

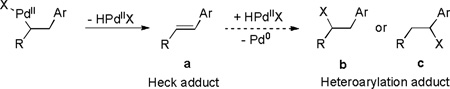
Direct functionalization of olefins provides a powerful tool for C-C bond formation.[1] As a prime example, Heck-type reactions have found broad utility in the synthesis of natural products and pharmaceuticals.[2] Typically, olefin insertion into the Pd-aryl bond is followed by β-H elimination to give Heck product a and also terminates the reaction (X = O, N or halogen, eq 1). However, further reaction of adduct a with the nascent HPdIIX species and subsequent loss of Pd(0) would constitute a cascade processes resulting in heteroarylation adduct b or c (eq 1).[3] The obvious value will be gained by extending the Heck process to achieve greater molecular diversity. Indeed, related efforts have been demonstrated by Yoshida,[3b,c] Sanford[3d,g,h] and Sigman.[3e,f] To our continuous interests in pursuing the functionalization of olefins,[4] herein we described unprecedented alternative pathways for Heck reaction: (i) homoallylic alcohol-directed oxidative Heck reactions and (ii) interception of the Heck intermediate via a novel intramolecular Pd-catalyzed olefinic oxy-annulation for the in situ construct of α-aryltetrahydrofurans.[5]
 |
(1) |
In the specific case of homoallylic alcohols, we envision the olefin would regioselectively insert into hydroxyl-coordinated Pd(II)-aryl complex to give the five-membered intermediate A (Scheme 1). Subsequent β-hydride elimination creates Heck intermediate B. Reductive elimination (Path A) leads to Heck adduct C.[6] However, an alternate fate is possible (Path B). It is known that β-hydride elimination is reversible and can lead to the relatively stable σ-alkyl palladium intermediate by a π-benzyl interaction.[3d–h] Once the intermediate HPdIIO species B adds intramolecularly to the olefin forming six-membered intermediate D, the reductive elimination yields tetrahydrofuran adduct E. Thus, the challenge would be how to control the eventual direction of intermediate B towards tetrahydrofuran.
Scheme 1.

Alternative Pathways for Heck Intermediate
As expected, in the presence of Pd(TFA)2 and the re-oxidant benzoquinone (BQ), the reaction readily yielded conventional Heck adducts (Table 1). A wide variance in electronic properties was tolerated on the boronic acid partner. Styrenyl olefins (3a–3c) as well as a heterocyclic-substituted olefin (3d) afforded good yields with a strong E-stereochemical preference. Notably, substantially bulky substrates (e.g. estrogen 4) survived in this oxidative version of the Heck reaction without compromising the chemical yield.
Table 1.
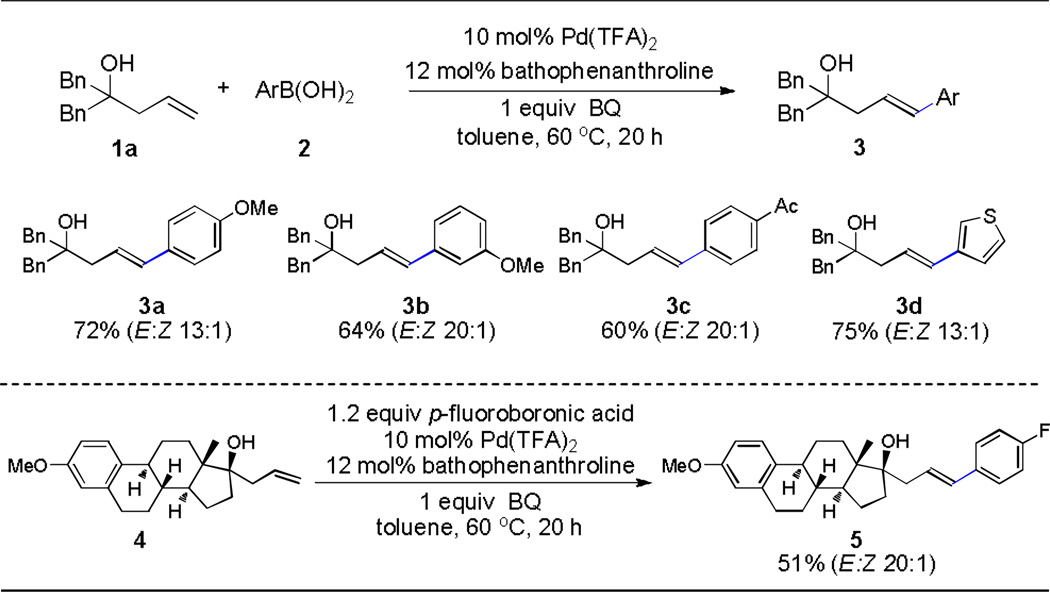 |
Isolated yield.
