

Abstract
Vertebrate vision is initiated by photoisomerization of the visual pigment chromophore, 11-cis-retinal, and is maintained by continuous regeneration of this retinoid through a series of reactions termed the retinoid cycle. However, toxic side reaction products, especially those involving reactive aldehyde groups of the photoisomered product, all-trans-retinal, can cause severe retinal pathology. Here we lowered peak concentrations of free all-trans-retinal with primary amine-containing FDA-approved drugs that did not inhibit chromophore regeneration in mouse models of retinal degeneration. Schiff base adducts between all-trans-retinal and these amines were identified by mass spectrometry. Adducts were observed in mouse eyes only when an experimental drug protected the retina from degeneration in both short-term and long-term treatment experiments. This study demonstrates a molecular basis of all-trans-retinal-induced retinal pathology and identifies an assemblage of FDA-approved compounds with protective effects against this pathology in a mouse model that displays features of Stargardt’s and age-related retinal degeneration.
Keywords: Photoreceptor cells, A2E, RPE, retina, Stargardt’s disease, age-related macular degeneration, retinal degeneration, retinal condensation products
Vision at the molecular level involves phototransduction, which initially propagates a light signal, and the retinoid (visual) cycle, a metabolic pathway that continuously regenerates a chromophore (Fig. 1a). Rhodopsin in rod photoreceptors and cone pigments in cone photoreceptors respond to light 1. Their common chromophore is the vitamin A metabolite 11-cis-retinal covalently linked via a protonated Schiff base to polypeptide chains embedded within the opsin transmembrane domain 2. Upon absorption of a photon, the chromophore undergoes photoisomerization to all-trans-retinylidene, changing the opsin from an inactive to an active conformation before all-trans-retinylidene is hydrolyzed and all-trans-retinal (atRAL) is released (Fig. 1a). Because of this isomerization, vertebrates evolved the retinoid cycle to provide the continuous supply of 11-cis-retinal needed to maintain vision and preserve retinal health 3. This pathway involves reactions in both photoreceptor cells and the retinal pigmented epithelium (RPE), where atRAL is converted back to 11-cis-RAL via several enzymatic transformations (Fig. 1a) 4.
Figure 1. Retinoid cycle and fate of atRAL in the retina.
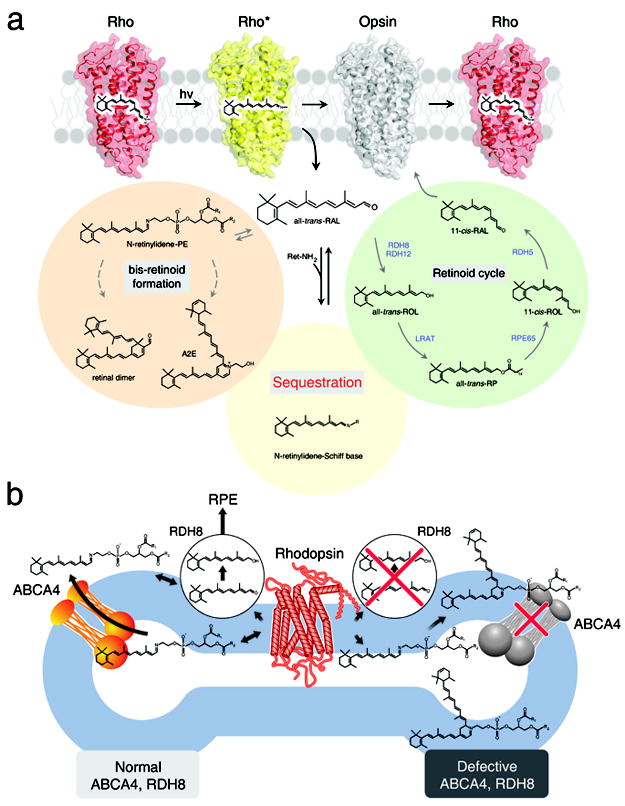
(a) The visual chromophore, 11-cis-retinal, is photoisomerized to atRAL and released after photoactivation. Possible fates of the released atRAL are to: (1) enter the retinoid cycle for recycling, (2) form bis-retinoids, and/or (3) persist as a free toxic aldehyde that directly impairs cell survival. Most of the atRAL is used to regenerate 11-cis-retinal through the retinoid cycle. But a fraction of atRAL forms bis-retinoid conjugate products such as A2E and atRAL dimer that eventually accumulate and contribute to damaging the RPE. Over-production of atRAL can induce retinal degeneration due to intense light illumination and genetic defects in RDH8 and ABCA4, which are critical for atRAL processing. These observations form the basis of our hypothesis that treatment with primary amines that form a Schiff base will lower excessive amounts of atRAL produced in mice by aging or lack of genes encoding ABCA4 and RDH8. (b) The biological role of ABCA4 and RDH8 in processing atRAL in ROS is illustrated. The left side of the diagram represents a ROS disk with a functional ABCA4 transporter and RDH8, whereas the right side portrays a ROS disk with an inactivated ABCA4 and RDH8. In rods, photoactivated rhodopsin releases atRAL outside (cytoplasm) and inside the disc lumen. Released atRAL in the disc lumen then reacts with PE producing N-retinylidene phosphatidylethanolamine (N-ret-PE). N-ret-PE is transported to the cytoplasm and releases atRAL that leaves the disc where it is converted to atROL.
Recently, we showed that mice carrying a double knockout of all-trans-retinol dehydrogenase 8 (Rdh8), one of the main enzymes that reduces atRAL in rod outer segments (ROS) and cone outer segments (COS) 5, and photoreceptor specific ATP-binding cassette transporter 4 (Abca4) (Fig. 1b) 6, which transports atRAL from the inside to the outside of disc membranes, rapidly accumulate atRAL condensation products and manifest RPE/photoreceptor dystrophy at an early age 7. Retinas from these mice exhibited many hallmarks of human age-related macular degeneration (AMD), namely lipofuscin, drusen, basal laminar deposit accumulation, Bruch’s membrane thickening, and choroidal neovascularization. Importantly, the severity of their visual dysfunction and retinopathy was exacerbated by light but attenuated by treatment with retinylamine (Ret-NH2 or A24; Supplementary Results; Supplementary Table 1), a retinoid cycle inhibitor that reduces levels of free atRAL in the retina 8-10. These findings provide direct evidence that aberrant production of toxic condensation byproducts of the retinoid cycle can lead to rapid, progressive retinal degeneration in mice. The similarity of this retinopathy to human AMD makes these Abca4−/−Rdh8−/− mice invaluable for research aimed at ameliorating this devastating blinding disease 11. Mutations in ABCA4 can cause human Stargardt’s macular degeneration 12, cone-rod dystrophy 13, or recessive retinitis pigmentosa (RP) 14,15. Moreover, heterozygous mutations in ABCA4 increase the risk of developing AMD 16.
Photoactivation of visual pigments leads to liberation of atRAL from its binding sites in rod and cone photoreceptors (Fig. 1a). The released atRAL is then reduced to all-trans-retinol (atROL) by RDHs before it is converted back to 11-cis-retinal through the retinoid cycle 4. However, a pool of atRAL that is trapped within the inner leaflet of rod photoreceptor outer segment disc membranes in form of N-retinylidenephosphatidyletanolamine (N-ret-PE) is not accessible for reduction 17-19. To ensure efficient clearance of this conjugate, ABCA4 translocates N-ret-PE to the cytoplasmic side of the disk membrane where RDH8 (the major RDH in rod photoreceptor cells 20) catalyzes conversion of atRAL to atROL (Fig. 1b). One of the major consequences of inefficient atRAL clearance is formation of the A2E fluorophore, believed to be toxic to the retina (Fig. 1b) 21. A2E is produced by the reaction of N-ret-PE with another molecule of atRAL followed by chemical rearrangements enhanced by phagocytosis of discs in the RPE 22. Because ABCA4 and RDH8 play indispensable roles in processing atRAL in the retina, Abca4−/−Rdh8−/− mice display a delayed clearance of atRAL after bright light illumination accompanied by age-related A2E accumulation in the RPE 7,11. Other condensation products between atRAL and endogenous amines were also reported 23. Free atRAL has been shown to be lethal to cultured human RPE (ARPE19) cells in a dose-dependent manner 11. Although atRAL has been suggested to be cytotoxic for decades, only recently has this been demonstrated experimentally in vivo 24. This concept is consistent with other medically-relevant approaches geared toward reducing the level of potentially toxic reactive carbonyl compounds by the use of scavengers 25,26.
