activator and calcium modulator and cyclophilin ligand
- Th cells
T helper type cells
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- TNFSF5
tumor necrosis factor super family 5
- TNFRSF13B
tumor necrosis factor receptor superfamily 13B
- VZV
varicella zoster virus
- WAS
Wiskott Aldrich Syndrome
- WASP
Wiskott Aldrich Syndrome protein
- WHIM
Warts, hypogammaglobulinemia, infections, myelokathexis
- WILD syndrome
Warts, immunodeficiency, lymphedema, and dysplasia syndrome
- XHIGM1
X-linked Hyper IgM Syndrome type 1
- XL
X-linked
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sterling JC, Handfield-Jones S, Hudson PM. Guidelines for the management of cutaneous warts. Br J Dermatol. 2001;144:4–11. doi: 10.1046/j.1365-2133.2001.04066.x. [DOI] [PubMed] [Google Scholar]
- 2.van Haalen FM, Bruggink SC, Gussekloo J, Assendelft WJ, Eekhof JA. Warts in primary schoolchildren: prevalence and relation with environmental factors. Br J Dermatol. 2009;161:148–52. doi: 10.1111/j.1365-2133.2009.09160.x. [DOI] [PubMed] [Google Scholar]
- 3.Williams HC, Pottier A, Strachan D. The descriptive epidemiology of warts in British schoolchildren. Br J Dermatol. 1993;128:504–11. doi: 10.1111/j.1365-2133.1993.tb00226.x. [DOI] [PubMed] [Google Scholar]
- 4.Rea JN, Newhouse ML, Halil T. Skin disease in Lambeth. A community study of prevalence and use of medical care. Br J Prev Soc Med. 1976;30:107–14. doi: 10.1136/jech.30.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kainz JT, Kozel G, Haidvogl M, Smolle J. Homoeopathic versus placebo therapy of children with warts on the hands: a randomized, double-blind clinical trial. Dermatology. 1996;193:318–20. doi: 10.1159/000246277. [DOI] [PubMed] [Google Scholar]
- 6.Massing AM, Epstein WL. Natural history of warts. A two-year study. Arch Dermatol. 1963;87:306–10. doi: 10.1001/archderm.1963.01590150022004. [DOI] [PubMed] [Google Scholar]
- 7.Leung L. Recalcitrant nongenital warts. Aust Fam Physician. 2011;40:40–2. [PubMed] [Google Scholar]
- 8.Lacey CJ. Therapy for genital human papillomavirus-related disease. J Clin Virol. 2005;32(Suppl 1):S82–90. doi: 10.1016/j.jcv.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 9.Winer RL, Kiviat NB, Hughes JP, Adam DE, Lee SK, Kuypers JM, et al. Development and duration of human papillomavirus lesions, after initial infection. J Infect Dis. 2005;191:731–8. doi: 10.1086/427557. [DOI] [PubMed] [Google Scholar]
- 10.Giuliano AR, Tortolero-Luna G, Ferrer E, Burchell AN, de Sanjose S, Kjaer SK, et al. Epidemiology of human papillomavirus infection in men, cancers other than cervical and benign conditions. Vaccine. 2008;26(Suppl 10):K17–28. doi: 10.1016/j.vaccine.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanigaki T, Endo H. A Case of Epidermodysplasia Verruciformis (Lewandowsky-Lutz, 1922) with Skin-Cancer - Histopathology of Malignant Cutaneous Changes. Dermatologica. 1984;169:97–101. doi: 10.1159/000249578. [DOI] [PubMed] [Google Scholar]
- 12.de Oliveira WRP, Neto CF, Rady PL, Tyring SK. Clinical aspects of epidermodysplasia verruciformis. Journal of the European Academy of Dermatology and Venereology. 2003;17:394–8. doi: 10.1046/j.1468-3083.2003.00703.x. [DOI] [PubMed] [Google Scholar]
- 13.Orth G. Genetics of epidermodysplasia verruciformis: Insights into host defense against papillomaviruses. Seminars in Immunology. 2006;18:362–74. doi: 10.1016/j.smim.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Antonsson A, Karanfilovska S, Lindqvist PG, Hansson BG. General acquisition of human papillomavirus infections of skin occurs in early infancy. J Clin Microbiol. 2003;41:2509–14. doi: 10.1128/JCM.41.6.2509-2514.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Astori G, Lavergne D, Benton C, Hockmayr B, Egawa K, Garbe C, et al. Human papillomaviruses are commonly found in normal skin of immunocompetent hosts. J Invest Dermatol. 1998;110:752–5. doi: 10.1046/j.1523-1747.1998.00191.x. [DOI] [PubMed] [Google Scholar]
- 16.Majewski S, Skopinska-Rozewska E, Jablonska S, Wasik M, Misiewicz J, Orth G. Partial defects of cell-mediated immunity in patients with epidermodysplasia verruciformis. J Am Acad Dermatol. 1986;15:966–73. doi: 10.1016/s0190-9622(86)70258-2. [DOI] [PubMed] [Google Scholar]
- 17.de Oliveira WRP, Carrasco S, Neto CF, Rady P, Tyring SK. Nonspecific cell-mediated immunity in patients with epidermodysplasia verruciformis. Journal of Dermatology. 2003;30:203–9. doi: 10.1111/j.1346-8138.2003.tb00372.x. [DOI] [PubMed] [Google Scholar]
- 18.Glinski W, Jablonska S, Langner A, Obalek S, Haftek M, Proniewska M. Cell-mediated immunity in epidermodysplasia verruciformis. Dermatologica. 1976;153:218–27. doi: 10.1159/000251060. [DOI] [PubMed] [Google Scholar]
- 19.Glinski W, Obalek S, Jablonska S, Orth G. T cell defect in patients with epidermodysplasia verruciformis due to human papillomavirus type 3 and 5. Dermatologica. 1981;162:141–7. doi: 10.1159/000250262. [DOI] [PubMed] [Google Scholar]
- 20.Haftek M, Jablonska S, Orth G. Specific cell-mediated immunity in patients with epidermodysplasia verruciformis and plane warts. Dermatologica. 1985;170:213–20. doi: 10.1159/000249535. [DOI] [PubMed] [Google Scholar]
- 21.Prawer SE, Pass F, Vance JC, Greenberg LJ, Yunis EJ, Zelickson AS. Depressed immune function in epidermodysplasia verruciformis. Arch Dermatol. 1977;113:495–9. [PubMed] [Google Scholar]
- 22.Pyrhonen S, Jablonska S, Obalek S, Kuismanen E. Immune reactions in epidermodysplasia verruciformis. Br J Dermatol. 1980;102:247–54. doi: 10.1111/j.1365-2133.1980.tb08136.x. [DOI] [PubMed] [Google Scholar]
- 23.Cooper KD, Androphy EJ, Lowy D, Katz SI. Antigen Presentation and T-Cell Activation in Epidermodysplasia Verruciformis. Journal of Investigative Dermatology. 1990;94:769–76. doi: 10.1111/1523-1747.ep12874631. [DOI] [PubMed] [Google Scholar]
