

Abstract
Flavoprotein autofluorescence imaging was used to examine auditory cortical synaptic responses in aged animals with behavioral evidence of tinnitus and hearing loss. Mice were exposed to noise trauma at 1–3 months of age and were assessed for behavioral evidence of tinnitus and hearing loss immediately after the noise trauma and again at ∼24–30 months of age. Within 2 months of the final behavioral assessment, auditory cortical synaptic transmission was examined in brain slices using electrical stimulation of putative thalamocortical afferents, and flavoprotein autofluorescence imaging was used to measure cortical activation. Noise-exposed animals showed a 68% increase in amplitude of cortical activation compared with controls (p = 0.008), and these animals showed a diminished sensitivity to GABAAergic blockade (p = 0.008, using bath-applied 200 nm SR 95531 [6-Imino-3-(4-methoxyphenyl)-1(6H)-p yridazinebutanoic acid hydrobromide]). The strength of cortical activation was significantly correlated to the degree of tinnitus behavior, assessed via a loss of gap detection in a startle paradigm. The decrease in GABAA sensitivity was greater in the regions of the cortex farther away from the stimulation site, potentially reflecting a greater sensitivity of corticocortical versus thalamocortical projections to the effects of noise trauma. Finally, there was no relationship between auditory cortical activation and activation of the somatosensory cortex in the same slices, suggesting that the increases in auditory cortical activation were not attributable to a generalized hyperexcitable state in noise-exposed animals. These data suggest that noise trauma can cause long-lasting changes in the auditory cortical physiology and may provide specific targets to ameliorate the effects of chronic tinnitus.
Introduction
Tinnitus is a common and often disabling symptom that increases in prevalence with age. It is characterized by the presence of abnormal spontaneous auditory percepts and is often associated with other symptoms, such as hyperacusis. Most population studies place the prevalence of tinnitus at 5–20%, with the proportion of those having moderate to severe symptoms being 20–30% (Møller, 2011). Tinnitus is almost always associated with peripheral hearing loss. This has led to the hypothesis that damage to peripheral auditory structures leads to central reorganization and that this maladaptive reorganization may lead to tinnitus. Several specific mechanisms for tinnitus have been proposed, including pathological synchronous cortical activation (Eggermont, 2007), homeostatic synaptic plasticity (Yang et al., 2011), or diminished synaptic inhibition (Wang et al., 2011).
The finding that tinnitus may be caused by damage to peripheral hearing structures has led to the development of several different animal models. These models typically involve a lesion of the peripheral hearing apparatus (Kaltenbach, 2011), and studies in these tinnitus models have revealed increases in spontaneous and driven activity seen in cochlear nucleus (Kaltenbach et al., 1998; Brozoski et al., 2002; Middleton et al., 2011), inferior colliculus (Willott and Lu, 1982; Salvi et al., 1990; Szczepaniak and Møller, 1995), and auditory cortex (Yang et al., 2011). These findings are consistent with the notion that peripheral hearing loss causes an increase in excitability in central auditory structures, possibly to enhance the processing of weakened peripheral sensory input (Wang et al., 2011). Unfortunately, most animal-based studies in the literature have used relatively young animals and studied the development of tinnitus in the acute or subacute phase after injury. However, tinnitus is most commonly encountered in the elderly population and frequently persists for many years. Therefore, in the current study, the functional consequences of chronic tinnitus in an aging population of mice were examined.
Although multiple central auditory structures have been implicated in tinnitus, recent findings point to a potential role for the auditory cortex (Mühlnickel et al., 1998; van der Loo et al., 2009; Noreña et al., 2010; Schecklmann et al., 2011; Stolzberg et al., 2011; Yang et al., 2011). Understanding the precise mechanisms underlying changes in auditory cortical processing mechanisms during tinnitus has important therapeutic implications because of recent attempts to treat tinnitus via direct modulation of auditory cortical excitability (Kleinjung et al., 2008). One difficulty in studying central mechanisms of tinnitus has been the separation of peripheral versus central changes in synaptic organization. Brain slice preparations may therefore be useful because they allow the isolation of potentially affected subcircuits within the auditory system.
Here, we examined the earliest level of cortical processing by studying the effects of stimulating afferents to the auditory cortex in animals with behavioral evidence of tinnitus. Auditory cortical activation was measured using flavoprotein autofluorescence, which permits the measurement of neural activity with high sensitivity and spatial resolution, without the potentially confounding factors present when using exogenous dyes, such as differential uptake or dye toxicity (Shibuki et al., 2003; Llano et al., 2009). In addition, given evidence for GABAergic (Wang et al., 2011) and NMDA (Lobarinas et al., 2006; Hwang et al., 2011) dysfunction in tinnitus, pharmacologic manipulations of these systems was also done.
Materials and Methods
Mice.
A total of 13 CBA/CAj male mice were used in this study. Six mice were exposed to closed-field unilateral noise stimulus, four mice were given sham treatment, and three mice did not receive sham treatment but were matched for age with the rest of the cohort. The data for the sham-treated and untreated older mice were similar and were pooled. All procedures used in the research protocol were approved by the Southern Illinois University School of Medicine and the University of Illinois Laboratory Animal Care and Use Committees (protocols #P190-06-005 and #10208, respectively), conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and followed the Society for Neuroscience Guidelines for the Use of Animals in Neuroscience Research.
Noise trauma and behavioral testing.
Mice were pretested using the gap prepulse inhibition (PPI) of acoustic startle procedure (Turner et al., 2006) and placed into either noise trauma or control (sham trauma) groups. The noise trauma consisted of a peak-calibrated level of 116 dB, centered on a 16 kHz octave band. The noise exposure was presented unilaterally to isoflurane-anesthetized mice for 1 h. Mice were of average age of 66 d at the time of noise exposure or sham.
Behavioral testing was conducted using startle reflex hardware and software, customized for this application by the manufacturer (Kinder Scientific). Background sounds in the startle chamber consisted of 60 dB SPL, 1000 Hz bandpass filtered noise (48 dB/octave roll off; Krohn-Hite model 3988) centered at 4, 8, 10, 12.5, 16, 20, 24, and 32 kHz and broadband noise. Gap and PPI testing used background sounds presented through one speaker (Vifa XT25TG30-04) and startle stimuli presented through a second speaker (Powerline CTS KSN-1005) located in the ceiling of the testing chamber, 15 cm above the animal's head. PPI was measured as a control condition to assess for hearing loss. Prepulse stimuli matched the backgrounds used in gap testing in both spectral and intensity dimensions. For schematic of PPI sound sequence, see Figure 1A. PPI responses were expressed as the ratio of the motor response to the prepulse to the motor response of the startle pulse, where 1.0 equals no PPI (suggesting loss of the ability to detect the prepulse). Gap testing was used to assess behavioral evidence of tinnitus. For schematic of the gap testing sequence, see Figure 1B. Gap ratio equals the ratio of the response to the startle stimulus without gap, to the startle stimulus with a preceding gap. Because animals with tinnitus perceive sound during the gap period, they will show reduction in the inhibition of the startle normally cued by the gap (i.e., the gap ratio will approach 1.0). Occasionally, values >1.0 are seen, and these are likely related to the fact that the technique has a floor of zero but no ceiling, coupled with behavioral variability seen in the technique. A specific value for the gap ratio was not used to assign mice to belonging to a “tinnitus” group (see below, Analysis). The use of gap-induced reduction of startle for the assessment of tinnitus has been validated against conditioning-based approaches to tinnitus and has been shown to be a reliable metric across several species (Turner, 2007; Longenecker and Galazyuk, 2011; Turner et al., 2012).
