

Summary
Understanding the genetic basis for tumor formation is crucial for treating cancer. Forward genetic screens using insertional mutagenesis technologies have identified many important tumor suppressor genes and oncogenes in mouse models of human cancer. Traditionally, retroviruses have been used for this purpose, allowing the identification of genes that can cause various forms of leukemia or lymphoma with murine leukemia viruses or mammary cancer with mouse mammary tumor viruses. Recently, the Sleeping Beauty transposon system has emerged as a tool for cancer gene discovery in mouse models of human cancer. Transposons mobilized in the mouse soma can insertionally mutate cancer genes, and the transposon itself serves as a molecular “tag,” which facilitates candidate cancer gene identification. We provide an overview of some general issues related to use of Sleeping Beauty for cancer genetic studies and present here the polymerase chain reaction-based method for cloning transposon-tagged sequences from tumors.
Keywords: Cancer genetics, linker-mediated PCR, mouse transgenesis, Sleeping Beauty, somatic mutagenesis, transposon
1. Introduction
The Sleeping Beauty (SB) transposon system is a useful tool for gene delivery or insertional mutagenesis in model genetic organisms (reviewed in refs. 1–3). The SB system consists of two parts—the transposon vector DNA and the enzyme that mobilizes this DNA, the transposase. Both the transposon and transposase must be in the same cell for transposition to occur. The original SB transposase enzyme is designated SB10, and was created by reconstruction of an active enzyme gene from defunct, mutated copies of Tc1/mariner family transposase genes cloned from various species of Salmonid fish (4). The minimal transposon vector DNA consists of a left and a right outer inverted repeat/direct repeat element (IRDR) that are the binding sites for the transposase enzyme. Transposons containing the original IRDRs built from sequences from Salmonid fish are designated “pT” vectors (4). Changes have been introduced into the IRDRs to increase transposition rates, and transposons based on this second generation of IRDRs are designated “pT2” vectors (5). A variety of insertional mutagenesis applications have been reported for SB including germline mutagenesis in the mouse and zebrafish (6–15), and most recently, somatic mutagenesis for cancer gene discovery in the mouse (16,17).
SB-based somatic mutagenesis for cancer gene discovery is accomplished by breeding transposon transgenic mice to transposase transgenic mice to generate transposon/transposase in doubly transgenic mice. Transposons are mobilizing in every somatic cell in which transposase is expressed, mutagenizing cancer genes and promoting tumor formation in these doubly transgenic mice. Once SB-induced tumors have been obtained, high-throughput methods for cloning transposon insertions are necessary to begin identifying candidate cancer genes. This chapter will detail the molecular biology protocols necessary for cloning transposon-DNA genomic junctions for SB transposons based on the “pT” or “pT2” versions of the IRDRs. Although beyond the scope of this chapter, methods must also be in place for automated “trimming” and genome mapping of these junctions. In addition, statistical analysis of insertions must be performed to identify common sites of insertion (CIS), which are regions of the genome that harbor multiple independent transposon insertions from multiple independent tumors. Although in a somatic cell, transposons are theoretically mobilizing to new locations throughout the genome, only those that land in or near a tumor suppressor gene or oncogene should promote tumorigenesis. Nevertheless, in any given tumor cell there will also be “passenger” insertions that have occurred stochastically and are not relevant to tumorigenesis. Statistical analysis must be performed to identify regions of the genome that are mutated by transposon integrations in multiple independent tumors more frequently than expected by random chance. These CISs indicate that a cancer gene is likely to be nearby because of the repetitive selection for transposon integrations in the chromosomal region in multiple different tumors. Statistical analysis of insertions and CIS identification is beyond the scope of this chapter, but we refer readers to several papers that address the topic (16–19). We describe here a protocol for cloning transposon-genomic DNA junctions that is based on our modifications of methods described by Karl Clark, Adam Dupuy, and the laboratory of Shawn M. Burgess (11,16,20). A brief description of the methods used to create cancer models using SB precedes this protocol.