Toxic effects of atRAL, including caspase activation and mitochondrial-associated cell death 11, further decrease clearance of atRAL by inactivating ABCA4 27. Even in the presence of the retinoid cycle, A2E, retinal dimer (RALdi) and other toxic atRAL condensation products 28-30 accumulate with age 31 (Fig. 1a) and serve as fluorescent biomarkers for burst of atRAL production. Patients affected by AMD, Stargardt’s disease or other retinal diseases associated with abnormal accumulation of these compounds all develop retinal degeneration16 (Fig. 1b). The 11-cis-retinal chromophore that responds to light by changing to its trans-isomer is highly light-sensitive, and it exists in a densely packed form bound to opsin (together called rhodopsin) that increases the probability of light absorption. Indeed, ROS contain 5 mM rhodopsin 32, which when bleached, yields an equivalent concentration of atRAL. Even a bleach less than 0.5% will generate toxic levels of atRAL if this retinoid is not rapidly cleared from the retina. Released atRAL needs to be quickly replaced by a new 11-cis-retinal molecule to perpetuate vision. The efficiency of the mammalian visual system and the health of photoreceptors and RPE diminish significantly with age, suggesting that there are biochemical changes that slowly poison retinal cells. These changes in turn trigger immune and other responses that culminate in retinal cell death 33.
We sought to trap free atRAL with primary amine-containing drugs to prevent this reactive compound from achieving toxic levels without inhibiting the retinoid cycle. Eventually, transient conjugates of atRAL with primary amine-containing drugs break down, and aldehydes are released at manageable levels such that photoreceptor cells can reduce them to non-toxic atROL. Herein we report testing this hypothesis in our experimental mouse models. We found that Food and Drug Administration (FDA)-approved compounds containing primary amine group protect against light-induced retinopathy in our mouse model of human blinding diseases.
RESULTS
Effects of amines on light-induced retinal degeneration
We tested a variety of small molecules that contain primary amine to determine their effect on acute light-induced retinal damage in 4-week-old Abca4−/−Rdh8−/− mice, an animal model of human Stargardt’s disease 7. These mice had been kept in the dark for 48 h, when a single dose of test amine was administered by oral gavage 2 h prior to light exposure at an intensity of 10,000 lux for 30 min. Mice then were maintained in the dark for 7 days until their final evaluation that included in vivo retinal imaging by optical coherence tomography (OCT) followed by euthanasia and retinal morphological and biochemical analyses (Supplementary Methods; Fig. 2a). Whereas no retinal degeneration was observed in 4-week-old Abca4−/−Rdh8−/− mice that were not exposed to intense bleaches, severe retinal degeneration was noted in cryosections and by OCT imaging 7 days after bright light exposure when dead cells were cleared (Fig. 2b). Indeed, OCT imaging revealed early retinal changes just one day after light exposure. Amounts of 11-cis-retinal, a surrogate for rhodopsin and photoreceptor number in light-exposed Abca4−/−Rdh8−/− mice, were reduced to 37% of levels in non-illuminated eyes, whereas these parameters were only minimally affected in illuminated WT mice (Fig. 2c).
Figure 2. Testing effects of amines on the development of acute light-induced retinal degeneration in Abca−/−Rdh8−/− mice.
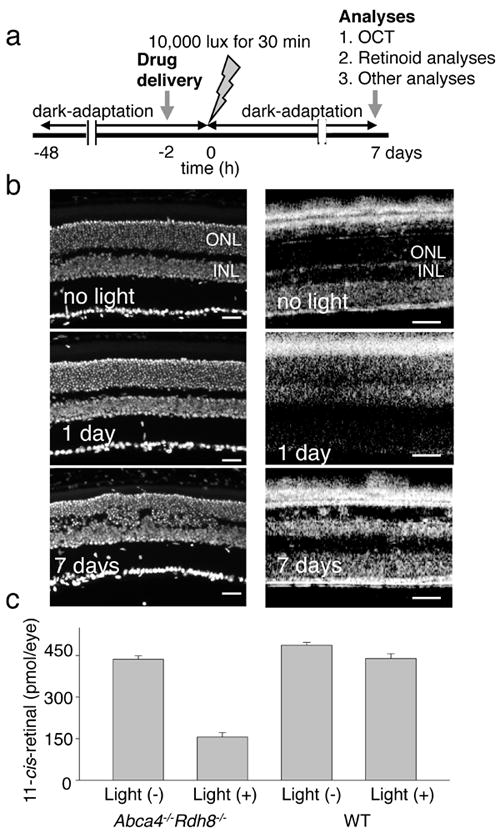
(a) Schematic representation of the experimental design.Four-week-old Abca4−/−Rdh8−/− mice were kept in the dark for 48 h when a single dose of drug was administered by oral gavage 2 h before light exposure at 10,000 lux for 30 min. Mice then were kept in the dark for 7 days when final retina evaluations were performed. (b) Four-week-old Abca4−/−Rdh8−/−mice evidenced severe retinal degeneration 7 days after bright light exposure whereas Abca4−/−Rdh8−/− mice maintained under regular laboratory lighting did not. Cryosections (left column) reveal retinal damage 7 days after bright light exposure whereas OCT images (right column) show hazy changes in the outer nuclear layer (ONL) one day after light exposure and reduced thickness of the ONL 7 days after light exposure. INL, inner nuclear layer. Bars indicate 20 μm in cryo-sections and 50 μm in OCT. (c) Amounts of 11-cis-retinal in the eye reflecting photoreceptor number were quantified by HPLC 7 days after 4-week-old Abca4−/−Rdh8−/− mice were exposed to 10,000 lux for 30 min. Light-illuminated Abca4−/−Rdh8−/− mice showed reduced amounts of 11-cis-retinal whereas 4-week-old WT mice did not.
The Physicians’ Desk Reference, a commercially published compilation of manufacturers’ information about prescription drugs, was used to select FDA-approved drugs with a documented safety record for evaluation as potential therapeutics for light-induced acute retinal degeneration. We chose 24 drugs that contain a primary amine. Drugs were tested in Abca4−/−Rdh8−/− mice at ~20% of their LD50 dose to determine if they could prevent light-induced acute retinal degeneration. A24, a potent, long-lasting inhibitor of retinoid isomerase, was used as a positive control because this retinoid is stored inside retinosomes 34 of the RPE and slowly released, inhibiting 11-cis-retinal production in vivo 10. Supplementary Table 1 summarizes the effects of the tested amines on light-induced acute retinal degeneration in Abca4−/−Rdh8−/− mice. The OCT results were arbitrarily scaled from Grade 0 (no degeneration) to Grade 4 (severe degeneration) as indicated in Supplementary Fig. 1, and levels of rhodopsin, as an indicator of photoreceptor health, were indirectly assessed by quantification of 11-cis-retinal in mouse eyes. Results are presented as the percent of 11-cis-retinal remaining after illumination, where 100% is the amount found in dark-adapted mice unexposed to light (~500 pmol/eye). The tested primary amines demonstrated significant protective effects against light-induced acute retinal degeneration in Abca4−/−Rdh8−/− mice. While vehicle-treated mice exhibited an average Grade 3.5 retinopathy by OCT imaging, 15 of the 24 A24-treated mice showed Grade 2 or milder degeneration and 16 of 24 evidenced greater than 30% protection against the light-induced decrease in rhodopsin (11-cis-retinal).
Those 16 drugs that maintained ocular 11-cis-retinal levels were next tested at 0.5 and 2 mg doses in our mouse protocol with OCT analyses performed as described in Supplementary Fig. 1a-b. These analyses confirmed significant protective effects of all 16 compounds (Supplementary Results; Supplementary Table 1). Among them, we then chose five: A4, A9, A13, A20, and A22, based on retinal protection measured with OCT images and retinoid analyses. Efficacy in preventing light-induced acute retinal degeneration was also compared between drug formulations used for humans and their chemically synthesized pure counterparts. In these tests, A4, A9, A20 and A22 displayed similar results with both the drug formulation and pure compound (data not shown).
Representative results for pretreatment with a racemic mixture of A20 (A20RS) are presented in Fig. 3. Preventative effects against light-induced retinal degeneration in Abca4−/−Rdh8−/− mice were dose-dependent with respect to both in vivo OCT imaging (Fig. 3a, c) and 11-cis-retinal quantification (Fig. 3b). Examination of plastic sections showed that treatment with 2 mg of A20 completely prevented retinal degeneration (Fig. 3d). ERG recordings of A20-treated mice showed higher a- and b-wave amplitudes under scotopic conditions and higher b-waves under photopic conditions (Fig. 3e), also supporting the protective effects of therapy with this drug.