The reaction conditions: 0.1 mmol homoallylic alcohol, 0.12 mmol boronic acid, 10 mol % Pd(TFA)2, 12 mol % bathophenanthroline and 0.1 mmol BQ in toluene at 60°C for 20 h.
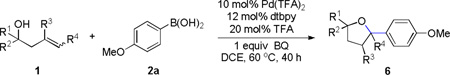
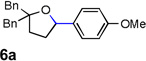
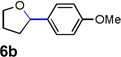
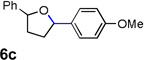
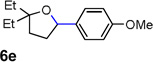
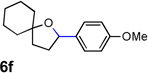
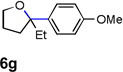
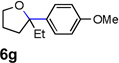
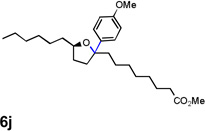
Encouraged by the above results, we investigated the intramolecular oxy-annulation of homoallylic alcohol 1a. After screening a variety of additives, to our delight, catalytic amounts (20 mol %) of trifluoroacetic acid (TFA) significantly accelerated addition of the HPdIIO species with the olefin to furnish tetrahydrofuran adducts.[7] Further optimization of conditions improved the chemical yield to 75% (Table 2, entry 1).[8] With the best conditions in hand, we applied them to diverse molecules containing homoallylic alcohols (Table 2). All the primary, secondary and tertiary alcohols were smoothly converted into corresponding tetrahydrofurans (entries 2–5). Interestingly, spiro-tetrahydrofuran 6f was formed in good yield from cyclic alcohol 1f (entry 6). Significantly, not only terminal olefins but also internal olefins were compatible with these conditions. For example, Z-1g as well as E-1g was transformed into the same product, 6g, in which a new quaternary carbon center was constructed (entries 7 and 8). Branched olefin 1h stereospecifically gave 6h despite the steric hindrance which might impact the olefin insertion into Pd-aryl bond during the transition state (entry 9).[9] Impressively, this methodology was also applicable with some complicated molecules. Regardless of their crowded skeletons, tetrahydrofuran 6i and 6j were readily obtained from estrogen 4 and fatty acid 1j, respectively (entries 10 and 11).
Table 2.
Oxyarylation of Homoallylic Alcohol [a]
 | |||
|---|---|---|---|
| Homoallylic alcohol | Tetrahydrofuran | Yield (%) [b] |
|
| 1 |  |
 |
75 |
| 2 |  |
 |
65 |
| 3 |  |
 |
74 [c] |
| 4 | 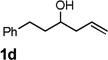 |
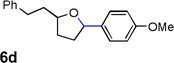 |
80 [d] |
| 5 | 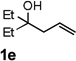 |
 |
72 |
| 6 | 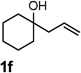 |
 |
70 |
| 7 | 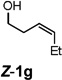 |
 |
52 |
| 8 | 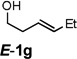 |
 |
61 |
| 9 | 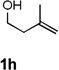 |
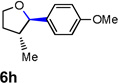 |
31 [e] |
| 10 | 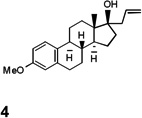 |
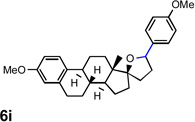 |
63 [f] |
| 11 | 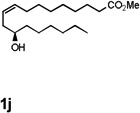 |
 |
40 [g] |
Entries 2,7,8 and 9 were conducted with 0.2 mmol homoallylic alcohol 1; other entries were conducted with 0.1 mmol homoallylic alcohol 1.
