

Abstract
Banana and its close relative, plantain are globally important crops and there is considerable interest in optimizing their cultivation. Plantain has superior cold tolerance compared with banana and a thorough understanding of the molecular mechanisms and responses of plantain to cold stress has great potential value for developing cold tolerant banana cultivars. In this study, we used iTRAQ-based comparative proteomic analysis to investigate the temporal responses of plantain to cold stress. Plantain seedlings were exposed for 0, 6, and 24 h of cold stress at 8 °C and subsequently allowed to recover for 24 h at 28 °C. A total of 3477 plantain proteins were identified, of which 809 showed differential expression from the three treatments. The majority of differentially expressed proteins were predicted to be involved in oxidation-reduction, including oxylipin biosynthesis, whereas others were associated with photosynthesis, photorespiration, and several primary metabolic processes, such as carbohydrate metabolic process and fatty acid beta-oxidation. Western blot analysis and enzyme activity assays were performed on seven differentially expressed, cold-response candidate plantain proteins to validate the proteomics data. Similar analyses of the seven candidate proteins were performed in cold-sensitive banana to examine possible functional conservation, and to compare the results to equivalent responses between the two species. Consistent results were achieved by Western blot and enzyme activity assays, demonstrating that the quantitative proteomics data collected in this study are reliable. Our results suggest that an increase of antioxidant capacity through adapted ROS scavenging capability, reduced production of ROS, and decreased lipid peroxidation contribute to molecular mechanisms for the increased cold tolerance in plantain. To the best of our knowledge, this is the first report of a global investigation on molecular responses of plantain to cold stress by proteomic analysis.
Plants constantly face environmental stresses, among which low temperature is often one of the most critical, and one that broadly affects growth and development, as well as the yield, quality, postharvest life and geographic distribution of crop plants (1, 2). Plants can be exposed to gradual temperature decreases during a fall-to-winter transition or sudden temperature drops associated with an early frost and so a range of adaptive mechanisms is essential. Most temperate plants can tolerant extracellular ice formation in their vegetative tissues, but many important crops, such as rice, maize, soybean, cotton, and tomato can not (3). Moreover, differences in cold tolerance can be exhibited by different varieties of the same species.
The mechanisms underlying cold tolerance have been studied in a range of species, including Arabidopsis thaliana, rice (Oryza sativa), barley (Hordeum vulgare), and wheat (Triticum aestivum), using microarray analysis to identify genes that are induced or repressed by cold stress (4–7). However, such quantitative mRNA data cannot necessarily be used to infer the effects of cold stress on protein expression (8, 9) because post-transcriptional regulation (10) can result in a poor correlation between levels of transcripts and their cognate proteins. For example, microRNAs (miRNAs) regulate gene expression post-transcriptionally by binding primarily to the 3′ untranslated region (3′UTR) of their target mRNAs, resulting in mRNA destabilization or translational repression (11, 12). Indeed, several cold stress-regulated miRNAs have been identified in Arabidopsis and rice (13, 14). Proteome studies focus on the complete set of proteins encoded by the genome and thus not only complement the transcriptome studies, but can also provide more direct insight into the signaling and metabolic processes associated with the perturbation conditions under study. Consequently, there is great potential value in studying cold stress responses on global protein dynamics, as reported for Arabidopsis thaliana, rice, chicory, meadow fescue, soybean, and pea, as well as for some woody plants such as poplar and peach (15).
Banana and plantain, which originated in the tropics are large herbaceous monocots from the genus Musa of the family Musaceae. The vast majority of cultivated bananas and plantains are hybrids derived from natural inter- and intraspecific crosses between two diploid wild species, Musa acuminata (designated by genome A) and Musa balbisiana (designated by genome B) (16). These diploid, triploid or tetraploid hybrids are of great economic importance in sub-Saharan Africa, South and Central America, and Asia, where they are a staple food for an estimated 400 million people (17). With an annual production of about 120 million tons, banana and plantain are among the most important food commodities following rice, wheat, and maize (18). Although they are mainly cultivated in tropical and subtropical regions, cold spells often occur during winter or early spring, which can have serious impact on fruit quality and yield. It is well known that plantain (Musa paradisiaca L.; ABB Group) has superior cold tolerance compared with banana (Musa acuminata AAA Group) (19) but, unlike banana, is able to tolerate temperatures of 0–4 °C, (20). When the temperature decreases to 8 °C banana growth is arrested in the interior of the pseudostem (21) and injury occurs (22), although application of certain chemicals such as low concentration H2O2, Ca2+, salicylic acid, brassinolide, or methyl jasmonate can increase cold tolerance and help avoid injury (23–26). Several cold stress related banana genes have been identified from in vitro cultures subjected to low temperature stress (27) and a cold-resistance-related plantain gene (MpRCI) has been identified that enhances low temperature-resistance when heterologously expressed in transgenic tobacco (28). However, despite its global importance, no systematic investigation of molecular responses in Musa to cold stress has been reported and the molecular mechanisms that underlie the superior cold tolerance of plantain remain poorly understood. Study of the plantain proteome profiles during cold stress therefore has the potential to yield new insight into the metabolic responses to cold stress and to identify cold-resistance candidate proteins which could be utilized for genetic manipulation and/or cross breeding in banana.
The rapid development of proteomic technologies, combined with vast amounts of plant genome sequence information, provides an unprecedented opportunity for plant proteomic profiling (29, 30). Comparative proteomic approaches employing solution-based “shotgun” techniques employing two-dimensional liquid chromatography (2D-LC) coupled directly with tandem MS (MS/MS) detection (31, 32) have been widely used. Two-dimensional liquid chromatography is highly compatible with multiplexed quantitative techniques involving isobaric tags for relative and absolute quantitation (iTRAQ)1 (33). As nonmodel organisms, both plantain and banana lack complete genomic sequence information, which has imposed obvious limits on protein identification through predictive matching or peptide sequences. However, in the past few years, next generation sequencing technologies have provided a new paradigm for genome and transcriptome (RNA-Seq) analysis for nonmodel organisms with unknown genomes (34). Recently we have completed a plantain RNA-Seq database (submitted to NCBI databases with submission number as SRA054939), facilitating iTRAQ-based quantitative proteomics to investigate the dynamic proteome response to cold stress. In this current study, plantain seedlings were exposed to low temperature over a time course and then returned to a normal nonstressing temperature.
We report the identification of proteomic responses to cold stress in plantain seedlings and compare the results to equivalent responses in banana. Together with supporting data from Western blot analyses and enzyme activity assays, these results provide insights into the complex molecular mechanisms associated with the response to cold stress in plantain, from which we were able to infer potential cold tolerance mechanisms.
EXPERIMENTAL PROCEDURES
Chemicals and Materials
Sequence-grade acetonitrile (ACN), trifluoroacetic acid (TFA), and formic acid (FA) were purchased from Fisher Scientific (Fair Lawn, NJ). The iTRAQ kit and strong cation exchange (SCX) cartridge were purchased from AB Sciex (Foster City, CA), the Sep-Pak solid-phase extraction (SPE) cartridges from Waters (Milford, MA), and modified trypsin from Promega (Madison, WI). The rest of the chemical reagents, unless otherwise noted, were obtained from Aldrich (Milwaukee, WI).
Plant Material and Stress Treatment
Seedlings of the cold-tolerant plantain cv. Dajiao (Musa paradisiaca L.; ABB Group cv. Dajiao) and the cold-sensitive banana cv. Brazil (Musa acuminata L.; AAA Group cv. Brazil) with a uniform growth stage were obtained from Institute of Fruit Tree Research, Guangdong Academy of Agricultural Sciences, Guangzhou, P. R. of China. Seedlings were grown in a growth chamber under a 30/28 °C (day/night) temperature regime, a photon flux density of 240 μmol m−2 s−1 throughout a 12-h photoperiod, and a relative humidity of 60–80%. Five-leaf stage seedlings were used in the experiment. Low temperature treatments were started at 12:00 am on the first day by setting the temperature to 8 °C, which was reached about 40 min later. The temperature was returned to 28 °C after 24 h of treatment. The first young leaf was detached from the top of each of the 5 plants at each time point (8 °C for 0, 6, 24 h and then returned to 28 °C for 24 h) for each biological replicate. The leaves from the five plants were cut into pieces (1.5 × 1.5 cm) and mixed well. Aliquots of the mixed tissues were frozen in liquid N2 and stored at −80 °C until use.
Measurements of Photosynthesis, Stomatal Conductance, and Transpiration
Leaf net photosynthetic rates, stomatal conductance and transpiration rates were measured using a portable gas analysis system, LI-COR 6400 with a light-emitting diode light source (LICOR Inc., Lincoln, NE). The measurement conditions were set as follows: leaf temperature 25 °C, photon flux density 800 μmol·m−2 s−1, humidity 65%, and CO2 concentration 0.038%.
Protein Extraction, Digestion, and iTRAQ Labeling
Plantain leaf proteins from two biological replicate samples were obtained by grinding tissues in liquid N2 followed by suspension in ice-cold sodium phosphate buffer (100 mm, pH 7.5) containing 1 mm EDTA, 100 μg/ml PMSF, and 0.1% Triton X-100. The homogenate was centrifuged at 12,000 × g for 15 min. The protein concentrations were determined by the Bradford assay using BSA as a standard (35). The proteins in the supernatant were lyophilized and then kept at −80 °C for further analysis.
