

Abstract
Calorie restriction (CR) promotes longevity. A prevalent mechanistic hypothesis explaining this effect suggests that protein degradation, including mitochondrial autophagy, is increased with CR, removing damaged proteins and improving cellular fitness. At steady state, increased catabolism must be balanced by increasing mitochondrial biogenesis and protein synthesis, resulting in faster protein replacement rates. To test this hypothesis, we measured replacement kinetics and relative concentrations of hundreds of proteins in vivo in long-term CR and ad libitum-fed mice using metabolic 2H2O-labeling combined with the Stable Isotope Labeling in Mammals protocol and LC-MS/MS analysis of mass isotopomer abundances in tryptic peptides. CR reduced absolute synthesis and breakdown rates of almost all measured hepatic proteins and prolonged the half-lives of most (∼80%), particularly mitochondrial proteins (but not ribosomal subunits). Proteins with related functions exhibited coordinated changes in relative concentration and replacement rates. In silico expression pathway interrogation allowed the testing of potential regulators of altered network dynamics (e.g. peroxisome proliferator-activated receptor gamma coactivator 1-alpha). In summary, our combination of dynamic and quantitative proteomics suggests that long-term CR reduces mitochondrial biogenesis and mitophagy. Our findings contradict the theory that CR increases mitochondrial protein turnover and provide compelling evidence that cellular fitness is accompanied by reduced global protein synthetic burden.
Calorie restriction (CR)1 is a dietary intervention in which calorie intake is reduced without malnutrition. CR is considered the most robust nongenetic method for increasing life span and has been shown to be effective in yeast (1), worms (2), flies (3), mice, rats (4), and nonhuman primates (5, 6). CR induces a host of physiological changes, including reduced reactive oxygen species (ROS) production (7), reduced core body temperature (8), and reduced global cell proliferation rates (9, 10). It is unknown whether these physiological effects are connected to the CR-dependent prevention of age-related diseases such as cancer, diabetes, hypertension, and cardiovascular disease in mammalian model systems (11). Beyond the prevention of disease, CR reduces age-related declines in cognitive function and the development of sarcopenia (5). Although CR-dependent reductions in disease and increased longevity were first demonstrated in rats 75 years ago (4), the underlying cellular mechanisms are not fully understood.
Two apparently contradictory mechanisms have been proposed to explain how CR reduces the accumulation of damaged proteins: 1) increased protein replacement resulting in less oxidative damage, or 2) decreased protein turnover due to lower demand for protein replacement. The first suggests that CR is similar to short-term starvation, which induces autophagy and lysosomal proteolysis in cells (12). Several researchers have reported that chronic CR increases protein degradation, including autophagic digestion of mitochondria, removing damaged proteins while increasing mitochondrial biogenesis and replacement with new proteins (13–18). Consistent with this hypothesis, CR-dependent increases in mitochondrial biogenesis (15, 18) and increased autophagy or protein catabolism (16, 19–21) have been reported. Yet, to date, no study has assessed the turnover of autophagic substrates (e.g. mitochondrial proteins) in calorie-restricted mammals.
The second mechanistic theory is supported by studies in yeast and C. elegans that demonstrated that a reduction in the rate of synthesis without a change in total abundance (i.e. reduced protein turnover) extends the maximal lifespan of these organisms (22–24). These reports are consistent with findings that the repression of protein synthetic signaling pathways leads to increased longevity in mice (25). It is hypothesized that reducing the rate of protein synthesis leads to improved translational fidelity, reduced proteolytic burden, increased chaperone capacity, and, ultimately, reduced generation of damaged proteins (26).
In this study, we directly measure how CR affects hepatic protein replacement (turnover). By measuring protein synthesis and degradation in vivo, we can directly address the core supposition of each mechanistic theory. To accomplish this, we combined stable isotope metabolic labeling (27) with exogenously labeled Stable Isotope Labeling in Mammals (SILAM) standards (28, 29) to concurrently measure fractional replacement rates (half-lives) and concentrations via LC-MS/MS. This combination of dynamic and quantitative proteomics allows, for the first time, absolute synthesis and breakdown rates of hundreds of proteins to be measured in vivo (Fig. 1). We compared age-matched long-term CR and ad libitum-fed (AL) C57BL6 mice from the CR colony maintained by the National Institutes on Aging (NIA). We found that CR reduced global protein synthesis and breakdown rates and prolonged the half-lives of the great majority of cellular proteins in the liver. Interestingly, we found that the turnover of mitochondrial proteins was disproportionately affected, with greatly increased half-lives. Concentrations of mitochondrial proteins were also reduced, resulting in dramatically slower absolute synthesis rates. The production of ribosomal proteins was also greatly affected, with markedly reduced concentrations, although these proteins had minimal changes in half-life. Selected functional groups of proteins had increased replacement rates, and others had the same concentrations in AL and CR conditions, but almost no protein measured showed increased absolute synthesis rates. Similar to an earlier report, we found that the turnover and concentration of functionally related proteins are regulated coordinately (30). Through the application of relational database software programs, perturbations in the dynamic proteome network were tested against expected patterns based on the published gene and protein regulatory literature. The role of selected transcription factors and co-activators was interrogated in this manner, and the changes were consistent with the previously proposed role for peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) in the hepatic response to CR (31). These findings suggest that protein categories are selectively regulated by coordinated mechanisms in CR to reduce absolute protein synthesis, reduce proteolytic flux rates, and prolong half-lives of proteins throughout the proteome. Proteolytic editing is not increased in either a relative or an absolute manner in long-term CR. These findings suggest that the reduced protein synthetic burden, rather than increased global proteolytic quality control, might play an important role in mediating the beneficial effects of CR.
Fig. 1.

Dynamic proteomics workflow. A, Mice were labeled with 2H2O via bolus injection followed by intake in drinking water. Tissues were harvested, and protein homogenates, with the addition of SILAM protein standards to selected samples, were separated via SDS-PAGE followed by in-gel trypsinization. LC-MS/MS was performed on tryptic peptides, and peptide isotopomer distributions were then analyzed using mass isotopomer distribution analysis to quantify the fractional replacement rates (f) of hundreds or thousands of newly synthesized peptides and, thus, their parent proteins. B, Two independent isotopic labeling approaches were used to concurrently measure protein concentration and protein turnover. Turnover was measured by utilizing AA metabolism to isotopically label (purple) the tRNA-amino acid precursor pool (tRNA-AA). Protein concentration was measured through the addition of exogenous labeled proteins (green). C, for each lysine containing peptide, ratios of SILAM-labeled peptides (green) to endogenous peptides (black) allowed differences in protein concentrations to be measured. For clarity, a curve joining the peak of each isotopomer is shown. D, Replacement of old, unlabeled proteins by newly synthesized proteins causes distinct changes in the isotope pattern of each peptide. These time-dependent changes allowed f and k to be measured for each protein from multiple peptides. For clarity, only curves joining the peak of each mass isotopomer are shown.
EXPERIMENTAL METHODS AND PROCEDURES
Animals
Eighteen-month-old CR male C57/BL6 mice (n = 12) and age-matched AL controls (n = 12) were purchased from Charles River (Wilmington, MA), where the NIA Caloric Restricted Mouse Colony is maintained (supplemental Fig. S1). Following 1 week of acclimation, animals in each group were labeled with an intraperitoneal injection of 100% 2H2O saline (0.35 ml/10 g body weight) and were subsequently provided with 8% 2H2O drinking water for the remainder of the study to maintain body 2H2O enrichments of ∼5%, as described elsewhere (32). Animals in the CR group were fed 3.0 g of the NIH-31/NIA fortified diet at 5:00 p.m. daily, and animals in the AL group were provided unrestricted access to the NIH-31 diet (supplemental Fig. S1). Animals were sacrificed following 0.5, 1, 4, 8, 15, or 32 days of heavy water labeling. Body weight and food intake were monitored on a weekly basis and at the time of euthanasia. Animals were anesthetized with isoflurane and euthanized by means of cardiac puncture. All experiments were performed under the approval of the Institutional Animal Care and Use Committees of the University of California at Berkeley.