Figure 1.

Behavioral assessment of hearing loss and tinnitus behavior in noise-exposed animals. The top images illustrate temporal sequences of sounds used to assess PPI (A) and gap detection (B). Bar graphs on the left illustrate the mean and SE of the PPI ratio (defined in Materials and Methods) of control animals (n = 4) and noise-exposed animals (n = 6). Bar graphs on the right illustrate mean and SE values for the gap detection ratio of control animals (n = 4) and noise-exposed animals (n = 6). p values are derived from Mann–Whitney U test.
Auditory brainstem response thresholds.
Auditory brainstem response (ABR) thresholds were obtained for each ear immediately before and after unilateral noise exposure. However, exit ABRs were not obtained on all animals because several animals died during ABR testing, likely related to their old age and use of anesthesia at the time of testing.
Brain slicing.
We used a slight modification of the thalamocortical brain slice preparation developed by Cruikshank et al. (2002). This preparation was used to increase the likelihood of capturing thalamocortical afferents to the auditory cortex. Chemoarchitectonic criteria (see Fig. 2), suggests that the imaging data were derived from primary auditory fields (primary auditory cortex and/or anterior auditory field). Use of the term “auditory cortex” within this report refers to primary auditory fields and makes no attempt to distinguish between primary auditory cortex and anterior auditory field.
Figure 2.
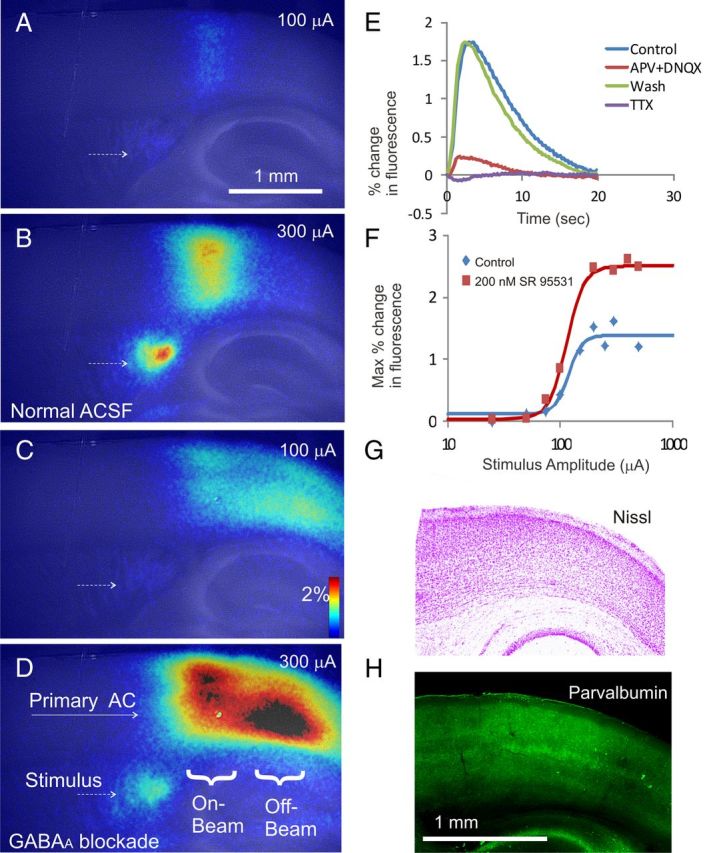
A–D, Δf/f maps of auditory cortical activations superimposed on raw fluorescence images under conditions of 100 μA stimulus and normal ACSF (A), 300 μA stimulus and normal ACSF (B), 100 μA stimulus and 100 nm SR 95331 (C), and 300 μA stimulus and 200 nm SR 95531 (D). Small dotted arrows show the stimulus site. R, Rostral; C, caudal; L, lateral; M, medial. Color bar shows percentage change in fluorescence relative to baseline. E, Percentage change in fluorescence over time (averaged over 5 runs, presented 20 s apart), before any drugs were presented (Control), after exposure to 100 μm APV and 80 μm DNQX (APV + DNQX), after washout of APV and DNQX (Wash), or after exposure to 1 μm tetrodotoxin (TTX). F, Amplitude, expressed as maximum percentage increase in fluorescence, at a series of stimulus amplitudes from 25 to 500 μA, for slice shown in A–D. Raw data were fit with a four-parameter sigmoid model. Nissl staining (G) and parvalbumin immunostaining (H) of sections taken from the same slice.
Brain slices were obtained containing thalamocortical projections contralateral to the ear that received noise trauma. In brief, mice were initially anesthetized with a ketamine (1 mg/g)/xylazine (10 mg/g). Mice were then perfused with chilled (4°C) sucrose-based slicing solution (in mm): 234 sucrose, 11 glucose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 10 MgCl2, and 0.5 CaCl2. The brain was blocked by removing the olfactory bulbs and the anterior 2 mm of frontal cortex with a razor blade. The brain was then placed on its cut surface, and an off-horizontal cut was made from the dorsal surface. This cut was angled at 20° from the horizontal plane such that a small dorsal flap was removed. The brain was then glued using the newly cut surface onto a cutting platform, and 600-μm-thick sections were cut. Sections were allowed to incubate at 32°C for 1 h before use in an incubation solution (26 NaHCO3, 2.5 KCl, 10 glucose, 126 NaCl, 1.25 NaH2PO4, 3 MgCl2, and 1 CaCl2). For flavoprotein experiments, slices were transferred to a chamber perfused with ACSF (26 NaHCO3, 2.5 KCl, 10 glucose, 126 NaCl, 1.25 NaH2PO4, 1 MgCl2, and 3 CaCl2) at room temperature, bubbled with 95% oxygen/5% carbon dioxide. The experimenter was blinded to the status of the animals until after the experiment was completed.
Stimulation and flavoprotein imaging.