The lyophilized protein powders from each sample were reconstituted in 0.2 m triethylammonium bicarbonate (pH 8.5). An aliquot (100 μg) of proteins in a total volume of 20 μl was denatured by adding 1 μl of 2% SDS and reduced with 2 μl of 50 mm Tris-(2-carboxyethyl)phosphine (TCEP). Cysteine residues were blocked with 1 μl of 200 mm methyl methanethiosulfonate (MMTS) using the iTRAQ Reagents kit (AB Sciex). The proteins were then digested using an enzyme to substrate ratio of 1:10 sequencing-grade trypsin at 36 °C overnight. The digested peptides were labeled with iTRAQ reagents following the manufacturer's (AB Sciex) instructions using 114-tag, 115-tag, 116-tag, and 117-tag for cold treated at 0, 6, 24 h and recovered for 24 h samples (named as C0h, C6h, C24h, and R24h), respectively. Efficiency of iTRAQ labeling was assessed by MALDI-TOF/TOF 4700 (AB Sciex) choosing five peptides from each sample randomly. After labeling, the four samples were combined and subjected to high pH reverse phase (hpRP) fractionation as described below.
High pH Reverse Phase Fractionation
The pooled iTRAQ labeled peptides were passed through SCX cartridges (AB Sciex, Framingham, MA) and desalted by Sep-Pak SPE cartridges (Waters) for subsequent hpRP separation. The hpRP chromatography was carried out using a Dionex UltiMate 3000 high performance LC system with a built-in microfraction collection option in its autosampler and UV detection (Thermo-Dionex, Sunnyvale, CA). The iTRAQ-tagged tryptic peptides were reconstituted in buffer A (20 mm ammonium formate, pH 9.5, in water) and loaded onto an XTerra MS C18 column (3.5 μm, 2.1 × 150 mm, Waters) with 20 mm ammonium formate (NH4HCO2, pH 9.5) as buffer A and 80% ACN/20% 20 mm NH4HCO2 as buffer B. The LC was performed using a gradient from 10 to 45% of buffer B in 30 min at a flow rate of 200 μl/min, as previously reported (36). Forty-eight fractions were collected at 1 min intervals and pooled into a total of 12 fractions, based on UV absorbance at 214 nm and with a multiple fraction concatenation strategy (37). The fractions were dried and reconstituted in 120 μl of 2% ACN/0.5% FA for nanoLC-MS/MS analysis.
NanoLC-MS/MS Analysis by LTQ-Orbitrap Velos
The nanoLC-MS/MS analysis was carried out using a LTQ-Orbitrap Velos (Thermo-Fisher Scientific, San Jose, CA) mass spectrometer equipped with a “CorConneX” nano-ion source (CorSolutions LLC, Ithaca, NY), containing a PepMap C18 RP nano column (3 μm, 75 μm × 15 cm, Dionex) connected to a 10 μm analyte emitter (NewObjective, Woburn, MA). The Orbitrap was interfaced with an UltiMate 3000 RSLC nano system (Thermo-Dionex, Sunnyvale, CA). Each reconstituted fraction (5 μl) was injected onto a PepMap C18 trapping column (5 μm, 300 μm × 5 mm, Dionex) at a 20 μl/min flow rate for loading, and then separated on the PepMap C18 RP nano column, using a 120 min gradient from 5 to 38% ACN in 0.1% FA at 300 nl/min, followed by a 5-min ramp to 95% ACN-0.1% FA and a 5-min hold at 95% ACN-0.1% FA. The column was re-equilibrated with 2% ACN-0.1% FA for 20 min before the next run. The eluted peptides were detected by the LTQ Orbitrap Velos, which was operated in positive ion mode with the nanospray voltage set at 1.5 kV and the source temperature at 275 °C. The instrument was externally calibrated using Ultramark 1621 for the FT mass analyzer. An internal calibration was performed using the background polysiloxane ion signal at m/z 445.120025 as the calibrant. The instrument was operated in data-dependent acquisition (DDA) mode. In all experiments, full MS scans were acquired over a mass range of m/z 400–1400, with detection in the Orbitrap mass analyzer at a resolution setting of 30,000. Fragment ion spectra produced via high energy collision dissociation (HCD) were acquired in the Orbitrap mass analyzer with a resolution setting of 7500 for the mass range of m/z 100–2000. In each cycle of DDA analysis, following each survey scan, the 10 most intense multiply charged ions above a threshold ion count of 5000 were selected for fragmentation at a normalized collision energy of 45%. Dynamic exclusion parameters were set at repeat count 1 with a 20 s repeat duration, an exclusion list size of 500, 30 s exclusion duration with ±10 ppm exclusion mass width. The activation time was 0.1 ms for HCD analysis. All data were acquired with Xcalibur 2.1 software (Thermo-Fisher Scientific).
Protein Identification and Quantitation
Raw data files acquired from the Orbitrap were converted into MGF files using Proteome Discoverer 1.2 (PD 1.2, Thermo). Subsequent database searches were carried out by Mascot Daemon (version 2.3, Matrix Science, Boston, MA) for both protein identifications and iTRAQ quantitation against a plantain protein database which was derived from RNA-Seq data (its submission number in NCBI databases is SRA054939) and annotated by Guangzhou iGenomics Co., Ltd. using the getorf program in EBOSS and BLAST software in NCBI. The plantain protein database consisting of 44,385 sequence entries and 10,792,935 amino acid residues, is a protein preferred database containing the combined proteins predicted by both longest six frame translations and expressed sequence tag (EST) scan. The default Mascot search settings were as follows: one missed cleavage site by trypsin allowed with fixed MMTS modification of cysteine, fixed four-plex iTRAQ modifications on Lys and N-terminal amines and variable modifications of methionine oxidation, deamidation of Asn and Gln residues, and 4-plex iTRAQ on Tyr. The peptide and fragment mass tolerance values were 20 ppm and 0.1 Da, respectively. To estimate the false discovery rate (FDR) for a measure of identification certainty in each replicate set, we employed the target-decoy strategy of Elias and Gygi (38). Specifically, an automatic decoy database search was performed in Mascot by choosing the decoy checkbox in which a random sequence of database is generated and tested for raw spectra as well as the real database. To reduce the probability of false peptide identification, only peptides with significance scores (≥20) at the 99% confidence interval by a Mascot probability analysis (www.matrixscience.com/help/scoring_help.html#PBM) greater than “identity” were counted as identified. It was required that each confident protein identification involve at least two unique peptide identifications indicated in Mascot. Proteins identified within same family were grouped in Mascot protein family summary. Furthermore, to be considered confidently quantified, we required that a protein produced at least two unique peptides that generate a complete iTRAQ reporter ion series. The quantitative protein ratios were weighted and normalized by the median ratio with outlier removal set automatic in Mascot for each set of experiments. The manufacturer's recommended isotope correction factors were applied. The functional annotation and classification of all proteins identified, and being a differential expression of over ±1.5-fold at C6h, C24h, and R24h over C0h control, were determined according to Blast2go (Bioinformatics Department, CIPF, Valencia, Spain) (39, 40).
Antibodies and Western Blot Analyses
Proteins were extracted from banana leaves as described above for plantain leaves. Protein samples from both plantain and banana containing aliquots (5, 20, 20, 30, 30, 15, and 30 μg) of banana and plantain leaf protein extracts were separated on 12.5% or 15% SDS-PAGE gels, and electroblotted onto a PVDF membrane using a Mini Trans-Blot cell (BioRad, Foster City, CA). Membranes were blocked, incubated with antisera that were raised to one of the following proteins: Cu/Zn superoxide dismutase (SOD), catalase, lipoxygenase, actin (each 1:1,000 dilution; all four antibodies were purchased from Agrisera, Sweden; product numbers: AS06 170, AS09 501, AS06 128, and AS10 702), or glutamate glyoxylate aminotransferase (GGAT), serine glyoxylate aminotransferase (SGAT), serine hydroxymethyltransferase (SHMT), and β-hexosaminidase (β-Hex) (each 1:1,000 dilution; polyclonal antisera raised in rabbit). GGAT, SGAT, SHMT, β-Hex proteins used for generating antibodies were expressed using their predicted full length rice cDNAs (with GenBank Accession numbers AY236227, XM_483211, NM_001057746 and NM_001062091, respectively), which were inserted into the pET-30a or pET-32a vector (Novagen, Madison, Wisconsin) and transformed into E. coli (RossettaII). The resulting proteins were purified on a gradient SDS-PAGE (4–20%) gel and injected into rabbits. Rabbit sera were collected and used as a source of polyclonal antibodies. Goat anti-rabbit IgG (1:10,000) conjugated with alkaline phosphatase was used as the secondary antibody and the bands were visualized with a premixed BCIP/NBT substrate solution (Sigma-Aldrich, St. Louis, MO).
Enzyme Activity Assay
Fresh plantain and banana leaves (0.5 g) from plants treated as described above (C0h, C6h, C24h, and R24h) were ground in a mortar and pestle in 1 ml of chilled 0.1 m phosphate buffer (pH 7.5) containing 1% (w/v) polyvinylpolypyrrolidone (PVP). The homogenates were centrifuged at 12,000 × g for 20 min at 4 °C and the supernatants used for enzyme activity assays. Protein concentrations were determined using the Bradford assay (35) with bovine serum albumin as the standard. Five technical replicates for each of the three biological replicates were performed for each enzyme assay.