Figure 3. Effects of pre-administered amine drugs on light-induced acute retinal degeneration in 4-week-old Abca4−/−Rdh8−/− mice.
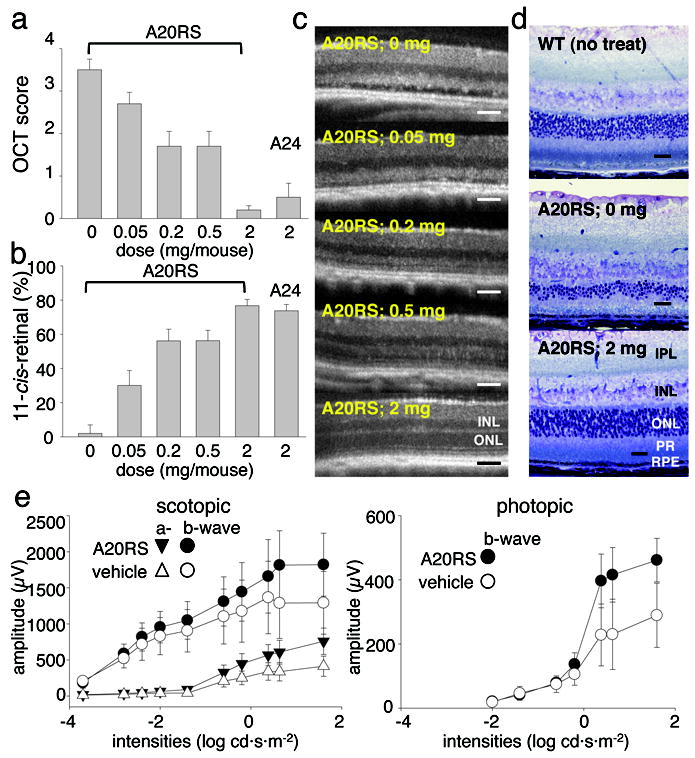
(a) Pretreatment with A20RS protected the retina from light induced damage in a dose-dependent manner as assessed by OCT, with 2 mg/mouse completely preserving retinal morphology. Included as a positive control, A24 (2 mg/mouse) also protected the retina 9. Experiments were performed with the protocol shown in Fig. 2a. (b) Amounts of 11-cis-retinal in the eye also were maintained in a dose-dependent manner by A20RS pretreatment, an effect replicated by pretreatment with A24 (2 mg/mouse). (c) Shown are representative OCT images from light-illuminated 4-week-old Abca4−/−Rdh8−/− mice pretreated with various doses of A20RS. Retinal morphology was preserved in a dose-dependent manner. INL, inner nuclear layer; ONL, outer nuclear layer. (d) Representative Epon-prepared retinal sections from light-illuminated 4-week-old Abca4−/−Rdh8−/− and WT mice are shown. Retinal histology was completely preserved by pretreatment with A20RS (2 mg/mouse). RPE, retinal pigmented epithelium; PR, photoreceptors; ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer. Bars indicate 20 μm in cryo-sections and 50 μm in OCT. (e) Full field ERG responses of A20RS- or vehicle-pretreated light-illuminated Abca4−/−Rdh8−/− mice at 4 weeks of age. ERG responses were recorded under scotopic (left) and photopic (right) conditions. Both scotopic and photopic ERG amplitudes plotted as a function of light intensity were better preserved in A20RS-pretreated Abca4−/−Rdh8−/− mice as compared with vehicle-pretreated animals. Bars indicate S.D. of the means (n > 3).
A20 has two stereoisomers (R and S) illustrated in Fig. 4a and Supplementary Table 1. The preventative effects of these stereoisomers were examined on light-induced retinal degeneration in Abca4−/−Rdh8−/− mice. Both A20 isomers preserved similar amounts of 11-cis-retinal 7 days after light exposure (Fig. 4b). In agreement with the retinoid results, retinal morphology was well maintained by both isomers as well (Fig. 4c). These results suggest that the R enantiomer, even though it has a much weaker affinity for its therapeutic target 35,36, could be useful in future studies.
Figure 4. A20 stereoisomers protect against light-induced acute retinal degeneration in Abca4−/−Rdh8−/− mice.
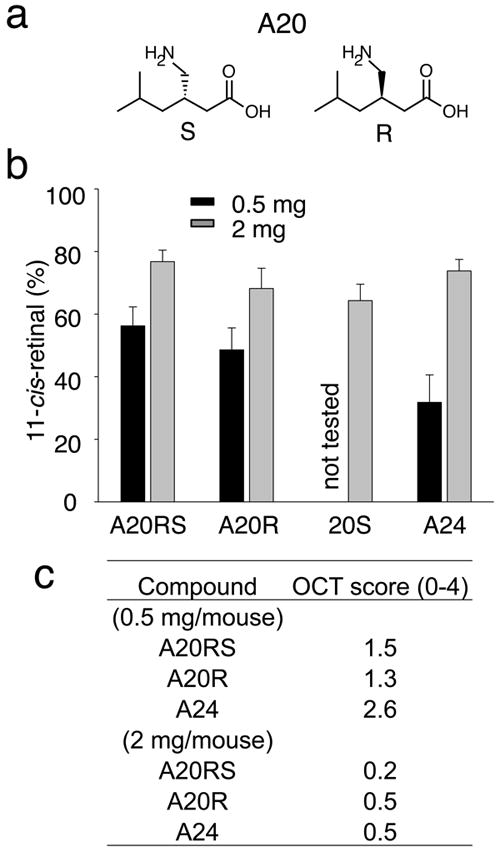
(a) Enantiomers of A20 are shown (R and S). The S-isomer of A20 is pregabalin, an analgesic that affects Ca2+ channels. (b) Amounts of 11-cis-retinal reflecting photoreceptor number were quantified by HPLC in the eyes of 4-week-old Abca4−/−Rdh8−/− mice 7 days after exposure to light at 10,000 lux for 30 min. Synthesized stereoisomers of A20 (A20RS and A20R), and a drug formulation A20S at 0.5 and 2 mg/mouse were orally gavaged 2 h before bright light exposure. All isomers of A20 showed similar protective effects against light-induced retinal degeneration. A24 was used as a positive control. Error bars indicate S.D. of the means (n > 3). (c) In vivo imaging by OCT was performed as described in Supplementary Fig. 1. OCT Grades for 0.5 and 2 mg of stereoisomer-treated mice are shown.
Tested primary amines did not affect retinoid cycle
Comparison of the 16 selected compounds that produced protective effects against light-induced acute retinal degeneration (Supplementary Table 1) shows a lack of structural similarity between them except for the primary amine. Therefore, it is unlikely that these compounds have common protein targets in vivo. To exclude the possibility that some of these compounds might directly affect visual chromophore regeneration by inhibiting retinyl ester or 11-cis-retinol production, we performed standard isomerization and LRAT activity assays in the presence of these test compounds. As summarized in Supplementary Fig. 2a, atROL esterification was inhibited less than 40% by all tested amines. Aromatic amines A1, A4, A7 and A23 inhibited retinoid isomerization significantly (Supplementary Fig. 2b). But, for example, A20R did not inhibit either reaction, even at a 50 mM concentration (Supplementary Fig. 2c, and d). Moreover, administration of these amines did not lower the rate of visual pigment regeneration in bright light exposed WT or Abca4−/−Rdh8−/− mice. Indeed, regeneration of 11-cis-retinal and the kinetics of all-trans-retinyl esters were not affected in A20- and A22-treated Abca4−/−Rdh8−/− mice, whereas persistent inhibition of the regeneration process was observed in A24-treated animals as previously reported 37 (Supplementary Fig. 3a, b). Thus, the protective effect of these primary amines, excluding A24, appears not to be caused by inhibition of 11-cis-retinoid production 8.
Because the etiology of light-induced acute retinal degeneration has been linked to the cytotoxicity of atRAL, we tested the effect of the primary amines on the rates of atRAL clearance after an extensive rhodopsin bleach in Abca4−/−Rdh8−/− mice. To assess the concentration of free atRAL in vivo, the rate of dark adaptation was examined by ERG, because atRAL is known to shut down rod cyclic nucleotide-gated channels and inhibit dark adaptation after bright light exposure 38. Importantly, administration of A20 significantly improved the rate of dark adaptation when mice were tested 2 h after its administration, suggesting that A20 reduced levels of free atRAL, probably by sequestering the aldehyde as a Schiff base (Supplementary Fig. 4a). Protective effects of A20 were no longer observed 3 days after drug administration when the rate of dark adaptation returned to that found for vehicle-treated Abca4−/−Rdh8−/− mice.