Measurement of 2H Enrichment in Body Water
Enrichment of 2H2O in body water (blood) was measured via chemical conversion to tetrabromoethane as described elsewhere (32, 33). Briefly, the hydrogen atoms in H2O were transferred to acetylene via the addition of 2 to 5 μl water via syringe to a chip of calcium carbide in a sealed vial equipped with a 3-ml syringe inserted into the septum. The resulting acetylene gas was drawn into the 3-ml syringe and expelled into another sealed vial containing 0.5 ml Br2 (0.1 mm) dissolved in CCl4. After 2 h of incubation at room temperature, the remaining Br2 was reacted with cyclohexene dissolved in CCl4 (10% solution). This solution was injected into the GC/MS for analysis. GC/MS analysis was performed with a DB-225 30-m column at 220 °C using methane chemical ionization with selected ion monitoring. The C2H2Br3+ fragment (m/z 265 and 266, representing the M0 and the M1 ions of the 79Br79Br81Br isotopologue) was used for calculating 2H enrichment by means of comparison to standard curves generated by mixing 100% 2H2O with natural abundance H2O in known proportions.
In Vivo Cell Proliferation Measurement
Liver cell proliferation was measured to confirm the previously established effects of shorter-term CR on the liver (9, 10). DNA was extracted from 30 to 50 μg liver tissue using the DNeasy kit (Qiagen, Valencia, CA) and was enzymatically hydrolyzed to free deoxyribonucleosides via overnight incubation at 37 °C with S1 nuclease and potato acid phosphatase. Hydrolysates were reacted with pentafluorobenzyl hydroxylamine and acetic acid and then acetylated with acetic anhydride and 1-methylimidazole. Dichloromethane extracts were dried, resuspended in ethyl acetate, and analyzed via GC/MS on a DB-17 column with negative chemical ionization, using He as a carrier and CH4 as the reagent gas. The fractional molar isotope abundances at m/z 435 (M0 mass isotopomer) and 436 (M1) of the pentafluorobenzyl triacetyl derivative of purine dR were quantified using ChemStation software. Excess fractional M+1 enrichment (EM1) is the normalized change in isotopomer intensity, calculated as
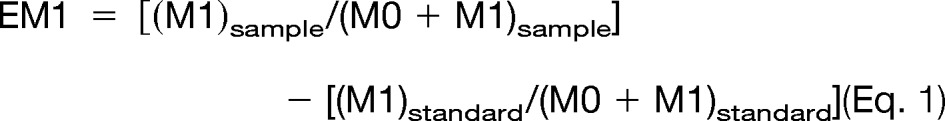 |
where “sample” and “standard” refer to the sample and an unenriched pentafluorobenzyl triacetyl purine derivative, respectively. The fractional replacement rate f was calculated based on a comparison of EM1 to the theoretical maximum EM1 of a fully turned over tissue at the measured body water enrichment according to the following equation: f = (EM1)sample/(EM1)max.
Protein Isolation and In-gel Trypsin Digestion
At each collection time point, two mice were anesthetized under isoflurane gas and were then euthanized via cardiac puncture and cervical dislocation. Tissues were harvested and snap-frozen in liquid nitrogen. Liver samples were thawed and homogenized for 75 s in PBS containing 1 mm PMSF and 5 mm EDTA using a Mini-BeadBeater 8 (BioSpec, Bartlesville, OK) placed on ice for 1 min. This procedure was repeated twice, and the resulting homogenate was diluted to 10% (w/v) in PBS containing 1 mm PMSF. Cultured cells were homogenized in 1 ml of M-PER reagent (Pierce, Rockford, IL) containing protease inhibitors. Protein from prepared homogenates was uniformly reduced via incubation in 10 mm DTT and SDS-PAGE sample loading buffer for 5 min at 95 °C. The reduced samples were then alkylated via incubation in 15 mm iodoacetamide for 1 h at room temperature. Proteins were then fractionated by SDS-PAGE (Invitrogen). Using in-gel molecular weight markers, each sample was divided into 10 molecular weight regions and subjected to overnight trypsin digestion at 37 °C (Trypsin Gold, Promega, Madison, WI). The peptides from the resulting 280 samples were extracted from the gel, dried, reconstituted in 5% acetonitrile/5% formic acid, and desalted using disposable C18 tips (spec-PT-C18, Fisher Scientific) according the manufacturer's recommendations. Briefly, the tips were activated using methanol and then washed three times with 5% acetonitrile/5% formic acid; the extracted peptide solution was then passed through the tip, washing three times with 5% acetonitrile/5% formic acid between each pass of the sample. The tip was then washed three times with 5% acetonitrile/5% formic acid. Desalted peptides were eluted from the tip using 80% acetonitrile/5% formic acid. The solvent was evaporated off from the sample in a SpeedVac, after which the sample was reconstituted in 25 μl 5% acetonitrile/5% formic acid for analysis via LC/MS.
LC/MS Data Acquisition
The isotopic distributions of peptides were measured using an Agilent 6520 QToF with Chip Nano source (Agilent, Santa Clara, CA). For optimal data acquisition, we sought to add approximately the same amount of peptide to the LC/MS column from each sample. To accomplish this, injection volumes of extracted peptides were normalized according to the staining density of the original regions of the gel. Stain density was quantitated using Image J (National Institutes of Health).
Each sample was injected two times per analysis. The mobile phase for the LC was 3% v/v acetonitrile and 0.1% formic acid in 18 mΩ water (Buffer A) and 95% acetonitrile and 0.1% formic acid in 18 MΩ water (Buffer B). Samples were eluted 0.25 μl/min over a 20-min gradient, starting at 5% B and increasing to 40% B over 11 min; the gradient then rapidly went to 80% B for the next 6 min, after which time it returned to 5% B. During the first injection, data-dependent MS/MS fragmentation spectra were collected with the instrument set to collect four MS scans per second with up to 6 MS/MS spectra from each scan. No MS/MS fragmentations were performed during the second injection, and a longer dwell time (one spectrum per second) was used in the full scan acquisition. The longer dwell time increased the signal-to-noise ratio for the observed isotopomer patterns. Raw data are available upon request.
Extraction of Kinetic Labeling Information
For analysis of the raw data, the mass tolerance for the precursor ion was initially set to 15 ppm, and the fragments were 30 ppm. MS/MS fragmentation data were analyzed using the Agilent software package Spectrum Mill (B0.3), and protein identifications were based on the Uniprot/Swissprot database (August 2010). We used species = mouse; trypsin digest; and carbamidomethylation of cysteine (fixed), oxidized methionine (variable), and pyroglutamic acid (variable) as restrictions on the search (16,201/510,076 entries searched). Up to two missed trypsin (Lys/Arg) cleavage sites were allowed in the search. Restricting the database to those proteins observed in the first Spectrum Mill search, a second search of the fragmentation data was performed, allowing for nonspecific cleavage of the protein. Following general recommendations from Agilent, peptides with a score greater than 7 and greater than 60% scored peptide intensity were used for further analysis (supplemental Table S2).
The kinetic information in the isotopomer patterns was extracted from the MS scan data using the Mass Hunter software package (B0.4) from Agilent. The peptide list with calculated neutral mass, elemental formula, and retention time was used to filter the observed isotope clusters. A visual basic application was used to calculate the peptide elemental composition from lists of peptide sequences and calculate isotopomer patterns over a range of precursor body 2H2O enrichments (p) for the number (n) of C-H positions actively incorporating (H/D proton or deuterium) from body water (see below). Subsequent data handling was performed using Microsoft Excel.
Measurement of Amino Acid Enrichments via GC/MS
To confirm the number (n) of C-H positions actively incorporating H/D from body water in peptides measured via LC/MS, we measured by means of GC/MS the 2H-content of individual amino acids (AAs) present in liver tissue. Perchloric acid (6% final) was added to liver tissue homogenate (10% w/v). AAs in the supernatant from this mixture were isolated using ion exchange (AG 50W-X8, Bio-Rad). The AA solution was dried under reduced pressure. Dried AAs were resuspended in 1 ml of 50% acetonitrile, 50 mm K2HPO4, pH 11. Pentafluorobenzyl bromide (20 μl) was added, and the sample was sealed and incubated at 100 °C for 1 h. After the sample had been cooled to room temperature, ethyl acetate (600 μl) was added to each sample, and this was followed by mixing. The top layer was then transferred to a fresh tube containing Na2SO4. The anhydrous organic solution was injected directly onto a DB-17MS (30 m × 0.25 mm inner diameter × 0.25 μm film thickness) (J&W Scientific, Santa Clara, CA). The data were acquired on an Agilent 6890N using a chemical ionization source maintained at 280 °C. The oven temperature was cycled from 140 °C to 280 °C over a 7.5-min run. Data were collected in selected ion monitoring mode with a 15-s dwell time using the ions listed in supplemental Table S1.