Total superoxide dismutase (SOD) activity was determined according to the method of Giannopolitis and Ries (41). Enzyme extracts of each sample (10 μl) were added to a 3 ml reaction solution containing 13 μm methionine, 63 μm ρ-nitro blue tetrazolium chloride (NBT), 1.3 μm riboflavin, and 50 mm phosphate buffer (pH 7.8). The reaction solution was incubated for 10 min under fluorescent light with 80 μmol m−2 s−1. Absorbance was determined at 560 nm with a spectrophotometer. One unit of SOD activity was defined as the amount of enzyme required for the inhibition of the photochemical reduction of NBT by 50%.
Catalase activity was determined spectrophotometrically by following the decrease of absorbance of H2O2 (extinction coefficient 0.0394 mm−1 cm−1) within 1 min at 240 nm according to the method of Aebi (42). Enzyme extracts of each sample (100 μl) were added into a 3 ml reaction solution containing 15 mm H2O2, 50 mm phosphate buffer (pH 7.0) to initiate the reaction.
For lipoxygenase activity assays (43), 0.5 g of lyophilized, powdered plantain or banana leaves was homogenized in 1 ml of extraction solution, (0.1 m phosphate, pH 7.5, 2 mm DTT, 1 mm EDTA, 0.1% (v/v) Triton X-100, and 1% (w/v) polyvinyl polypyrrolidone (PVPP)). The resulting homogenates were centrifuged at 12,000 × g for 20 min at 4 °C and the supernatant was recovered and stored on ice. Protein concentrations were determined by the Bradford assay (35) with bovine serum albumin as the standard. Lipoxygenase activity was assayed by mixing 2.5 ml of 0.1 m phosphate, pH 6.0, 0.4 ml substrate solution (8.6 mm linoleic acid, 0.25% (v/v) Tween-20, in 0.1 m phosphate, pH 8.0), and 100 μl enzyme extract. Activity was measured spectrophotometrically by following the increase in absorbance at 234 nm over time, as a result of formation of hydrogen peroxide from linoleic acid during the catalytic reaction. One activity unit (U) was defined as the increase in one unit of absorbance at 234 nm min−1, and the results were expressed as specific activity (U mg protein−1).
RESULTS
Experimental Design and Proteomics Workflow
The main objective in this study was to identify cold-response proteins in cold-tolerant plantain to develop a better understanding of the underlying metabolic processes and the molecular mechanisms of cold stress tolerance. An iTRAQ-based shotgun quantitation approach was utilized to obtain a global view of the proteome dynamics and the changes associated with the cold response of plantain seedlings. An overview of the iTRAQ experimental design and workflow conducted is shown in Fig. 1 for cold-treatment plantain samples at 0, 6, and 24 h of cold exposure, as well as a 24-hour post-treatment “recovery” sample. Following the large scale identification and functional categorization of the differentially expressed plantain proteins, Western blot analyses and enzyme activity assays were carried out in both cold-tolerant plantain and cold-sensitive banana.
Fig. 1.

Experimental design and schematic diagram of the workflow used in this study. Two sets of biological replicate samples were analyzed by iTRAQ, using the hpRP-nanoLC-MS/MS workflow for examining proteome changes in plantain seedlings in response to cold stress (8 °C for 0, 6, 24 h and then recovery at 28 °C for 24 h). Proteins were identified using Mascot software.
Physiological Responses of Plantain to Cold Stress
Mature plantain plants can tolerate temperatures of 0–4 °C, but plantain seedlings display severe necrosis and wilting symptoms below 4 °C and do not recover after being returned to normal temperature (28 °C; data not shown), so 8 °C was selected as the cold treatment for these studies. The leaves drooped and pseudostem tilted after 24 h of cold treatment and mostly recovered after being returned to 28 °C for 24 h (Fig. 2A). To evaluate the adverse effects of cold stress quantitatively and determine the best time for sample collection after cold stress for subsequent proteomics analysis, photosynthetic characteristics were measured. Photosynthesis in warm climate plants is substantially affected by cold stress through a process called low temperature photoinhibition (44). In response to the cold treatment, the net photosynthetic rate (Pn) decreased to 53 and 26% of pre-treatment levels after 6 and 24 h, respectively, but returned to 76% after 24 h of recovery. The changes of stomatal conductance (Gs), a rate of passage of either water vapor or carbon dioxide through the stomata followed similar trends to those shown for the Pn, but the intercellular CO2 concentration (Ci) showed little change (Fig. 2B). These findings are consistent with the report for rice under cold stress, a species that also originated in the tropics (8).
Fig. 2.

Physiological responses of plantain to cold stress. Five-leaf stage seedlings were treated at 8 °C for 0, 6, and 24 h (C0h, C6h, and C24h) and then allowed to recover for 24 h (R24h). Scale bars = 5 cm (A). The net photosynthetic rate (Pn), stomatal conductance (Gs), and the intercellular CO2 concentration (Ci) are shown in (B). The values of relative % for each column are means ± S.D. of three biological replicates with each having three to five technical replicate measurements. The different lowercase letters labeled above columns indicate a significant difference at p ≤ 0.05 between the columns by Duncan's test using SPSS statistical software (version 16.0, SPSS Inc. Chicago, IL). The columns with the same letters mean no significant difference (p > 0.05) between each other.
Detection of Cold Stress Induced Plantain Proteins
To investigate the temporal changes of protein profiles in response to moderate cold stress treatment at C6h, C24h and the subsequent R24h treatment, compared with the pre-treatment control (C0h), proteins were extracted from plantain leaves and analyzed using an iTRAQ-based shotgun proteomics strategy. After nanoLC-MS/MS analysis, all protein ratios were obtained using the Mascot search engine. The change in relative concentration of any given protein for C6h and C24h cold stress and R24h recovery was obtained from the iTRAQ 4-plex reporter ion ratios (115/114, 116/114, and 117/114, respectively) by a weighted average of all confidently identified peptides, requiring that at least two were uniquely assigned to any given protein in order for it to be classified as “identified.” The detailed protein identifications and quantitations from two independent biological replicates are listed in Supplemental Table S1. A summary of the results of the protein identifications is presented in Table I. A total of 3477 distinct were identified with estimated ∼2% FDR, of which 2682 (77%) were identified in both sets, and 2658 proteins were quantified, of which 2075 (78%) overlapped in the replicate data sets (Table I).
Table I. Comparison of results for two sets of biological replicate samples.
| Summary data | Set 1 | Set 2 |
|---|---|---|
| Total number of protein IDsa | 3092 | 3067 |
| Number of unique proteins from each setb | 410 | 385 |
| Combined distinct protein IDs (total/overlap) from two sets | 3477/2682 | |
| Reproducibility of protein IDs in two sets | 86.7% | 87.4% |
| Total proteins with iTRAQ ratio | 2360 (76.3%) | 2373 (77.4%) |
| Unique proteins with iTRAQ ratio from each set | 285 | 298 |
| Combined distinct protein IDs with iTRAQ ratio (total/overlap) from two sets | 2658/2075 | |
| Reproducibility of protein IDs with iTRAQ ratio in two sets | 87.9% | 87.8% |
| Total peptides/unique peptides | 63974/13192 (20.6%) | 65243/13289 (20.4%) |
| Peptides only identified in one set | 2221 | 2318 |
| Combined unique peptides (total/overlap) from two sets | 15510/10971 | |
a The total number of protein IDs indicates the total protein IDs identified by two sets of biological replicate samples.
b The number of unique proteins from each sets denotes the number of protein IDs exclusively identified from each of the two sets.
An analysis of variance of the two sets of data was conducted to determine the quantitative precision and reproducibility. The proteins identified and quantified by both set 1 and set 2 were used to assess the quantitative variations from both biological and analytical replicates. As shown in Fig. 3, plots of 115/114, 116/114, and 117/114 ratios for each of quantified proteins between the two sets generated comparable quantification results, as determined by a linear regression analysis that revealed a slope of ≈0.86 at an R2 of 0.87 for 115/114, ≈0.98 at an R2 of 0.88 for 116/114, and ≈0.89 at an R2 of 0.85 for 117/114, suggesting the quantitative iTRAQ data from replicates is quite reproducible. The three plots were used to further compute the internal error, which was defined as the value of the log2 iTRAQ ratio at which 95% of all proteins had no deviation from each other, in which the deviation was the absolute value of the difference in iTRAQ log2 ratios between the x- and y axis (45, 46). The internal errors for the three plots at log2 scale were 0.583, 0.443, and 0.502, which correspond to 1.50-fold, 1.36-fold, and 1.42-fold internal variation respectively, representing a combined technical and biological deviation. The largest deviation ±0.583 at log2 115/114 (corresponding to a ±1.50-fold change) was then used as a threshold for determining protein changes in the present work.
Fig. 3.

Comparison of log2 iTRAQ ratio (115/114, 116/114, and 117/114) for 2075 proteins identified in both biological replicates: Set 1 and Set 2. The largest internal errors were 0.582, corresponding to a 1.50-fold change, which was used as a threshold of significant change in response to cold stress or recovery.