1.1. Creating Cancer Models Using SB
1.1.1. Transposon Design
Mobilization of highly mutagenic transposons from chromosomally resident concatomers in the mouse soma has been used to initiate or accelerate tumor formation (16,17). The transposon used for these experiments, T2/onc (Fig. 1), was introduced into mice by standard pronuclear injection techniques, resulting in a multicopy array, or concatomer, integrated into one place in the genome of the transgenic mice produced. T2/onc is designed with splice acceptors (SA)/polyadenylation sequences in both orientations. On mobilization by transposase, if T2/onc lands in a tumor suppressor gene these elements should disrupt splicing, and therefore generate a loss-of-function mutation. T2/onc also contains sequences from the murine stem cell virus (MSCV) LTR, which contains promoter/enhancer elements followed by a splice donor that facilitates splicing of transcripts initiated in the MSCV into downstream endogenous exons. Therefore, if T2/onc lands within or near a proto-oncogene it can promote its overexpression. Transgenic lines harboring approx 25 copies of T2/onc in their chromosomal concatomers have been generated and are referenced to as “low copy lines” (16). Dupuy et al. (17) modified T2/onc to contain a longer version of one SA and the resulting vector is named T2/onc2 to designate this difference. The transgenic lines that were generated using T2/onc2 have over 100 copies of transposons in their chromosomal concatomers and are therefore referred to as “high copy lines” (17).
Fig. 1.

T2/onc. T2/onc has SA/polyadenylation (pA) sequences in both orientations to generate loss-of-function mutations in genes in which it lands. Between the two SA are sequences from the long terminal repeat of the MSCV LTR, which contains promoter/enhancer elements to promote overexpression of genes near which it lands. Also present is a splice donor (SD) so that transcripts initiated in the MSCV LTR can splice into downstream endogenous exons.
1.1.2. Transposase Design
Two different sources of SB transposase have been used in published work on SB-induced tumors. A CAGGS-SB10 transgene (10) was used to accelerate sarcomagenesis in p19Arf−/−mice (16). This transgene is expressed from a multicopy concatomer created by standard transgenesis. Although the CAGGS promoter has been described as being active in all cells (21), we have recently found that the CAGGS-SB10 line expresses transposase primarily in mesenchymal cells. Furthermore, the expression is highly variegated and absent from most epithelial cell types (unpublished data). This may explain why CAGGS-SB10 was able to accelerate sarcoma development in T2/onc, p19Arf−/−mice (16). In a second paper, a transgene for the SB11 version of the transposase was introduced into the Rosa26 locus (22) using homologous recombination in mouse embryonic cells (17). When mice carrying this Rosa26-SB11 allele were bred to high copy T2/onc2 transgenic mice, non-Mendelian inheritance was observed as most doubly transgenic mice died in utero. Those that survived to birth developed tumors, primarily lymphocytic leukemia, that was caused by T2/onc insertional mutagenesis (17).
In the future, modifications to the system will likely improve the utility of SB for cancer genetics studies in a wide variety of tissue types. Although beyond the scope of this chapter, we refer readers to several reviews that discuss how the system may be modified for future studies (1,23,24).
1.1.3. Considerations for Cloning and Sequence Analysis of Tumor-Associated Transposon Insertion Sites
Once the SB system has been used to create a cancer model the next step is to clone, sequence, and analyze a large number of transposon insertion sites. As in studies using slow transforming retroviruses, the goal is to identify CIS. The greater the number of transposon-induced or transposon-accelerated tumors available, the greater the power to identify CISs that are infrequently mutated by transposon insertion. In addition, the closer one can approach “saturation” cloning of all the transposon insertion sites within a tumor, the better the chances are that no CIS will be missed. These considerations apply to both retroviral mutagenesis using murine leukemia virus (MLV), or mammary cancer with mouse mammary tumor virus (MMTV) and to transposon mutagenesis for cancer gene discovery. In order to process a large number of tumor genomic DNAs and to approach saturation for insertion site recovery, we use polymerase chain reaction (PCR)-based methods for amplifying insertion sites. Several reviews discuss high-throughput proviral insertion site cloning and analysis using MLV (1,25–27).