Calculation of Turnover Rate
The fractional replacement (f) is the proportion of newly synthesized proteins in a population, expressed as a fraction of the total pool. The kinetic interpretation of the time-dependent replacement of preexisting protein molecules by newly synthesized molecules requires knowledge of the mass isotopomer pattern of newly synthesized species as compared with unlabeled species (27, 34). The mass isotopomer pattern of peptides synthesized in the presence of a stable-isotope enriched precursor pool can be calculated based on the elemental composition of the peptide and combinatorial probabilities. Each protein (and, by extension, each tryptic peptide) acquires isotopic enrichment from the precursor 2H2O pool at the rate of protein replacement (k), the 2H-isotopic enrichment in the body water (p), and the number of sites in the peptide capable of incorporating H/D from water (n). Accordingly, p and n must both be known in order for one to calculate k. In these experiments, we measured p directly (supplemental Fig. S2). At the 2H enrichments used in this study, the mass spectra of newly synthesized protein occupy the same general m/z range as the unlabeled species, but if one knows or estimates n and p, one can deconvolute the isotopomer patterns to calculate f and k.
Previous estimates of n have been calculated for each AA from 3H2O administered in utero through adult life (35). We confirmed these literature values of nAA for each AA in two ways: first by directly measuring the relative deuterium incorporation in soluble AAs, and then by comparing measured mass isotopomer patterns of peptides isolated from the labeled tissue to theoretical patterns for different values of n and establishing the best fit value (supplemental Fig. S3).
Because of the rapid equilibration of water in the body (supplemental Fig. S2A), p can be measured from any accessible bodily fluid. In this study we used blood plasma collected from each mouse at each time point. Because the body water enrichment was slightly different for each animal (supplemental Fig. S2B), the amino acid precursor pool 2H-enrichment used to calculate the fractional replacement was different in each animal.
For values of f between 0% and 100% (i.e. a mixed protein pool), deconvoluting the two subpopulations is carried out by treating each peptide as a biochemical polymer and calculating quantitative changes in the relative isotopic abundance pattern using mass isotopomer distribution analysis (MIDA) (34). As described elsewhere (27), abundances of individual mass isotopomers in each peptide were normalized to the total intensity of the isotopomer envelope (typically four masses) (M0–M3; Fig. 2). Peptides with a mass greater than ∼2,400 Da exhibit a larger isotopomer envelope, in which cases five masses (M0–M4) were used. We calculated f based on the change in intensity of the normalized monoisotopic peak (EM0), as the signal-to-noise is most favorable for EM0 because of the larger change in fractional abundance for this isotopomer (each 2H-label incorporated decreases the relative abundance of EM0 but may distribute from EM1 to EM4). Peptides that met our criteria for inclusion required a signal intensity > 30,000 counts, a root mean square error against the theoretical natural abundance spectra of less than 1.5% for the day 0 (D0) sample, and an LC elution time within 30 s of the unlabeled control. The f of each peptide was calculated using the n specific for that sequence and the p measured for the mouse. Each peptide was considered as an internal replicate measurement of the fractional replacement for the parent protein of origin. Therefore, the protein f in each mouse was calculated as the median f of the peptide population from that protein. A time-dependent fractional replacement curve was constructed for each protein by plotting the median protein f for each mouse in each feeding regime against the time of exposure to 2H2O (supplemental Fig. S4). Proteins that were observed in fewer than three mice were removed from the data set. The k for each protein was calculated using a regression fit for the single pool model (f = 1 − e(−kt)) in the Prism software package (Graph Pad, La Jolla, CA). A coefficient of variation (CV) was calculated to assess analytical and biological variability for each protein as the ratio of standard deviation reported for the regression over the rate constant. Proteins that had a CV of more than 30% for the fit in either the CR or AL feeding regime were removed from the data set.
Fig. 2.

Endogenous kinetic labeling. Sample spectral data showing the incorporation of 2H over time in glutathione S-transferase Mu 1 tryptic peptides: A, FEDGDLTLYQSNAILR; B, ADIVENQVMDTR. At each time point the spectral contribution from the population of newly synthesized peptide (fraction new peptide, or f) can be calculated. This isotopically labeled population of peptide has a very different isotope pattern, with the intensity of heavy isotopic masses increasing relative to the monoisotopic peak (M0) intensity.
SILAM Quantitation
The relative concentration of proteins in the tissue was measured via comparison to exogenous labeled standards mixed into tissues (SILAM) (28, 29, 36). Importantly, the exogenous labeled peptides optimally do not have overlapping isotopomer distributions with the metabolically labeled isotopomers (Fig. 1C). 13C6-lysine labeled mouse liver (MouseExpress Liver, Cambridge Isotope, Andover, MA) was used as the heavy standard for LC-MS/MS quantitation. Chronic CR mice (18 months old) were used to test the effects of adapted, weight stable CR versus age matched AL controls. Because a single tissue standard was used in the quantitation of both experimental groups, the SILAM sample simply acts as a standard for comparison, making it unnecessary to age-match the SILAM standard. In order to validate the LC-MS/MS-based quantitation method, test the range of quantifiable relative concentration differences, and assess the variability within the measurements, a standard curve of labeled cell lysate was also constructed. Rat neural stem cells (EMD Millipore, Billerica, MA) were grown as a mono-layer in flasks coated with poly-ornithine and laminin. Two different media containing DMEM/F-12 medium with B-27 serum-free supplement and 20 ng/ml FGF (EMD Millipore, Billerica, MA) were used—either unlabeled lysine and arginine (light) or 13C6 lysine and 13C6 arginine (heavy), as described elsewhere (37). The total protein content of cell lysates from these cultures was measured using bicinchoninic acid (BCA) (Pierce, Rockford, IL). Light and heavy cell lysates were mixed to form a curve of protein isotopic ratios at 1:1, 1:3, 1:6, and 1:10 light:heavy protein. The mixtures of crude cell lysates were fractionated using SDS-PAGE. The 37–50 kDa molecular weight range of the gel was digested using trypsin and analyzed via LC/MS as described above. The same peptide identification and isotopomer extraction methods described for the kinetic analysis were used in this experiment. Relative protein concentrations were calculated as the average values of ratios of light/heavy isotopomer intensity measured for all peptides belonging to the same protein.
For relative liver tissue protein concentration measurements, the protein was quantified in day 0 (D0) experimental tissue from each feeding group. Total protein contents of the experimental and SILAM tissue lysates were measured using BCA (Pierce, Rockford, IL). Sample and standard proteins were mixed in a 1:1 ratio of total protein content prior to SDS-PAGE and trypsinization. Our protocol was patterned after literature reports (28, 29). Ratios of light to heavy peptides were calculated independently using two different software packages: (a) MassHunter Qualitative Analysis (Agilent) and (b) the Trans-Proteomic Pipeline (Seattle Proteome Center, Seattle, WA).