Isolated yield.
d.r. 1.2:1.
d.r. 1.4:1.
trans: cis = 16:1.
d.r. 1.3:1.
d.r. 1.3:1.
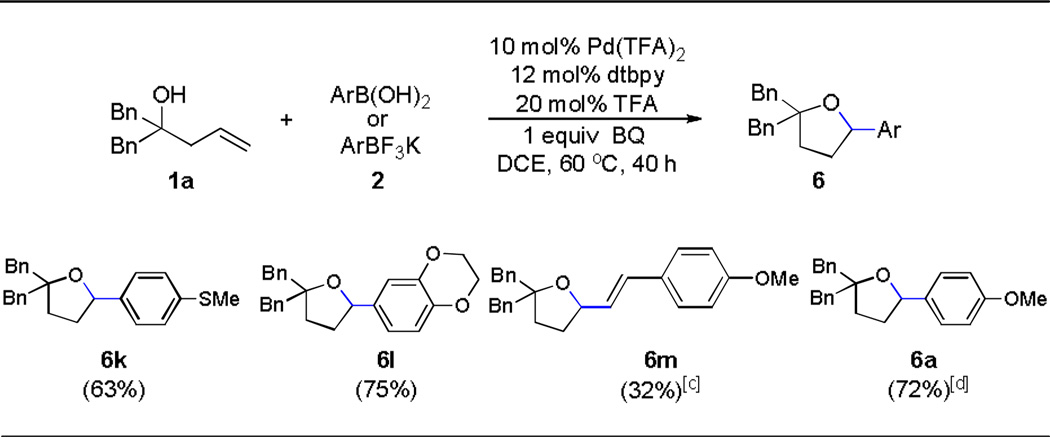
Besides boronic acid 2a, other boronic acids were also surveyed and furnished corresponding product 6k–6m (Table 3). The electron rich arene on olefin was found to be critical for the insertion of olefin into HPdIIO species. Importantly, potassium trifluoroborate was also a competent transmetallating reagent which afforded comparable results to that given by a related boronic acid 2a (Table 3, 6a and Table 2, entry 1).
Table 3.
 |
The reactions were conducted with 0.1 mmol homoallylic alcohol 1a.
Isolated yield.
The reaction was performed at 90 °C for 40 h.
1.2 equiv ArBF3K was used instead of boronic acid 2a.
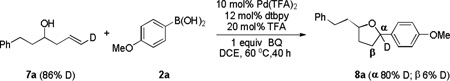
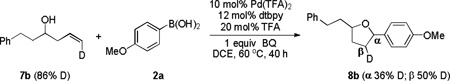
We next turned our efforts to investigate the mechanism of these cascade transformations. First, experiments were carried out by using either trans or cis terminally deuterium-labelled olefin to understand the details of stereochemistry. When trans deuterium olefin 7a was utilized, the retention of deuterium in 8a was observed on the same carbon (α-carbon) (eq 2). In contrast, most of the deuterium in 8b migrated to the adjacent carbon (β-carbon) when using cis deuterium olefin 7b (eq 3).[10] These results suggested that cis hydrogen was preferable in the step of β-hydride elimination and then migrated to the adjacent carbon via consequent Pd-H insertion.
 |
(2) |
 |
(3) |
Two other pathways to the tetrahydrofuran adducts: acid-promoted cyclization (eq 4) and Wacker-type cyclization (eq 5), might be possible starting from the Heck adducts. In both cases, the proton on the β-carbon comes from the acidic medium. In order to exclude these possibilities, control experiment was conducted by performing the reaction in acetic acid-d4 as co-solvent (eq 6). Introduction of a deuterium on the β-carbon was not observed in the reaction. Furthermore, exposing the Heck adduct 3a to 20 mol % TFA or 10 mol % Pd(TFA)2, decomposition occurred rather than forming annulated product 6a (eq 7).