Glass micropipettes were broken back to a diameter of 50 μm, filled with ACSF, and gently placed onto a fiber bundle ∼100–200 μm ventral to the auditory cortex. The stimulating electrode was placed in the subcortical white matter rather than the thalamus because, in our experience and that of others (Broicher et al., 2010), there is a great deal of slice-to-slice variability in the ability to capture intact projections from thalamus to cortex. This would make it difficult in a relatively small sample size to see differences between noise-exposed and control animals. In several experiments, the stimulating electrode was moved 2 mm rostrally to the white matter beneath the secondary somatosensory cortex (Franklin and Paxinos, 2007) to determine the specificity of the findings to the auditory cortex. Trains of electrical pulses were delivered using PowerLab software and hardware and a World Precision Instruments stimulus isolator. Trains were 1 s in duration, with 2 ms pulses, delivered at 40 pulses/s, and trains were delivered every 20 s, with five repetitions total.
Flavoprotein autofluorescence was used to measure cortical activation. This technique takes advantage of the endogenous fluorescence of mitochondrial flavoproteins. These molecules fluoresce when oxidized and will cause neuronal tissue to fluoresce when active. Flavoprotein autofluorescence has been shown to be a highly stable, sensitive, and specific marker for neuronal activation, both in vivo and in vitro (Shibuki et al., 2003; Husson et al., 2007; Llano et al., 2009; Reinert et al., 2011). A fluorescence illuminator (either a Prior Lumen 200 or Olympus X-Cite Series 120 PC Q) and a UMNIB Olympus filter cube (470–490 nm excitation, 505 nm dichroic, 515 nm emission long pass) was used for fluorescence optics. A coverslip was placed over the slice to provide a stable imaging plane. All data were collected at four frames per second using an infinity-corrected 4× objective (NA 0.13) and a Retiga EXi camera using 4 × 4 binning and StreamPix software.
Drugs.
Two hundred nanomolar SR 95531 [2-(3-carboxypropyl)-3-amino-6-(4-methoxyphenyl)pyridazinium bromide], 50 μm APV, 100 μm DNQX, and 1 μm tetrodotoxin were all purchased from Tocris Bioscience. In all cases, drugs were dissolved in ACSF just before the experiment, and 1 h wash-in and 1 h washout times were used.
Histology.
Cytochrome oxidase staining, parvalbumin immunohistochemistry, and Nissl staining were used to assist in identifying imaged regions in brain slices. Previous work suggested that these markers separate primary auditory fields (primary auditory cortex and anterior auditory field) from nonprimary fields (Caviness, 1975; Wallace et al., 1991; Molinari et al., 1995; Budinger et al., 2000; Cruikshank et al., 2001; Llano and Sherman, 2008) Slices were stored in 4% paraformaldehyde in PBS after each experiment. They were then placed in an ascending sucrose gradient up to 30% sucrose in PBS, frozen, and resectioned on a freezing microtome at 75 μm. For immunostaining, sections were washed in PBS, placed in a microwave for 15 s to increase antigenicity, and then incubated for 30 min in PBS containing 0.3% Triton X-100, followed by 3% normal goat serum (NGS). Sections were then incubated with primary antibody solution consisting of monoclonal anti-parvalbumin produced in mouse [1:1000, catalog #P3088 (Sigma), in 0.3% Triton X-100 and 3% NGS in PBS]. Secondary antibody consisted of 1:100 goat anti-mouse IgG coupled to Alexa Fluor 488 (catalog #A-11001; Invitrogen), to produce green fluorescence. For cytochrome oxidase staining, the method of Wong-Riley (1979) was used. Sections were incubated with a solution of 0.05% DAB, 4% sucrose, and 20% cytochrome c (Sigma) in PBS in the dark at 37°C for 4 h and quenched with PBS.
Analyses.
For behavioral data, two complementary types of analysis were done. First, a categorical analysis based on noise-exposure or no-exposure/controls was done. No attempt was made to segregate animals based on the presence or absence of tinnitus. Second, a correlation analysis was done to assess whether a quantitative relationship exists between tinnitus-related deficits and cortical activation measurements. This was done because it is likely that some noise-exposed animals may not develop tinnitus and because some older mice (like some older humans) may spontaneously develop hearing loss and tinnitus. Here, all animals were pooled, and no assumptions were made about the development of tinnitus based on group assignment.
For imaging data, in each run, a total of 420 images were collected, and image size was 260 × 348 pixels (using 4 × 4 hardware binning). The first 5 s (20 frames) were used as a baseline for the subsequent images. All image analysis, except curve fitting, was done using custom software written in MATLAB. Time-based analyses were done by computing change in fluorescence as a function of baseline fluorescence. Time series for each pixel were corrected for quenching by high-pass filtering each time series. Several regions of interest were used for analysis. An “on-beam” region of interest was constructed centered on the column of activation seen without blockade of GABAA receptors (see Fig. 2). For this, a 1 mm (rostral–caudal) region of interest from the pial surface to the gray–white junction was used, and a change in fluorescence was computed, relative to the first 5 s (20 frames) of the run. An additional region of interest, termed the “off-beam” region, was of same width of the above-described on-beam region (1 mm) but was displaced caudally from the on-beam region and did not overlap the on-beam region. This off-beam regions corresponds to most of the remainder of the primary auditory cortex not encompassed in the on-beam region (Cruikshank et al., 2002; Hackett et al., 2011). For each stimulation site, a series of stimulation amplitudes were used, ranging from 5 to 500 μA, and an input–output curve was constructed. In all cases, input–output curves resembled a sigmoidal function, and therefore a traditional four-parameter dose–response curve using ReaderFit software was used to fit the data. From the fit curves, the minimum, maximum, midpoint (EC50), and slope could be extracted.
To compute the width of activation, a region of interest comprising the whole cortex was used. Images from runs at saturation stimulus amplitudes were imported into NIH ImageJ (http://rsbweb.nih.gov/ij/), and the curvature of the cortex was corrected using the “Straighten” function in NIH ImageJ. NIH ImageJ was then used to collapse all pixel values from pia to the white matter into a linear array of bins, and this histogram was used to compute a full-width at half-maximum.
For image display, spatial averaging was done using a 3 × 3 kernel. Change in fluorescence over baseline fluorescence maps (Δf/f maps) were overlaid on raw fluorescence images for localization of anatomic structures.
Statistics.
Given the relatively small numbers of animals in each group, nonparametric statistics were used. To compare across groups (control vs noise-exposed), a Mann–Whitney U test statistic was used, and, for correlation analysis, a Spearman's correlation was calculated. p < 0.05 was taken as an indicator of statistical significance. Two-tailed testing was done for all analysis except for behavioral testing (PPI ratio and gap ratio), in which one-tailed testing was done. This is because responses are indexed against a theoretical minimum of zero (for PPI) or theoretical maximum of 1.0 (for gap), and only deviations upward (for PPI) or downward (for gap) are of relevance. Mann–Whitney test and Spearman's correlations were run using GraphPad software. To assess interactions between noise-exposure and location in auditory cortex (on-beam vs off-beam) or layer of cortex (supragranular vs infragranular), a mixed-effects model was used. Data were log transformed to maintain normality, and post hoc analysis was performed to assess for an interaction of condition by brain region. Mixed-effects modeling was done in R (http://www.r-project.org/).