To further confirm the statistical analysis of the data from the two biological replicates, we applied another commercially available software package for statistical distribution studies. The relative expression data (treated/control) for all proteins identified for each of the individual treatments were simultaneously fitted to 50 standard data distribution models using EasyFit (MathWave Technologies, http://www.mathwave.com). Overall, the data were judged to be best fit by the Johnson SU distribution (47) using the Kolmogrov/Sminov, Aderson/Darling and Chi-Squared tests for goodness of fit (48). The data were found to be normally distributed, as indicated in Supplemental Figs. S1–S4, which shows graphs of the probability density function, the cumulative distribution function and both the P-P and Q-Q plots of each of the data sets. Additional parameters describing the fit of the empirical data to the statistical model are given in Supplemental Table S3.
In combining the results of differentially expressed proteins obtained from biological duplicate samples, 219, 222, and 180 of the up-regulated proteins were found in both sets at C6h, C24h cold treatment, and R24h recovery, respectively (Fig. 4), whereas 424, 402, 336, respectively, of the down-regulated proteins were detected in both sets. These numbers represent ∼80% of the total differentially expressed proteins found in each group, suggesting excellent reproducibility between the two sets and agreement with previous reports (36, 49). As shown in Figs. 4A–4F, the relative standard deviations (RSD) for up-regulated proteins at three time points (C6h, C24h and R24h) between two sets were less than 26%, 20%, and 20%, respectively. The RSD for down-regulated proteins were less than 35%, 25%, and 21%, respectively. These results provided additional evidence for the good quantitative reproducibility and accuracy obtained in this study. Further comparison of up-regulated proteins or down-regulated proteins identified at the three time points indicates a significantly higher percentage of identified proteins were in common between C6h and C24h than between either of C6h or C24h and R24h (Fig. 5).
Fig. 4.

Venn diagrams of differentially expressed identified proteins in duplicated biological samples. After 6 h of cold stress the maximum RSDs for up-regulated (A) and down-regulated (B) proteins were less than 26% (A) and 35% (B) with average RSDs = 9 and 11% respectively. After 24 h cold stress, the maximum RSDs for up-regulated (C) and down-regulated (D) proteins were less than 20%, with average RSDs = of ∼8 and 7% respectively. After 24 h of recovery, the maximum RSDs for up-regulated (E) and down-regulated (F) proteins were less than 20 and 21%, respectively, with average RSDs = 8% for both.
Fig. 5.

Venn diagram of differentially expressed proteins identified in three different treatment time points. The numbers of up-regulated proteins (A) and down-regulated proteins (B) at each particular time point are shown in the different segments.
Identities and Functional Classification of Differentially Expressed Proteins
All differential proteins identified from the three time points were functionally categorized using Blast2go, a web-based bioinformatic tool that groups proteins based on their GO annotations. In this study, proteins were classified based on biological process (as opposed to cellular compartment or molecular function) at a GO annotation level of 3, and 10 functional categories were selected to cover the data sets. GO Term Scores are computed at each node according to the formula: , where seq is the number of different sequences annotated as a child GO term and dist the distance to the node of the child (39). Fig. 6 shows the GO prediction for both up and down-regulated proteins. The majority of differentially expressed proteins identified in this study were in the categories oxidation-reduction process, cellular metabolic process, response to stress and primary metabolic process, regardless of whether they were up-regulated (Fig. 6A) or down-regulated (Fig. 6B).
Fig. 6.
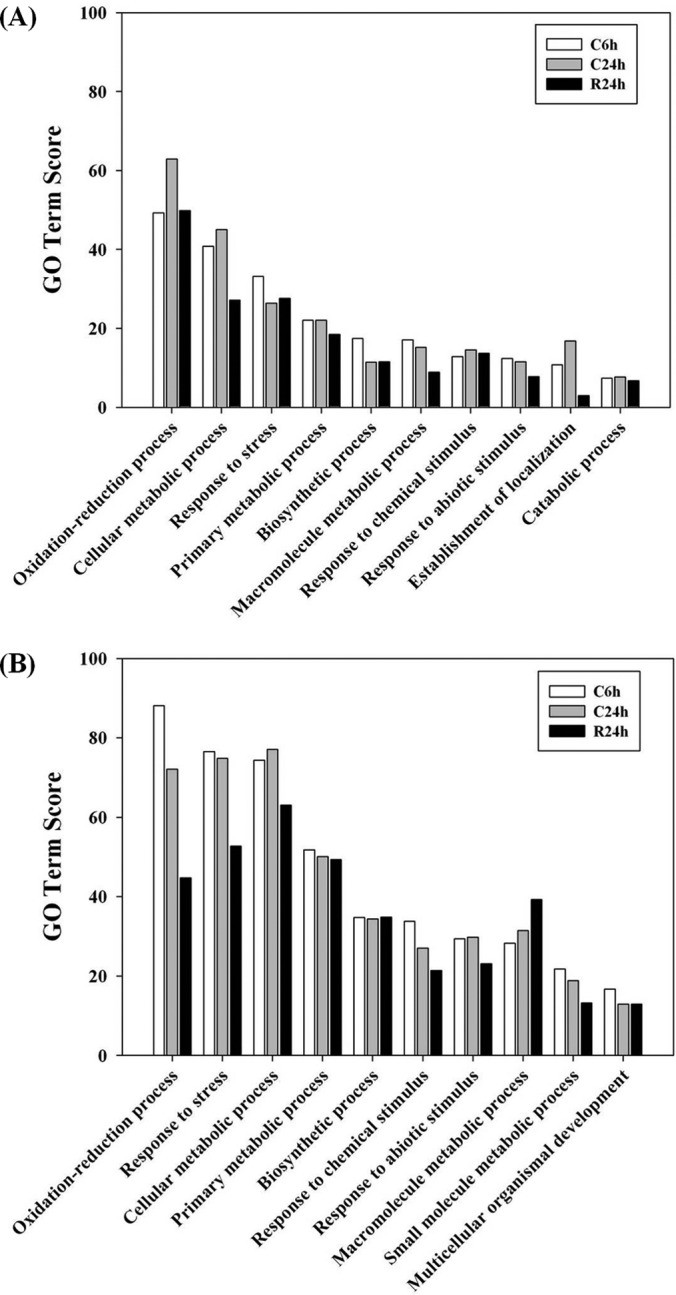
Functional categorization of differentially expressed proteins at each time point(s). GO categories of proteins up-regulated (A) or down-regulated (B) after cold stress.
To help focus on potentially more differentially expressed protein, we used a database for the monocotyledonous plant rice to filter out some possible redundant hits in the plantain database and the change threshold was set at 1.7-fold. After filtering, the remaining 116 differential proteins were classified into the following 8 functional categories: oxidation reduction (including oxylipin biosynthesis, photosynthesis, photorespiration, glycolysis, tricarboxylic acid cycle, carbohydrate metabolic process, fatty acid biosynthetic process, and fatty acid beta-oxidation (Table II).
Table II. Primary functional classification on differential proteins with iTRAQ ratios > or <1.7-fold.