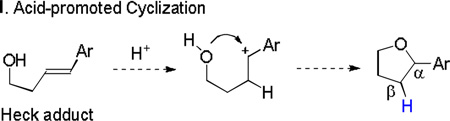 |
(4) |
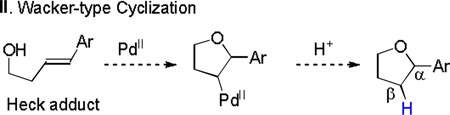 |
(5) |
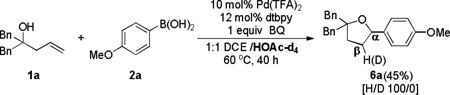 |
(6) |
 |
(7) |
Another significant issue observed in the reaction was that BQ was uniquely effective oxidant for affording the desired oxy-arylated product. Replacement of BQ with O2 would result in 3a, not 6a, as major product (eq 8). It was reasoned that: a) exposure to O2 would quench intermediate B (Scheme 1) resulting in PdII-hydroperoxide and stop Path B,[11] b) BQ could promote the formation of C-O bond via reductive elimination from PdII in intermediate D (Scheme 1).[12] The role of TFA is ambiguous. However, at this preliminary stage, we suppose it suppresses the reductive elimination of intermediate B and regeneration of PdII-H from Pd0.[13]
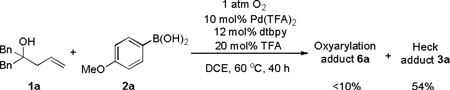 |
(8) |
In summary, we described two variants of the oxidative Heck reaction in which the hydroxyl of homoallylic alcohols coordinates with palladium resulting in good yields and regioselectivities in the adducts. Moreover, addition of a catalytic amount of TFA mediates the oxy-arylation of the penultimate Heck intermediate giving rise to a wide range of tetrahydrofurans including highly functionalized scaffolds. Further mechanistic study and other applications are ongoing in our lab.
Experimental Section
General Procedure for Oxidative Heck Reaction
Homoallylic alcohol 1a (25.2 mg, 0.1 mmol), boronic acid 2a (18.2 mg, 0.12 mmol), benzoquinone (11 mg, 0.1 mmol), Pd(TFA)2 (3.3 mg, 0.01 mmol) and bathophenanthroline (4.0 mg, 0.012 mmol) were loaded into a dry vial which was subjected to evacuation/flushing with dry argon three times. Anhydrous toluene (0.8 mL) was syringed into the mixture which was then stirred at 60 °C for 20 h or until the starting material had been consumed as determined by TLC. Upon cooling to room temperature, all volatiles were evaporated and the residue was purified by preparative TLC to give 3a as yellow oil in 72% yield. E:Z = 13:1. E-isomer: 1H NMR (500 MHz) δ 2.30 (d, J =7.0 Hz, 2H), 2.87 (s, 4H), 3.83 (s, 3H), 6.15 (ddd, J = 7.5, 7.5, 15.5 Hz, 1H), 6.37 (d, J = 16.0 Hz, 1H), 6.88 (d, J = 9.0 Hz, 2H), 7.19–7.35 (m, 12H); 13C NMR (125 MHz) δ 42.6, 46.1, 55.56, 55.59, 74.5, 114.2, 123.4, 126.8, 127.5, 128.5, 130.4, 131.1, 133.6, 137.5, 159.2. FT-IR (CH2Cl2) 3559, 3028, 2919, 2836, 2361, 2343, 1607, 1511, 1495, 1454, 1298, 1248, 1175, 1113, 1086, 1033, 970, 888, 834, 753, 725, 702 cm−1. HRMS calcd for C25H27O2 [M+1]+ 359.2006, found 359.2000.