To further evaluate time-dependence of amine treatment, 4-week-old Abca4−/−Rdh8−/− mice gavaged with A20 were subjected to the acute light-induced retinal degeneration experimental protocol. Whereas significant protection (OCT Grade 0.75) was achieved 2 h after treatment, severe light-induced retinal degeneration (OCT Grade 3.75) was observed 18 h after A20 administration (Supplementary Fig. 4b). This result is in good agreement with the ~6 h half-life of A20 in vivo. Thus, a lower tissue concentration of A20 was accompanied by failure to protect against atRAL-dependent retinal degeneration.
Reduction of atRAL cytotoxicity by Schiff base formation
The predominant reaction of aldehydes with primary and secondary amines in biological systems is condensation resulting in Schiff base formation. Aldehydes are also readily reduced to corresponding alcohols. We found that atRAL caused cytotoxicity to ARPE19 cells that was reversed by A22 treatment (Supplementary Methods; Supplementary Fig. 5a). The likely mechanism involved transient conjugates between atRAL and amines. Retinyl imines were identified based on their characteristic UV/Vis spectra upon protonation (460 nm and 500 nm for alkyl and aromatic imines, respectively). When a source of RDH activity was provided (bROS), the Schiff base disappered with time (Supplementary Fig. 5b). When production of atROL was monitored, depletion of atRAL corresponded to increase in atROL. The rates for both the Schiff base depletion and atRAL reduction were comparable (Supplementary Fig. 5c). Consistently, we found that the cytotoxicity of atRAL could be neutralized by either transient formation of retinyl Schiff base or by supplementation with RDH activity (Supplementary Fig. 5d and 5e). These results indicate that Schiff bases formed between atRAL and amine drugs are not cytotoxic and that the transient nature of this conjugate does not affect the reduction of atRAL.
Quantification of the Schiff base in vivo
Compounds that contain primary amines and penetrate the eye could react with an excess of free atRAL to form a reversible Schiff base, thus lowering the effective concentration of atRAL without causing its permanent depletion or inhibiting the regeneration of visual pigments. To evaluate the role of transient conjugates between atRAL and amines in light-induced retinal degeneration, we examined the retinoid composition of eyes from Abca4−/−Rdh8−/− mice treated with selected amines by gavage and exposed to light according to the acute retinal degeneration protocol (Fig. 2a). Retinoids and some retinyl imines separated by HPLC were identified based on both their characteristic UV/Vis spectra upon protonation and spectra obtained by tandem mass spectrometry (Supplementary Fig. 6a-d). Fig. 5 illustrates two examples of retinyl imine detection in mice treated with A2 and A20, indicating that retinylidene Schiff base formation with externally administered amines can be directly detected in the eyes of treated animals. To quantify the amounts of Schiff base formed, samples were spiked with deuterated synthetic standards during tissue homogenization and then examined by mass spectrometry. The condensation products between all-trans-retinal and amines resulted in the presence of 9-cis-, 13-cis- and all-trans-isomers (Supplementary Fig. 7). Retinal adducts were only observed in mice treated with tested primary amines that showed protective effects against retinal degeneration. Moreover, retinyl imines formed directly after light exposure of treated mice but were absent in dark-adapted animals. They also were detectable in eye homogenates for more than one hour after the bleach. The time course of retinylidene Schiff base decay closely resembled atRAL clearance in mouse retina (Supplementary Fig. 6e)7. The amount of retinyl imines detected immediately after light exposure was 55 pmol and one hour later it varied between 10 and 20 pmol per eye depending on the chemical structure of the tested primary amine and the stability of the resulting Schiff base (Supplementary Fig. 6f). The hallmark of atRAL excess in retina is production of A2-PE that rearranges to form the A2E chromophore in the RPE. Therefore, formation of a transient Schiff base with external amines that reduces the pool of free atRAL should diminish A2E accumulation. Indeed, deuterated synthetic standard-based quantification of A2E in the eyes of Abca4−/−Rdh8−/− mice revealed significant decreases in A2E levels upon long term treatment with either A24, A20 or A22 (Supplementary Methods; Supplementary Fig. 8).
Figure 5. Identification of retinal conjugates in retinas of Abca4−/−Rdh8−/− mice treated with A2 and A20.
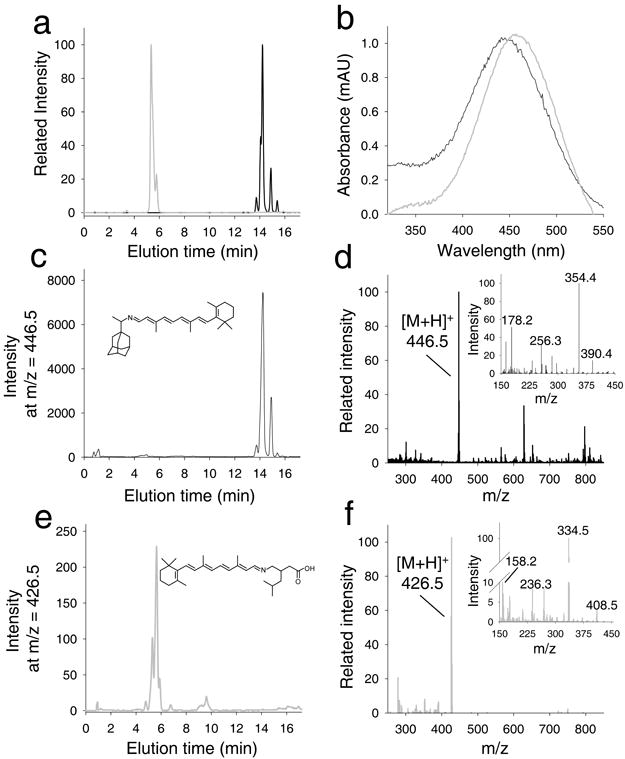
These mice were gavaged with test amines 2 h before exposure to intense light at 10,000 lux for 20 min; one hour later they were euthanized and retinoids extracted from whole eyeballs were analyzed. (a) Chromatographic separation of synthetic standards represented by selected ion chromatograms for A2 (gray) and A20 (black) retinyl imines (at m/z = 446.5 [MH]+ and m/z = 426.5 [MH]+, respectively). Panel b shows corresponding UV/Vis spectra of their protonated Schiff bases. (c) Sample extracted from mice treated with A4. Extracted ion chromatogram at m/z = 446.5 indicates a peak with an elution time, molecular mass (d) and ion fragmentation pattern (d, inset) that is identical with that of the synthetic standard A2 retinyl imine. Importantly, samples extracted from control untreated mice lacked a distinct peak at m/z = 446.5. Panels e and f represent an analogous set of data for samples extracted from the eyes of mice treated with A20. Thus, oral administration of these tested primary amines led to formation of their corresponding Schiff bases detected in organic extracts of the eyes.
Together, our observations suggest that by producing Schiff base conjugates primary amines can sequester excess atRAL that is not efficiently processed by the retinoid cycle.
Attenuation of age-related retinal degeneration
Amines were tested for their effect on age-related retinal degeneration observed in Abca4−/−Rdh8−/− mice under laboratory lighting conditions (12 h at ~ 10 lux light/12 h dark). These mice did not show significant retinal degeneration at 1 month of age but did develop progressive retinal degeneration by 4 months of age that progressed to severe loss of rod and cone photoreceptors by 8 months of age. At 4 months of age, the thickness of the photoreceptor layer in Abca4−/−Rdh8−/− inferior retina was only 58% that of WT retina 7,11.
Because A20 and A22 demonstrated good bioavailability and excellent protective effects against light-induced acute retinal degeneration in Abca4−/−Rdh8−/− mice, these two amines were examined further for their effects on age-related retinal degeneration in these animals. Because of their relatively short half-lives of 5-6 h, A20 and A22 (each at 2 mg/mouse) were administered daily by gavage. A24 (2 mg/mouse), which has a much longer half-life, 9 was given weekly. Abca4−/−Rdh8−/− mice were treated for 3 months starting at 1 month of age. As previously reported 7, treatment with A24 for 3 months prevented age-related retinal degeneration with decreased fundus autofluorescence as detected by in vivo scanning laser ophthalmoscopy (SLO) imaging (Fig. 6a). The intensity of fundus autofluorescence was measured by SLO at 1, 2, and 3 months after initiation of primary amine treatment. All amine-treated mice showed reduced retinal autofluorescence (Supplementary Fig. 9a). The distribution of SLO values after 3-month-treatment is summarized in Supplementary Table 2. The intensity of fundus autofluorescence measured by SLO correlated well with amounts of A2E measured in the eye. Eyes from amine-treated mice contained less A2E than those from vehicle-treated animals after 3 months of treatment (Supplementary Fig. 8c and Fig 6b).