| Protein name | Protein accession | Oryza sativa homology | Trendsa | C6hb | C24hb | R24hb |
|---|---|---|---|---|---|---|
| Oxidation reduction | ||||||
| Catalase isozyme 2 | Locus_1320_Contig9 | NP_001048861.1 | + + + | 2.778 ± 0.209 | 2.450 ± 0.036 | 1.678 ± 0.010 |
| Acyl-CoA dehydrogenase | Locus_319_Contig1 | NP_001060619.1 | + + + | 2.224 ± 0.235 | 1.675 ± 0.271 | 1.508 ± 0.211 |
| Probable tocopherol cyclase | Locus_6948_Contig1 | NP_001046545.1 | + + + | 2.196 ± 0.082 | 1.768 ± 0.032 | 1.841 ± 0.207 |
| 60S ribosomal protein L35a-3 | Locus_835_Contig4 | NP_001062631.2 | + + + | 2.089 ± 0.147 | 1.536 ± 0.048 | 2.041 ± 0.350 |
| Zeaxanthin epoxidase | Locus_12183_Contig1 | NP_001059681.2 | + + = | 2.044 ± 0.239 | 1.673 ± 0.099 | 1.091 ± 0.103 |
| Short-chain alcohol dehydrogenase | Locus_1804_Contig1 | NP_001051666.1 | + + + | 1.985 ± 0.292 | 2.326 ± 0.587 | 1.730 ± 0.180 |
| Superoxide dismutase [Cu-Zn] | Locus_2385_Contig4 | NP_001062514.1 | + + + | 1.981 ± 0.345 | 2.014 ± 0.389 | 1.784 ± 0.304 |
| Nucleoside diphosphate kinase | Locus_6933_Contig1 | NP_001056515.1 | + + + | 1.964 ± 0.620 | 1.765 ± 0.132 | 1.557 ± 0.565 |
| Catalase isozyme A | Locus_1788_Contig4 | NP_001045673.1 | + = = | 1.963 ± 0.431 | 1.354 ± 0.153 | 1.090 ± 0.143 |
| Probable cinnamyl alcohol dehydrogenase 6 | Locus_5573_Contig5 | NP_001052290.1 | + + + | 1.854 ± 0.109 | 1.634 ± 0.154 | 1.634 ± 0.190 |
| Formate dehydrogenase 1 | Locus_708_Contig2 | NP_001057666.1 | + + = | 1.824 ± 0.077 | 1.550 ± 0.009 | 1.203 ± 0.014 |
| 40S ribosomal protein S16 | Locus_8212_Contig1 | NP_001048958.1 | + + + | 1.789 ± 0.048 | 1.793 ± 0.181 | 1.773 ± 0.104 |
| Tropinone reductase 1 | Locus_1025_Contig1 | NP_001067819.1 | + + = | 1.634 ± 0.364 | 1.735 ± 0.021 | 1.350 ± 0.211 |
| Oryzain alpha chain | Locus_6578_Contig1 | NP_001054211.2 | + + + | 1.617 ± 0.026 | 1.608 ± 0.063 | 1.828 ± 0.101 |
| Bacterial-induced peroxidase precursor | Locus_15360_Contig1 | NP_001057822.1 | + + + | 1.604 ± 0.074 | 2.201 ± 0.059 | 2.157 ± 0.142 |
| l-idonate 5-dehydrogenase | Locus_829_Contig3 | NP_001062412.1 | + + + | 1.599 ± 0.041 | 1.592 ± 0.009 | 2.072 ± 0.020 |
| Protease Do-like 1, chloroplastic | Locus_10265_Contig3 | NP_001056360.1 | + + = | 1.598 ± 0.050 | 1.878 ± 0.053 | 1.332 ± 0.109 |
| ABC transporter C family member 14 | Locus_350_Contig9 | NP_001052252.1 | + + = | 1.500 ± 0.212 | 2.062 ± 0.270 | 0.980 ± 0.130 |
| Fructose-1,6-bisphosphatase, cytosolic | Locus_5417_Contig1 | NP_001044906.1 | = = + | 1.303 ± 0.011 | 1.271 ± 0.038 | 1.725 ± 0.070 |
| Tropine dehydrogenase | Locus_2854_Contig7 | NP_001049675.1 | = = + | 1.276 ± 0.097 | 1.042 ± 0.005 | 2.546 ± 0.248 |
| Glutamate dehydrogenase | Locus_2830_Contig2 | NP_001051540.1 | = + + | 1.170 ± 0.045 | 1.950 ± 0.323 | 1.668 ± 0.123 |
| Myosin-J heavy chain | Locus_330_Contig3 | NP_001064913.1 | = − + | 1.062 ± 0.231 | 0.304 ± 0.023 | 5.026 ± 0.428 |
| Primary amine oxidase | Locus_21191_Contig1 | NP_001052338.2 | = + = | 1.037 ± 0.079 | 2.130 ± 0.285 | 1.409 ± 0.390 |
| Aci-reductone dioxygenase-like protein | Locus_8396_Contig1 | NP_001049046.1 | = = + | 0.908 ± 0.181 | 0.680 ± 0.035 | 2.863 ± 0.400 |
| Geranylgeranyl-diphosphate synthase | Locus_233_Contig1 | NP_001060102.1 | = − − | 0.721 ± 0.046 | 0.583 ± 0.038 | 0.611 ± 0.019 |
| Putative 12-oxophytodienoate reductase 11 | Locus_7578_Contig1 | NP_001057140.1 | = = + | 0.699 ± 0.010 | 0.742 ± 0.070 | 2.442 ± 0.314 |
| Subtilisin-like protease | Locus_3032_Contig1 | NP_001044124.1 | = = − | 0.677 ± 0.177 | 1.157 ± 0.103 | 0.563 ± 0.126 |
| Auxin-induced protein PCNT115 | Locus_3905_Contig3 | NP_001064615.2 | = + = | 0.675 ± 0.016 | 2.160 ± 0.054 | 1.499 ± 0.057 |
| Zeta-carotene desaturase, chloroplastic/chromoplastic | Locus_137_Contig3 | NP_001059145.1 | − − = | 0.648 ± 0.021 | 0.583 ± 0.003 | 0.733 ± 0.002 |
| Germin-like protein 5–1 | Locus_1579_Contig1 | NP_001055081.1 | − − + | 0.646 ± 0.064 | 0.491 ± 0.037 | 1.522 ± 0.253 |
| Glutathione S-transferase DHAR2 | Locus_5821_Contig1 | NP_001054470.1 | − − = | 0.579 ± 0.007 | 0.557 ± 0.063 | 0.918 ± 0.342 |
| Mn-superoxide dismutase | Locus_7041_Contig2 | NP_001055195.1 | − = = | 0.576 ± 0.091 | 0.909 ± 0.006 | 1.025 ± 0.155 |
| Ferritin-3, chloroplastic | Locus_24284_Contig1 | NP_001065936.1 | − + = | 0.573 ± 0.027 | 4.016 ± 0.644 | 0.777 ± 0.089 |
| 3-isopropylmalate dehydrogenase 2, chloroplastic | Locus_5880_Contig1 | NP_001050807.1 | − − = | 0.569 ± 0.000 | 0.627 ± 0.038 | 0.820 ± 0.018 |
| Trans-cinnamate 4-monooxygenase | Locus_3077_Contig3 | NP_001055190.1 | − = − | 0.567 ± 0.021 | 0.842 ± 0.114 | 0.295 ± 0.023 |
| Peroxiredoxin-2E, chloroplastic | Locus_13207_Contig1 | NP_001058113.1 | − − − | 0.565 ± 0.029 | 0.552 ± 0.001 | 0.599 ± 0.031 |
| TPR repeat-containing thioredoxin TDX | Locus_1796_Contig4 | NP_001045581.1 | − − − | 0.565 ± 0.002 | 0.483 ± 0.027 | 0.618 ± 0.002 |
| UDP-glucose dehydrogenase | Locus_38_Contig1 | NP_001066705.1 | − − − | 0.555 ± 0.044 | 0.605 ± 0.001 | 0.494 ± 0.042 |
| Tryptophan synthase beta chain 2, chloroplastic | Locus_7244_Contig1 | NP_001060945.1 | − − = | 0.554 ± 0.042 | 0.500 ± 0.008 | 0.680 ± 0.062 |
| Naringenin,2-oxoglutarate 3-dioxygenase (Fragment) | Locus_1924_Contig1 | NP_001048799.1 | − + = | 0.543 ± 0.038 | 1.811 ± 0.021 | 0.912 ± 0.011 |
| Delta(24)-sterol reductase | Locus_3349_Contig3 | NP_001064534.1 | − − − | 0.494 ± 0.058 | 0.622 ± 0.036 | 0.632 ± 0.011 |
| Protein disulfide isomerase-like 1–4 | Locus_8562_Contig1 | NP_001045579.1 | − = − | 0.493 ± 0.010 | 0.755 ± 0.026 | 0.562 ± 0.032 |
| Peroxidase PX5 | Locus_1311_Contig4 | NP_001045481.1 | − = = | 0.477 ± 0.007 | 0.865 ± 0.027 | 1.070 ± 0.002 |
| Probable iron/ascorbate oxidoreductase DDB_G0283291 | Locus_21211_Contig1 | NP_001062658.1 | − = = | 0.472 ± 0.080 | 0.673 ± 0.033 | 0.759 ± 0.013 |
| Hypoxia up-regulated protein 1 (Fragment) | Locus_15808_Contig1 | NP_001047898.1 | − − − | 0.463 ± 0.051 | 0.655 ± 0.022 | 0.619 ± 0.085 |
| Ketol-acid reductoisomerase, chloroplastic | Locus_4019_Contig6 | NP_001056384.1 | − − − | 0.458 ± 0.001 | 0.474 ± 0.016 | 0.651 ± 0.097 |
| Luminal-binding protein 4 | Locus_992_Contig1 | NP_001045675.1 | − − − | 0.438 ± 0.042 | 0.647 ± 0.005 | 0.468 ± 0.024 |
| Ascorbate peroxidase | Locus_4933_Contig1 | NP_001060741.1 | − − − | 0.399 ± 0.023 | 0.478 ± 0.049 | 0.595 ± 0.048 |
| Probable calcium-binding protein CML7 | Locus_7153_Contig1 | NP_001060862.1 | − − − | 0.394 ± 0.069 | 0.618 ± 0.006 | 0.527 ± 0.038 |
| Calmodulin | Locus_1814_Contig1 | NP_001042747.1 | − = = | 0.375 ± 0.083 | 0.756 ± 0.022 | 0.871 ± 0.096 |
| Probable prenylcysteine oxidase | Locus_6821_Contig1 | NP_001054372.2 | − − − | 0.371 ± 0.028 | 0.604 ± 0.008 | 0.558 ± 0.032 |
| Putative anthocyanin synthase | Locus_1730_Contig1 | NP_001043065.1 | − + = | 0.364 ± 0.072 | 3.396 ± 0.139 | 1.215 ± 0.008 |
| Calreticulin | Locus_6308_Contig3 | NP_001051799.1 | − − − | 0.339 ± 0.014 | 0.580 ± 0.046 | 0.321 ± 0.005 |
| Peroxidase 5 | Locus_2279_Contig5 | NP_001065172.1 | − − − | 0.249 ± 0.029 | 0.574 ± 0.012 | 0.608 ± 0.051 |