The MassHunter quantitation relied upon the peptide identifications from the SpectrumMill analysis described above. Paired light/heavy isotopomer envelopes were extracted from the same elution time frame of the raw data. The sum of extracted ion intensities was calculated for each molecule, and relative quantitation was calculated as the light-to-heavy ratio of the summed intensity in Microsoft Excel. Protein quantitation ratios were calculated as the average ratio for the peptide population. In the TPP (Trans-Proteomic Pipeline) analysis, raw data files were searched against two databases, the X!Tandem and SpectraST databases. The X!Tandem database uses a spectrum interpretation algorithm that attempts to match sample spectra with hypothetical peptide sequences predicted from a sequence database. Next, the data were searched against the SpectraST database, which contains previously identified MS/MS spectra contained within a spectral library. Searches were performed using carbamidomethylation as a fixed modification and oxidized methionine pyroglutamic acid and 13C6-lysine as variable modifications. Search results were then validated using the PeptideProphet algorithm, which performs statistical validation of each peptide spectrum match by assigning a probability to each identified match. The results of each database search were then combined using iProphet and finally quantified using the XPRESS algorithm to identify SILAC (stable isotope labeling in cell culture) peptide pairs and determine the relative expression levels of peptides from 13C6-Lysine isotopically labeled samples in comparison with unlabeled controls. Peptide quantification results were then combined into parent proteins using ProteinProphet.
The relative protein concentration was calculated as the weighted average of the measurements from both software packages, and the number of peptides measured for a given protein was used as the weighting factor.
Calculation of Absolute Synthesis Rates
In the kinetic labeling experiment we calculated the turnover rate constant (k) from the change in f over time using the relationship f = 1 − e(−kt). In order to directly compare the CR and AL experiments, we also calculated the absolute synthesis and breakdown rates (flux in and out) of each specific protein pool. Protein absolute synthesis and breakdown were calculated as the turnover rate constant (k) multiplied by the pool size (V): Flux = kV. For this calculation we assumed steady-state in protein concentrations in these 18-month-old mice during the ∼1-month labeling period (i.e. although V might be different between the AL and CR experiments, it is constant over the duration of the experiment). In keeping with this assumption, the mass of each mouse (supplemental Fig. S1B) and presumably the mass of the organ were essentially unchanged over the course of the experiment. Flux into each protein (synthesis) is therefore calculated to be equal to the flux out (degradation). For these calculations, we normalized all concentration measurements to the AL pool size. This resulted in a unitless (relative mass per day) synthesis rate that was directly comparable between experimental groups.
Gene Ontology and Pathway Analysis
Gene annotation, gene ontology information, and biochemical pathway information were obtained from the Database for Annotation, Visualization and Integrated Discovery, v6.7, from the National Institute of Allergy and Infectious Diseases at the National Institutes of Health (NIH) (38, 39). Mitochondrial proteins were cross-referenced against the Mitocarta database (40). The network analysis testing expression control of protein dynamics was done using both the Ingenuity IPA (version 12402621) and the GeneGo Metacore (version 6.11) databases. Parallel pathway analysis methods were used to assess the correlation between the predicted transcriptional control of protein and measured changes in the replacement rate and relative concentration. For this analysis we considered a 5% difference in replacement rate and 10% in concentration necessary in order for a change to be considered significant. Special emphasis was placed on testing the recently proposed hypothesis that PGC-1α is a central regulator of the mitochondrial adaptation to CR (31).
RESULTS
CR Model and Confirmation of Reduced Liver Cell Proliferation
CR mice gained weight at a rate similar to that of the AL controls maintaining the ∼30% difference in total mass (supplemental Fig. S1B). AL mice consumed an average of 5 g, whereas CR mice were administered 3 g daily, as per the NIA protocol.
Liver cell proliferation was measured to confirm the previously established effects of short-term CR on the liver (9, 10). Cell proliferation rates were measured following 15 and 32 days of 2H2O labeling (supplemental Fig. S1C). The rate of new cell production was reduced by ∼50% in the CR animals relative to the AL group, resulting in proliferation rates of 0.0023 ± 0.0003 and 0.0047 ± 0.0004 d−1, respectively.
Body Water Labeling
The bolus injection protocol used to initiate isotopic labeling results in a rapid equilibration of body water (BW) at the target isotopic enrichment of ∼5% (supplemental Fig. S2). In order to measure the rate of isotopic equilibration, in a separate experiment we serially sampled the BW enrichment in C57BL6 mice every 60 min for a total of 240 min following a fixed volume bolus injection of H218O. We confirmed that in vivo labeled H2O isotopic equilibrium was established within 60 min, and isotopic enrichment was dependent on body weight (supplemental Fig. S2A). Assuming approximately five half-lives are required in order for a kinetic process to achieve equilibrium, this suggests that the body water equilibration has a half-life of at most 12 min in a mouse, or a BW equilibration rate constant of >86 d−1 in our experiment. A similar bolus injection was used to initiate labeling of the CR and AL mice. BW enrichments were measured at the time of tissue collection and throughout the duration of the experiment (supplemental Fig. S2B). The BW enrichments observed were comparable between CR and AL mice. We also observed that the enrichment was very stable within the CR group, whereas the AL group enrichment declined slightly as the experiment progressed, perhaps reflecting the greater food consumption (metabolic water production) or lower lean body mass in the AL mice.
Validation of Literature n
Each peptide n is the sum of its individual AA n values (nAA). As previously described in humans (27), we used literature values for 3H2O labeled AAs (35) to calculate n as a ratio of the validated n for alanine (4). We tested the validity of these n values in two different ways. First, we isolated free AAs from tissue that had been labeled for 32 d to measure maximal deuterium incorporation (supplemental Table S1). We observed a strong linear correlation between the measured isotopic enrichment and the theoretical value from the literature n at corrected values for the measured p (supplemental Fig. S3A).
Next, we tested theoretical estimates of n by comparing experimental peptide spectra to theoretical predictions for mass isotopomer patterns from protein pools expected to be fully replaced by newly synthesized protein molecules (supplemental Fig. S3B). Measured mass isotopomer patterns for tryptic peptides from these proteins were compared with a family of theoretical spectra, for which n was varied around the literature value. We found that for most peptides the best fit n for measured patterns of isotopomer abundances closely matched the summed literature value (35) for nAA (supplemental Table S1). Some random variation was observed, but the minimum root mean square error (RMSE) typically occurred at between 90% and 100% of the expected value for n.
Rate of Precursor Pool Enrichment
AA metabolism and the intracellular tRNA-AA pool are the link between the BW isotopic enrichment and peptide isotopic enrichment (Fig. 1). In order to determine whether the isotopic content and enrichment of the true biosynthetic precursor pool were changing over time, we measured the stability of peptide n over time. To do this, the best fit n values for several different peptides were calculated at each time point of the labeling experiment (supplemental Fig. S3C). We targeted peptides from multiple proteins with measurable labeled peptide populations beginning at the 9-h time point (k > 30% d−1), and which were observed in both AL and CR animals. Comparing each peptide at each time point against a family of theoretical spectra (as per supplemental Fig. S3B), we found that the observed best fit n value was within 10% of the literature value throughout the time course. Observing the stable n at 9 h suggests that the true AA precursor pool had achieved equilibrium. If, as suggested by these data, 9 h is >∼5 half-lives for precursor pool turnover, the half-life of the precursor pool is at most 1.8 h. These results are consistent with literature values for intracellular AA turnover (41).
Liver Proteome Dynamics
Accurate and reproducible quantitation of relative mass isotopomer abundances (isotope ratios) is required to measure the dynamics of proteins via LC-MS/MS of tryptic peptides. This represents a much higher bar analytically than the simple identification of peptides via LC-MS/MS. Ion trap instruments that are commonly used in proteomic studies are not optimal for the study of isotope labeling on multiple analytes (42, 43). As a practical consequence, data acquisition using a TOF-based instrument followed by application of strict analytic criteria is required to filter static proteome identifications into reliable dynamic measurements. These filtering criteria reduce the number of usable peptides. We used four selection criteria to remove low confidence protein information from the kinetic analyses: peptide measurement signal intensity (>30,000 counts), RMSE for peptide mass isotopomer abundance measurements (unlabeled sample, RMSE < 1.5% compared with natural abundance values), observation of the protein in at least three mice per experimental group, and a rate constant that could be defined with less than 30% coefficient of variation from the incorporation curve. We identified 2,769 peptides in AL and 3,939 peptides in CR after filtering. These peptides were heterogeneously distributed among proteins, with up to 120 peptides measured for individual proteins. The AL group had kinetic information for somewhat fewer proteins (384) than CR (447). There were 288 shared proteins that could be compared from both groups (supplemental Fig. S5).