- 24.Kaminski M, Pawinska M, Jablonska S, Szmurlo A, Majewski S, Orth G. Increased natural killer cell activity in patients with epidermodysplasia verruciformis. Arch Dermatol. 1985;121:84–6. doi: 10.1001/archderm.1985.01660010088025. [DOI] [PubMed] [Google Scholar]
- 25.Majewski S, Malejczyk J, Jablonska S, Misiewicz J, Rudnicka L, Obalek S, et al. Natural cell-mediated cytotoxicity against various target cells in patients with epidermodysplasia verruciformis. J Am Acad Dermatol. 1990;22:423–7. doi: 10.1016/0190-9622(90)70058-p. [DOI] [PubMed] [Google Scholar]
- 26.de Oliveira WR, Rady PL, Grady J, Hughes TK, Festa Neto C, Rivitti EA, et al. Polymorphisms of the interleukin 10 gene promoter in patients from Brazil with epidermodysplasia verruciformis. J Am Acad Dermatol. 2003;49:639–43. doi: 10.1067/s0190-9622(03)01567-6. [DOI] [PubMed] [Google Scholar]
- 27.Androphy EJ, Dvoretzky I, Lowy DR. X-Linked Inheritance of Epidermodysplasia Verruciformis - Genetic and Virologic Studies of a Kindred. Archives of Dermatology. 1985;121:864–8. [PubMed] [Google Scholar]
- 28.McDermott DF, Gammon B, Snijders PJ, Mbata I, Phifer B, Hartley AH, et al. Autosomal Dominant Epidermodysplasia Verruciformis Lacking a Known EVER1 or EVER2 Mutation. Pediatric Dermatology. 2009;26:306–10. doi: 10.1111/j.1525-1470.2008.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramoz N, Rueda LA, Bouadjar B, Montoya LS, Orth G, Favre M. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nature Genetics. 2002;32:579–81. doi: 10.1038/ng1044. [DOI] [PubMed] [Google Scholar]
- 30.Lazarczyk M, Cassonnet P, Pons C, Jacob Y, Favre M. The EVER Proteins as a Natural Barrier against Papillomaviruses: a New Insight into the Pathogenesis of Human Papillomavirus Infections. Microbiology and Molecular Biology Reviews. 2009;73:348. doi: 10.1128/MMBR.00033-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vohra S, Sharma NL, Shanker V, Mahajan VK, Jindal N. Autosomal dominant epidermodysplasia verruciformis: A clinicotherapeutic experience in two cases. Indian Journal of Dermatology Venereology & Leprology. 2010;76:557–61. doi: 10.4103/0378-6323.69092. [DOI] [PubMed] [Google Scholar]
- 32.Berthelot C, Dickerson MC, Rady P, He Q, Niroomand F, Tyring SK, et al. Treatment of a patient with epidermodysplasia verrucifomis carrying a novel EVER2 mutation with imiquimod. Journal of the American Academy of Dermatology. 2007;56:882–6. doi: 10.1016/j.jaad.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 33.Hayashi J, Matsui C, Mitsuishi T, Kawashima M, Morohashi M. Treatment of localized epidermodysplasia verruciformis with tacalcitol ointment. International Journal of Dermatology. 2002;41:817–20. doi: 10.1046/j.1365-4362.2002.01642.x. [DOI] [PubMed] [Google Scholar]
- 34.Anadolu R, Oskay T, Erdem C, Boyvat A, Terzi E, Gurgey E. Treatment of epidermodysplasia verruciformis with a combination of acitretin and interferon alfa-2a. Journal of the American Academy of Dermatology. 2001;45:296–9. doi: 10.1067/mjd.2001.114575. [DOI] [PubMed] [Google Scholar]
- 35.Micali G, Nasca MR, Dall’Oglio F, Musumeci ML. Cimetidine therapy for epidermodysplasia verruciformis. Journal of the American Academy of Dermatology. 2003;48:S9–S10. doi: 10.1067/mjd.2003.111. [DOI] [PubMed] [Google Scholar]
- 36.Karrer S, Szeimies RM, Abels C, Wlotzke U, Stolz W, Landthaler M. Epidermodysplasia verruciformis treated using topical 5-aminolaevulinic acid photodynamic therapy. British Journal of Dermatology. 1999;140:935–8. doi: 10.1046/j.1365-2133.1999.02830.x. [DOI] [PubMed] [Google Scholar]
- 37.Lutzner MA. Epidermodysplasia Verruciformis - Autosomal Recessive Disease Characterized by Viral Warts and Skin Cancer - Model for Viral Oncogenesis. Bulletin Du Cancer. 1978;65:169–82. [PubMed] [Google Scholar]
- 38.Jablonsk S, Jakubowi K, Dabrowsk J. Epidermodysplasia Verruciformis as a Model in Studies on Role of Papovaviruses in Oncogenesis. Cancer Research. 1972;32:583. [PubMed] [Google Scholar]
- 39.Zuelzer WW. “Myelokathexis”--a New Form of Chronic Granulocytopenia. Report of a Case. N Engl J Med. 1964;270:699–704. doi: 10.1056/NEJM196404022701402. [DOI] [PubMed] [Google Scholar]
- 40.Krill CE, Jr, Smith HD, Mauer AM. Chronic Idiopathic Granulocytopenia. N Engl J Med. 1964;270:973–9. doi: 10.1056/NEJM196405072701902. [DOI] [PubMed] [Google Scholar]
- 41.Kawai T, Malech HL. WHIM syndrome: congenital immune deficiency disease. Curr Opin Hematol. 2009;16:20–6. doi: 10.1097/MOH.0b013e32831ac557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarzi MD, Jenner M, Hattotuwa K, Faruqi AZ, Diaz GA, Longhurst HJ. Sporadic case of warts, hypogammaglobulinemia, immunodeficiency, and myelokathexis syndrome. J Allergy Clin Immunol. 2005;116:1101–5. doi: 10.1016/j.jaci.2005.08.040. [DOI] [PubMed] [Google Scholar]
- 43.Balabanian K, Lagane B, Pablos JL, Laurent L, Planchenault T, Verola O, et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood. 2005;105:2449–57. doi: 10.1182/blood-2004-06-2289. [DOI] [PubMed] [Google Scholar]
- 44.Chae KM, Ertle JO, Tharp MD. B-cell lymphoma in a patient with WHIM syndrome. J Am Acad Dermatol. 2001;44:124–8. doi: 10.1067/mjd.2001.111337. [DOI] [PubMed] [Google Scholar]
- 45.Imashuku S, Miyagawa A, Chiyonobu T, Ishida H, Yoshihara T, Teramura T, et al. Epstein-Barr virus-associated T-lymphoproliferative disease with hemophagocytic syndrome, followed by fatal intestinal B lymphoma in a young adult female with WHIM syndrome. Warts, hypogammaglobulinemia, infections, and myelokathexis. Ann Hematol. 2002;81:470–3. doi: 10.1007/s00277-002-0489-9. [DOI] [PubMed] [Google Scholar]
- 46.Diaz GA. CXCR4 mutations in WHIM syndrome: a misguided immune system? Immunol Rev. 2005;203:235–43. doi: 10.1111/j.0105-2896.2005.00226.x. [DOI] [PubMed] [Google Scholar]