| Monocopper oxidase-like protein SKU5 | Locus_2588_Contig1 | NP_001056550.1 | − − − | 0.188 ± 0.058 | 0.369 ± 0.017 | 0.389 ± 0.033 |
| Peroxidase | Locus_275_Contig1 | NP_001060737.2 | − − − | 0.095 ± 0.053 | 0.112 ± 0.010 | 0.137 ± 0.029 |
| l-ascorbate oxidase homolog | Locus_10392_Contig1 | NP_001058365.1 | − − − | 0.083 ± 0.010 | 0.207 ± 0.034 | 0.270 ± 0.012 |
| Cytochrome P450 CYP97A16 | Locus_18922_Contig1 | NP_001048526.1 | − − − | 0.083 ± 0.004 | 0.299 ± 0.009 | 0.158 ± 0.000 |
| Oxylipin biosynthetic process | ||||||
| Allene oxide synthase | Locus_3322_Contig1 | NP_001051386.1 | − = + | 0.597 ± 0.051 | 0.686 ± 0.009 | 2.956 ± 0.102 |
| Lipoxygenase 2.3, chloroplastic | Locus_7458_Contig3 | NP_001046180.1 | − − = | 0.551 ± 0.024 | 0.464 ± 0.000 | 0.782 ± 0.016 |
| Lipoxygenase 2 | Locus_119_Contig1 | NP_001050995.2 | − − = | 0.346 ± 0.017 | 0.284 ± 0.035 | 1.115 ± 0.069 |
| Lipoxygenase | Locus_440_Contig2 | NP_001050993.1 | − − − | 0.329 ± 0.019 | 0.247 ± 0.016 | 0.556 ± 0.044 |
| Photosynthesis | ||||||
| Chloroplast Rubisco small subunit | Locus_14638_Contig3 | NP_001066559.1 | + + = | 4.581 ± 1.195 | 3.264 ± 0.030 | 0.814 ± 0.144 |
| Photosystem I reaction center subunit III, chloroplastic | Locus_1142_Contig7 | NP_001051444.2 | + + = | 2.006 ± 0.344 | 2.434 ± 0.083 | 0.870 ± 0.145 |
| Photosystem I reaction center subunit VI, chloroplastic | Locus_7873_Contig1 | NP_001056304.1 | + + − | 1.801 ± 0.342 | 1.994 ± 0.242 | 0.517 ± 0.062 |
| Rubisco large subunit | Locus_1035_Contig1 | NP_039391.1 | + = + | 1.726 ± 0.069 | 1.404 ± 0.092 | 1.809 ± 0.095 |
| Ribosomal protein S4 | Locus_936_Contig5 | NP_039383.1 | + + = | 1.725 ± 0.334 | 1.673 ± 0.192 | 0.977 ± 0.066 |
| Photosystem I reaction center subunit X psaK | Locus_1830_Contig1 | NP_001058895.1 | + + = | 1.558 ± 0.120 | 1.894 ± 0.056 | 0.883 ± 0.000 |
| Photosystem II 22 kDa protein, chloroplastic | Locus_2131_Contig6 | NP_001044929.1 | = + = | 1.133 ± 0.103 | 1.802 ± 0.135 | 1.309 ± 0.159 |
| Oxygen-evolving enhancer protein 3–2, chloroplastic | Locus_734_Contig5 | NP_001059911.1 | = − = | 1.105 ± 0.042 | 0.562 ± 0.023 | 1.345 ± 0.059 |
| Magnesium-chelatase subunit chlI, chloroplastic | Locus_3670_Contig1 | NP_001050493.1 | = − − | 0.787 ± 0.039 | 0.655 ± 0.031 | 0.536 ± 0.040 |
| Magnesium-protoporphyrin IX monomethyl ester [oxidative] cyclase, chloroplastic | Locus_2737_Contig2 | NP_001042745.1 | = − − | 0.779 ± 0.030 | 0.467 ± 0.032 | 0.330 ± 0.004 |
| Magnesium-protoporphyrin O-methyltransferase | Locus_9910_Contig3 | NP_001056701.2 | = − − | 0.704 ± 0.013 | 0.508 ± 0.007 | 0.560 ± 0.002 |
| Pyrophosphate-fructose 6-phosphate 1-phosphotransferase subunit beta | Locus_6638_Contig2 | NP_001057284.1 | − − = | 0.658 ± 0.014 | 0.545 ± 0.001 | 0.712 ± 0.077 |
| Magnesium-chelatase subunit chlD, chloroplastic | Locus_4380_Contig1 | NP_001051668.1 | − − − | 0.555 ± 0.039 | 0.409 ± 0.008 | 0.591 ± 0.008 |
| Geranylgeranyl diphosphate reductase, chloroplastic | Locus_2055_Contig1 | NP_001048106.2 | − − − | 0.500 ± 0.056 | 0.426 ± 0.032 | 0.302 ± 0.007 |
| Protoporphyrinogen oxidase, chloroplastic | Locus_1112_Contig1 | NP_001042777.1 | − − − | 0.479 ± 0.022 | 0.456 ± 0.006 | 0.423 ± 0.035 |
| Protochlorophyllide reductase B, chloroplastic | Locus_962_Contig4 | NP_001054274.1 | − − − | 0.375 ± 0.063 | 0.263 ± 0.006 | 0.428 ± 0.006 |
| Coproporphyrinogen-III oxidase, chloroplastic | Locus_3893_Contig1 | NP_001053833.1 | − − = | 0.353 ± 0.014 | 0.398 ± 0.027 | 0.686 ± 0.063 |
| Photorespiration | ||||||
| Alanine aminotransferase 2 | Locus_1285_Contig2 | NP_001058716.1 | + + + | 1.906 ± 0.035 | 1.648 ± 0.013 | 1.738 ± 0.113 |
| Glycine dehydrogenase [decarboxylating] 2 | Locus_282_Contig2 | NP_001044046.1 | + + + | 1.848 ± 0.064 | 2.126 ± 0.027 | 1.646 ± 0.004 |
| Putative serine-glyoxylate aminotransferase | Locus_775_Contig6 | NP_001062170.1 | + + + | 1.696 ± 0.094 | 1.661 ± 0.085 | 2.099 ± 0.067 |
| Mitochondrial serine hydroxymethyltransferase | Locus_304_Contig7 | NP_001051211.1 | + + + | 1.531 ± 0.107 | 1.983 ± 0.141 | 1.555 ± 0.072 |
| Glycolysis | ||||||
| Glyceraldehyde-3-phosphate dehydrogenase B, chloroplastic | Locus_2438_Contig3 | NP_001048847.1 | + + + | 2.169 ± 0.046 | 2.126 ± 0.100 | 1.686 ± 0.227 |
| Fructose-bisphosphate aldolase, cytoplasmic isozyme 1 | Locus_7311_Contig1 | NP_001058042.1 | + = + | 2.164 ± 0.069 | 1.450 ± 0.069 | 1.944 ± 0.044 |
| Glyceraldehyde-3-phosphate dehydrogenase A, chloroplastic | Locus_1999_Contig4 | NP_001052984.1 | + + = | 1.782 ± 0.025 | 1.748 ± 0.038 | 1.187 ± 0.044 |
| Glyceraldehyde-3-phosphate dehydrogenase, cytosolic | Locus_512_Contig1 | NP_001053139.1 | = + + | 1.354 ± 0.125 | 1.585 ± 0.246 | 1.946 ± 0.171 |
| 6-phosphofructokinase 3 | Locus_8951_Contig1 | NP_001042273.1 | = = + | 1.166 ± 0.045 | 1.040 ± 0.016 | 1.946 ± 0.218 |
| Pyruvate kinase, cytosolic isozyme | Locus_1808_Contig4 | NP_001065749.1 | − − − | 0.619 ± 0.017 | 0.532 ± 0.026 | 0.658 ± 0.035 |
| Putative uncharacterized protein YCR013C | Locus_12650_Contig1 | NP_001058317.1 | − − = | 0.562 ± 0.039 | 0.603 ± 0.058 | 0.859 ± 0.079 |
| Enolase 1 | Locus_4262_Contig4 | NP_001064223.1 | − − = | 0.529 ± 0.009 | 0.607 ± 0.043 | 1.107 ± 0.067 |
| Pyruvate kinase isozyme A, chloroplastic | Locus_793_Contig2 | NP_001059042.1 | − − − | 0.428 ± 0.043 | 0.434 ± 0.006 | 0.598 ± 0.035 |
| Tricarboxylic acid cycle | ||||||
| Malate dehydrogenase | Locus_4351_Contig1 | NP_001067346.2 | + + = | 2.558 ± 0.032 | 1.947 ± 0.061 | 1.305 ± 0.100 |
| Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex 2, mitochondrial | Locus_6_Contig4 | NP_001052652.1 | + + = | 1.602 ± 0.036 | 1.714 ± 0.111 | 1.134 ± 0.144 |
| Phosphoenolpyruvate carboxylase | Locus_8734_Contig8 | NP_001061646.1 | = = + | 1.302 ± 0.266 | 1.124 ± 0.320 | 2.254 ± 0.449 |
| Isocitrate dehydrogenase | Locus_7158_Contig1 | NP_001056381.1 | − − − | 0.382 ± 0.031 | 0.510 ± 0.017 | 0.509 ± 0.050 |
| Carbohydrate metabolic process | ||||||
| Beta-galactosidase 15 | Locus_11681_Contig1 | NP_001066673.1 | = = + | 1.227 ± 0.183 | 1.259 ± 0.181 | 1.807 ± 0.330 |
| Glucan endo-1,3-beta-glucosidase 7 | Locus_5546_Contig2 | NP_001051111.1 | = + + | 1.181 ± 0.099 | 2.671 ± 0.387 | 1.834 ± 0.310 |
| Cell wall invertase | Locus_695_Contig1 | NP_001052747.1 | = = + | 1.149 ± 0.058 | 0.984 ± 0.031 | 1.822 ± 0.062 |
| Heparanase-like protein 2 | Locus_7612_Contig1 | NP_001067122.1 | − = = | 0.585 ± 0.062 | 0.720 ± 0.046 | 0.712 ± 0.066 |
| Alpha-glucan phosphorylase, H isozyme | Locus_206_Contig2 | NP_001044823.1 | − = = | 0.560 ± 0.098 | 1.219 ± 0.007 | 0.867 ± 0.032 |
| Probable beta-d-xylosidase 6 | Locus_20345_Contig1 | NP_001053392.1 | − − = | 0.460 ± 0.008 | 0.515 ± 0.026 | 0.828 ± 0.048 |
| Beta-galactosidase | Locus_1556_Contig2 | NP_001043405.1 | − − − | 0.454 ± 0.002 | 0.375 ± 0.001 | 0.472 ± 0.001 |
| Beta-hexosaminidase subunit B2 | Locus_1367_Contig4 | NP_001055557.1 | − − − | 0.387 ± 0.044 | 0.579 ± 0.017 | 0.635 ± 0.126 |
| 1,4-alpha-glucan-branching enzyme 2, chloroplastic/amyloplastic | Locus_23620_Contig1 | NP_001052723.2 | − − = | 0.276 ± 0.084 | 0.499 ± 0.028 | 0.972 ± 0.090 |