The proteomic data demonstrate that CR induced a general increase in half-life (reduction in k) of hepatic proteins (Figs. 3 and 4). Of proteins observed in both AL and CR, ∼85% had longer half-lives in CR. We calculated the half-life ratio (CR/AL) and divided proteins into three broad categories (supplemental Table S2): shorter (CR/AL < 0.90), unchanged (0.90 < CR/AL < 1.10), and longer half-lives (CR/AL > 1.10).
Fig. 3.

CR results in broad and substantial reductions in hepatic protein synthesis and breakdown rates and prolongation of protein half-lives. A, Direct comparison of half-lives for all observed proteins shows that CR (black) prolonged the half-lives relative to AL (open) for 85% of proteins. B, Absolute synthesis rates for each protein were calculated from the replacement rate and the SILAM concentration measurements. Differences between CR and AL mice were enhanced on this absolute scale, with CR mice producing substantially less of almost all proteins.
Fig. 4.

Protein half-lives are longer (turnover is slower) throughout the cell in CR mice than in AL. A, Selected functional classes like the 40S ribosomal subunit and some classes of cytosolic proteins are less perturbed than the average, whereas the mitochondrial proteins are affected to a greater degree. B, Comparison of the half-life (t ½, calculated as ln(2)/k) for proteins associated with specific organelles. Boxes show the interquartile range for protein turnover in the organelle. Bars extend to 5% and 95% of the data, with outliers as points. Only proteins that were solely and unambiguously assigned to the indicated organelle were used. Although the median half-life was systematically longer in CR, only the mitochondrial proteins were statistically significant (Student's two-tailed heteroscedastic t test, ** p < 0.005).
Various regions of the cell were enriched for proteins from these categories (Fig. 4). Proteins associated with the mitochondria were changed most significantly with 70% (58/84) proteins having half-lives increased more than 20% (p < 0.0005). In keeping with this observation, we measured significantly lower replacement rates (longer t ½) for proteins associated with TCA, fatty acid metabolism, and ATP synthesis. Although the median replacement rates of proteins from the endoplasmic reticulum and the nucleus were also slower in CR, neither shift was statistically significant (Fig. 4B, supplemental Table S3). Interestingly, although fewer than 15% of observed proteins had replacement rates that were faster in CR (i.e. shorter half-lives; supplemental Table S2), half of the observed 40S ribosomal proteins had either the same or a faster replacement rate (Figs. 4 and 5, supplemental Table S3).
Fig. 5.

A, Distribution and comparison of change in half-lives to the change in concentration for each protein (open circles). The great majority of proteins detected exhibited lower concentrations in liver from CR than from AL mice, and these were almost all associated with longer half-lives, not shorter half-lives. There was no correlation between t ½ and protein concentrations. B, Distribution and comparison of change in total synthesis rates (calculated as the product of k and concentration) to the change in concentration for each protein (open circles). As noted in A, the great majority of proteins detected exhibited lower concentrations in liver from CR than from AL mice, and almost all were associated with lower total synthesis rates rather than increased degradation. Thus, most proteins were in the lower left quadrant (lower concentration, lower total synthesis rate). C, Proteins belonging to selected Gene Ontology (GO) categories showed clustering, with similar changes in both concentration and half-life. D, Changes in total synthesis rates for CR/AL mice for proteins within functionally related selected GO categories. Proteins within a pathway tended to exhibit coordinated changes in total synthesis rates in response to CR.
SILAM Quantitation
A standard curve of heavy to light cell lysates was used to validate the quantitation method. We measured the ratios for ∼50 peptides belonging to seven different proteins throughout a series of dilutions from 1:1 to 1:10 light-to-heavy ratios (supplemental Table S4). Every protein and all associated peptides showed a highly linear relation between the measured and expected ratios.
Initial analysis of the experimental liver samples used the Agilent Mass Hunter software program, which identified 2,832 peptides with high confidence; of these only 36% contained lysine, as the majority were arginine terminal peptides. This resulted in SILAM ratios for 223 proteins in CR and 203 in AL. These data were then analyzed again using the Trans Proteomic Pipeline software package, which measured SILAM ratios for 2,010 peptides yielding 163 proteins observed in both AL and CR groups. There were 123 proteins that were reported in both analysis methods, with 41 unique to the TPP analysis and 72 to the Agilent analysis. For proteins common to both analyses, there was good agreement of the measurements, with a median deviation of 10%. For the shared proteins, an average quantitation measurement was calculated, weighted by the number of peptides observed in each method. To facilitate data analysis, all CR protein measurements were normalized to the AL pool. This resulted in quantitation ratios (CR/AL) for 236 proteins in total.
We observed a general shift toward lower relative protein concentrations in the CR mice, with 82% (194/236) of the proteins measured exhibiting a >20% decrease in pool size (supplemental Table S2). Based on the 10% deviation observed in the standard curve, any ratios between 0.9 and 1.1 were designated as unchanged. Within this group of unchanged proteins, there was a significant enrichment (p = 0.008) for the proteins involved in carboxylic acid and valine metabolism (supplemental Table S3). There were a number of proteins (19% of total) with markedly reduced relative concentrations (<50% CR/AL), and ribosomal proteins were enriched (p = 0.04) among this group.
Calculation of Absolute Synthesis Rates
Having measured fractional replacement (measured here by means of heavy water labeling) and relative concentration (measured via SILAM), we were able to calculate absolute synthesis and breakdown rates. Flux calculations could be performed, however, only for proteins that were observed in both the kinetic and SILAM proteomic analyses (Figs. 1C and 1D, supplemental Table S2). The calculation of protein absolute synthesis rates magnified the contrasts between the AL and the CR groups (Fig. 3B, supplemental Table S2). We observed that the CR concentrations were on average 70% of those of the AL pool. When half-life (degradation efficiency) was plotted against relative protein concentrations, there was no pattern or significant correlation (Fig. 5A). For the same subset of proteins, the average replacement rate constant in CR was 81% of the rate observed in the AL group, resulting in an average flux for the CR group that was 55% of the flux observed in the AL group. When the changes in absolute synthesis rates were plotted against changes in pool size in a four-quadrant graph (Fig. 5B), the great majority of proteins fell in the same quadrant (low production rate, low concentration), indicating that the decline in relative concentration for most proteins could be explained by reduced production, not increased degradation. Conversely, proteins belonging to specific ontological groups tended to have similar changes in half-life and relative concentration (Fig. 5C).
Gene Ontology
In order to identify protein networks with differentially regulated flux, we analyzed both functional and spatial interconnectivity between proteins in groups. Changes in relative concentration and replacement rate clustered at three distinct levels: the organelle, the multi-protein complex, and the protein itself (Figs. 3, 4, and 5).
Expression Pathway Analysis
We adopted a hypothesis-driven approach to the initial interrogation of the proteome dynamic network using relational database software programs. First we asked whether, as proposed (31), PGC-1α control of transcription could account for many of the proteome dynamic changes observed in CR. Both pathway analysis databases suggested that four transcription factors (MYC, HNF4A, PPARγ, and MYCN) were connected to approximately half of the proteins for which we measured replacement rates (Fig. 6). The predicted directionality of the expression profiles (supplemental Table S5) was significantly negatively correlated for MYC (z = −2.3), HNF4A (z = −2.1), MYCN (z = −3.0), and PPARγ (z = −3.1). MYC, MYCN, and PPARγ also showed negative correlations in terms of changes in relative protein concentrations (z = −2.9, −2.2, and −2.1, respectively) and absolute synthesis (z = −2.7, −2.3, and −2.1). HNF4A and PPARγ are known targets of PGC-1α, whereas MYC and MYCN are known to be under the control of HNF4A. PGC-1α is also involved in transcriptional regulation of proteins known to alter mitochondrial biogenesis, including ERR1, ERR3, NRF1, and NRF2 (supplemental Fig. S6). These findings are consistent with the model (31) stating that reduced PGC-1α activity might be linked to or participate in the reduced general protein synthesis and mitochondrial protein flux in CR (Fig. 6). Because the intervention tested here (CR) resulted in a relatively monotonic reduction in global protein synthesis rates rather than a variety of specific increases or decreases, an unsupervised approach to identifying underlying causal factors for the proteome dynamic network was unlikely a priori to revealing fingerprints of specific underlying factors and is not reported here.