- 47.Gorlin RJ, Gelb B, Diaz GA, Lofsness KG, Pittelkow MR, Fenyk JR., Jr WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet. 2000;91:368–76. [PubMed] [Google Scholar]
- 48.Gulino AV, Moratto D, Sozzani S, Cavadini P, Otero K, Tassone L, et al. Altered leukocyte response to CXCL12 in patients with warts hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome. Blood. 2004;104:444–52. doi: 10.1182/blood-2003-10-3532. [DOI] [PubMed] [Google Scholar]
- 49.Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34:70–4. doi: 10.1038/ng1149. [DOI] [PubMed] [Google Scholar]
- 50.Lagane B, Chow KY, Balabanian K, Levoye A, Harriague J, Planchenault T, et al. CXCR4 dimerization and beta-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. 2008;112:34–44. doi: 10.1182/blood-2007-07-102103. [DOI] [PubMed] [Google Scholar]
- 51.Balabanian K, Levoye A, Klemm L, Lagane B, Hermine O, Harriague J, et al. Leukocyte analysis from WHIM syndrome patients reveals a pivotal role for GRK3 in CXCR4 signaling. J Clin Invest. 2008;118:1074–84. doi: 10.1172/JCI33187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pablos JL, Amara A, Bouloc A, Santiago B, Caruz A, Galindo M, et al. Stromal-cell derived factor is expressed by dendritic cells and endothelium in human skin. Am J Pathol. 1999;155:1577–86. doi: 10.1016/S0002-9440(10)65474-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tassone L, Moratto D, Vermi W, De Francesco M, Notarangelo LD, Porta F, et al. Defect of plasmacytoid dendritic cells in warts, hypogammaglobulinemia, infections, myelokathexis (WHIM) syndrome patients. Blood. 2010;116:4870–3. doi: 10.1182/blood-2010-03-272096. [DOI] [PubMed] [Google Scholar]
- 54.Weston B, Axtell RA, Todd RF, 3rd, Vincent M, Balazovich KJ, Suchard SJ, et al. Clinical and biologic effects of granulocyte colony stimulating factor in the treatment of myelokathexis. J Pediatr. 1991;118:229–34. doi: 10.1016/s0022-3476(05)80488-3. [DOI] [PubMed] [Google Scholar]
- 55.Wetzler M, Talpaz M, Kellagher MJ, Gutterman JU, Kurzrock R. Myelokathexis: normalization of neutrophil counts and morphology by GM-CSF. JAMA. 1992;267:2179–80. [PubMed] [Google Scholar]
- 56.McDermott DH, Liu Q, Ulrick J, Kwatemaa N, Anaya-O’Brien S, Penzak SR, et al. The CXCR4 antagonist plerixafor corrects panleukopenia in patients with WHIM syndrome. Blood. 2011;118:4957–62. doi: 10.1182/blood-2011-07-368084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chu EY. Cutaneous Manifestations of DOCK8 Deficiency Syndrome. Arch Dermatol. 2011 doi: 10.1001/archdermatol.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124:1289–302. e4. doi: 10.1016/j.jaci.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Q, Davis JC, Dove CG, Su HC. Genetic, clinical, and laboratory markers for DOCK8 immunodeficiency syndrome. Dis Markers. 2010;29:131–9. doi: 10.3233/DMA-2010-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Su HC. Combined immunodeficiency associated with DOCK8 mutations and related immunodeficiencies. Dis Markers. 2010;29:121–2. doi: 10.3233/DMA-2010-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lambe T, Crawford G, Johnson AL, Crockford TL, Bouriez-Jones T, Smyth AM, et al. DOCK8 is essential for T-cell survival and the maintenance of CD8+ T-cell memory. Eur J Immunol. 2011;41:3423–35. doi: 10.1002/eji.201141759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chatila T, Al Khatib S, Keles S, Garcia-Lioret M, Koc-Aydiner EK, Reisli I, et al. Defects along the T(H)17 differentiation pathway underlie genetically distinct forms of the hyper IgE syndrome. Journal of Allergy and Clinical Immunology. 2009;124:342–8. doi: 10.1016/j.jaci.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Su HC. Dedicator of cytokinesis 8 (DOCK8) deficiency. Curr Opin Allergy Clin Immunol. 2010;10:515–20. doi: 10.1097/ACI.0b013e32833fd718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gatz SA, Benninghoff U, Schutz C, Schulz A, Honig M, Pannicke U, et al. Curative treatment of autosomal-recessive hyper-IgE syndrome by hematopoietic cell transplantation. Bone Marrow Transplant. 2011;46:552–6. doi: 10.1038/bmt.2010.169. [DOI] [PubMed] [Google Scholar]
- 65.Unexplained CD4+ T-lymphocyte depletion in persons without evident HIV infection--United States. MMWR Morb Mortal Wkly Rep. 1992;41:541–5. [PubMed] [Google Scholar]
- 66.Zonios DI, Falloon J, Bennett JE, Shaw PA, Chaitt D, Baseler MW, et al. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood. 2008;112:287–94. doi: 10.1182/blood-2007-12-127878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gubinelli E, Posteraro P, Girolomoni G. Idiopathic CD4+ T lymphocytopenia associated with disseminated flat warts and alopecia areata. J Dermatol. 2002;29:653–6. doi: 10.1111/j.1346-8138.2002.tb00197.x. [DOI] [PubMed] [Google Scholar]
- 68.Hansen ER, Lisby S, Baadsgaard O, Ho VC, de Villiers EM, Vejlsgaard GL. Abnormal function of CD4+ helper/inducer T lymphocytes in a patient with widespread human papillomavirus type 3-related infection. Arch Dermatol. 1990;126:1604–8. [PubMed] [Google Scholar]
- 69.Manchado Lopez P, Ruiz de Morales JM, Ruiz Gonzalez I, Rodriguez Prieto MA. Cutaneous infections by papillomavirus, herpes zoster and Candida albicans as the only manifestation of idiopathic CD4+ T lymphocytopenia. Int J Dermatol. 1999;38:119–21. doi: 10.1046/j.1365-4362.1999.00364.x. [DOI] [PubMed] [Google Scholar]
- 70.Requena L, Sarasa JL, Terai M, Sata T, Matsukura T. Lifelong severe verrucosis associated with human papillomavirus type 2: report of a case with a 38-year follow-up. Br J Dermatol. 1998;139:1081–6. doi: 10.1046/j.1365-2133.1998.02571.x. [DOI] [PubMed] [Google Scholar]
- 71.Stetson CL, Rapini RP, Tyring SK, Kimbrough RC. CD4+ T lymphocytopenia with disseminated HPV. J Cutan Pathol. 2002;29:502–5. doi: 10.1034/j.1600-0560.2002.290809.x. [DOI] [PubMed] [Google Scholar]