| Starch branching enzyme II | Locus_1381_Contig5 | NP_001047009.1 | − − = | 0.256 ± 0.050 | 0.468 ± 0.003 | 1.132 ± 0.053 |
| Beta-mannosidase 2 | Locus_792_Contig2 | NP_001043156.1 | − − − | 0.076 ± 0.005 | 0.196 ± 0.013 | 0.189 ± 0.002 |
| Fatty acid biosynthetic process | ||||||
| 3-oxoacyl-[acyl-carrier-protein] reductase, chloroplastic | Locus_21034_Contig1 | NP_001066481.1 | − = = | 0.558 ± 0.043 | 0.673 ± 0.034 | 0.736 ± 0.072 |
| Triosephosphate isomerase, cytosolic | Locus_13069_Contig3 | NP_001042016.1 | − − = | 0.453 ± 0.146 | 0.597 ± 0.054 | 0.708 ± 0.044 |
| Acyl-ACP thioesterase | Locus_1123_Contig1 | NP_001063601.1 | − − − | 0.357 ± 0.054 | 0.437 ± 0.010 | 0.585 ± 0.044 |
| Acyl-[acyl-carrier-protein] desaturase, chloroplastic | Locus_2497_Contig2 | NP_001045215.1 | − − − | 0.324 ± 0.061 | 0.340 ± 0.022 | 0.402 ± 0.012 |
| Acyl carrier protein 1, chloroplastic | Locus_6396_Contig1 | NP_001066930.1 | − − − | 0.232 ± 0.034 | 0.376 ± 0.011 | 0.420 ± 0.001 |
| Fatty acid beta-oxidation | ||||||
| 3-ketoacyl-CoA thiolase 2, peroxisomal precursor | Locus_4508_Contig3 | NP_001048523.1 | + + = | 1.915 ± 0.054 | 1.922 ± 0.057 | 1.364 ± 0.045 |
| Acyl-CoA oxidase homolog | Locus_7611_Contig1 | NP_001057591.1 | + = + | 1.858 ± 1.016 | 1.300 ± 0.336 | 2.073 ± 0.517 |
a +: up-regulated; −: down-regulated; = : not significantly changed.
b C6h: the ratios for 115-tag (cold treated at 6h sample)/114-tag(cold treated at 0h sample); C24h: the ratios for 116-tag (cold treated at 24h sample)/114-tag(cold treated at 0h sample); R24h: the ratio for 117-tag (recovered for 24 h sample)/114-tag(cold treated at 0h sample).
Confirmation of Cold Induced Candidate Proteins by Western Blot and Enzyme Activity Assay
To validate the identifications of the cold-responsive proteins and to compare the cold tolerance mechanisms of plantain and banana, we used immunoblot analysis (Fig. 7A) to assess the expression of seven proteins whose abundance changed in response to cold stress and recovery as determined by iTRAQ. Three of the seven proteins are involved in oxidation reduction and oxylipin biosynthesis, including [Cu-Zn] SOD, catalase (CAT), and lipoxygenase (LOX), whereas the other four targeted proteins are involved in photorespiration and carbohydrate metabolism: glutamate glyoxylate aminotransferase (GGAT), glyoxylate aminotransferase (SGAT), serine hydroxymethyltransferase (SHMT), and β-hexosaminidase (β-Hex).
Fig. 7.

Comparison and validation of seven selected, cold responsive candidate proteins by iTRAQ profiling, Western blot analysis and enzyme activity assays for plantain and banana. Western blot analysis showed temporal changes of Cu/Zn SOD, CAT, GGAT, SGAT, SHMT, LOX, and β-Hex in the cold-sensitive banana and cold-tolerant plantain in response to cold stress (at C0h, C6h, and C24h) and after a 24 h recovery (R24h) (A). iTRAQ analysis showed temporal and quantitative changes of GGAT, SGAT, SHMT, and β-Hex (B); and Cu/Zn SOD, CAT, and LOX (C) in plantain in response to 6 h and 24 h cold stress, and after 24 h recovery. Enzyme activity assays revealed the temporal changes of SOD, CAT, and LOX activities for both cold-sensitive banana and cold-tolerant plantain under 6 h, 24 h cold stress and 24 h recovery (D). The values of relative activity for each column indicate means ±S.D. of three biological replicates with each having five technical replicate measurements. The different lowercase letters labeled above columns indicate a significant difference at p ≤ 0.05 between the columns by Duncan's test using SPSS statistical software (version 16.0, SPSS Inc. Chicago, IL). The columns with the same letters mean no significant difference (p > 0.05) between each other.
As shown in Fig. 7A, Cu/Zn SOD (Locus_2385_Contig4) in both plantain and banana were induced by cold stress and remained up-regulated in 24 h recovery, in good agreement with the plantain data from iTRAQ analysis (Fig. 7B). Two isoforms of CAT (catalase isozyme A, Locus_1788_Contig4; and catalase isozyme 2, Locus_1320_Contig9) were detected in both plantain and banana. In plantain, catalase isozyme 2 was induced and remained at a higher level than the control, whereas catalase isozyme A levels decreased in response to cold treatments, but returned to normal after recovery. However, levels of both catalase isozymes in banana were lower in C6h and C24h and significantly elevated at R24h, revealing a major difference in the response to the cold stress for catalase isozyme 2 between plantain and banana. For LOX, two different isoform bands (corresponding to Locus_7458_Contig3, Locus_119_Contig1, Locus_440_Contig2) were also detected in both plantain and banana. In plantain, consistent with the iTRAQ data (Fig. 7B) both isoforms displayed a continuous decrease during the cold stress time course and returned to almost normal levels during the 24 h recovery phase. A substantially different LOX profile in response to cold stress was observed in banana as both LOX isoforms showed a small increase at C6h, followed by a significant increase at C24h and a return to the normal levels at R24h. Notable differences between plantain and banana in SGAT (Locus_775_Contig6) and GGAT (a highly homologous enzyme with alanine aminotransferase, Locus_1285_Contig2) expression in response to the cold treatments were also detected. The abundance of both proteins was up-regulated in plantain over the three different time points, but consistently decreased during the cold stress and recovery in banana. In addition, the immunoblot analysis showed that SHMT (Locus_304_Contig7) protein levels were up-regulated in plantain following cold stress and partially returned to normal during the 24 h recovery, but no substantial change was induced by the cold treatment in banana. β-Hex (Locus_1367_Contig4) also showed a diametrically opposite response to cold stress in plantain and banana: levels steadily decreased in plantain at C6h and C24h, but slowly increased in banana over the time period of the cold stress and recovery point. We note that in general there was a good correlation between the Western blot analysis iTRAQ-based data sets (Fig. 7C).
Enzyme activity assays were performed to further validate these results. Because the iTRAQ and Western data indicated that oxidation-reduction is one of the most responsive pathways to the cold stress, assays were performed of SOD, CAT, and LOX in both plantain and banana that had been subjected to the cold stress and recovery. As shown in Fig. 7D, total SOD activities increased by 36% in plantain and decreased by 27% in banana at C24h, and returned to normal levels at R24h for both species. At C6h, C24h, and R24h, the total CAT activities increased by 12%, 31%, and 14%, respectively, in plantain, and decreased by 9%, 2%, and 26% respectively, in banana. Under the same conditions, the activities of LOX decreased by 29%, 49%, and 11% in plantain, but increased by 40%, 110%, and 80% in banana, respectively, this showing a strong correlation with the immunoblot and iTRAQ data.