Two general methods for PCR amplification have been used for cloning transposable element-genomic DNA junction fragments, namely inverse PCR and linker-mediated PCR (reviewed in ref. 28). For inverse PCR, a transposon/cellular genomic DNA junction fragment is first generated by restriction enzyme digestion, then is circularized by ligation of the restricted tumor DNA, and two rounds of PCR using outward facing primers from the transposon sequence are used to amplify the junction between the transposon and adjacent cellular DNA. The resulting PCR fragment is then cloned into a plasmid vector for sequencing. The circularization step may be inefficient and may bias against recovery of some insertions. Linker-mediated PCR is the most common technique currently used to clone viral or transposon integration sites from tumors. In this method, shown in Fig. 2, specially designed linkers are ligated onto restricted tumor genomic DNA, then subjected to two rounds of PCR using transposable element-specific and linker-specific primers before cloning into a plasmid vector for sequencing.
Fig. 2.

Outline of linker-mediated PCR for cloning proviral- or transposon-genomic DNA junctions. Linker-mediated PCR involves the generation of transposon (or proviral)-genomic DNA fragments by restriction digest of tumor genomic DNA. Linkers with overhangs complementary to the restriction fragments used are then ligated onto the genomic DNA. Two rounds of PCR with linker and IRDR (or proviral) specific primers are used to amplify the transposon-genomic DNA junction.
For either inverse PCR or linker-mediated PCR approaches, more complete saturation of insertion site cloning can be achieved by performing reactions that allow the cloning from both the left IRDR (5′ end) as well as the right IRDR (3′ end) of the transposable element. The protocol described here includes instructions for cloning from both ends of the transposon. Using multiple frequent-cutting restriction enzymes to digest the genomic DNA can also increase the number of transposon-genomic DNA junctions by assuring that a restriction enzyme site in adjacent cellular DNA creates a small enough junction fragment to be PCR amplified. Enzymes that frequently cut mammalian genomic DNA (e.g., enzymes that recognize 4 bp sites that lack CpG dinucleotides) are often used for this purpose. In addition, the restriction enzymes that are chosen for this approach must meet certain criteria including cutting close to the end of the transposon, cutting genomic DNA often enough to generate PCR-amplifiable junction fragments, and producing a sticky overhang that can be ligated to the designed double-stranded oligonucleotide linker efficiently. BfaI (for cloning from the left IRDR) and NlaIII (for cloning from the right IRDR) are two enzymes that we frequently use for this protocol.
Although the basic steps described above are common to methods for cloning MLV, MMTV, and transposon insertions, there are features unique to cloning SB transposon insertions as compared with MLV or MMTV. Most importantly, SB transposon insertions that cause cancer are derived by the mobilization of transposon vectors from a chromosomally resident multicopy concatomer. Therefore, in an SB-induced tumor there are tumor-specific, clonal transposon-insertion sites (anywhere from 5 to 30+ have been observed) and the remaining copies of the transposon vector that reside in the donor concatomer. A method to avoid repeatedly cloning the junction fragment between adjacent copies of the SB transposon vectors in the concatomers is needed in this situation. To achieve this, we have used “blocking” primers (with blocked 3′ ends that prevent polymerase extension) that are complementary to sequences from plasmid DNA that flanks each copy of the SB transposon in the concatomer. However, this method often has limited success. Alternately, after ligation of the linkers to the restricted DNA one can redigest with another restriction enzyme that cuts at least once in the plasmid DNA that flanks each copy of the SB transposon in the concatomers, but does not cut in the SB transposon sequence itself. Thus, the SB transposon junction fragments generated from within the concatomers cannot be amplified as the binding sites for the primers are now separated on two different DNA molecules. Using enzymes that cut more rarely in genomic DNA (e.g., restriction enzymes that have a 6-bp recognition sequence) reduce the possibility that the enzyme will also cut the genomic DNA between the end of the IRDR and the enzyme used for the first digest of genomic DNA. We currently use BamHI (for cloning from the left IRDR) and XhoI (for cloning from the right IRDR) for this purpose. Although complex, the basic method described in Subheadings 3.2. to 3.4. can be used to clone insertion sites from any application using a SB vector that uses “pT” or “pT2”-based IRDR. However, we recommend that the reader verify that the IRDR primer sequences provided are present in the transposon vector used. In addition, the restriction enzyme used for the second digestion (to prevent amplification of transposon junction fragments from within the donor concatomers) will depend on the sequence of the plasmid backbone sequences that were left linked to the SB transposon vector when the transgenic line was made. The protocol described below will work for tumors generated using the T2/onc (16) and T2/onc2 (17) transgenic lines. Whatever the method used to PCR amplify the junction fragments, by “shot-gun” cloning these PCR products and sequencing 96 or more clones per tumor, it is likely that even rare insertion sites will be identified, again contributing to saturation cloning.