- 72.Pasic S, Minic P, Dzudovic S, Minic A, Slavkovic B. Idiopathic CD4+ lymphocytopenia and juvenile laryngeal papillomatosis. Pediatric Pulmonology. 2005;39:281–3. doi: 10.1002/ppul.20173. [DOI] [PubMed] [Google Scholar]
- 73.Salit RB, Hankey KG, Yi R, Rapoport AP, Mann DL. Detection of CD4(+) T-cell antibodies in a patient with idiopathic CD4 T lymphocytopenia and cryptococcal meningitis. Br J Haematol. 2007;139:133–7. doi: 10.1111/j.1365-2141.2007.06781.x. [DOI] [PubMed] [Google Scholar]
- 74.Vicente C, Conchillo A, Garcia-Sanchez MA, Odero MD. The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit Rev Oncol Hematol. 2011 doi: 10.1016/j.critrevonc.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 75.Rodrigues NP, Janzen V, Forkert R, Dombkowski DM, Boyd AS, Orkin SH, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood. 2005;106:477–84. doi: 10.1182/blood-2004-08-2989. [DOI] [PubMed] [Google Scholar]
- 76.Bigley V, Haniffa M, Doulatov S, Wang XN, Dickinson R, McGovern N, et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med. 2011;208:227–34. doi: 10.1084/jem.20101459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–7. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vinh DC, Patel SY, Uzel G, Anderson VL, Freeman AF, Olivier KN, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood. 2010;115:1519–29. doi: 10.1182/blood-2009-03-208629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Emberger JM, Navarro M, Dejean M, Izarn P. Deaf-mutism, lymphedema of the lower limbs and hematological abnormalities (acute leukemia, cytopenia) with autosomal dominant transmission. J Genet Hum. 1979;27:237–45. [PubMed] [Google Scholar]
- 80.Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118:2656–8. doi: 10.1182/blood-2011-06-360313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118:2653–5. doi: 10.1182/blood-2011-05-356352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome) Nat Genet. 2011;43:929–31. doi: 10.1038/ng.923. [DOI] [PubMed] [Google Scholar]
- 83.Attal M, Huguet F, Nouvel C, Dastugue N, Laurent G, Duchayne E, et al. Idiopathic Lymphoedema Associated with Familial Acute-Leukemia - a New Case. Presse Medicale. 1985;14:600. [PubMed] [Google Scholar]
- 84.Drony S, Bauters F, Barthel B, Moriceau M, Sarrazin C. 2 Family Cases of Congenital Elephantiasis with Acute Myeloid-Leukemia. Nouvelle Revue Francaise D Hematologie. 1983;25:184. [Google Scholar]
- 85.Gaillard S, Bordessoule D, Malinvaud G, Verger C. Preleukemic Dyshematopoiesis and Congenital Lymphedema. Nouvelle Revue Francaise D Hematologie. 1983;25:184. [Google Scholar]
- 86.Mansour S, Connell F, Steward C, Ostergaard P, Brice G, Smithson S, et al. Emberger syndrome-primary lymphedema with myelodysplasia: report of seven new cases. Am J Med Genet A. 2010;152A:2287–96. doi: 10.1002/ajmg.a.33445. [DOI] [PubMed] [Google Scholar]
- 87.Cuellar-Rodriguez J, Gea-Banacloche J, Freeman AF, Hsu AP, Zerbe CS, Calvo KR, et al. Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood. 2011;118:3715–20. doi: 10.1182/blood-2011-06-365049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Calvo KR, Vinh DC, Maric I, Wang W, Noel P, Stetler-Stevenson M, et al. Myelodysplasia in autosomal dominant and sporadic monocytopenia immunodeficiency syndrome: diagnostic features and clinical implications. Haematologica. 2011;96:1221–5. doi: 10.3324/haematol.2011.041152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ostrow RS, Manias D, Mitchell AJ, Stawowy L, Faras AJ. Epidermodysplasia verruciformis. A case associated with primary lymphatic dysplasia, depressed cell-mediated immunity, and Bowen’s disease containing human papillomavirus 16 DNA. Arch Dermatol. 1987;123:1511–6. doi: 10.1001/archderm.123.11.1511. [DOI] [PubMed] [Google Scholar]
- 90.Kreuter A, Hochdorfer B, Brockmeyer NH, Altmeyer P, Pfister H, Wieland U. A human papillomavirus-associated disease with disseminated warts, depressed cell-mediated immunity, primary lymphedema, and anogenital dysplasia: WILD syndrome. Arch Dermatol. 2008;144:366–72. doi: 10.1001/archderm.144.3.366. [DOI] [PubMed] [Google Scholar]
- 91.Wilkinson RD, Curtis GH, Hawk WA. Netherton’s Disease; Trichorrhexis Invaginata (Bamboo Hair), Congenital Ichthyosiform Erythroderma and the Atopic Diathesis. A Histopathologic Study. Archives of Dermatology. 1964;89:46–54. doi: 10.1001/archderm.1964.01590250052010. [DOI] [PubMed] [Google Scholar]
- 92.Judge MR, Morgan G, Harper JI. A clinical and immunological study of Netherton’s syndrome. Br J Dermatol. 1994;131:615–21. doi: 10.1111/j.1365-2133.1994.tb04971.x. [DOI] [PubMed] [Google Scholar]
- 93.Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nature Genetics. 2000;25:141–2. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 94.Komatsu N, Saijoh K, Jayakumar A, Clayman GL, Tohyama M, Suga Y, et al. Correlation between SPINK5 gene mutations and clinical manifestations in Netherton syndrome patients. Journal of Investigative Dermatology. 2008;128:1148–59. doi: 10.1038/sj.jid.5701153. [DOI] [PubMed] [Google Scholar]
- 95.Greene SL, Muller SA. Netherton’s syndrome. Report of a case and review of the literature. Journal of the American Academy of Dermatology. 1985;13:329–37. [PubMed] [Google Scholar]
- 96.Schnyder UW, Wiegand K. Hair anomalies in Comel’s linear circumflex ichthyosis. Hautarzt. 1968;19:494–9. [PubMed] [Google Scholar]
- 97.Weber F, Fuchs PG, Pfister HJ, Hintner H, Fritsch P, Hoepfl R. Human papillomavirus infection in Netherton’s syndrome. Br J Dermatol. 2001;144:1044–9. doi: 10.1046/j.1365-2133.2001.04196.x. [DOI] [PubMed] [Google Scholar]
- 98.Sedlacek V, Krenar J. Symptomatology of Comel’s linear circumflex ichthyosis (a case associated with genito-anal papillomatosis) Hautarzt. 1971;22:390–7. [PubMed] [Google Scholar]
- 99.Hintner H, Jaschke E, Fritsch P. Netherton syndrome: weakened immunity, generalized verrucosis and carcinogenesis. Hautarzt. 1980;31:428–32. [PubMed] [Google Scholar]