General Procedure for Oxy-arylation of Olefins
Homoallylic alcohol 1a (25.2 mg, 0.1 mmol), boronic acid 2a (18.2 mg, 0.12 mmol), benzoquinone (11 mg, 0.1 mmol), Pd(TFA)2 (3.3 mg, 0.01 mmol) and 4,4'-di-tert-butyl-2,2'-bipyridyl (dtbpy, 3.2 mg, 0.012 mmol) were loaded into a dry vial which was subjected to evacuation/flushing with dry argon three times. Anhydrous dichloroethane (0.8 mL) followed by trifluoroacetic acid (1.5 µL, 0.02 mmol) were syringed into the mixture which was then stirred at 60 °C for 40 h or until the starting material had been consumed as determined by TLC. Upon cooling to room temperature, all volatiles were evaporated and the residue was purified by preparative TLC to give 6a as pale yellow oil in 75% yield. 1H NMR (400 MHz) δ 1.37–1.43 (m, 1H), 1.85–1.90 (m, 1H), 1.90–2.01 (m, 2H), 2.86 (d, 2.93, J = 13.6 Hz, 1H), 2.92 (d, J = 13.6 Hz, 1H), 2.99 (d, J = 13.2 Hz, 1H), 3.01 (d, J = 13.6 Hz, 1H), 3.80 (s, 3H), 4.47 (dd, J = 5.2, 9.6 Hz, 1H), 6.84 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.8 Hz, 2H), 7.23–7.34 (m, 10H); 13C NMR (100 MHz) δ 33.5, 34.6, 46.1, 47.1, 55.5, 81.1, 85.7, 113.8, 126.4, 126.5, 128.0, 128.1, 128.2, 131.2, 131.3, 134.6, 138.3, 138.4, 159.2. FT-IR (CH2Cl2) 3060, 3027, 2936, 2835, 1613, 1513, 1494, 1454, 1302, 1246, 1173, 1082, 1035, 942, 827, 754, 736, 701 cm−1. HRMS calcd for C25H27O2 [M+1]+ 359.2006, found 359.2016.
Supplementary Material
Footnotes
We thank the NIH (GM31278) and the Robert A. Welch Foundation (GL625910) for financial support.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For reviews, see: Jia C, Kitamura T, Fujiwara Y. Acc. Chem. Res. 2001;34:633. doi: 10.1021/ar000209h. Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem. Rev. 2007;107:5318. doi: 10.1021/cr068006f. Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273. Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e.
- 2.For reviews, see: Heck RF. Chapter 4.3. In: Trost BM, editor. Comprehensive Organic Synthesis. Vol. 4 New York: Pergamon; 1991. Cabri W, Candiani I. Acc. Chem. Res. 1995;28:2. Crisp GT. Chem. Soc. Rev. 1998;27:427. Whitcombe NJ, Hii KK, Gibson SE. Tetrahedron. 2001;57:7449. Dounay AB, Overman LE. Chem. Rev. 2003;103:2945. doi: 10.1021/cr020039h. Alonso F, Beletskaya IP, Yus M. Tetrahedron. 2005;61:11771. Karimi B, Behzadnia H, Elhamifar D, Akhavan PF, Esfahani FK, Zamani A. Synthesis. 2010:1399.
- 3.Other examples involving trapping σ-alkyl PdII Heck intermediate, see: Nicolaou KC, Edmonds DJ, Bulger PG. Angew. Chem., Int. Ed. 2006;45:7134. doi: 10.1002/anie.200601872. and references cited therein; Tamaru Y, Hojo M, Higashimura H, Yoshida Z. Angew. Chem., Int. Ed. 1986;25:735. Tamaru Y, Hojo M, Kawamura S, Yoshida Z. J. Org. Chem. 1986;51:4089. Kalyani D, Sanford MS. J. Am. Chem. Soc. 2008;130:2150. doi: 10.1021/ja0782798. Urkalan KB, Sigman MS. Angew. Chem., Int. Ed. 2009;48:3146. doi: 10.1002/anie.200900218. Werner EW, Urkalan KB, Sigman MS. Org. Lett. 2010;12:2848. doi: 10.1021/ol1009575. Kalyani D, Satterfield AD, Sanford MS. J. Am. Chem. Soc. 2010;132:8419. doi: 10.1021/ja101851v. Satterfield AD, Kubota A, Sanford MS. Org. Lett. 2011;13:1076. doi: 10.1021/ol103121r.