2. Materials
2.1. DNA Preparation
1 M Tris-HCl (pH 8.0).
0.5 M Ethylenediamine tetra acetic acid (EDTA) (pH 8.0).
10% Sodium dodecyl sulfate in distilled water (caution—dust is an irritant).
5 M NaCl.
TE buffer: 4 mL of 1 M Tris-HCl (pH 8.0), 0.08 mL of 0.5 M EDTA (pH 8.0) to 400 total in distilled water.
-
Proteinase K: dissolved in TE at 10 mg/mL, incubated at 37°C for 1 h before storing in aliquots at −20°C.
Oligo name Oligo sequence Modifications Notes
Bfa linker+ GTAATACGACTCACTATAGGGCTCCGCTTAAGGGAC None – Bfa linker− TAGTCCCTTAAGCGGAG 5′ Phosphate, 3′ amino group See Note 12 NlaIII linker+ GTAATACGACTCACTATAGGGCTCCGCTTAAGGGACCATG None – NlaIII linker− GTCCCTTAAGCGGAGCC 5′ Phosphate, 3′ amino group See Note 12 IR/DR(R)KJC1 CCACTGGGAATGTGATGAAAGAAATAAAAGC None – Long IR/DR (R) GCTTGTGGAAGGCTACTCGAAATGTTTGACCC None – Long IR/DR (L2) CTGGAATTTTCCAAGCTGTTTAAAGGCACAGTCAAC None – New L1 GACTTGTGTCATGCACAAAGTAGATGTCC None – Linker primer GTAATACGACTCACTATAGGGC None – Linker nested primer AGGGCTCCGCTTAAGGGAC None –
STE buffer: 1.6 mL of 0.5 M EDTA, 8 mL of 1 M Tris-HCl (pH 8.0), 16 mL of 5 M NaCl to 800 mL then autoclaved.
Tissue lysis buffer (for one tumor): 4.6 mL of STE, 100 μL of 0.5 M EDTA, 100 μL of proteinase K, and 200 μL of 10% sodium dodecyl sulfate made fresh before using.
RNase A: dissolved in TE at 2 mg/mL, incubated at 37°C for 1 h before storing in aliquots at −20°C.
pH 7.9-buffer saturated phenol (caution—hazardous).
Chloroform (caution—hazardous).
95% Ethanol.
Glass rods for spooling DNA (we use Kimble melting point capillary tube part no. 34505-99).
2.2. DNA Digestion and Linker Annealing
Restriction enzymes: NlaIII, XhoI, BfaI, and BamHI (New England Biolabs Ipswich, MA) (see Note 1). The appropriate 10× buffer and 100× bovine serum albumin, if required, for each enzyme is provided with the enzyme.
PCR reaction purification kit (QIAquick PCR Purification Kit by Qiagen, Maryland).
Linker oligonucleotides (see Table 1 for sequences). Dissolve primers in Millipore purified or PCR grade water at 100 μM and store at −20°C.
T4 DNA ligase supplied with 5× ligase buffer (Invitrogen, Carlsbad, CA) stored at −20°C.
2.3. PCR Reactions
Platinum Taq supplied with 10× PCR buffer and 50 mM MgCl2 (Invitrogen).
dNTPs: 2.5 mM each for a total concentration of 10 mM in PCR grade water. Aliquot and store at −20°C.
PCR primers (see Table 1) dissolved in Millipore purified or PCR grade water at 25 μM.
Agarose.
Ethidium bromide.
50× TAE: 242.2 g of Tris Base, 100 mL of 0.5 M EDTA (pH 8.0), 57.1 mL of glacial acetic acid (caution—hazardous, use in fume hood) brought up to 1 L with Millipore water.