- 100.Nehme NT, Pachlopnik Schmid J, Debeurme F, Andre-Schmutz I, Lim A, Nitschke P, et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T cells survival. Blood. 2011 doi: 10.1182/blood-2011-09-378364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schaffer AA, et al. The phenotype of human STK4 deficiency. Blood. 2012 doi: 10.1182/blood-2011-09-378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 103.Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. 2001;2:223–8. doi: 10.1038/85277. [DOI] [PubMed] [Google Scholar]
- 104.Tobin E, Rohwedder A, Holland SM, Philips B, Carlson JA. Recurrent ‘sterile’ verrucous cyst abscesses and epidermodysplasia verruciformis-like eruption associated with idiopathic CD4 lymphopenia. Br J Dermatol. 2003;149:627–33. doi: 10.1046/j.1365-2133.2003.05543.x. [DOI] [PubMed] [Google Scholar]
- 105.Hanson EP, Monaco-Shawver L, Solt LA, Madge LA, Banerjee PP, May MJ, et al. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. 2008;122:1169–77. e16. doi: 10.1016/j.jaci.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Glanzmann E, Riniker P. Essential lymphocytophthisis; new clinical aspect of infant pathology. Ann Paediatr. 1950;175:1–32. [PubMed] [Google Scholar]
- 107.Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet. 1972;2:1067–9. doi: 10.1016/s0140-6736(72)92345-8. [DOI] [PubMed] [Google Scholar]
- 108.Cossu F. Genetics of SCID. Ital J Pediatr. 2010;36:76. doi: 10.1186/1824-7288-36-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Griffith LM, Cowan MJ, Notarangelo LD, Puck JM, Buckley RH, Candotti F, et al. Improving cellular therapy for primary immune deficiency diseases: recognition, diagnosis, and management. J Allergy Clin Immunol. 2009;124:1152–60. e12. doi: 10.1016/j.jaci.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Antony FC, Webster AD, Bain MD, Harland CC. Recalcitrant palmoplantar warts associated with adult-onset adenosine deaminase deficiency. Br J Dermatol. 2002;147:182–3. doi: 10.1046/j.1365-2133.2002.47562.x. [DOI] [PubMed] [Google Scholar]
- 111.Fairbanks LD, Shovlin CL, Webster AD, Hughes JM, Simmonds HA. Adenosine deaminase deficiency with altered biochemical parameters in two sisters with late-onset immunodeficiency. J Inherit Metab Dis. 1994;17:135–7. doi: 10.1007/BF00735418. [DOI] [PubMed] [Google Scholar]
- 112.Shovlin CL, Hughes JM, Simmonds HA, Fairbanks L, Deacock S, Lechler R, et al. Adult presentation of adenosine deaminase deficiency. Lancet. 1993;341:1471. doi: 10.1016/0140-6736(93)90910-9. [DOI] [PubMed] [Google Scholar]
- 113.Laffort C, Le Deist F, Favre M, Caillat-Zucman S, Radford-Weiss I, Debre M, et al. Severe cutaneous papillomavirus disease after haemopoietic stem-cell transplantation in patients with severe combined immune deficiency caused by common gammac cytokine receptor subunit or JAK-3 deficiency. Lancet. 2004;363:2051–4. doi: 10.1016/S0140-6736(04)16457-X. [DOI] [PubMed] [Google Scholar]
- 114.Marini ANT, Stege H, Ruzicka T, Hengge UR. Plantar warts in twins after successful bone marrow transplantation for severe combined immunodeficiency. Journal of the German Society of Dermatology. 2006;4:417–20. doi: 10.1111/j.1610-0387.2006.05944.x. [DOI] [PubMed] [Google Scholar]
- 115.Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–86. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 116.Reid TM, Fraser NG, Kernohan IR. Generalized warts and immune deficiency. Br J Dermatol. 1976;95:559–64. doi: 10.1111/j.1365-2133.1976.tb00870.x. [DOI] [PubMed] [Google Scholar]
- 117.Uluhan A, Sager D, Jasin HE. Juvenile rheumatoid arthritis and common variable hypogammaglobulinemia. J Rheumatol. 1998;25:1205–10. [PubMed] [Google Scholar]
- 118.Lynn J, Knight AK, Kamoun M, Levinson AI. A 55-year-old man with hypogammaglobulinemia, lymphopenia, and unrelenting cutaneous warts. J Allergy Clin Immunol. 2004;114:409–14. doi: 10.1016/j.jaci.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 119.Lin JH, Wang KY, Kraft S, Roberts RL. Resolution of warts in association with subcutaneous immunoglobulin in immune deficiency. Pediatr Dermatol. 2009;26:155–8. doi: 10.1111/j.1525-1470.2009.00874.x. [DOI] [PubMed] [Google Scholar]
- 120.Hayward AR, Harvey BA, Leonard J, Greenwood MC, Wood CB, Soothill JF. Delayed separation of the umbilical cord, widespread infections, and defective neutrophil mobility. Lancet. 1979;1:1099–101. doi: 10.1016/s0140-6736(79)91786-0. [DOI] [PubMed] [Google Scholar]
- 121.Crowley CA, Curnutte JT, Rosin RE, Andre-Schwartz J, Gallin JI, Klempner M, et al. An inherited abnormality of neutrophil adhesion. Its genetic transmission and its association with a missing protein. N Engl J Med. 1980;302:1163–8. doi: 10.1056/NEJM198005223022102. [DOI] [PubMed] [Google Scholar]
- 122.Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175–94. doi: 10.1146/annurev.me.38.020187.001135. [DOI] [PubMed] [Google Scholar]
- 123.Arnaout MA. Leukocyte adhesion molecules deficiency: its structural basis, pathophysiology and implications for modulating the inflammatory response. Immunol Rev. 1990;114:145–80. doi: 10.1111/j.1600-065x.1990.tb00564.x. [DOI] [PubMed] [Google Scholar]
- 124.Uzel G, Kleiner DE, Kuhns DB, Holland SM. Dysfunctional LAD-1 neutrophils and colitis. Gastroenterology. 2001;121:958–64. doi: 10.1053/gast.2001.28022. [DOI] [PubMed] [Google Scholar]
- 125.Etzioni A. Leukocyte adhesion deficiencies: molecular basis, clinical findings, and therapeutic options. Adv Exp Med Biol. 2007;601:51–60. doi: 10.1007/978-0-387-72005-0_5. [DOI] [PubMed] [Google Scholar]