- 4.For the functionalization of olefins via C-H bond activation, see: Zhu C, Falck JR. Org. Lett. 2011;13:1214. doi: 10.1021/ol200093f.
- 5.For another strategy to construct tetrahydrofuran via Pd(0)-mediated olefin insertion to Pd-O bond, see: Wolfe JP. Synlett. 2008:2913. Wolfe JP, Rossi MA. J. Am. Chem. Soc. 2004;126:1620. doi: 10.1021/ja0394838. Hay MB, Wolfe JP. J. Am. Chem. Soc. 2005;127:16468. doi: 10.1021/ja054754v. Hay MB, Hardin AR, Wolfe JP. J. Org. Chem. 2005;70:3099. doi: 10.1021/jo050022+. Nakhla JS, Kampf JW, Wolfe JP. J. Am. Chem. Soc. 2006;128:2893. doi: 10.1021/ja057489m. Ward AF, Wolfe JP. Org. Lett. 2009;11:2209. doi: 10.1021/ol900594h. Ward AF, Wolfe JP. Org. Lett. 2010;12:1268. doi: 10.1021/ol1001472.
- 6.For examples of oxidative Heck reaction involving chelating groups, see: Delcamp JH, White MC. J. Am. Chem. Soc. 2006;128:15076. doi: 10.1021/ja066563d. Ruan J, Li X, Saidi O, Xiao J. J. Am. Chem. Soc. 2008;130:2424. doi: 10.1021/ja0782955. Pan D, Chen A, Su Y, Zhou W, Li S, Jia W, Xiao J, Liu Q, Zhang L, Jiao N. Angew. Chem., Int. Ed. 2008;47:4729. doi: 10.1002/anie.200800966. Delcamp JH, Brucks AP, White MC. J. Am. Chem. Soc. 2008;130:11270. doi: 10.1021/ja804120r. Su Y, Jiao N. Org. Lett. 2009;11:2980. doi: 10.1021/ol9009865.
- 7.Excess TFA dehydrated the starting material resulting in diminished yields.
- 8.For details, see Supporting Information.
- 9.The relative configuration was determined by 1H NMR, see: Dana G, Touboul E, Convert O, Pascal YL. Tetrahedron. 1988;44:429. Schmitt A, Reissig H. Eur. J. Org. Chem. 2000:3893.
- 10.Due to the higher dissociation energy of C-D bond than C-H bond, the β-hydride elimination becomes the competitive reaction to the β-deuteride elimination which results in the retention of 36% deuterium at α-carbon.
- 11.For examples, see: Konnick MM, Gandhi BA, Guzei IA, Stahl SA. Angew. Chem., Int. Ed. 2006;45:2904. doi: 10.1002/anie.200600532. Denney MC, Smythe NA, Cetto KL, Kemp RA, Goldberg KI. J. Am. Chem. Soc. 2006;128:2508. doi: 10.1021/ja0562292. Popp BV, Stahl SS. J. Am. Chem. Soc. 2007;129:4410. Konnick MM, Stahl SS. J. Am. Chem. Soc. 2008;130:5753. doi: 10.1021/ja7112504.
- 12.For examples, see: Chen MS, Prabagaran N, Labenz NA, White MC. J. Am. Chem. Soc. 2005;127:6970. doi: 10.1021/ja0500198. Campbell AN, White PB, Guzei IA, Stahl SS. J. Am. Chem. Soc. 2010;132:15116. doi: 10.1021/ja105829t.
- 13.Mueller JA, Goller CP, Sigman MS. J. Am. Chem. Soc. 2004;126:9724. doi: 10.1021/ja047794s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.