100-bp DNA ladder.
2.4. Cloning of PCR Products
TA-based cloning vector kit (see Note 2).
Bacteria for transformation (store at −70 to −80°C) (see Note 3).
Luria Bertani (LB): 10 g of tryptone, 5 g of yeast extract, and 5 g of sodium chloride brought up to a liter in distilled water and autoclaved.
LB plates containing the appropriated antibiotic for selection for the TA cloning vector.
3. Methods
3.1. Preparation of Genomic DNA (See Notes 4 and 5)
Tumors isolated from mice are snap frozen in liquid nitrogen and stored at −70 to −80°C until ready to be processed.
Approximately 100–200 mg of tumor tissue is dissociated by douncing in 5 mL of tissue lysis buffer. Samples are incubated overnight in 15-mL conical polypropylene tubes (see Note 6) at 55°C with shaking to allow complete proteinase K digestion of samples. If smaller tumor samples are available, decrease digestion volume.
Allow tubes to cool slightly before addition of 50 μL of RNase A. Incubate at 37°C for 1 h to remove contaminating RNA. After this stage, samples may be stored at −20°C for several days.
Add an equal amount of buffered phenol (caution—hazardous, use in fume hood) to samples. Samples should be vortexed before centrifugation in a clinical centrifuge for 5 min.
The aqueous layer (top layer) is removed into a new polypropylene conical tube. Take care not to remove the fluffy precipitate that often forms at the junction of the aqueous and organic layers. An equal volume of 1:1 phenol:chloroform (caution— hazardous, use in fume hood) is then added, the sample is vortexed and centrifuged as earlier.
The aqueous layer should be essentially clear at this stage. If it is not, repeat step 5. If clear, remove aqueous layer to new tube taking care not to disturb the organic/aqueous interface. Add an equal volume of chloroform (caution—hazardous, use in fume hood), vortex and centrifuge.
Remove aqueous phase to a new tube and precipitate DNA by adding approx 3 vol 95% ethanol and mixing gently but well by inverting tube several times.
(Optional): store samples for a few hours at −20°C to enhance DNA precipitation.
For most samples a stringy or fluffy white pellet of precipitated DNA will be visible. This precipitate can be spooled on a glass rod and placed into a 1.5-mL centrifuge tube and allowed to air-dry. If no precipitate is visible, DNA can be precipitated by centrifugation in a clinical centrifuge. The supernatant should be removed carefully to prevent dislodging of the pellet and the pellet should be air-dried.
When dry, pellet should be resuspended in TE. For small or not easily visible pellets, use 50 μL of TE, large pellets may require 200–300 μL of TE.
Incubate at 37°C overnight to allow DNA to go into solution. If still viscous, additional TE should be added and sample should be reincubated. Vortexing is not recommended as it may shear DNA.
DNA concentration is determined using a spectrophotometer and DNA should be stored long-term at −20°C. Aliquoting is also recommended if the DNA is to be used for additional purposes such as Southern blotting.
3.2. DNA Digestion and Linker Ligation (See Note 7)
Linker annealing. Mix in an Eppendorf tube 50 μL each of Bfa linker+ and linker− (for left) or Nla linker+ and linker− (for right), then add 2 μL of 5 M NaCl. Incubate the tubes in a heating block at 95°C for 5 min, then turn the heating block off and allow them to anneal by slow cooling. Annealed linkers can be stored at −20°C.
First restriction digest. Approximately 2 μg of DNA is digested with BfaI (for cloning off the left IRDR) or NlaIII (for cloning off the right IRDR). Digests are performed at 37°C with 10 U of enzyme in the appropriate buffer in 50 μL reaction volume (to ensure appropriate dilution of the glycerol that the enzymes are stored in).
DNA purification. DNA is purified from the reaction buffer using the QIAquick PCR purification kit from Qiagen following the directions provided by the kit. There is one modification to the protocol—DNA is eluded from column using 30 μL of Millipore purified water.
Linker ligation. Linkers are ligated onto the DNA fragments in a 20 μL reaction that contains: 6 μL of annealed linkers (Bfa for left or NlaIII for right), 8 μL of digested DNA (Bfa for left or NlaIII for right), 4 μL of 5× ligase buffer, and 2 μL of T4 DNA ligase (Invitrogen). Incubate at 16°C for 6 h to overnight.