Fig. 6.

Significant expression networks within the dynamic proteome data set. Predicted expression patterns for proteins (colored icons) regulated by MYC, HNF4A, PPARγ, and MYCN (white) were significantly negatively correlated with measured directional changes, consistent with reduced effects (inhibition) of these transcription factors. These transcription factors are also linked (highlighted connection), and PGC-1α is known as an upstream regulator influencing all of these expression patterns. Green indicates longer measured t ½, red indicates shorter measured t ½, and white indicates an absence of half-life information.
DISCUSSION
The cell regulates protein metabolism through changes in synthesis rates, proteolysis, and concentrations of proteins. In order to understand the effect of CR on protein metabolism in vivo, we have used a new quantitative technology, dynamic proteomics, wherein for the first time absolute protein synthesis and breakdown rates, as well as half-lives and relative concentrations, are measured across the proteome. Using this methodology, we directly tested a whether CR-dependent mitochondrial fitness (44) occurs through stimulation of mitochondrial biogenesis (1, 15, 18) and mitophagy (14, 16, 20, 21). We show here clearly that absolute rates of general proteolysis throughout the cell, including mitophagy, are markedly reduced in CR relative to AL feeding. In addition, the pattern of changes in protein turnover was consistent with a hypothesized role for PGC-1α (31).
One of the assumptions in our analysis is that protein concentrations and kinetics are stable during the experiment (steady-state). To ensure that the experimental groups were at a condition of protein steady-state, our study used healthy adult mice of stable weight, and the CR mice had been maintained for 14 months on the diet at the NIA (supplemental Fig. S1). The stable body mass indicates that the mice were in a condition of protein steady-state over the labeling period (homeostasis). Individual proteins might fluctuate briefly due to diurnal cycling and/or feeding, but our kinetic values reflect the entire labeling time course, and therefore fluctuations due to short-term cycling will be measured as the average rate. The heavy water labeling period spanned 32 d, with tissue collection starting at <0.5 d. This protocol was chosen to detect kinetics of proteins with half-lives ranging from hours to months. Proteins with half-lives of <12 h will not yield an accurate characterization of kinetics with this approach, but maximally labeled proteins at the first collection time point were very unusual.
Proper interpretation of kinetic processes such as protein turnover requires the measurement of the isotopic enrichment of the true precursor pool and the protein concentration. The methods described here provide some important technical advantages over previous approaches for measuring protein synthesis and breakdown with isotopes (27, 30, 42, 45, 46). The technique is operationally simple, involving the administration of 2H2O-enriched water, and provides a rigorous solution to the major technical barrier that has long confounded isotopic measurement of protein synthesis (estimation of precursor pool enrichment (34, 41)). We have recently shown that by using this technique we can measure protein turnover in humans (27). Now, by using both exogenously labeled proteins (13C6-lysine, SILAM mouse) and metabolically labeled proteins (2H2O), relative protein concentrations and replacement rates were measured for the first time in a single protocol. Metabolic labeling with water has a number of advantages compared with diets incorporating a heavy AA labeled diet for long-term experiments, particularly for translation into humans. Labeled drinking water is inexpensive, stable, and very simple to formulate; also, we avoid manipulation of the experimental diet, an important consideration in CR studies. Deuterated water labeling also allows the independent direct measurement of label enrichment via the sampling of accessible body fluids (47), as well as the stability in the true biosynthetic precursor pool in vivo, by use of mass isotopomer patterns (MIDA) (34) in the peptides themselves (supplemental Fig. S3). Here, we confirm literature values for 2H-labeling of amino acids from labeled H2O in vivo (supplemental Table S1) showing that each AA has its unique value for n (the number of 2H atoms incorporated from BW) (27, 35, 42, 45). Moreover, the LC/MS results show nicely that the true precursor pool for protein synthesis (the charged intracellular tRNA-AA pool) rapidly achieves plateau values during heavy water labeling in mouse liver. The long-term n value was established by 9 h, the earliest point within our experimental time course (supplemental Fig. S3). This fast rate of precursor enrichment is a distinct advantage over the use of 15N-labeled algae proteins, for example, with which a lag in the delivery of isotopic label has been observed (48).
The effect of CR on protein metabolism and mitochondrial biogenesis has been controversial. Several studies propose that CR induces autophagy and mitochondrial biogenesis (13–21), whereas others suggest that total cell metabolism is slower (22–24). In a study conducted in vitro and in vivo, CR induced proliferation of mitochondria with reduced membrane potential and rates of ROS production (49), whereas in another investigation CR promoted increases in oxidative capacity concomitant with decreases in triglyceride content in rat skeletal muscle (13). A third study reported that CR promoted mitochondrial biogenesis in mice by activating endothelial nitric oxide synthase (18), whereas in humans CR promoted increased mitochondrial DNA (15). These investigations appear to support the hypothesis that CR promotes bioenergetic efficiency via increases in mitochondrial biogenesis.
Other investigators, however, have challenged the view that CR increases mitochondrial biogenesis (50). In our study, mitochondrial proteins showed a significant decrease in absolute synthesis and absolute proteolytic rates, resulting in consistently longer half-lives (58/84 mitochondrial proteins measured). This provides strong evidence that mitochondrial biogenesis, mitochondrial protein turnover, and mitochondrial protein concentration are reduced in response to CR. Interestingly, multiple groups of functionally related proteins showed coordinated changes in turnover rate and pool size (Fig. 5C), suggesting that a broader investigation of the proteome might elucidate mechanisms of CR-induced benefits.
Finally, we explored the use of network relational database expression pathway approaches to interrogate these kinetic data sets. Our initial approach has been to apply a hypothesis-driven interrogation of the proteome dynamic network. We chose this approach because the intervention (CR) resulted in a relatively monotonic reduction in protein synthesis rates, rather than heterogeneous changes, so that an unsupervised approach to identifying underlying causal factors for the proteome dynamic network changes was unlikely to reveal a priori specific fingerprints for underlying factors.
The first question we asked was whether PGC-1α control of transcription could account for many of the proteome dynamic changes observed in CR (31) and, more specifically, whether PGC-1α was involved in the transcriptional regulation of proteins involved in mitochondrial biogenesis. It should be recognized that catabolic control of half-life might be equally as important as synthesis. With that caveat, our data suggest that HNF4A, MYC, PPARγ, and MYCN, which are all associated with cellular proliferation, exhibit reduced activity in long-term CR. These factors were connected to approximately half of the proteins with measured replacement rates (Fig. 6), and the predicted directionalities of the expression profiles (supplemental Table S5) were significantly negatively correlated for all four factors. PGC-1α activation of HNF4A and PPARγ is known, which in turn activates MYC and MYCN (Fig. 6). PGC-1α also activates transcription factors responsible for mitochondrial biogenesis (supplemental Fig. S6), and proteins whose expression is known to be stimulated by these transcription factors showed significantly down-regulated synthesis in CR (Fig. 6). In contrast, protein synthetic patterns expected for other putative targets of PGC-1α (such as PPARα) were not strongly correlated with our protein kinetic data. Taken as a whole, these proteome dynamic results can suggest testable hypotheses regarding the control of selected or general proteins of interest.
The literature suggests that CR-extended life span results from multiple interacting processes. CR has been shown to protect from oxidative stress, but in mice overexpression of antioxidant proteins is insufficient to extend lifespan (51). CR slows cell proliferation (9) and protein turnover (this study) and lowers core body temperature (8), all of which suggest that general cellular metabolism is slower. Yet studies have shown that CR improves the liver's ability to recover from injury, showing significantly increased cell proliferation during tissue repair relative to AL controls (52, 53). We have shown here that hepatic protein turnover is slower, whereas in vitro activity assays of proteasome activity show that CR increases the activity of the proteasome relative to age matched controls (54). These dissonant findings suggest that although we have shown that the global cellular metabolism is slower in CR, the capacity for dynamic adaptation of cellular response to outside stimuli might be more sensitive.