Figure 6. Effects of treatment with amine drugs on age-related retinal degeneration in Abca4−/−Rdh8−/− mice.
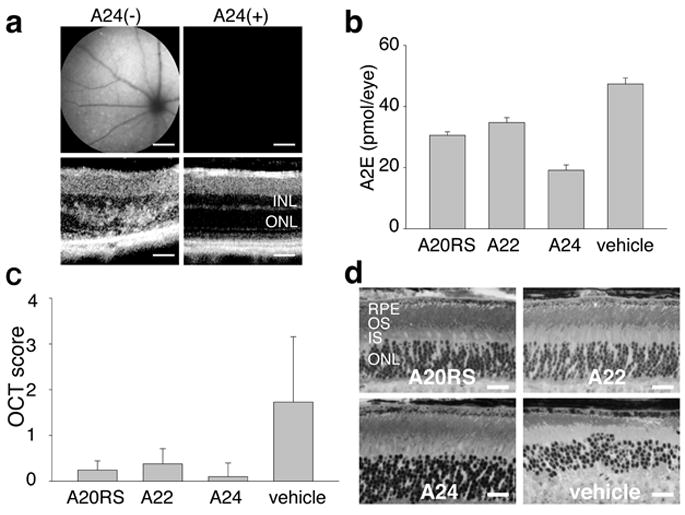
Abca4−/−Rdh8−/− mice were gavaged daily with either A20RS (2 mg/mouse) or A22 (2 mg/mouse) and weekly with A24 (2 mg/mouse) for 3 months beginning at 1 month of age. (a) Representative SLO (upper) and OCT (lower) images of A24-pretreated Abca4−/−Rdh8−/− mice are shown. INL, inner nuclear layer; ONL, outer nuclear layer. A24 prevented both an increase of fundus autofluorescence intensity and retinal degeneration. Bars indicate 20 μm in cryo-sections and 50 μm in OCT. (b) Amounts of A2E were quantified by reverse phase HPLC after 3 months of treatment of Abca4−/−Rdh8−/− mice by oral gavage. A20RS, A22, and A24 treatment reduced accumulation of A2E relative to vehicle-treated animals. Error bars indicate S.D. of the means (n = 3 for A20RS, 3 for A22, 3 for A24 and 4 for vehicle). (c) OCT analyses of Abca4−/−Rdh8−/− mouse retinas performed 3 months after treatment of one-month-old animals with either A20RS, A22 or A24 reveal maintenance of retinal structure. Error bars indicate S.D. of the means (n = 15 for A20RS, 15 for A22, 10 for A24 and 22 for vehicle). (d) Morphology of retinas from 4-month-old Abca4−/−Rdh8−/− mice treated with A20RS, A22, A24 or vehicle for 3 months. Retinas from drug-treated mice evidenced preserved structures compared with vehicle-pretreated animals. RPE, retinal pigmented epithelium; OS, outer segment; IS, inner segment; ONL, outer nuclear layer. Bars indicate 20 μm.
In addition to SLO and A2E examinations, in vivo OCT imaging, ERG and histological assessment after Epon-embedment were also performed after drug-treatment of Abca4−/−Rdh8−/− mice for 3 months. Mice treated with A20, A22, and A24 showed much milder retinal degeneration with OCT Grades of 0.2, 0.33, and 0.1, respectively, whereas Grade 1.73 changes were observed in vehicle-treated mice (Fig. 6c and Supplementary Fig. 1b-c). A20 and A22 treated mice also demonstrated higher ERG amplitudes under scotopic conditions (Supplementary Fig. 9b-c), but improvement in their photopic ERGs was not evident (Supplementary Fig. 9d). Epon-prepared retinal sections demonstrated preservation of retinal structures in amine-treated mice (Fig. 6d). Overall, treatment with A20, A22 or A24 largely rescued Abca4−/−Rdh8−/− mice from age-related retinal degeneration.
Protection in mice with diverse genetic backgrounds
The amine treatment paradigm was further examined for its effect on other mouse models that display light-induced retinal degeneration. First, we tested light-induced retinal damage in WT (BALB/c) mice. Light-induced degeneration in these mice can be induced by illumination at 5,000 lux for 2 h. A20 or A24 (each at 2 mg/mouse) were administered 2 h or 16 h, respectively, before the illumination. Representative Epon-prepared retinal sections demonstrated partially preserved retinal structures in A20-treated mice, and, as previously reported, A24 prevented light damage (Fig. 7a). None of the 7 mice pre-treated with A24 displayed light-induced retinal degeneration. BALB/c mice with A20 pretreatment demonstrated milder retinal degeneration. This indicates that an increased concentration of atRAL after illumination is involved in the pathogenesis of light damage in WT animals too. Mice treated with A20 and A24 showed much milder retinal degeneration with OCT scores of 0.78 and 0, whereas an OCT score of 1.93 was observed in vehicle-treated mice (Fig. 7b). Both A20- and A24-treated mice demonstrated higher 11-cis-retinal levels than vehicle-treated mice, which also indicates more preserved photoreceptors in amine-treated mice (Fig. 7c). Thus, treatment with A20 or A24 also protected WT mice from light-induced retinal degeneration.
Figure 7. Effects of pre-administered amine drugs on light-induced retinal degeneration in BALB/c mice.
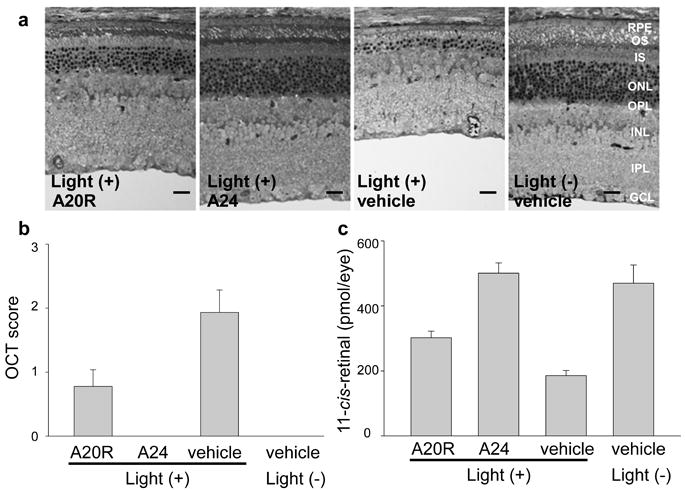
(a) Representative epon-prepared retinal sections from light-illuminated 6-week-old WT (BALB/c) mice are shown. Mice were illuminated with 5,000 lux for 2 h, and analysis was performed 7 days later. Pretreatment with A20R (2 mg/mouse, 2 h before light) partially protected the retina from light induced damage, and A24, included as a positive control (2 mg/mouse, 16 h before light) protected the retina as previously reported 9. RPE, retinal pigmented epithelium; OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Bars indicate 10 μm. (b) OCT analyses of WT mouse retinas were performed. Score 0, no degeneration; score 1, reduced ONL thickness with more than 50% of ONL remaining as compared to no-illuminated animals; score 2, reduced ONL thickness with less than 50% of ONL for non-illuminated animals; score 3, severe degeneration without reflection from the ONL layer. A20R provided partial protection of retinal structure, whereas A24 protected retinal degeneration completely. Error bars indicate S.D. of the means (n = 12 for A20R, 7 for A24 and 15 for vehicle). (c) Amounts of 11-cis-retinal in the eye quantified by HPLC also were partially preserved by A20R (2 mg/mouse) and completely protected by A24 pretreatment. Bars indicate S.D. of the means (n > 3).