DNA purification is the same as step 2 above.
Second restriction digest. Linker-ligated DNA fragments are digested with BamH1 (left) or XhoI (right) in 50 μL of total reaction buffer. Use 22 μL of purified DNA from step 5 (essentially all the eluate that is recovered), 10 U enzyme, and the appropriate buffers. Incubate at 37°C for 3 h to overnight (see Note 8).
DNA purification is the same as step 2 above.
3.3. Polymerase Chain Reaction (See Note 9)
-
Primary PCR reaction conditions (see Notes 10 and 11):
For one reaction:
10× PCR buffer 5 μL 50 mM MgCl2 2 μL 10 mM dNTPs 1 μL Primary IRDR primer (long IRDR [L2] for left, long IRDR [R] for right) 0.5 μL Linker primer 0.5 μL Platinum Taq 0.25 μL PCR grade water 37.75 μL Purified, linker-ligated DNA 3 μL
-
Primary PCR thermocycler conditions:
94°C for 2 min.
25 cycles of 94°C for 15 s, 60°C for 30 s, and 72°C for 90 s.
72°C for 5 min.
4°C hold.
For secondary PCR, the primary PCR product is first diluted 1:75 with water. In addition, the volume for secondary PCR is doubled compared with primary PCR to allow half the reaction to be visualized on an agarose gel and half to be saved for cloning.
-
Secondary PCR reaction conditions:
10× PCR buffer 10 μL 50 mM MgCl2 4 μL 10 mM dNTPs 2 μL Nested IRDR primer (new L1 for left, KJC1 for right) 1 μL Linker nested primer 1 μL Platinum Taq 0.5 μL PCR grade water 75.5 μL Diluted primary PCR product 6 μL
Primary PCR thermocycler conditions are the same as for primary PCR (step 2 above).
Half of each PCR reaction is run on an approx 2% of agarose gel in 1× TAE containing ethidium bromide for visualization (caution ethidium bromide is a suspected carcinogen) using a 100-bp ladder for visualization. Linker-mediated PCR usually results in a smear or products, with occasional distinct bands visible (see Fig. 3).
The remaining half of each reaction is purified using QIAquick PCR columns for cloning. Again, purified DNA is eluted into 30 μL of Millipore purified water.
Fig. 3.
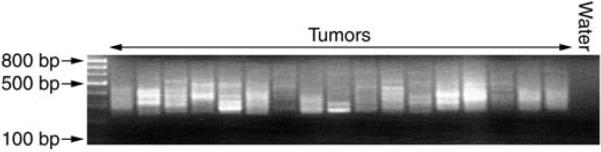
A typical linker-mediated PCR result on leukemias induced by T2/onc insertional mutagenesis. Most successful linker-mediated PCR reactions produce a “smear” of PCR products, although some individual bands can be discerned.
3.4. Cloning of PCR Products
Ligate purified PCR products into the appropriate TA cloning vector following manufacturers directions. We use the maximum volume of purified PCR in the ligation reaction as possible. For example, for pGEMT Teasy, the ligation reaction consists of 1 μL vector, 2 μL of 5× ligase buffer (Invitrogen), 6 μL of purified PCR product, and 1 μL of T4 DNA ligase (Invitrogen) and is allowed to incubate overnight at 16°C.
Transform bacteria with approximately half of the ligation (the rest can be stored at −20°C for later use if necessary) and plate onto LB plates containing the appropriate antibiotic for the cloning vector used.
For sequencing of a small number of PCR products, mini-preps can be performed by hand to isolate plasmid DNA for sequencing. For high-throughput sequencing, colonies are often grown in 96-well plates in preparation for automated plasmid purification.
4. Notes
The manufacturer recommends storage of NlaIII and BfaI at −70°C. We also recommend aliquoting these enzymes to prevent multiple rounds of freeze/thaw.