Results
Thirteen aged mice were used in this study. Six animals were exposed to noise trauma, four animals were exposed to sham conditions, and three additional age-matched animals were included in the control dataset. The mean ± SD age at the time that thalamocortical slices were cut did not significantly differ in the two groups (controls, 26.7 ± 3.6 months; tinnitus animals, 28.6 ± 2.6 months; p = 0.285).
Animals were assessed for hearing loss at an average ± SD of 24.7 ± 1.7 months after the noise exposure or sham. At 16 kHz, noise-exposed mice demonstrated an increase in PPI ratio relative to controls (Fig. 1A), suggesting that these noise-exposed animals had hearing loss at this frequency. There was no increase in PPI ratio at 12.5 kHz; therefore, 12.5 kHz frequency was used for assessment of gap detection. An increase in gap ratio was observed in the noise-exposed animals at 12.5 kHz (Fig. 1B), suggesting that these animals had behavioral evidence of tinnitus. No significant differences in gap ratio were seen between groups when tested at frequencies from 4 to 10 kHz or from 20 to 31 kHz.
Cortical responses were measured in all animals. In all cases, electrical stimulation of the white matter ventral to the cortex produced a robust change in fluorescence in a column of the overlying auditory cortex (Fig. 2A,B). Exposure of the slices to 200 nm SR 95331 caused a significant increase in the magnitude and breadth of the cortical response to axonal stimulation (Fig. 2C,D). The concentration of 200 nm SR 95331 was chosen because, in this preparation, we have found that concentrations higher than 500 nm often lead to paroxysmal activity. The increase in breadth of the response in all tissues was anisotropic, that is, was only seen extending into the caudal direction.
Under conditions of normal ACSF, the peak of activation occurred within ∼2 s of the stimulus onset and then gradually returned to baseline over the subsequent 10–20 s. As shown in Figure 2E, the peak in the response dropped by >85% in response to blockade of ionotropic glutamate receptors using 100 μm APV and 80 μm DNQX and dropped by >95% with the use of tetrodotoxin, suggesting that the changes in cortical fluorescence are caused by axonal stimulation and that the majority of the response is driven by orthodromic and likely thalamocortical stimulation. In all cases, gradual increases of stimulation amplitude from 5 to 500 μA produced a nonlinear increase in response amplitude that resembled sigmoidal response function (Fig. 2F). Nissl staining (Fig. 2G) and immunostaining for parvalbumin (Fig. 2H) of different sections taken from the same slice reveal that the area of flavoprotein activation corresponds to an area of cortex with granularity in layer 4 and strong parvalbumin immunopositivity relative to adjacent regions, respectively. Based on these findings and previous work (Cruikshank et al., 2002; Hackett et al., 2011), this area likely corresponds to the primary auditory cortex.
Cortical response magnitude differed between the control animals and the noise-exposed animals. Examples of cortical activation are shown in Figure 3, A and B, demonstrating a substantially greater response in the noise-exposed animal. As shown in Figure 4A, the mean ± SD on-beam response to afferent stimulation in control animals was 1.9 ± 0.5% (n = 7) above baseline, whereas in noise-exposed animals, this increase was 3.2 ± 0.8% (n = 6; p = 0.008, Mann–Whitney U test), yielding a mean increase in response amplitude of 68%. Blockade of GABAA receptors with 20 nm SR 95531 increased the strength of the responses in both groups, such that the responses under conditions of GABAA blockade were not significantly different (mean ± SD, 4.29 ± 1.36% vs 3.48 ± 0.96%; p = 0.29, Mann–Whitney U test). Sensitivity of this response to GABAA blockade was assessed using the ratio of the cortical response under conditions of GABAA blockade to conditions when normal ACSF was used. For control animals, the mean ± SD ratio was 1.86 ± 0.33 (n = 7), whereas in noise-exposed animals, this ratio decreased to a mean ± SD value of 1.32 ± 0.22 (n = 6; p = 0.008, Mann–Whitney U test; Fig. 4B). Full-width at half-maximum trended higher in noise-exposed animals than control animals (mean ± SD, 1215 ± 371 vs 1500 ± 223 μm; p = 0.13) but did not reach the 0.05 threshold.
Figure 3.

Examples of Δf/f cortical activation maps for a control animal (A) and a noise-exposed animal (B). Stimulus amplitudes/parameters were identical: 300 μA, 2 ms duration pulses at 40 pulses/s for 1 s. Color bar corresponds to the percentage increase in fluorescence over baseline.
Figure 4.

Comparison of strength of cortical activation and GABAAergic sensitivity in the on-beam and off-beam regions of the auditory cortex (left and right, respectively). Top row shows the maximum increase in fluorescence. Bottom row shows the ratio of the response under conditions of exposure to 200 nm SR 95331 to normal ACSF. Each diamond or triangle represents a single slice. Horizontal lines represent mean value. P values are derived from Mann–Whitney U test.
The off-beam regions of cortex were analyzed for strength of activation and sensitivity to SR 95531. Analysis of the strength of activation in these zones in animals revealed that noise-exposed animals had a significantly greater activation by greater than twofold compared with control animals (Fig. 4C; mean ± SD, control, 0.62 ± 0.23%; tinnitus, 2.1 ± 0.9%; p = 0.003, Mann–Whitney U test). In addition, noise-exposed animals demonstrated significantly less sensitivity to SR 95531 than controls (Fig. 4D; mean ± SD ratio of response under SR 95531 conditions vs normal ACSF in control animals, 5.45 ± 2.4 and tinnitus animals, 1.82 ± 0.49; p = 0.003, Mann–Whitney U test). In addition, in a mixed-effects model analysis, condition × brain region interaction showed a significant effect of brain region on the difference between tinnitus and control animals for both amplitude of activation (p = 0.01) and sensitivity to SR 95531 (p = 0.01).
In addition, increases in cortical activation were seen in noise-exposed animals in both the infragranular (1.69 vs 2.67%; p = 0.022) and supragranular (2.11 vs 4.43%; p = 0.001) layers (Fig. 5). An interaction between noise exposure and cortical layers was observed, such that there was a greater increase in the supragranular layers (mixed-effect model, p = 0.01).
Figure 5.

Comparison of strength of cortical activation in the infragranular and supragranular layers of the auditory cortex (A and B, respectively). Each diamond or triangle represents a single slice. Horizontal lines represent mean value. P values are derived from Mann–Whitney U test.
Slices from 11 animals (six controls and five noise-exposed) were exposed to 100 μm APV to assess the effects of NMDA blockade on cortical activation. APV induced a modest drop in the cortical response that did not differ between control versus noise-exposed animals (mean drop of 27 vs 22% in control vs noise-exposed; p = 0.646, Mann–Whitney U test; Fig. 6).