Finally, light-sensitive mice were employed to test the effect of amine treatment. Rdh12−/− mice and Palm−/− (palmitoylation-deficient rhodopsin) mice were used for this purpose. Structurally unstable rhodopsin due to absence of palmitoylation in Palm−/− mice 39 or altered retinoid metabolism seen in the photoreceptor inner segments of Rdh12−/− mice are suggested to contribute to retinal degeneration in these animals 40. Light-induced retinal degeneration was initiated by illumination at 10,000 lux for 15 min for Palm−/− mice or by 10,000 lux for 60 min for Rdh12−/− mice. A20 (2 mg/mouse) was administrated 2 h before the light exposure. Untreated Palm−/− mice showed severe retinal damage under these conditions with a 72% loss of 11-cis-retinal (Supplementary Fig. 10a, b) whereas Rdh12−/− mice displayed light-induced retinal degeneration with an irregular ONL arrangement as revealed by OCT examination with 44% loss of 11-cis-retinal. Pretreatment with A20 prevented light-induced retinal degeneration in both models with a greater protective effect found in Rdh12−/− mice (Supplementary Fig. 10a, b). Taken together, our results with these different genetic mouse models suggest that atRAL mediates photo-damage in the retina and that sequestration of free atRAL by primary amine drugs protects the retina from light-induced disorders.
DISCUSSION
At the heart of the visual system is a small molecular chromophore that is uniquely suited for absorbing light, yet is intrinsically toxic. The oxidation-prone polyene chain and chemically-reactive aldehyde groups of 11-cis-retinal and its trans isomer are protected by opsins in the photoreceptor outer segments and by retinoid-binding proteins as they move between different compartments of the retina. Nonetheless, toxic side reactions, especially those involving the reactive aldehyde group of atRAL, still occur and can cause severe retinal dysfunction and pathology. Here we tested the efficacy of several FDA-approved primary amine-containing drugs and their chemical analogues in lowering the toxicity of elevated levels of atRAL accumulated in native and genetically-engineered mouse models of human retinal disease. We chose primary amines because they transiently and reversibly react with aldehydes and can directly compete with PE to prevent A2E formation. We selected 24 drugs that: (a) contain a primary amine, (b) display a hydrophobic character without containing a membrane impermeable substituent such as a phosphate or sulfo group, and (c) exhibit a good safety profile at high doses. Such compounds were primarily those directed against infectious diseases, because these are less likely to target human proteins. Selection of compounds other than primary amines, such as thiols and hydrazines, was also considered and some thiols were tested. However, thiols react with retinal producing a complex mixture of products. These have foul odors and could disturb the red-ox potential of cells. Hydrazine-containing drugs are uncommon because they react readily with ketones and often display toxicity. Moreover, products of hydrazine condensation hydrolyze slowly back to free substrates 41 and could cause problems with dark adaptation.
The primary goal of this study was to identify effective and safe therapy to prevent or ameliorate retinal pathology in an animal model that mimics human Stargardt’s disease, with a subset of phenotypical changes also found in AMD. Lacking transport of atRAL from the inside to the outside of disc membranes, Abca4−/− mice accumulated atRAL condensation products, including A2E and RALdi 7,42. Nonetheless, despite the increase of pigmented granules in the RPE apparent in EM micrographs, we saw no retinal degeneration in Abca4−/− mice with a pigmented background by the age of 15 months 43, probably because clearance by RDHs maintained atRAL below toxic levels. However, mice carrying a double knockout of Rdh8, one of the main enzymes that reduces atRAL in ROS and COS, and Abca4 7 rapidly accumulated atRAL condensation products and exhibited RPE/photoreceptor dystrophy at an early age 7,11. Importantly, these young Abca4−/−Rdh8−/− mice displayed a retinal phenotype similar to both Stargardt’s disease and dry-AMD that included accumulation of lipofuscin, drusen and basal laminar deposits as well as thickening of Bruch’s membrane. Choroidal neovascularization, a hallmark of wet-AMD, was also observed in some older mice (> 6 months). The severity of visual dysfunction and retinopathy was exacerbated by light 7,11. Retinal degeneration in Abca4−/−Rdh8−/− mice started as a punctate process at the age of 4-6 weeks. By the age of 3 months, degeneration had progressed to the entire inferior retina. These changes were accompanied by decreases in 11-cis-retinal and increases in atRAL and its condensation products. Thus, we propose that aberrant reactions of the retinoid cycle leading to increased levels of atRAL are responsible for the initial insult to the retinas of these animals 7,11.
Light exposure is critical for A2E accumulation and induction of cytotoxicity associated with atRAL 44. High levels of A2E are reported in patients with Stargardt’s disease and AMD and age-dependent retinal accumulation of A2E also is evident in healthy individuals. To determine if retinal morphology in mice with delayed atRAL clearance is influenced by light, we exposed 4-week-old Abca4−/−Rdh8−/−, 3-month-old Abca4−/−, and 3-month-old Rdh8−/− mice without obvious retinal damage to 10,000 lux of fluorescent light for 60 min. Retinal degeneration with retinal folds was found in all genotypes tested but was most severe in Abca4−/−Rdh8−/− mice. A high incidence (8 of 8 mice) of mild retinal changes was also induced by this light in Rdh8−/− mice, further indicating that accumulation of atRAL during light exposure promoted retinal changes 7. Fifteen-month-old pigmented Abca4−/− mice failed to exhibit retinal degeneration under ordinary lighting conditions 43, but exposure to 10,000 lux for 30 min did induce mild retinal degeneration in 42% (n = 12) of these animals. WT mice showed no retinal changes under these conditions 7,11. Thus, short-term experiments conducted within one week after intense illumination of a large number of Abca4−/−Rdh8−/− mice can be used to identify successful therapeutics for prevention of retinal degeneration. This would not be practical if a 6- to 9-month treatment regimen was required.
We suggest that A2E 22 and other condensation products 23,45 formed via reactions with endogenous amines can lower levels of atRAL in the eye and that this natural process reduces atRAL toxicity 11. These condensation products likely are surrogate markers for aberrations in atRAL clearance and that accumulate during our life-time 22. The unique structure of photoreceptor outer segments perhaps limits the number of eligible amines from the total repertoire available in our body. We propose that excess atRAL is temporarily sequestered by naturally occurring amines thereby preventing toxicity from the free aldehyde.
This constitutes an unusual pharmacological approach because the target here is a toxic small molecule rather than an enzyme or receptor. Aldehyde-selective chemistry is employed to reversibly bind atRAL as a Schiff base and lower the level of this toxic intermediate in the retinoid cycle. Slow release of atRAL from the Schiff base should allow the retinoid to flow back into the retinoid cycle without affecting phototransduction. This hypothesis was also tested in cultured cells with atRAL, amines and bovine ROS as a source of RDH. Producing reversible Schiff base conjugates prevented the cytoxicity of atRAL without changing RDH activity that converted atRAL to atROL. This experiment also provides evidence that the Schiff base conjugate itself does not impair cell survival (Supplementary Fig. 5). This platform can also be used to design and synthesize similar compounds for the next stage of investigation and pharmacological development.
In summary, we tested the efficacy of several FDA-approved amine-containing drugs and their chemical analogues for their ability to lower elevated levels of atRAL and accumulation of its conjugation products in the retina and thereby decrease these compounds’ toxicity in WT and genetically-engineered mouse models of human retinal disease. Comparison of more than 20 FDA-approved drugs and a subset of analogues selected from chemical libraries allowed identification of efficacious, bioavailable and safe candidates that preserved the retina without inhibiting chromophore regeneration. Further elucidation of the mechanisms and physiological consequences of atRAL toxicity will lead to improved pharmacological treatment of patients suffering from blinding diseases. Finally, despite their promise, these compounds should not be used to treat human blinding diseases until shown safe and effective in controlled clinical trials.
METHODS
Animals and treatments
Abca4−/−Rdh8−/− double knockout mice were generated as previously described and all knockout mice were genotyped by established methods 7,46. Only mice with the leucine variation at amino acid 450 of RPE65 were used 47. Mice were housed in the animal facility at the School of Medicine, Case Western Reserve University, where they were maintained either under complete darkness or a 12 h light (~10 lux) /12 h dark cycle. All primary amines tested were dissolved or suspended in 100 μl of water with less than 10% DMSO and were administered by oral gavage with a 22G feeding needle. Experimental manipulations in the dark were done under dim red light transmitted through a Kodak No. 1 safelight filter (transmittance > 560 nm). All animal procedures and experiments were approved by the Case Western Reserve University Animal Care Committees and conformed to both recommendations of the American Veterinary Medical Association Panel on Euthanasia and the Association of Research for Vision and Ophthalmology.
Induction of light-induced retinal degeneration in Abca4−/−Rdh8−/− mice
After dark adaptation for 48 h, one or two Abca4−/−Rdh8−/− mice with pupils dilated by 1% tropicamide were exposed to fluorescent light (10,000 lux) (150 W spiral lamp, Commercial Electric) for 30 min in a white plastic bucket (Papersmith) with food and water and then kept in the dark. Histological and biochemical experiments were performed one week after exposure.