Any vector that allows the ligation of the 3′ A overhangs generated by Taq can be used. pCR®4-TOPO® (Invitrogen) is advantageous because self-ligated vectors restore a lethal Escherichia coli gene and therefore, cannot be recovered. We have also had success with cloning into pGEMT-easy (Promega, Madison, WI) followed by performing blue–white selection on 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside plates to detect plasmid-containing ligated PCR product inserts. In either case, ligation of insert and vector should be performed according to manufacturer's recommendations.
Some TA-cloning kits include bacteria for transformation. For example, TOPO cloning kits from Invitrogen include TOP10 competent cells. When not supplied, we use chemically competent DH5-α for routine cloning of PCR products. When large-scale sequencing of cloned products is required, we use electrocompetent cells such as ElectroMAX™ DH10B™ from Invitrogen as they have higher transformation efficiency. Follow the transformation directions supplied by the manufacturer.
Traditionally, our laboratory has used phenol/chloroform extraction followed by ethanol precipitation to purify genomic DNA from snap frozen tumor tissues digested with proteinase K as described here. We find that the protocol described here is the most consistent at isolating high-quality DNA. All steps utilizing phenol and chloroform must be carried out in an appropriately ventilated fume hood with appropriate safety precautions. Waste must be treated according to local regulations. However, for DNA extraction we have also had success using protocols that use “salting out” methods for protein precipitation followed by isopropanol precipitation of DNA from the resulting supernatant. We have not had great success using commercially available “spin column” type methods for preparing genomic DNA for linker-mediated PCR.
We take great care in handling reagents and samples to prevent cross-contamination. This involves using disposable single use plastics or glass pipets when possible, and using aerosol-resistant pipet tips (also known as “filter” tips).
Use polypropylene tubes and not polystyrene as polystyrene will not be resistant to the organic phenol used in later stages.
Cloning of junctions from both the right and left sides of the transposon requires two separate reactions for each tumor sample. Although the reactions are technically similar, they require different restriction enzymes, linkers, and PCR primers. Details are given for both left and right reactions.
It is very important that this digest goes to completion to prevent the amplification of the T2-vector junction that is present in every transposon remaining in the concatomer. Therefore, overnight incubations are recommended.
To prevent contamination of PCR with T2/onc plasmids from the laboratory, we perform PCR in a dedicated PCR hood equipped with an ultraviolet lamp. We have dedicated PCR pipets, filter tips, and water that do not leave the PCR hood. In addition, we expose our PCR tubes, PCR buffer, and MgCl2 solutions to ultraviolet light for 30 min before PCR.
It is often convenient to make a PCR “master-mix” that contains everything but the DNA, which is then aliquoted into the individual PCR tubes before DNA addition.
In addition to the DNA samples, we always include a PCR reaction that contains only water to control that no plasmid contamination is occurred.
Linker-primers are 5′ phosphorylated and 3′ amino modified. The phosphate modification is necessary for ligation onto digested genomic DNA. The 3′ amino group prevents Taq from extending off the 3′ end of the linker and copying the region of nonhomology in linker+.
Acknowledgments
This work was supported by grants R21 CA118600 and R01 CA113636 from the National Cancer Institute (to DAL) and K01 CA122183 from the National Cancer Institute (to LSC).