- 126.Uzel G, Tng E, Rosenzweig SD, Hsu AP, Shaw JM, Horwitz ME, et al. Reversion mutations in patients with leukocyte adhesion deficiency type-1 (LAD-1) Blood. 2008;111:209–18. doi: 10.1182/blood-2007-04-082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Uzel GK, Douglas, Hsu Amy P, Stoddard Jennifer, Holland Steven M. Clinical Immunology Society. Chicago, IL: 2011. The New Face of Leukocyte Adhesion Deficiency Type 1 (LAD-1) pp. S20–1. [Google Scholar]
- 128.Bauer TR, Jr, Allen JM, Hai M, Tuschong LM, Khan IF, Olson EM, et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat Med. 2008;14:93–7. doi: 10.1038/nm1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kapoor N. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. 2009;15:84–90. doi: 10.1016/j.bbmt.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 130.Wiskott A. Familiarer, angeobren Morbus Werlhofi? Monatschrift Kinderheilkd. 1937;68:212–6. [Google Scholar]
- 131.Aldrich RA, Steinberg AG, Campbell DC. Pedigree Demonstrating a Sex-Linked Recessive Condition Characterized by Draining Ears, Eczematoid Dermatitis and Bloody Diarrhea. Pediatrics. 1954;13:133–9. [PubMed] [Google Scholar]
- 132.Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrich syndrome. Curr Opin Hematol. 2008;15:30–6. doi: 10.1097/MOH.0b013e3282f30448. [DOI] [PubMed] [Google Scholar]
- 133.Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. 1994;125:876–85. doi: 10.1016/s0022-3476(05)82002-5. [DOI] [PubMed] [Google Scholar]
- 134.Bouma G, Burns SO, Thrasher AJ. Wiskott-Aldrich Syndrome: Immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation. Immunobiology. 2009;214:778–90. doi: 10.1016/j.imbio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jin Y, Mazza C, Christie JR, Giliani S, Fiorini M, Mella P, et al. Mutations of the Wiskott-Aldrich Syndrome Protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood. 2004;104:4010–9. doi: 10.1182/blood-2003-05-1592. [DOI] [PubMed] [Google Scholar]
- 136.Villa A, Notarangelo L, Macchi P, Mantuano E, Cavagni G, Brugnoni D, et al. X-linked thrombocytopenia and Wiskott-Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nature Genetics. 1995;9:414–7. doi: 10.1038/ng0495-414. [DOI] [PubMed] [Google Scholar]
- 137.Notarangelo LD, Mazza C, Giliani S, D’Aria C, Gandellini F, Ravelli C, et al. Missense mutations of the WASP gene cause intermittent X-linked thrombocytopenia. Blood. 2002;99:2268–9. doi: 10.1182/blood.v99.6.2268. [DOI] [PubMed] [Google Scholar]
- 138.Ochs HD, Slichter SJ, Harker LA, Von Behrens WE, Clark RA, Wedgwood RJ. The Wiskott-Aldrich syndrome: studies of lymphocytes, granulocytes, and platelets. Blood. 1980;55:243–52. [PubMed] [Google Scholar]
- 139.Orange JS, Ramesh N, Remold-O’Donnell E, Sasahara Y, Koopman L, Byrne M, et al. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc Natl Acad Sci U S A. 2002;99:11351–6. doi: 10.1073/pnas.162376099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dupuis-Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M, Le Deist F, et al. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. 2003;111:e622–7. doi: 10.1542/peds.111.5.e622. [DOI] [PubMed] [Google Scholar]
- 141.Ormerod AD, Finlay AY, Knight AG, Mathews N, Stark JM, Gough J. Immune deficiency and multiple viral warts: a possible variant of the Wiskott-Aldrich syndrome. Br J Dermatol. 1983;108:211–5. doi: 10.1111/j.1365-2133.1983.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 142.Stevens DA, Ferrington RA, Merigan TC, Marinkovich VA. Randomized trial of transfer factor treatment of human warts. Clin Exp Immunol. 1975;21:520–4. [PMC free article] [PubMed] [Google Scholar]
- 143.Kim JK, Yoon MS, Huh JY, Kim HJ, Kim DH. A novel mutation of the WAS gene in a patient with Wiskott-Aldrich syndrome presenting with recalcitrant viral warts. J Dermatol Sci. 2010;60:120–2. doi: 10.1016/j.jdermsci.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 144.Filipovich AH, Stone JV, Tomany SC, Ireland M, Kollman C, Pelz CJ, et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry and the National Marrow Donor Program. Blood. 2001;97:1598–603. doi: 10.1182/blood.v97.6.1598. [DOI] [PubMed] [Google Scholar]
- 145.Kobayashi R, Ariga T, Nonoyama S, Kanegane H, Tsuchiya S, Morio T, et al. Outcome in patients with Wiskott-Aldrich syndrome following stem cell transplantation: an analysis of 57 patients in Japan. Br J Haematol. 2006;135:362–6. doi: 10.1111/j.1365-2141.2006.06297.x. [DOI] [PubMed] [Google Scholar]
- 146.Pai SY, DeMartiis D, Forino C, Cavagnini S, Lanfranchi A, Giliani S, et al. Stem cell transplantation for the Wiskott-Aldrich syndrome: a single-center experience confirms efficacy of matched unrelated donor transplantation. Bone Marrow Transplant. 2006;38:671–9. doi: 10.1038/sj.bmt.1705512. [DOI] [PubMed] [Google Scholar]
- 147.Charrier S, Dupre L, Scaramuzza S, Jeanson-Leh L, Blundell MP, Danos O, et al. Lentiviral vectors targeting WASp expression to hematopoietic cells, efficiently transduce and correct cells from WAS patients. Gene Ther. 2007;14:415–28. doi: 10.1038/sj.gt.3302863. [DOI] [PubMed] [Google Scholar]
- 148.Dupre L, Marangoni F, Scaramuzza S, Trifari S, Hernandez RJ, Aiuti A, et al. Efficacy of gene therapy for Wiskott-Aldrich syndrome using a WAS promoter/cDNA-containing lentiviral vector and nonlethal irradiation. Hum Gene Ther. 2006;17:303–13. doi: 10.1089/hum.2006.17.303. [DOI] [PubMed] [Google Scholar]
- 149.Galy A, Thrasher AJ. Gene therapy for the Wiskott-Aldrich syndrome. Current Opinion in Allergy and Clinical Immunology. 2011;11:545–50. doi: 10.1097/ACI.0b013e32834c230c. [DOI] [PubMed] [Google Scholar]
- 150.Crawford TO. Ataxia telangiectasia. Semin Pediatr Neurol. 1998;5:287–94. doi: 10.1016/s1071-9091(98)80007-7. [DOI] [PubMed] [Google Scholar]
- 151.Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- 152.Boder E, Sedgwick RP. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics. 1958;21:526–54. [PubMed] [Google Scholar]
- 153.Ammann AJ. Respiratory complications of ataxia-telangiectasia. N Engl J Med. 1969;281:1019. [PubMed] [Google Scholar]