Figure 6.

A, Amplitude of the on-beam cortical response in the animals that were tested for APV sensitivity. B, Amplitude of the cortical activation in the same slices shown in A after exposure to 100 μm APV. C, The ratio of the cortical response under conditions of APV exposure to conditions of normal ACSF exposure. Each diamond or triangle represents a single slice. Horizontal lines represent mean value. p values are derived from Mann–Whitney U test.
To assess whether the increase in auditory cortical responsiveness was attributable to a generalized increase in excitability versus a specific change in the auditory cortical system, responses in the auditory cortex were compared with responses in the somatosensory cortex. Identical analysis protocols were used for the two cortical areas. The stimulation site for the somatosensory cortex was ∼2 mm rostral to the site for the auditory cortex (Fig. 7A). This area of cortex does not show barrel architecture, confirming that this is non-barrel cortex (Fig. 7B). An example of a flavoprotein response in the somatosensory cortex is shown in Figure 7C. In general, somatosensory responses were smaller than those in the auditory cortex, but the response magnitudes were uncorrelated (Fig. 7D; R2 = 0.51, p = 0.154, n = 6), suggesting that changes described above in the auditory cortex were not attributable to a generalized change in cortical excitability.
Figure 7.

A, Diagram illustrating the relative locations of stimulating electrodes for auditory cortex (AC) stimulation (left) and secondary somatosensory cortex (S2) stimulation (right). B, Cytochrome oxidase-stained section from same slice, demonstrating lack of barrel architecture in the region of cortical activation. SCWM, Subcortical white matter; R, rostral; C, caudal; L, lateral; M, medial. C, Example of a response in secondary somatosensory cortex to 200 μA stimulus to the subcortical white matter. D, Correlation between the magnitude of activation in the auditory cortex to the magnitude of activation in the somatosensory cortex in the same slices. Black circles, Noise-exposed animals; black triangles, sham-treated animals. R2 and p values generated from Spearman's correlations.
Correlations between behavioral measures of hearing loss and tinnitus, quantified using a paired-pulse protocol and gap detection protocol, respectively, and cortical responses, were assessed. There was a significant (p = 0.024) trend for a positive association between gap detection ratio at 12.5 kHz and cortical response magnitude (Fig. 8A; R2 = 0.52, p = 0.024). There was a nonsignificant trend for a positive correlation between the PPI ratio at 12.5 kHz and cortical response magnitude (Fig. 8B; R2 = 0.31, p = 0.081).
Figure 8.

A, Correlation between maximum increase in fluorescence versus gap startle ratio at 12.5 kHz. B, Correlation between maximum increase in fluorescence versus PPI ratio at 16 kHz. Black diamonds, Noise-exposed animals; black triangles, sham-treated animals. R2 and p values generated from Spearman's correlations.
Discussion
In the current study, we show that aged animals with noise exposure in young adulthood and subsequent behavioral evidence of hearing loss and tinnitus demonstrate changes in auditory cortical responses to afferent stimulation. Specifically, noise-exposed animals show a greater magnitude of auditory cortical activation and show a diminished sensitivity to GABAA blockade than control animals. These findings appeared to be more prominent in the upper cortical layers, specific to the auditory cortex (rather than the somatosensory cortex), and other receptor systems, such as the NMDA system, did not appear to be altered in tinnitus animals in this preparation.
Methodological considerations
The current study is limited by the blind approach to stimulation and imaging in the auditory thalamocortical slice preparation. Although noise damage was induced at a particular range of frequencies (1 octave band centered at 16 kHz), the frequency representation of the stimulated area of cortex in the thalamocortical slice preparation is not known. Although it is possible to map frequency representations before slicing using either electrophysiological means (Hackett et al., 2011) or non-invasive imaging (Takahashi et al., 2006), this was not attempted here given the overall frailty of these aged animals. It should be noted that, despite this limitation, we did observe significant changes in the blind activations of the auditory cortex, similar to changes seen with blind stimulation of the afferents to the cochlear nucleus (Middleton et al., 2011). Future work will involve auditory cortical mapping before slice preparation to better understand the relationship between the spectral characteristics of noise exposure and alterations in cortical activations in relation to the tonotopic map of the auditory cortex.
It should be noted that the specific circuitry driving the cortical activation signal is not known. Given the substantial drop (>85%) in cortical activation after blockade of NMDA and AMPA receptors (Fig. 1E), similar to our previous work in the somatosensory thalamocortical slice (Llano et al., 2009), we assume that most of the signal is driven by orthrodromic activation of thalamocortical afferents. However, there are other circuit elements, such as short-range corticocortical activations, that may be responsible for the fluorescence signal and may contribute to the differences seen between control and tinnitus animals.
Mechanisms underlying noise-induced changes
The specific behavioral consequence of noise exposure that is associated with the changes in cortical responses described here is not known. Animals in this study demonstrated evidence for both hearing loss (evidenced by the elevation of PPI ratios at 16 kHz) and tinnitus (evidenced by the elevation of the gap ratio at 12.5 kHz). Therefore, it is not clear whether or not the physiological changes in auditory cortical activation that were observed are driven by hearing loss, tinnitus, or other factors not tested, such as hyperacusis (Eggermont, 2012). In this case, the tinnitus behavior and hearing loss were partially separable based on sound frequency. It is not uncommon for the tinnitus frequency to differ from the center frequency of noise trauma (Longenecker and Galazyuk, 2011) and may often be lower than the traumatic acoustic stimulus in chronic stages, especially in older animals (Wang et al., 2008). It should be noted that the frequency at which tinnitus-related deficits were most prominent in the current study, 12.5 kHz, is well within the bandwidth of the trauma stimulus (1 octave band centered at 16 kHz). There are multiple studies showing that hearing loss induced by noise or other mechanisms other than acoustic trauma (e.g., salicylate), with or without a behavioral assessment of tinnitus, can induce similar changes in the auditory cortex (Wang et al., 2006; Sarro et al., 2008; Sun et al., 2009, 2012; Xu et al., 2010a,b). In addition, a recent report demonstrates that early conductive hearing loss can lead to long-lasting decreases in cortical inhibition (Takesian et al., 2012). These findings suggest that hearing loss may play a significant role in the results described here. Future studies that relate the spectral characteristics of the animals' behavior to the spectral representations in the auditory cortex will help to clarify the relative contributions of hearing loss versus tinnitus in changes in auditory cortical activation.