It is of interest to speculate based on our findings that dramatically reduced global protein synthesis rates represent an indirect “quality control” mechanism. A global reduction in protein synthesis rates might permit more efficient chaperone function, less generation of unfolded proteins, and reduced induction of the unfolded protein response. These alterations might then explain the longer half-lives of proteins that are present. The dynamics of ribosomal proteins appear to represent a different strategy. The reduced relative concentrations and total synthesis rates of ribosomal proteins are consistent with slower operation of the protein synthetic machinery, but S40 ribosomal subunits had essentially unchanged half-life in CR. The ribosome is a first response element in the production of new proteins, and the relatively short half-lives of ribosomal subunits suggest that the pool is prepared to expand rapidly under the right conditions to ensure a rapid response to newly expressed mRNA. A similar strategy has been shown to regulate the hypoxia response through the degradation of the HIF-1α transcription factor (55, 56).
CONCLUSION
In summary, we investigated the effect of CR on the absolute rates of protein synthesis and breakdown in vivo and provide compelling evidence that hepatic mitochondrial biogenesis and mitophagy, as well as general protein metabolic rates, are dramatically reduced in response to chronic CR. Global interrogation of proteome dynamics also revealed that the cell modulates protein concentration, protein half-life, and protein synthesis at the level of the functional group, as well as of the individual protein, and that the role of specific transcription factors can be quantitatively evaluated in the context of broad proteostasis. This approach to the identification of dynamic proteomic signatures and regulatory nodes in disease states holds great promise for wider application.
Acknowledgments
We thank Joan Protasio and Chancy Fessler for their assistance in collecting the gas chromatography data. We thank Antonio Moreno and the UC Berkeley Laboratory Animal Research and Care facility, including Bob Williams, Julio Gonzalez, and Joan Wallace, for assistance with labeling and animal maintenance. We also thank Leonie Waanders for her inspiration and generous assistance in the cellular representation of our data.
Footnotes
 This article contains supplemental Figs. S1 to S6 and Tables S1 to S5.
This article contains supplemental Figs. S1 to S6 and Tables S1 to S5.
Author Contributions: J. C. P., C. F. K., M. D. B., and M. K. H. designed experiments; C. F. K., M. D. B., M. D., N. A. F., and L. S. R. executed the experiments; J. C. P., K. W. L., M. S., W. E. H., and S. M. T. provided new reagents or tools; J. C. P., C. F. K., K. W. L., and W. E. H. analyzed data; J. C. P., C. F. K., and M. K. H. wrote the paper.
Conflict of Interest: J. C. P., N. A. F., K. W. L., M. S., W. E. H., S. M. T., and M. K. H. have a financial interest in KineMed Inc.
1 The abbreviations used are:
- AA
- amino acid
- AAPP
- amino acid precursor pool
- AL
- ad libitum
- BCA
- bicinchoninic acid
- BW
- body water
- CR
- calorie restriction
- CV
- coefficient of variation
- D0
- time point at day 0
- EM0
- absolute value of change in M0 intensity
- f
- fractional replacement
- k
- turnover rate constant
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- MIDA
- mass isotopomer distribution analysis
- MPE
- molar percent excess
- mtDNA
- mitochondrial DNA
- m/z
- mass-to-charge ratio
- M0
- monoisotopic mass
- n
- number of sites within peptide capable of incorporating label
- nAA
- number of sites within amino acid capable of incorporating label
- NIA
- National Institute on Aging
- NIH
- National Institutes of Health
- p
- precursor pool enrichment
- PGC-1α
- peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- RMSE
- root mean square error
- ROS
- reactive oxygen species
- SILAM
- Stable Isotope Labeling in Mammals.
REFERENCES
- 1. Guarente L. (2005) Calorie restriction and SIR2 genes—towards a mechanism. Mech. Ageing Dev. 126, 923–928 [DOI] [PubMed] [Google Scholar]
- 2. Houthoofd K., Vanfleteren J. R. (2006) The longevity effect of dietary restriction in Caenorhabditis elegans. Exp. Gerontol. 41, 1026–1031 [DOI] [PubMed] [Google Scholar]
- 3. Partridge L., Piper M. D., Mair W. (2005) Dietary restriction in Drosophila. Mech. Ageing Dev. 126, 938–950 [DOI] [PubMed] [Google Scholar]
- 4. McCay C. M., Crowell M. F., Maynard L. A. (1935) The effect of retarded growth upon the length of life span and upon the ultimate body size. Nutrition 5, 155–171, discussion 172 [PubMed] [Google Scholar]
- 5. Colman R. J., Anderson R. M., Johnson S. C., Kastman E. K., Kosmatka K. J., Beasley T. M., Allison D. B., Cruzen C., Simmons H. A., Kemnitz J. W., Weindruch R. (2009) Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325, 201–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Das S. K., Gilhooly C. H., Golden J. K., Pittas A. G., Fuss P. J., Cheatham R. A., Tyler S., Tsay M., McCrory M. A., Lichtenstein A. H., Dallal G. E., Dutta C., Bhapkar M. V., Delany J. P., Saltzman E., Roberts S. B. (2007) Long-term effects of 2 energy-restricted diets differing in glycemic load on dietary adherence, body composition, and metabolism in CALERIE: a 1-y randomized controlled trial. Am. J. Clin. Nutr. 85, 1023–1030 [DOI] [PubMed] [Google Scholar]
- 7. Caro P., Gomez J., Lopez-Torres M., Sanchez I., Naudi A., Jove M., Pamplona R., Barja G. (2008) Forty percent and eighty percent methionine restriction decrease mitochondrial ROS generation and oxidative stress in rat liver. Biogerontology 9, 183–196 [DOI] [PubMed] [Google Scholar]
- 8. Soare A., Cangemi R., Omodei D., Holloszy J. O., Fontana L. (2011) Long-term calorie restriction, but not endurance exercise, lowers core body temperature in humans. Aging 3, 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruss M. D., Thompson A. C., Aggarwal I., Khambatta C. F., Hellerstein M. K. (2011) The effects of physiological adaptations to calorie restriction on global cell proliferation rates. Am. J. Physiol. Endocrinol. Metab. 300, E735-E745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shaddock J. G., Chou M. W., Casciano D. A. (1996) Effects of age and caloric restriction on cell proliferation in hepatocyte cultures from control and hepatectomized Fischer 344 rats. Mutagenesis 11, 281–284 [DOI] [PubMed] [Google Scholar]
- 11. Omodei D., Fontana L. (2011) Calorie restriction and prevention of age-associated chronic disease. FEBS Lett. 585, 1537–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boya P., Gonzalez-Polo R. A., Casares N., Perfettini J. L., Dessen P., Larochette N., Metivier D., Meley D., Souquere S., Yoshimori T., Pierron G., Codogno P., Kroemer G. (2005) Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 25, 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barazzoni R., Zanetti M., Bosutti A., Biolo G., Vitali-Serdoz L., Stebel M., Guarnieri G. (2005) Moderate caloric restriction, but not physiological hyperleptinemia per se, enhances mitochondrial oxidative capacity in rat liver and skeletal muscle—tissue-specific impact on tissue triglyceride content and AKT activation. Endocrinology 146, 2098–2106 [DOI] [PubMed] [Google Scholar]
- 14. Bergamini E., Cavallini G., Donati A., Gori Z. (2007) The role of autophagy in aging: its essential part in the anti-aging mechanism of caloric restriction. Ann. N. Y. Acad. Sci. 1114, 69–78 [DOI] [PubMed] [Google Scholar]
- 15. Civitarese A. E., Carling S., Heilbronn L. K., Hulver M. H., Ukropcova B., Deutsch W. A., Smith S. R., Ravussin E., Team C. P. (2007) Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 4, e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee C. K., Klopp R. G., Weindruch R., Prolla T. A. (1999) Gene expression profile of aging and its retardation by caloric restriction. Science 285, 1390–1393 [DOI] [PubMed] [Google Scholar]
- 17. Lin S. J., Kaeberlein M., Andalis A. A., Sturtz L. A., Defossez P. A., Culotta V. C., Fink G. R., Guarente L. (2002) Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418, 344–348 [DOI] [PubMed] [Google Scholar]