Ultra-high resolution spectral-domain OCT (SD-OCT) and scanning laser ophthalmoscopy (SLO) imaging
Ultra-high resolution SD-OCT (Bioptigen, Research Triangle Park, NC) and HRAII SLO (Heidelberg Engineering, Germany) were employed for in vivo imaging of mouse retinas. Mice were anesthetized by intraperitoneal injection of a cocktail (20 μl/g body weight) containing ketamine (6 mg/ml) and xylazine (0.44 mg/ml) in 10 mM sodium phosphate, pH 7.2, containing 100 mM NaCl. Pupils were dilated with 1% tropicamide. Four pictures acquired in the B-scan mode were used to construct each final averaged SD-OCT image.
RPE microsomal preparations
Bovine RPE microsomes, isolated from RPE homogenates by differential centrifugation as previously described48, were resuspended in 10 mM Tris/HCl, Ph 7.2, 1 μM leupeptin and 1 mM DTT to achieve a total protein concentration of ~5 mg/ml. To destroy endogenous retinoids, 200 μl aliquots of RPE microsomes placed in a quartz cuvette were irradiated for 5 min at 0°C with a ChromatoUVE-transilluminator (model TM-15 from UVP, Upland, CA).
Retinoid cycle enzyme assays: LRAT and retinoid isomerase activity
All assays were performed under dim red light. LRAT reactions were carried out in 25 mM Tris/HCl buffer, pH 8.0, 1% BSA, and 1 mM 1,2-diheptanoyl-sn-glicerol-3-phosphocholine. AtROL was delivered in 1 μl of dimethylformamide (DMF) to reach a final concentration of 10 μM. Tested amines were added in DMF or 50% DMF in 0.67 mM phosphate buffer, pH 7.2, to reach a final concentration of 1 mM. Reactions were initiated by addition of RPE microsomes (2.5 μg of total protein) to achieve a total volume of 0.1 ml. Reactions proceeded for 2 min at room temperature and then were stopped by addition of 0.3 ml of methanol followed by an equal volume of hexane. Retinyl heptanoate was extracted and quantified by normal phase HPLC (Agilent, Si, 5 μm, 4.6 × 250 mm) in 20% ethyl acetate in hexane. Flow rate was 1.4 ml/min. The retinoid isomerase reaction was carried out in 10 mM Bis-Tris propane buffer, pH 7.5, 1% BSA, containing 1 mM ATP and 6 μM apo-CRALBP 49. AtROL and tested amines were delivered in DMF or 50% DMF in 0.67 mM phosphate buffer, pH 7.2, to a final concentration of 10 μM and 1 mM, respectively. Reactions were initiated by adding RPE microsomes (500 μg of total protein), and then incubated at 30°C for 10 min. Reactions were stopped by addition of 0.3 ml of methanol and the reaction products were extracted with 0.3 ml of hexane.
Lactate dehydrogenase (LDH) activity
ARPE19 cells (human RPE cells purchased from ATCC) were cultured in 96 well plates (2 × 104 cells well) at 37°C for 16 h and then further incubated with 30 μM of atRAL and 30 μM of A22 for 6 or 24 h. Activity of LDH released from dead cells was measured with an LDH assay kit from BioVision (Mountain View, CA). The percentage of cytotoxicity was calculated as [(test sample – cell negative control) / (lysis control – cell negative control)] × 100.
Quantification of retinylidene condensation products in mouse eyes
Product extraction from eyes of mice treated with compounds containing primary amines was performed by the procedure described for A2E. Extracted compounds were separated by reverse phase HPLC chromatography (Agilent Zorbax Eclipse XBD C18, 5 μm, 4.6 × 150 mm column) with a linear gradient of water in acetonitrile (50 – 100%) for 20 min at a fixed flow rate of 1.5 ml/min. Retinal conjugates were identified with a LXQ mass spectrometer equipped with an atmospheric pressure chemical ionization (APCI) source (Thermo Finnigan) operating in a positive ionization mode. MS scans were recorded in the SIM mode for each individual compound. Identities of detected adducts were confirmed based on their MS/MS spectra. Amounts of imides were quantified with the aid of isotopically-labeled synthetic standards added prior to extraction.
Electroretinograms (ERGs)
All ERG procedures were performed by published methods 46. For single-flash recording, the duration of white light flash stimuli (from 20 μs to 1 ms) was adjusted to provide a range of illumination intensities (from -3.7 to 1.6 log cd·s/m2). Three to five recordings were made at sufficient intervals between flash stimuli (from 3 s to 1 min) to allow recovery from any photo-bleaching effects.
Statistical analyses
Data representing the means ± S.D. for the results of at least three independent experiments were compared by the one way ANOVA-test.
Supplementary Material
Acknowledgments
We would like to thank Z. Dong for expert handling of mice, Satsumi Roos for block preparation and plastic sectioning, and Melissa Matosky for retinoid analyses. We also thank Dr. Leslie Webster Jr., Dr. Jack Saari, Dr. Michael E. Maguire, and members of Palczewski’s laboratory for critical comments on the manuscript. This research was supported in part by grants EY009339, EY021126, EY019031, EY019880 and P30 EY11373 from the National Institutes of Health, TECH 09 004 from the State of Ohio Department of Development and Third Frontier Commission. KP is John H. Hord Professor of Pharmacology.
Abbreviations used
- A 2 E
2–[2,6–dimethyl–8–(2,6,6–trimethyl–1–cyclohexen–1–yl)–1E,3E,5E,7E–octatetraenyl]–1–(2–hydroxyethyl)–4–[4–methyl–6–(2,6,6–trimethyl–1–cyclohexen–1–yl)–1E,3E,5E–hexatrienyl]–pyridinium (or N-retinyl-N-retinylidene-ethanolamine)
- ABCA4/ABCR
photoreceptor specific ATP–binding cassette transporter
- AMD
age–related macular degeneration
- atRAL
all-trans-retinal
- atROL
vitamin A, all-trans-retinol
- bROS
bovine rod outer segments
- DMF
dimethylformamide
- GA
geographic atrophy
- LRAT
lecithin:retinol acyl transferase
- PE
phosphatidylethanolamine
- RPE65
retinoid isomerase
- TFA
trifluoroacetic acid
- TPM
two–photon microscopy
- SLO
scanning laser ophthalmoscope
- WT
wild type
Footnotes
Contributions A.M., M.G. and K.P. conceived and directed the project. A.M., M.G., T.M., Y.C., K.O., S.S. and K.I. designed and conducted experiments. G. P. and K.P. performed pharmacological analyses of the data, A.M., M.G. and K.P. prepared the manuscript. A.M., M.G., G.P., WH. and T.M. analyzed the data and edited the manuscript.
Competing financial interests Case Western Reserve University, and Visum Inc. may commercialize some of the technology described in this work. AM, MG and TM are consultants for Visum Inc. and KP and WH are co-founders of Visum Inc.
Contributor Information
Akiko Maeda, Department of Ophthalmology and Visual Sciences, Case Western Reserve University, Cleveland, USA; Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio, USA.
Marcin Golczak, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio, USA.
Yu Chen, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio, USA.
Kiichiro Okano, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio, USA.
Hideo Kohno, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio, USA.
Satomi Shiose, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio, USA.
Kaede Ishikawa, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio, USA.
William Harte, Visum Inc.; Cleveland, Ohio 44106, USA.
Grazyna Palczewska, Polgenix Inc., Cleveland, Ohio 44106, USA.
Tadao Maeda, Department of Ophthalmology and Visual Sciences, Case Western Reserve University, Cleveland, USA; Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio, USA.
Krzysztof Palczewski, Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio, USA.