References
- 1.Collier LS, Largaespada DA. Hopping around the tumor genome: transposons for cancer gene discovery. Cancer Res. 2005;65:9607–9610. doi: 10.1158/0008-5472.CAN-05-3085. [DOI] [PubMed] [Google Scholar]
- 2.Ivics Z, Izsvak Z. Transposons for gene therapy! Curr. Gene Ther. 2006;6:593–607. doi: 10.2174/156652306778520647. [DOI] [PubMed] [Google Scholar]
- 3.Miskey C, Izsvak Z, Kawakami K, Ivics Z. DNA transposons in vertebrate functional genomics. Cell. Mol. Life Sci. 2005;62:629–641. doi: 10.1007/s00018-004-4232-7. [DOI] [PubMed] [Google Scholar]
- 4.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 5.Geurts AM, Yang Y, Clark KJ, et al. Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol. Ther. 2003;8:108–117. doi: 10.1016/s1525-0016(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 6.Geurts AM, Collier LS, Geurts JL, et al. Gene Mutations and Genomic Rearrangements in the Mouse as a Result of Transposon Mobilization from Chromosomal Concatemers. PLoS Genet. 2006;2:E156. doi: 10.1371/journal.pgen.0020156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keng VW, Yae K, Hayakawa T, et al. Region-specific saturation germline mutagenesis in mice using the Sleeping Beauty transposon system. Nat. Methods. 2005;2:763–769. doi: 10.1038/nmeth795. [DOI] [PubMed] [Google Scholar]
- 8.Balciunas D, Davidson AE, Sivasubbu S, et al. Enhancer trapping in zebrafish using the Sleeping Beauty transposon. BMC Genomics. 2004;5:62. doi: 10.1186/1471-2164-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davidson AE, Balciunas D, Mohn D, et al. Efficient gene delivery and gene expression in zebrafish using the Sleeping Beauty transposon. Dev. Biol. 2003;263:191–202. doi: 10.1016/j.ydbio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Dupuy AJ, Fritz S, Largaespada DA. Transposition and gene disruption in the male germline of the mouse. Genesis. 2001;30:82–88. doi: 10.1002/gene.1037. [DOI] [PubMed] [Google Scholar]
- 11.Dupuy AJ, Clark K, Carlson CM, et al. Mammalian germ-line trans-genesis by transposition. Proc. Natl. Acad. Sci. USA. 2002;99:4495–4499. doi: 10.1073/pnas.062630599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlson CM, Dupuy AJ, Fritz S, Roberg-Perez KJ, Fletcher CF, Largaespada DA. Transposon mutagenesis of the mouse germline. Genetics. 2003;165:243–256. doi: 10.1093/genetics/165.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horie K, Kuroiwa A, Ikawa M, et al. Efficient chromosomal transposition of a Tc1/mariner-like transposon Sleeping Beauty in mice. Proc. Natl. Acad. Sci. USA. 2001;98:9191–9196. doi: 10.1073/pnas.161071798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horie K, Yusa K, Yae K, et al. Characterization of Sleeping Beauty transposition and its application to genetic screening in mice. Mol. Cell. Biol. 2003;23:9189–9207. doi: 10.1128/MCB.23.24.9189-9207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sivasubbu S, Balciunas D, Davidson AE, et al. Gene-breaking transposon mutagenesis reveals an essential role for histone H2afza in zebrafish larval development. Mech. Dev. 2006;123:513–529. doi: 10.1016/j.mod.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–276. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 17.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–226. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 18.Mikkers H, Allen J, Knipscheer P, et al. High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat. Genet. 2002;32:153–159. doi: 10.1038/ng950. [DOI] [PubMed] [Google Scholar]
- 19.Wu X, Luke BT, Burgess SM. Redefining the common insertion site. Virology. 2006;344:292–295. doi: 10.1016/j.virol.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 20.Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- 21.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. `Green mice' as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–339. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 22.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 23.Dupuy AJ, Jenkins NA, Copeland NG. Sleeping beauty: a novel cancer gene discovery tool. Hum. Mol. Genet. 2006;15(Spec No. 1):R75–R79. doi: 10.1093/hmg/ddl061. [DOI] [PubMed] [Google Scholar]
- 24.Starr TK, Largaespada DA. Cancer gene discovery using the Sleeping Beauty transposon. Cell Cycle. 2005;4:1744–1748. doi: 10.4161/cc.4.12.2223. [DOI] [PubMed] [Google Scholar]
- 25.Uren AG, Kool J, Berns A, van Lohuizen M. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005;24:7656–7672. doi: 10.1038/sj.onc.1209043. [DOI] [PubMed] [Google Scholar]
- 26.Neil JC, Cameron ER. Retroviral insertion sites and cancer: fountain of all knowledge? Cancer Cell. 2002;2:253–255. doi: 10.1016/s1535-6108(02)00158-7. [DOI] [PubMed] [Google Scholar]
- 27.Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32:D523–D527. doi: 10.1093/nar/gkh013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hui EK, Wang PC, Lo SJ. Strategies for cloning unknown cellular flanking DNA sequences from foreign integrants. Cell. Mol. Life Sci. 1998;54:1403–1411. doi: 10.1007/s000180050262. [DOI] [PMC free article] [PubMed] [Google Scholar]