- 18. Nisoli E., Tonello C., Cardile A., Cozzi V., Bracale R., Tedesco L., Falcone S., Valerio A., Cantoni O., Clementi E., Moncada S., Carruba M. O. (2005) Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310, 314–317 [DOI] [PubMed] [Google Scholar]
- 19. Matecic M., Smith D. L., Pan X., Maqani N., Bekiranov S., Boeke J. D., Smith J. S. (2010) A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet. 6, e1000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petrovski G., Das D. K. (2010) Does autophagy take a front seat in lifespan extension? J. Cell. Mol. Med. 14, 2543–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salminen A., Kaarniranta K. (2009) Regulation of the aging process by autophagy. Trends Mol. Med. 15, 217–224 [DOI] [PubMed] [Google Scholar]
- 22. Chen D., Thomas E. L., Kapahi P. (2009) HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 5, e1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jia K., Chen D., Riddle D. L. (2004) The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131, 3897–3906 [DOI] [PubMed] [Google Scholar]
- 24. Kaeberlein M., Powers R. W., 3rd, Steffen K. K., Westman E. A., Hu D., Dang N., Kerr E. O., Kirkland K. T., Fields S., Kennedy B. K. (2005) Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310, 1193–1196 [DOI] [PubMed] [Google Scholar]
- 25. Sharp Z. D., Bartke A. (2005) Evidence for down-regulation of phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR)-dependent translation regulatory signaling pathways in Ames dwarf mice. J. Gerontol. A Biol. Sci. Med. Sci. 60, 293–300 [DOI] [PubMed] [Google Scholar]
- 26. Kapahi P., Chen D., Rogers A. N., Katewa S. D., Li P. W., Thomas E. L., Kockel L. (2010) With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 11, 453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Price J. C., Holmes W. E., Li K. W., Floreani N. A., Neese R. A., Turner S. M., Hellerstein M. K. (2012) Measurement of human plasma proteome dynamics with 2H2O and liquid chromatography tandem mass spectrometry. Anal. Biochem. 420, 73–83 [DOI] [PubMed] [Google Scholar]
- 28. Pandey A., Mann M. (2000) Proteomics to study genes and genomes. Nature 405, 837–846 [DOI] [PubMed] [Google Scholar]
- 29. Wu C. C., MacCoss M. J., Howell K. E., Matthews D. E., Yates J. R., 3rd (2004) Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal. Chem. 76, 4951–4959 [DOI] [PubMed] [Google Scholar]
- 30. Price J. C., Guan S., Burlingame A., Prusiner S. B., Ghaemmaghami S. (2010) Analysis of proteome dynamics in the mouse brain. Proc. Natl. Acad. Sci. U.S.A. 107, 14508–14513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anderson R. M., Barger J. L., Edwards M. G., Braun K. H., O'Connor C. E., Prolla T. A., Weindruch R. (2008) Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 7, 101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neese R. A., Misell L. M., Turner S., Chu A., Kim J., Cesar D., Hoh R., Antelo F., Strawford A., McCune J. M., Christiansen M., Hellerstein M. K. (2002) Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc. Natl. Acad. Sci. U.S.A. 99, 15345–15350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neese R. A., Siler S. Q., Cesar D., Antelo F., Lee D., Misell L., Patel K., Tehrani S., Shah P., Hellerstein M. K. (2001) Advances in the stable isotope-mass spectrometric measurement of DNA synthesis and cell proliferation. Anal. Biochem. 298, 189–195 [DOI] [PubMed] [Google Scholar]
- 34. Hellerstein M. K., Neese R. A. (1999) Mass isotopomer distribution analysis at eight years: theoretical, analytic, and experimental considerations. Am. J. Physiol. 276, E1146–E1170 [DOI] [PubMed] [Google Scholar]
- 35. Commerford S. L., Carsten A. L., Cronkite E. P. (1982) Histone turnover within nonproliferating cells. Proc. Natl. Acad. Sci. U.S.A. 79, 1163–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 37. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 38. Dennis G., Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 39. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 40. Pagliarini D. J., Calvo S. E., Chang B., Sheth S. A., Vafai S. B., Ong S. E., Walford G. A., Sugiana C., Boneh A., Chen W. K., Hill D. E., Vidal M., Evans J. G., Thorburn D. R., Carr S. A., Mootha V. K. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Waterlow J. C., Garlick P. J., Millward D. J. (1978) Protein Turnover in Mammalian Tissues and in the Whole Body, CAB International, Oxfordshire, UK [Google Scholar]
- 42. Kasumov T., Ilchenko S., Li L., Rachdaoui N., Sadygov R. G., Willard B., McCullough A. J., Previs S. (2011) Measuring protein synthesis using metabolic (2)H labeling, high-resolution mass spectrometry, and an algorithm. Anal. Biochem. 412, 47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. MacCoss M. J., Wu C. C., Matthews D. E., Yates J. R., 3rd (2005) Measurement of the isotope enrichment of stable isotope-labeled proteins using high-resolution mass spectra of peptides. Anal. Chem. 77, 7646–7653 [DOI] [PubMed] [Google Scholar]
- 44. Mammucari C., Rizzuto R. (2010) Signaling pathways in mitochondrial dysfunction and aging. Mech. Ageing Dev. 131, 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Riva A., Deery M. J., McDonald S., Lund T., Busch R. (2010) Measurement of protein synthesis using heavy water labeling and peptide mass spectrometry: discrimination between major histocompatibility complex allotypes. Anal. Biochem. 403, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Busch R., Kim Y. K., Neese R. A., Schade-Serin V., Collins M., Awada M., Gardner J. L., Beysen C., Marino M. E., Misell L. M., Hellerstein M. K. (2006) Measurement of protein turnover rates by heavy water labeling of nonessential amino acids. Biochim. Biophys. Acta 1760, 730–744 [DOI] [PubMed] [Google Scholar]
- 47. Lis G., Wassenaar L. I., Hendry M. J. (2008) High-precision laser spectroscopy D/H and 18O/16O measurements of microliter natural water samples. Anal. Chem. 80, 287–293 [DOI] [PubMed] [Google Scholar]
- 48. Guan S., Price J. C., Prusiner S. B., Ghaemmaghami S., Burlingame A. L. (2011) A data processing pipeline for mammalian proteome dynamics studies using stable isotope metabolic labeling. Mol. Cell. Proteomics 10, M111.010728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lopez-Lluch G., Hunt N., Jones B., Zhu M., Jamieson H., Hilmer S., Cascajo M. V., Allard J., Ingram D. K., Navas P., de Cabo R. (2006) Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. U.S.A. 103, 1768–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hancock C. R., Han D. H., Higashida K., Kim S. H., Holloszy J. O. (2011) Does calorie restriction induce mitochondrial biogenesis? A reevaluation. FASEB J. 25, 785–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perez V. I., Van Remmen H., Bokov A., Epstein C. J., Vijg J., Richardson A. (2009) The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 8, 73–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chou M. W., Shaddock J. G., Kong J., Hart R. W., Casciano D. A. (1995) Effect of dietary restriction on partial hepatectomy-induced liver regeneration of aged F344 rats. Cancer Lett. 91, 191–197 [DOI] [PubMed] [Google Scholar]
- 53. Himeno Y., Engelman R. W., Good R. A. (1992) Influence of calorie restriction on oncogene expression and DNA synthesis during liver regeneration. Proc. Natl. Acad. Sci. U.S.A. 89, 5497–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Selsby J. T., Judge A. R., Yimlamai T., Leeuwenburgh C., Dodd S. L. (2005) Life long calorie restriction increases heat shock proteins and proteasome activity in soleus muscles of Fisher 344 rats. Exp. Gerontol. 40, 37–42 [DOI] [PubMed] [Google Scholar]
- 55. Ivan M., Kondo K., Yang H., Kim W., Valiando J., Ohh M., Salic A., Asara J. M., Lane W. S., Kaelin W. G., Jr. (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468 [DOI] [PubMed] [Google Scholar]
- 56. Jaakkola P., Mole D. R., Tian Y.-M., Wilson M. I., Gielbert J., Gaskell S. J., von Kriegsheim A., Hebestreit H. F., Mukherji M., Schofield C. J., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 [DOI] [PubMed] [Google Scholar]