The data presented here are consistent with the hypothesis that tinnitus may be associated with cortical hyperexcitability caused by a downregulation of cortical GABAA synaptic transmission. Although a substantial body of previous work has suggested that cortical changes may be found in tinnitus (Arnold et al., 1996; Mühlnickel et al., 1998; Andersson et al., 2000), the current study, by disconnecting the auditory forebrain from its peripheral inputs, demonstrates that at least a portion of the cortical changes seen in tinnitus are generated locally. The data demonstrating that noise-exposed animals show greater activations and lower GABAA sensitivity in the off-beam regions of cortex also suggest that the cortical changes may not be restricted to a particular part of the auditory cortex and may be greater in the corticocortical projections than the thalamocortical projections. These data are consistent with previous work demonstrating diminished cortical GABAergic transmission (Wang et al., 2011), although these changes are likely dependent on the spectral content of the noise trauma and pitch of tinnitus percept (Yang et al., 2011).
GABAergic inhibition appeared sensitive to noise exposure. It is known that feedforward inhibition can critically shape cortical information processing. For example, thalamocortical projections are known to make powerful feedforward synaptic connections onto intrinsic inhibitory interneurons (Cruikshank et al., 2007; Schiff and Reyes, 2012). In addition, GABAergic interneurons are interposed between corticocortical projections, often have their synapses positioned at proximal locations on the dendritic shaft (Gonchar and Burkhalter, 1999), and therefore have significant effect on corticocortical projections (Swadlow, 2003). There are multiple types of intrinsic GABAergic neurons in the cortex (Porter et al., 2001; Dumitriu et al., 2007; Gonchar et al., 2007), and it is not known which of these may be downregulated after noise damage. It is possible that thalamocortical, corticocortical, or both may be effected by tinnitus, and the identification of the subtype of downregulated GABAergic neurons may potentially provide a specific therapeutic target for the treatment of tinnitus. Indeed, drugs that enhance GABAAergic synaptic transmission, such as the benzodiazepines, have been shown to provide some relief for the symptoms of tinnitus but come at the cost of sedation (Bahmad et al., 2006; Darlington and Smith, 2007). Therefore, more specific targeting of subpopulations of involved neurons may prove to have therapeutic value. Other therapies that dampen synaptic transmission, such as transcranial magnetic stimulation, may also has been used to target the cortical networks underlying tinnitus (Kleinjung et al., 2008), and the mechanism for the potential efficacy of this approach may lie in the hyperexcitability described herein.
Finally, this work may shed light on the influence of aging on tinnitus. In this study, tinnitus behavior and changes in cortical responsiveness were observed well over 1 year after the noise trauma and were observed in a population of animals of an age range known to be associated with significant hearing loss (Zheng et al., 1999). In addition, previous work has suggested that aging is associated with drops in cortical GABAergic transmission (Ling et al., 2005; Caspary et al., 2008; Burianova et al., 2009; Hughes et al., 2010), such that there may be interactions between the drops in cortical GABAAergic transmission associated with normal aging and with tinnitus. It is not known whether aging plus noise exposure depress auditory cortical GABA levels decreased beyond what would be expected from either insult on its own. However, such a hypothesized “double-hit” mechanism may help explain the high levels of tinnitus seen with aging (Møller, 2011). In addition, given the known differences in the physiology and GABA sensitivity seen in the auditory cortex with aging, it is likely that any potential new therapeutic mechanism for tinnitus may work differently in aged versus young subjects. Therefore, it will be important to use both aged and young animals in the translational approaches to the development of new therapeutic modalities for tinnitus.
Footnotes
This work was supported by the National Institute on Deafness and Other Communication Disorders (D.A.L., D.M.C.) and the Tinnitus Research Consortium (J.T.). We thank Deb Larsen and Alexandria Lesicko for their expert technical assistance.
References
- Andersson G, Lyttkens L, Hirvel ä C, Furmark T, Tillfors M, Fredrikson M. Regional cerebral blood flow during tinnitus: a PET case study with lidocaine and auditory stimulation. Acta Otolaryngol. 2000;120:967–972. doi: 10.1080/00016480050218717. [DOI] [PubMed] [Google Scholar]
- Arnold W, Bartenstein P, Oestreicher E, Römer W, Schwaiger M. Focal metabolic activation in the predominant left auditory cortex in patients suffering from tinnitus: a PET study with [18F]deoxyglucose. ORL J Otorhinolaryngol Relat Spec. 1996;58:195–199. doi: 10.1159/000276835. [DOI] [PubMed] [Google Scholar]
- Bahmad FM, Jr, Venosa AR, Oliveira CA. Benzodiazepines and GABAergics in treating severe disabling tinnitus of predominantly cochlear origin. Int Tinnitus J. 2006;12:140–144. [PubMed] [Google Scholar]
- Broicher T, Bidmon HJ, Kamuf B, Coulon P, Gorji A, Pape HC, Speckmann EJ, Budde T. Thalamic afferent activation of supragranular layers in auditory cortex in vitro: a voltage sensitive dye study. Neuroscience. 2010;165:371–385. doi: 10.1016/j.neuroscience.2009.10.025. [DOI] [PubMed] [Google Scholar]
- Brozoski TJ, Bauer CA, Caspary DM. Elevated fusiform cell activity in the dorsal cochlear nucleus of chinchillas with psychophysical evidence of tinnitus. J Neurosci. 2002;22:2383–2390. doi: 10.1523/JNEUROSCI.22-06-02383.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budinger E, Heil P, Scheich H. Functional organization of auditory cortex in the Mongolian gerbil (Meriones unguiculatus). III. Anatomical subdivisions and corticocortical connections. Eur J Neurosci. 2000;12:2425–2451. doi: 10.1046/j.1460-9568.2000.00142.x. [DOI] [PubMed] [Google Scholar]