- 154.Sedgwick RP, Boder E. Ataxia-telangiectasia. In: Vinken PJ, B G, Klawans HL, et al., editors. Handbook of Clinical Neurology. Amsterdam: Elsevier; 1991. pp. 347–423. [Google Scholar]
- 155.Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr. 2004;144:505–11. doi: 10.1016/j.jpeds.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 156.Ammann AJ, Cain WA, Ishizaka K, Hong R, Good RA. Immunoglobulin E deficiency in ataxia-telangiectasia. N Engl J Med. 1969;281:469–72. doi: 10.1056/NEJM196908282810904. [DOI] [PubMed] [Google Scholar]
- 157.Oxelius VA, Berkel AI, Hanson LA. IgG2 deficiency in ataxia-telangiectasia. N Engl J Med. 1982;306:515–7. doi: 10.1056/NEJM198203043060905. [DOI] [PubMed] [Google Scholar]
- 158.Sanal O, Ersoy F, Yel L, Tezcan I, Metin A, Ozyurek H, et al. Impaired IgG antibody production to pneumococcal polysaccharides in patients with ataxia-telangiectasia. J Clin Immunol. 1999;19:326–34. doi: 10.1023/a:1020599810261. [DOI] [PubMed] [Google Scholar]
- 159.Sadighi Akha AA, Humphrey RL, Winkelstein JA, Loeb DM, Lederman HM. Oligo-/monoclonal gammopathy and hypergammaglobulinemia in ataxia-telangiectasia. A study of 90 patients. Medicine (Baltimore) 1999;78:370–81. doi: 10.1097/00005792-199911000-00002. [DOI] [PubMed] [Google Scholar]
- 160.Paganelli R, Scala E, Scarselli E, Ortolani C, Cossarizza A, Carmini D, et al. Selective deficiency of CD4+/CD45RA+ lymphocytes in patients with ataxia-telangiectasia. J Clin Immunol. 1992;12:84–91. doi: 10.1007/BF00918137. [DOI] [PubMed] [Google Scholar]
- 161.Carbonari M, Cherchi M, Paganelli R, Giannini G, Galli E, Gaetano C, et al. Relative increase of T cells expressing the gamma/delta rather than the alpha/beta receptor in ataxia-telangiectasia. N Engl J Med. 1990;322:73–6. doi: 10.1056/NEJM199001113220201. [DOI] [PubMed] [Google Scholar]
- 162.Waldmann TA, Broder S, Goldman CK, Frost K, Korsmeyer SJ, Medici MA. Disorders of B cells and helper T cells in the pathogenesis of the immunoglobulin deficiency of patients with ataxia telangiectasia. J Clin Invest. 1983;71:282–95. doi: 10.1172/JCI110768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Crawford TO, Skolasky RL, Fernandez R, Rosquist KJ, Lederman HM. Survival probability in ataxia telangiectasia. Arch Dis Child. 2006;91:610–1. doi: 10.1136/adc.2006.094268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rosen FS, Janeway CA. The gamma globulins. 3. The antibody deficiency syndromes. N Engl J Med. 1966;275:709–15. doi: 10.1056/NEJM196609292751307. contd. [DOI] [PubMed] [Google Scholar]
- 165.Notarangelo LD, Hayward AR. X-linked immunodeficiency with hyper-IgM (XHIM) Clin Exp Immunol. 2000;120:399–405. doi: 10.1046/j.1365-2249.2000.01142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Herve M, Isnardi I, Ng YS, Bussel JB, Ochs HD, Cunningham-Rundles C, et al. CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J Exp Med. 2007;204:1583–93. doi: 10.1084/jem.20062287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Facchetti F, Appiani C, Salvi L, Levy J, Notarangelo LD. Immunohistologic analysis of ineffective CD40-CD40 ligand interaction in lymphoid tissues from patients with X-linked immunodeficiency with hyper-IgM. Abortive germinal center cell reaction and severe depletion of follicular dendritic cells. J Immunol. 1995;154:6624–33. [PubMed] [Google Scholar]
- 168.Yilmaz GG, Yilmaz E, Coskun M, Karpuzoglu G, Gelen T, Yegin O. Cutaneous histoplasmosis in a child with hyper-IgM. Pediatr Dermatol. 1995;12:235–8. doi: 10.1111/j.1525-1470.1995.tb00166.x. [DOI] [PubMed] [Google Scholar]
- 169.Chang MW, Romero R, Scholl PR, Paller AS. Mucocutaneous manifestations of the hyper-IgM immunodeficiency syndrome. J Am Acad Dermatol. 1998;38:191–6. doi: 10.1016/s0190-9622(98)70239-7. [DOI] [PubMed] [Google Scholar]
- 170.Leone V, Tommasini A, Andolina M, Runti G, De Vonderweid U, Campello C, et al. Elective bone marrow transplantation in a child with X-linked hyper-IgM syndrome presenting with acute respiratory distress syndrome. Bone Marrow Transplant. 2002;30:49–52. doi: 10.1038/sj.bmt.1703581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Thivolet J, Hegazy MR, Viac J, Chardonnet Y. An in vivo study of cell-mediated immunity in human warts. Preliminary results. Acta Derm Venereol. 1977;57:317–9. [PubMed] [Google Scholar]
- 172.Viac J, Thivolet J, Hegazy MR, Chardonnet Y, Dambuyant C. Comparative study of delayed hypersensitivity skin reactions and antibodies to human papilloma virus (HPV) Clin Exp Immunol. 1977;29:240–6. [PMC free article] [PubMed] [Google Scholar]
- 173.Fruchter RG, Maiman M, Sedlis A, Bartley L, Camilien L, Arrastia CD. Multiple recurrences of cervical intraepithelial neoplasia in women with the human immunodeficiency virus. Obstet Gynecol. 1996;87:338–44. doi: 10.1016/0029-7844(95)00408-4. [DOI] [PubMed] [Google Scholar]
- 174.Fennema JS, van Ameijden EJ, Coutinho RA, van den Hoek AA. HIV, sexually transmitted diseases and gynaecologic disorders in women: increased risk for genital herpes and warts among HIV-infected prostitutes in Amsterdam. AIDS. 1995;9:1071–8. [PubMed] [Google Scholar]
- 175.Moscicki AB, Ellenberg JH, Farhat S, Xu J. Persistence of human papillomavirus infection in HIV-infected and -uninfected adolescent girls: risk factors and differences, by phylogenetic type. J Infect Dis. 2004;190:37–45. doi: 10.1086/421467. [DOI] [PubMed] [Google Scholar]
- 176.Palefsky J. Human papillomavirus-related disease in people with HIV. Curr Opin HIV AIDS. 2009;4:52–6. doi: 10.1097/COH.0b013e32831a7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Coleman N, Stanley MA. Characterization and functional analysis of the expression of vascular adhesion molecules in human papillomavirus-related disease of the cervix. Cancer. 1994;74:884–92. doi: 10.1002/1097-0142(19940801)74:3<884::aid-cncr2820740315>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 178.Stanley MA, S C, Coleman N. Cell mediated immunity and lower genital tract neoplasia. 2003 [Google Scholar]