References
- 1.Palczewski K. G protein-coupled receptor rhodopsin. Annu Rev Biochem. 2006;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 3.Travis GH, Golczak M, Moise AR, Palczewski K. Diseases caused by defects in the visual cycle: retinoids as potential therapeutic agents. Annu Rev Pharmacol Toxicol. 2007;47:469–512. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von Lintig J, Kiser PD, Golczak M, Palczewski K. The biochemical and structural basis for trans-to-cis isomerization of retinoids in the chemistry of vision. Trends Biochem Sci. 2010;35:400–410. doi: 10.1016/j.tibs.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rattner A, Smallwood PM, Nathans J. Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J Biol Chem. 2000;275:11034–11043. doi: 10.1074/jbc.275.15.11034. [DOI] [PubMed] [Google Scholar]
- 6.Molday RS, Beharry S, Ahn J, Zhong M. Binding of N-retinylidene-PE to ABCA4 and a model for its transport across membranes. Adv Exp Med Biol. 2006;572:465–470. doi: 10.1007/0-387-32442-9_64. [DOI] [PubMed] [Google Scholar]
- 7.Maeda A, Maeda T, Golczak M, Palczewski K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J Biol Chem. 2008;283:26684–26693. doi: 10.1074/jbc.M804505200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Golczak M, Kuksa V, Maeda T, Moise AR, Palczewski K. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci U S A. 2005;102:8162–8167. doi: 10.1073/pnas.0503318102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maeda A, et al. Effects of potent inhibitors of the retinoid cycle on visual function and photoreceptor protection from light damage in mice. Mol Pharmacol. 2006;70:1220–1229. doi: 10.1124/mol.106.026823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Golczak M, et al. Lecithin:retinol acyltransferase is responsible for amidation of retinylamine, a potent inhibitor of the retinoid cycle. J Biol Chem. 2005;280:42263–42273. doi: 10.1074/jbc.M509351200. [DOI] [PubMed] [Google Scholar]
- 11.Maeda A, et al. Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J Biol Chem. 2009;284:15173–15183. doi: 10.1074/jbc.M900322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allikmets R, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 13.Cremers FP, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7:355–362. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 14.Martinez-Mir A, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1998;18:11–12. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Q, et al. Severe autosomal recessive retinitis pigmentosa maps to chromosome 1p13.3-p21.2 between D1S2896 and D1S457 but outside ABCA4. Hum Genet. 2005;118:356–365. doi: 10.1007/s00439-005-0054-4. [DOI] [PubMed] [Google Scholar]
- 16.Allikmets R. Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am J Hum Genet. 2000;67:487–491. doi: 10.1086/303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsybovsky Y, Molday RS, Palczewski K. The ATP-Binding Cassette Transporter ABCA4: Structural and Functional Properties and Role in Retinal Disease. Adv Exp Med Biol. 2010;703:105–125. doi: 10.1007/978-1-4419-5635-4_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molday RS, Zhang K. Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog Lipid Res. 2010 doi: 10.1016/j.plipres.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong M, Molday LL, Molday RS. Role of the C terminus of the photoreceptor ABCA4 transporter in protein folding, function, and retinal degenerative diseases. J Biol Chem. 2009;284:3640–3649. doi: 10.1074/jbc.M806580200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maeda A, et al. Redundant and unique roles of retinol dehydrogenases in the mouse retina. Proc Natl Acad Sci U S A. 2007;104:19565–19570. doi: 10.1073/pnas.0707477104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sparrow JR, Wu Y, Kim CY, Zhou J. Phospholipid meets all-trans-retinal: the making of RPE bisretinoids. J Lipid Res. 2010;51:247–261. doi: 10.1194/jlr.R000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parish CA, Hashimoto M, Nakanishi K, Dillon J, Sparrow J. Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc Natl Acad Sci U S A. 1998;95:14609–14613. doi: 10.1073/pnas.95.25.14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murdaugh LS, et al. Compositional studies of human RPE lipofuscin. J Mass Spectrom. 2010;45:1139–1147. doi: 10.1002/jms.1795. [DOI] [PubMed] [Google Scholar]
- 24.Rozanowska M, Sarna T. Light-induced damage to the retina: role of rhodopsin chromophore revisited. Photochem Photobiol. 2005;81:1305–1330. doi: 10.1562/2004-11-13-IR-371. [DOI] [PubMed] [Google Scholar]
- 25.Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol. 2008;153:6–20. doi: 10.1038/sj.bjp.0707395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ellis EM. Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacol Ther. 2007;115:13–24. doi: 10.1016/j.pharmthera.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 27.Sun H, Nathans J. ABCR, the ATP-binding cassette transporter responsible for Stargardt macular dystrophy, is an efficient target of all-trans-retinal-mediated photooxidative damage in vitro. Implications for retinal disease. J Biol Chem. 2001;276:11766–11774. doi: 10.1074/jbc.M010152200. [DOI] [PubMed] [Google Scholar]
- 28.Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A. 2000;97:7154–7159. doi: 10.1073/pnas.130110497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim YK, et al. Retinyl ester formation by lecithin: retinol acyltransferase is a key regulator of retinoid homeostasis in mouse embryogenesis. J Biol Chem. 2008;283:5611–5621. doi: 10.1074/jbc.M708885200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fishkin N, Yefidoff R, Gollipalli DR, Rando RR. On the mechanism of isomerization of all-trans-retinol esters to 11-cis-retinol in retinal pigment epithelial cells: 11-fluoro-all-trans-retinol as substrate/inhibitor in the visual cycle. Bioorg Med Chem. 2005;13:5189–5194. doi: 10.1016/j.bmc.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Yannuzzi LA, et al. Ophthalmic fundus imaging: today and beyond. Am J Ophthalmol. 2004;137:511–524. doi: 10.1016/j.ajo.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 32.Nickell S, Park PS, Baumeister W, Palczewski K. Three-dimensional architecture of murine rod outer segments determined by cryoelectron tomography. J Cell Biol. 2007;177:917–925. doi: 10.1083/jcb.200612010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holz FG, et al. Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am J Ophthalmol. 2007;143:463–472. doi: 10.1016/j.ajo.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 34.Imanishi Y, Batten ML, Piston DW, Baehr W, Palczewski K. Noninvasive two-photon imaging reveals retinyl ester storage structures in the eye. J Cell Biol. 2004;164:373–383. doi: 10.1083/jcb.200311079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor CP, et al. Potent and stereospecific anticonvulsant activity of 3-isobutyl GABA relates to in vitro binding at a novel site labeled by tritiated gabapentin. Epilepsy Res. 1993;14:11–15. doi: 10.1016/0920-1211(93)90070-n. [DOI] [PubMed] [Google Scholar]
- 36.Field MJ, Oles RJ, Singh L. Pregabalin may represent a novel class of anxiolytic agents with a broad spectrum of activity. Br J Pharmacol. 2001;132:1–4. doi: 10.1038/sj.bjp.0703794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Golczak M, et al. Metabolic basis of visual cycle inhibition by retinoid and nonretinoid compounds in the vertebrate retina. J Biol Chem. 2008;283:9543–9554. doi: 10.1074/jbc.M708982200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dean DM, Nguitragool W, Miri A, McCabe SL, Zimmerman AL. All-trans-retinal shuts down rod cyclic nucleotide-gated ion channels: a novel role for photoreceptor retinoids in the response to bright light? Proc Natl Acad Sci U S A. 2002;99:8372–8377. doi: 10.1073/pnas.122681899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maeda A, et al. Palmitoylation stabilizes unliganded rod opsin. Proc Natl Acad Sci U S A. 2010;107:8428–8433. doi: 10.1073/pnas.1000640107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeda A, et al. Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. J Biol Chem. 2006;281:37697–37704. doi: 10.1074/jbc.M608375200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalia J, Raines RT. Hydrolytic stability of hydrazones and oximes. Angew Chem Int Ed Engl. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weng J, et al. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 43.Mata NL, et al. Delayed dark-adaptation and lipofuscin accumulation in abcr+/- mice: implications for involvement of ABCR in age-related macular degeneration. Investigative ophthalmology & visual science. 2001;42:1685–1690. [PubMed] [Google Scholar]
- 44.Sparrow JR, Nakanishi K, Parish CA. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Investigative ophthalmology & visual science. 2000;41:1981–1989. [PubMed] [Google Scholar]
- 45.Vollmer-Snarr HR, et al. Amino-retinoid compounds in the human retinal pigment epithelium. Adv Exp Med Biol. 2006;572:69–74. doi: 10.1007/0-387-32442-9_11. [DOI] [PubMed] [Google Scholar]
- 46.Maeda A, et al. Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J Biol Chem. 2005;280:18822–18832. doi: 10.1074/jbc.M501757200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim SR, et al. Rpe65 Leu450Met variant is associated with reduced levels of the retinal pigment epithelium lipofuscin fluorophores A2E and iso-A2E. Proc Natl Acad Sci U S A. 2004;101:11668–11672. doi: 10.1073/pnas.0403499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stecher H, Palczewski K. Multienzyme analysis of visual cycle. Methods Enzymol. 2000;316:330–344. doi: 10.1016/s0076-6879(00)16733-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stecher H, Gelb MH, Saari JC, Palczewski K. Preferential release of 11-cis-retinol from retinal pigment epithelial cells in the presence of cellular retinaldehyde-binding protein. J Biol Chem. 1999;274:8577–8585. doi: 10.1074/jbc.274.13.8577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.