- Burianova J, Ouda L, Profant O, Syka J. Age-related changes in GAD levels in the central auditory system of the rat. Exp Gerontol. 2009;44:161–169. doi: 10.1016/j.exger.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Caspary DM, Ling L, Turner JG, Hughes LF. Inhibitory neurotransmission, plasticity and aging in the mammalian central auditory system. J Exp Biol. 2008;211:1781–1791. doi: 10.1242/jeb.013581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness VS., Jr Architectonic map of neocortex of the normal mouse. J Comp Neurol. 1975;164:247–263. doi: 10.1002/cne.901640207. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Killackey HP, Metherate R. Parvalbumin and calbindin are differentially distributed within primary and secondary subregions of the mouse auditory forebrain. Neuroscience. 2001;105:553–569. doi: 10.1016/s0306-4522(01)00226-3. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Rose HJ, Metherate R. Auditory thalamocortical synaptic transmission in vitro. J Neurophysiol. 2002;87:361–384. doi: 10.1152/jn.00549.2001. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, Connors BW. Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat Neurosci. 2007;10:462–468. doi: 10.1038/nn1861. [DOI] [PubMed] [Google Scholar]
- Darlington CL, Smith PF. Drug treatments for tinnitus. In: Langguth B, Hajak G, Kleinjung T, Cacace A, Møller AR, editors. Progress in brain research. Amsterdam: Elsevier; 2007. pp. 249–262. [DOI] [PubMed] [Google Scholar]
- Dumitriu D, Cossart R, Huang J, Yuste R. Correlation between axonal morphologies and synaptic input kinetics of interneurons from mouse visual cortex. Cereb Cortex. 2007;17:81–91. doi: 10.1093/cercor/bhj126. [DOI] [PubMed] [Google Scholar]
- Eggermont JJ. Pathophysiology of tinnitus. In: Langguth B, Hajak G, Kleinjung T, Cacace A, Møller AR, editors. Progress in brain research. Amsterdam: Elsevier; 2007. pp. 19–543. [Google Scholar]
- Eggermont JJ. Hearing loss, hyperacusis, or tinnitus: what is modeled in animal research? Hear Res. 2012 doi: 10.1016/j.heares.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Franklin K, Paxinos G. The mouse brain in stereotaxic coordinates. Ed 3. Amsterdam: Elsevier Science; 2007. [Google Scholar]
- Gonchar Y, Burkhalter A. Differential subcellular localization of forward and feedback interareal inputs to parvalbumin expressing GABAergic neurons in rat visual cortex. J Comp Neurol. 1999;406:346–360. doi: 10.1002/(sici)1096-9861(19990412)406:3<346::aid-cne4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Gonchar Y, Wang Q, Burkhalter AH. Multiple distinct subtypes of GABAergic neurons in mouse visual cortex identified by triple immunostaining. Front Neuroanat. 2007;1:3. doi: 10.3389/neuro.05.003.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett TA, Barkat TR, O'Brien BM, Hensch TK, Polley DB. Linking topography to tonotopy in the mouse auditory thalamocortical circuit. J Neurosci. 2011;31:2983–2995. doi: 10.1523/JNEUROSCI.5333-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes LF, Turner JG, Parrish JL, Caspary DM. Processing of broadband stimuli across A1 layers in young and aged rats. Hear Res. 2010;264:79–85. doi: 10.1016/j.heares.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husson TR, Mallik AK, Zhang JX, Issa NP. Functional imaging of primary visual cortex using flavoprotein autofluorescence. J Neurosci. 2007;27:8665–8675. doi: 10.1523/JNEUROSCI.2156-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JH, Chen JC, Yang SY, Wang MF, Liu TC, Chan YC. Expression of COX-2 and NMDA receptor genes at the cochlea and midbrain in salicylate-induced tinnitus. Laryngoscope. 2011;121:361–364. doi: 10.1002/lary.21283. [DOI] [PubMed] [Google Scholar]
- Kaltenbach JA. Tinnitus: models and mechanisms. Hear Res. 2011;276:52–60. doi: 10.1016/j.heares.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbach JA, Godfrey DA, Neumann JB, McCaslin DL, Afman CE, Zhang J. Changes in spontaneous neural activity in the dorsal cochlear nucleus following exposure to intense sound: relation to threshold shift. Hear Res. 1998;124:78–84. doi: 10.1016/s0378-5955(98)00119-1. [DOI] [PubMed] [Google Scholar]
- Kleinjung T, Vielsmeier V, Landgrebe M, Hajak G, Langguth B. Transcranial magnetic stimulation: a new diagnostic and therapeutic tool for tinnitus patients. Int Tinnitus J. 2008;14:112–118. [PubMed] [Google Scholar]
- Ling LL, Hughes LF, Caspary DM. Age-related loss of the GABA synthetic enzyme glutamic acid decarboxylase in rat primary auditory cortex. Neuroscience. 2005;132:1103–1113. doi: 10.1016/j.neuroscience.2004.12.043. [DOI] [PubMed] [Google Scholar]
- Llano DA, Sherman SM. Evidence for nonreciprocal organization of the mouse auditory thalamocortical-corticothalamic projection systems. J Comp Neurol. 2008;507:1209–1227. doi: 10.1002/cne.21602. [DOI] [PubMed] [Google Scholar]
- Llano DA, Theyel BB, Mallik AK, Sherman SM, Issa NP. Rapid and sensitive mapping of long-range connections in vitro using flavoprotein autofluorescence imaging combined with laser photostimulation. J Neurophysiol. 2009;101:3325–3340. doi: 10.1152/jn.91291.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobarinas E, Yang G, Sun W, Ding D, Mirza N, Dalby-Brown W, Hilczmayer E, Fitzgerald S, Zhang L, Salvi R. Salicylate- and quinine-induced tinnitus and effects of memantine. Acta Otolaryngol. 2006;(Suppl)(556):13–19. doi: 10.1080/03655230600895408. [DOI] [PubMed] [Google Scholar]
- Longenecker RJ, Galazyuk AV. Development of tinnitus in CBA/CAj mice following sound exposure. J Assoc Res Otolaryngol. 2011;12:647–658. doi: 10.1007/s10162-011-0276-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton JW, Kiritani T, Pedersen C, Turner JG, Shepherd GM, Tzounopoulos T. Mice with behavioral evidence of tinnitus exhibit dorsal cochlear nucleus hyperactivity because of decreased GABAergic inhibition. Proc Natl Acad Sci U S A. 2011;108:7601–7606. doi: 10.1073/pnas.1100223108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari M, Dell'Anna ME, Rausell E, Leggio MG, Hashikawa T, Jones EG. Auditory thalamocortical pathways defined in monkeys by calcium-binding protein immunoreactivity. J Comp Neurol. 1995;362:171–194. doi: 10.1002/cne.903620203. [DOI] [PubMed] [Google Scholar]
- Møller AR. Epidemiology of tinnitus in adults. In: Møller AR, Langguth B, Ridder D, Kleinjung T, editors. Textbook of tinnitus. New York: Springer; 2011. pp. 29–37. [Google Scholar]
- Mühlnickel W, Elbert T, Taub E, Flor H. Reorganization of auditory cortex in tinnitus. Proc Natl Acad Sci U S A. 1998;95:10340–10343. doi: 10.1073/pnas.95.17.10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noreña AJ, Moffat G, Blanc JL, Pezard L, Cazals Y. Neural changes in the auditory cortex of awake guinea pigs after two tinnitus inducers: salicylate and acoustic trauma. Neuroscience. 2010;166:1194–1209. doi: 10.1016/j.neuroscience.2009.12.063. [DOI] [PubMed] [Google Scholar]