- 179.Welters MJ, de Jong A, van den Eeden SJ, van der Hulst JM, Kwappenberg KM, Hassane S, et al. Frequent display of human papillomavirus type 16 E6- specific memory t-Helper cells in the healthy population as witness of previous viral encounter. Cancer Res. 2003;63:636–41. [PubMed] [Google Scholar]
- 180.Welters MJ, van der Logt P, van den Eeden SJ, Kwappenberg KM, Drijfhout JW, Fleuren GJ, et al. Detection of human papillomavirus type 18 E6 and E7- specific CD4+ T-helper 1 immunity in relation to health versus disease. Int J Cancer. 2006;118:950–6. doi: 10.1002/ijc.21459. [DOI] [PubMed] [Google Scholar]
- 181.Evans EM, Man S, Evans AS, Borysiewicz LK. Infiltration of cervical cancer tissue with human papillomavirus-specific cytotoxic T-lymphocytes. Cancer Res. 1997;57:2943–50. [PubMed] [Google Scholar]
- 182.Nakagawa M, Stites DP, Farhat S, Sisler JR, Moss B, Kong F, et al. Cytotoxic T lymphocyte responses to E6 and E7 proteins of human papillomavirus type 16: relationship to cervical intraepithelial neoplasia. J Infect Dis. 1997;175:927–31. doi: 10.1086/513992. [DOI] [PubMed] [Google Scholar]
- 183.Nakagawa M, Stites DP, Palefsky JM, Kneass Z, Moscicki AB. CD4-positive and CD8-positive cytotoxic T lymphocytes contribute to human papillomavirus type 16 E6 and E7 responses. Clin Diagn Lab Immunol. 1999;6:494–8. doi: 10.1128/cdli.6.4.494-498.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lee SJ, Cho YS, Cho MC, Shim JH, Lee KA, Ko KK, et al. Both E6 and E7 oncoproteins of human papillomavirus 16 inhibit IL-18-induced IFN-gamma production in human peripheral blood mononuclear and NK cells. J Immunol. 2001;167:497–504. doi: 10.4049/jimmunol.167.1.497. [DOI] [PubMed] [Google Scholar]
- 185.Bonagura VR, Hatam LJ, Rosenthal DW, de Voti JA, Lam F, Steinberg BM, et al. Recurrent respiratory papillomatosis: a complex defect in immune responsiveness to human papillomavirus-6 and -11. APMIS. 2010;118:455–70. doi: 10.1111/j.1600-0463.2010.02617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Carter JJ, Koutsky LA, Wipf GC, Christensen ND, Lee SK, Kuypers J, et al. The natural history of human papillomavirus type 16 capsid antibodies among a cohort of university women. J Infect Dis. 1996;174:927–36. doi: 10.1093/infdis/174.5.927. [DOI] [PubMed] [Google Scholar]
- 187.Fausch SC, Da Silva DM, Rudolf MP, Kast WM. Human papillomavirus virus-like particles do not activate Langerhans cells: a possible immune escape mechanism used by human papillomaviruses. J Immunol. 2002;169:3242–9. doi: 10.4049/jimmunol.169.6.3242. [DOI] [PubMed] [Google Scholar]
- 188.Tindle RW. Immune evasion in human papillomavirus-associated cervical cancer. Nat Rev Cancer. 2002;2:59–65. doi: 10.1038/nrc700. [DOI] [PubMed] [Google Scholar]
- 189.Stanley MA. Immune responses to human papilloma viruses. Indian J Med Res. 2009;130:266–76. [PubMed] [Google Scholar]
- 190.Kanodia S, Fahey LM, Kast WM. Mechanisms used by human papillomaviruses to escape the host immune response. Curr Cancer Drug Targets. 2007;7:79–89. doi: 10.2174/156800907780006869. [DOI] [PubMed] [Google Scholar]
- 191.Li M, Lee H, Guo J, Neipel F, Fleckenstein B, Ozato K, et al. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor. J Virol. 1998;72:5433–40. doi: 10.1128/jvi.72.7.5433-5440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mahr JA, Gooding LR. Immune evasion by adenoviruses. Immunol Rev. 1999;168:121–30. doi: 10.1111/j.1600-065x.1999.tb01287.x. [DOI] [PubMed] [Google Scholar]
- 193.Symons JA, Alcami A, Smith GL. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell. 1995;81:551–60. doi: 10.1016/0092-8674(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 194.Woodworth CD, Lichti U, Simpson S, Evans CH, DiPaolo JA. Leukoregulin and gamma-interferon inhibit human papillomavirus type 16 gene transcription in human papillomavirus-immortalized human cervical cells. Cancer Res. 1992;52:456–63. [PubMed] [Google Scholar]
- 195.Recommendations on the use of quadrivalent human papillomavirus vaccine in males--Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep. 2011;60:1705–8. [PubMed] [Google Scholar]
- 196.Markowitz LE, Dunne EF, Saraiya M, Lawson HW, Chesson H, Unger ER. Quadrivalent Human Papillomavirus Vaccine: Recommendations of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm Rep. 2007;56:1–24. [PubMed] [Google Scholar]
- 197.Palefsky JM, Giuliano AR, Goldstone S, Moreira ED, Jr, Aranda C, Jessen H, et al. HPV vaccine against anal HPV infection and anal intraepithelial neoplasia. N Engl J Med. 2011;365:1576–85. doi: 10.1056/NEJMoa1010971. [DOI] [PubMed] [Google Scholar]
- 198.Giuliano AR, Palefsky JM, Goldstone S, Moreira ED, Jr, Penny ME, Aranda C, et al. Efficacy of quadrivalent HPV vaccine against HPV Infection and disease in males. N Engl J Med. 2011;364:401–11. doi: 10.1056/NEJMoa0909537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lehtinen M, Paavonen J, Wheeler CM, Jaisamrarn U, Garland SM, Castellsague X, et al. Overall efficacy of HPV-16/18 AS04-adjuvanted vaccine against grade 3 or greater cervical intraepithelial neoplasia: 4-year end-of-study analysis of the randomised, double-blind PATRICIA trial. Lancet Oncol. 2012;13:89–99. doi: 10.1016/S1470-2045(11)70286-8. [DOI] [PubMed] [Google Scholar]
- 200.Wheeler CM, Castellsague X, Garland SM, Szarewski A, Paavonen J, Naud P, et al. Cross-protective efficacy of HPV-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by non-vaccine oncogenic HPV types: 4-year end-of-study analysis of the randomised, double-blind PATRICIA trial. Lancet Oncol. 2012;13:100–10. doi: 10.1016/S1470-2045(11)70287-X. [DOI] [PubMed] [Google Scholar]