- Porter JT, Johnson CK, Agmon A. Diverse types of interneurons generate thalamus-evoked feedforward inhibition in the mouse barrel cortex. J Neurosci. 2001;21:2699–2710. doi: 10.1523/JNEUROSCI.21-08-02699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert KC, Gao W, Chen G, Wang X, Peng YP, Ebner TJ. Cellular and metabolic origins of flavoprotein autofluorescence in the cerebellar cortex in vivo. Cerebellum. 2011;10:585–599. doi: 10.1007/s12311-011-0278-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi RJ, Saunders SS, Gratton MA, Arehole S, Powers N. Enhanced evoked response amplitudes in the inferior colliculus of the chinchilla following acoustic trauma. Hear Res. 1990;50:245–257. doi: 10.1016/0378-5955(90)90049-u. [DOI] [PubMed] [Google Scholar]
- Sarro EC, Kotak VC, Sanes DH, Aoki C. Hearing loss alters the subcellular distribution of presynaptic GAD and postsynaptic GABAA receptors in the auditory cortex. Cereb Cortex. 2008;18:2855–2867. doi: 10.1093/cercor/bhn044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schecklmann M, Landgrebe M, Poeppl TB, Kreuzer P, Männer P, Marienhagen J, Wack DS, Kleinjung T, Hajak G, Langguth B. Neural correlates of tinnitus duration and distress: a positron emission tomography study. Hum Brain Mapp. 2011 doi: 10.1002/hbm.21426. Advanced online publication. Retrieved September 28, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff ML, Reyes AD. Characterization of thalamocortical responses of regular-spiking and fast-spiking neurons of the mouse auditory cortex in vitro and in silico. J Neurophysiol. 2012;107:1476–1488. doi: 10.1152/jn.00208.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuki K, Hishida R, Murakami H, Kudoh M, Kawaguchi T, Watanabe M, Watanabe S, Kouuchi T, Tanaka R. Dynamic imaging of somatosensory cortical activity in the rat visualized by flavoprotein autofluorescence. J Physiol. 2003;549:919–927. doi: 10.1113/jphysiol.2003.040709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolzberg D, Chen GD, Allman BL, Salvi RJ. Salicylate-induced peripheral auditory changes and tonotopic reorganization of auditory cortex. Neuroscience. 2011;180:157–164. doi: 10.1016/j.neuroscience.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Lu J, Stolzberg D, Gray L, Deng A, Lobarinas E, Salvi RJ. Salicylate increases the gain of the central auditory system. Neuroscience. 2009;159:325–334. doi: 10.1016/j.neuroscience.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Deng A, Jayaram A, Gibson B. Noise exposure enhances auditory cortex responses related to hyperacusis behavior. Brain Res. 2012 doi: 10.1016/j.brainres.2012.02.008. Advance online publication. Retrieved September 28, 2012. [DOI] [PubMed] [Google Scholar]
- Swadlow HA. Fast-spike interneurons and feedforward inhibition in awake sensory neocortex. Cereb Cortex. 2003;13:25–32. doi: 10.1093/cercor/13.1.25. [DOI] [PubMed] [Google Scholar]
- Szczepaniak WS, Møller AR. Evidence of decreased GABAergic influence on temporal integration in the inferior colliculus following acute noise exposure: a study of evoked potentials in the rat. Neurosci Lett. 1995;196:77–80. doi: 10.1016/0304-3940(95)11851-m. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Hishida R, Kubota Y, Kudoh M, Takahashi S, Shibuki K. Transcranial fluorescence imaging of auditory cortical plasticity regulated by acoustic environments in mice. Eur J Neurosci. 2006;23:1365–1376. doi: 10.1111/j.1460-9568.2006.04662.x. [DOI] [PubMed] [Google Scholar]
- Takesian AE, Kotak VC, Sanes DH. Age-dependent effect of hearing loss on cortical inhibitory synapse function. J Neurophysiol. 2012;107:937–947. doi: 10.1152/jn.00515.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J. Behavioral measures of tinnitus in laboratory animals. In: Langguth B, Hajak G, Kleinjung T, Cacace A, Møller AR, editors. Progress in brain research. Amsterdam: Elsevier; 2007. pp. 147–549. [DOI] [PubMed] [Google Scholar]
- Turner J, Larsen D, Hughes L, Moechars D, Shore S. Time course of tinnitus development following noise exposure in mice. J Neurosci Res. 2012;90:1480–1488. doi: 10.1002/jnr.22827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JG, Brozoski TJ, Bauer CA, Parrish JL, Myers K, Hughes LF, Caspary DM. Gap detection deficits in rats with tinnitus: a potential novel screening tool. Behav Neurosci. 2006;120:188–195. doi: 10.1037/0735-7044.120.1.188. [DOI] [PubMed] [Google Scholar]
- van der Loo E, Gais S, Congedo M, Vanneste S, Plazier M, Menovsky T, Van de Heyning P, De Ridder D. Tinnitus intensity dependent gamma oscillations of the contralateral auditory cortex. PLoS One. 2009;4:e7396. doi: 10.1371/journal.pone.0007396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MN, Kitzes LM, Jones EG. Chemoarchitectonic organization of the cat primary auditory cortex. Exp Brain Res. 1991;86:518–526. doi: 10.1007/BF00230525. [DOI] [PubMed] [Google Scholar]
- Wang H, Ling L, Brozoski T, Turner J, Parrish J, Hughes LF, Caspary DM. Altered markers of glycinergic function in the DCN of a rat model of tinnitus. Assoc Res Otolaryngol. 2008;31:117. [Google Scholar]
- Wang H, Brozoski TJ, Caspary DM. Inhibitory neurotransmission in animal models of tinnitus: maladaptive plasticity. Hear Res. 2011;279:111–117. doi: 10.1016/j.heares.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HT, Luo B, Zhou KQ, Xu TL, Chen L. Sodium salicylate reduces inhibitory postsynaptic currents in neurons of rat auditory cortex. Hear Res. 2006;215:77–83. doi: 10.1016/j.heares.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Willott JF, Lu SM. Noise-induced hearing loss can alter neural coding and increase excitability in the central nervous system. Science. 1982;216:1331–1334. doi: 10.1126/science.7079767. [DOI] [PubMed] [Google Scholar]
- Wong-Riley M. Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res. 1979;171:11–28. doi: 10.1016/0006-8993(79)90728-5. [DOI] [PubMed] [Google Scholar]
- Xu J, Yu L, Cai R, Zhang J, Sun X. Early continuous white noise exposure alters auditory spatial sensitivity and expression of GAD65 and GABAA receptor subunits in rat auditory cortex. Cereb Cortex. 2010a;20:804–812. doi: 10.1093/cercor/bhp143. [DOI] [PubMed] [Google Scholar]
- Xu J, Yu L, Zhang J, Cai R, Sun X. Early continuous white noise exposure alters l-α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunit glutamate receptor 2 and γ-aminobutyric acid type a receptor subunit β3 protein expression in rat auditory cortex. J Neurosci Res. 2010b;88:614–619. doi: 10.1002/jnr.22232. [DOI] [PubMed] [Google Scholar]
- Yang S, Weiner BD, Zhang LS, Cho SJ, Bao S. Homeostatic plasticity drives tinnitus perception in an animal model. Proc Natl Acad Sci U S A. 2011;108:14974–14979. doi: 10.1073/pnas.1107998108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]