DISCUSSION
Research Challenges for Plantain and Banana as Nonmodel Organisms
Banana and plantain are grown in more than one hundred tropical and subtropical countries and collectively are the fourth most consumed staple food worldwide (17). However, despite their major societal and economic importance, the lack of fully sequenced genome for banana and plantains has limited the application of the high-throughput “omics” technologies. Moreover, there are currently only ∼11,000 plantain ESTs and a few hundreds of full length gene coding sequences from plantain and banana, respectively, in public databases. This represents a major bottleneck for understanding molecular mechanisms underlying stress responses, such as cold tolerance genes, and so strategies to date have been based on low throughput case-by-case approaches (27, 28). However, next generation sequencing technologies have provided a new paradigm for genome and transcriptome (RNA-Seq) analysis of nonmodel organisms with undetermined genome sequences, opening the door for “omics” studies (50–52). As a prelude to this current study, we generated 54 million sequence reads (with a mean length 106 bp), corresponding to 5.8 Gb of raw sequence data, by Illumina sequencing of mRNA pooled from different plantain tissues. These were assembled into 57,845 contigs, of which 43,313 were annotated based on sequence homology to functionally classified proteins in public databases. Given a recent report showing that RNA-Seq data sets and a highly curated DNA database can provide comparable platforms for proteome studies (34), we hypothesized that the plantain RNA-Seq database would allow the definition of gene expression units and consequently enable protein identification, and this indeed proved to be the case.
Statistical Analysis and Reproducibility of Plantain Proteome to the Cold Stress
One of the challenges for global quantitative proteomics analysis using existing high throughput shotgun technologies is the potential for low data reproducibility resulting from biological and technical variation. We therefore performed a careful statistical analysis of the data to ensure reproducibility and reliability. Two biological sets of data generated a combined 3477/2682 of distinct/common proteins from 3092 and 3067 protein IDs in set 1 and set 2, respectively (Table I). As a result, the reproducibility of protein IDs defined as the percentage of identified proteins identified in common over the total protein IDs from each replicate set, was 87% for each of the two sets. In addition, the reproducibility of the protein IDs with quantification ratios was ∼88% for each of the two sets. More importantly, we performed analyses of variance to determine the quantitative precision and reproducibility for two levels of quantitative data sets. In the first phase of analysis, all proteins quantified in both set 1 and set 2 in response to each of the three treatment conditions were compared against each other, as we reported recently (36), to assess the quantitative variations from both biological replicates and to obtain internal errors in establishing a cutoff for cold responsive changes. As shown in Fig. 3, all three plots gave a slope ranged from 0.86 to 0.98 at an R2 ranged from 0.85 to 0.88, indicating the quantitative iTRAQ data from replicates is statistically reproducible. The internal errors for the three plots showed the cutoff of 1.50-fold, 1.36-fold, and 1.42-fold internal variation for C6h, C24h, and R24h respectively, representing a combined technical and biological deviation. These results suggest a slightly higher variation found for the C6h sample than the rest two conditions, which may suggest more variable changes in response to the early cold stress. In addition, the distribution fitting with goodness of fit tests for all proteins quantified for each of the individual treatments were found to be normally distributed, thereby confirming that the relative expression data is statistically reproducible in the two biological replicates (Supplemental Figs. S1–S4 and Supplemental Table S3). In the second phase of analysis, we focused on the subsets of quantitative data for only differential proteins (both up-regulated and down-regulated proteins) commonly quantified in the biological duplicates at each cold treatment condition (Fig. 4). The RSDs for up-regulated proteins at three time points between two sets were ranged from 20% to 26%. The RSD for down-regulated proteins were ranged from 21% to 35%, again consistent with the finding in the first phase of analysis, the largest RSDs found for up-regulated and down-regulated proteins were from the C6h samples (Fig. 4), indicating the quantitative proteomics data in this study are reproducible.
Identities and Functional Characterization of Candidate Proteins
Previous proteomic analyses of cold susceptible crops such as rice, or freezing tolerant species, such as A. thaliana and the woody species P. obovata and P. persica (53) have revealed a complex set of cold responses. In this study, 2658 proteins were confidently quantified in cold stressed plantain, of which 983 were differentially expressed (>1.5-fold changes against C0h sample) at a 95% confidence interval. A BLAST analysis of these 983 proteins versus the rice genome sequence removed some redundancy, resulting in a final list of 809 unique differentially expressed proteins (Supplemental Table S2). Based on the annotations, the predominant functional groups were oxidation-reduction process, cellular metabolic process, response to stress and primary metabolic process, for both up and down-regulated proteins. Supplemental Fig. S5 shows functional classifications of all 3477 proteins identified in this study, with the top four categories of the identified proteins being metabolic process (44.06%), cellular process (36.2%), localization (5.32%) and response to stimuli (4.33%). This information should provide a valuable platform for future plantain proteomics studies.
Biological Implications of Plantain Proteome Dynamics Following Cold Stress and Recovery
Table II shows the 116 differentially expressed proteins that provided the focus of this study, and which are classified into eight molecular function categories: oxidation reduction (including oxylipin biosynthetic process), photosynthesis, photorespiration, glycolysis, tricarboxylic acid cycle, carbohydrate metabolic process, fatty acid biosynthetic process, and fatty acid beta-oxidation. The biological significance of key differentially proteins and their associated metabolic pathways is discussed below.
Proteins Involved in Photosynthesis
Photosynthesis converts light energy into ATP and redox equivalents (NADPH). For optimal photosynthesis, the balance between light absorbed by the primary photochemical reactions and its use must be fine-tuned in response to fluctuations in the environment, a process termed photostasis (54). Photosynthesis is greatly inhibited by low temperature in various crops (44), but the mechanism is still not fully understood. Our proteomic analysis showed that cold stress affects different aspects of photosynthesis in plantain. The net photosynthetic rate and stomatal conductance were dramatically reduced under cold stress (Fig. 2), but our data showed that many photosynthesis related proteins, including Rubisco large and small subunits, Photosystem I reaction center subunit III, VI, X, and Photosystem II 22 kDa protein are up-regulated. We interpret this to reflect an adaptive response to maintain chloroplast function under cold stress. An increase in the abundance of Photosystem II and I proteins in response to salt stress was also previously reported, and it was proposed that this may protect the reaction center from stromal proteases and may modulate electron transfer efficiency (55). Many other important photosynthesis proteins, including magnesium-chelatase subunit chlI and subunit chlD, magnesium-protoporphyrin IX monomethyl ester cyclase, magnesium-protoporphyrin, O-methyltransferas, protoporphyrinogen oxidase, protochlorophyllide reductase B, and coproporphyrinogenIII oxidase, were found to be substantially down-regulated by cold stress (Table II). Given that the photosynthetic components are functionally linked, damage of any one component may lead to an overall reduction in photosynthetic activity (8), in which appears to be the case for plantain.
Proteins Involved in Photorespiration
Photorespiration is a process whereby ribulose-1,5-bisphosphate (RuBP) uses oxygen rather than carbon dioxide during photosynthesis. This represents the first step of the Calvin-Benson cycle and reduces photosynthetic efficiency in C3 plants. It is widely accepted that this pathway influences a wide range of processes from bioenergetics, photosystem II function, carbon metabolism to nitrogen assimilation, and respiration. During the course of photorespiration, two aminotransferases are responsible for the conversion of glyoxylate to glycine (Gly) in the peroxisomes: SGAT (EC 2.6.1.45), catalyzing the conversion of Ser plus glyoxylate to hydroxypyruvate plus Gly; and GGAT (EC 2.6.1.4), catalyzing the synthesis of α-ketoglutarate plus Gly, or pyruvate plus Gly. The Gly traffics to the mitochondria, where it is partially decomposed to CO2 and NH3 by glycine decarboxylase (GDC) and partially converted to Ser by SHMT (56). Photorespiratory metabolism in the chloroplast and peroxisome is known to reduce the production of H2O2 (57). Mutations in individual SGAT, GGAT, or SHMT genes increases H2O2 levels and results in plants that are more susceptible to pathogen infection and abiotic stress than wild-type, and the resulting constitutive over accumulation of reactive oxygen species (ROS) impairs defense responses (58). Our proteome data show the up-regulation of GDC, SGAT, GGAT, and SHMT in response to cold stress (Table II and Fig. 7C), providing the evidence that cold stress in plantain can stimulate the photorespiration pathway and potentially reduce the production of H2O2, thereby suppressing the subsequent oxidation of membrane lipids, proteins or nucleotides and enhancing cold-resistance. In contrast, the expression of GGAT, SGAT, and SHMT were significantly suppressed or showed not altered (for SHMT) in banana under cold stress conditions (Fig. 7A), which we infer would lead to oxidative stress as described below. This result explains the previous observation that higher levels of accumulation of H2O2 and superoxide radicals were found in banana than plantain after cold stress (19).
Proteins Involved in ROS Scavenging System
Along with other abiotic stress, cold stress in plants induces the accumulation of ROS, including superoxide radicals, H2O2 and hydroxyl radicals (59, 60), which can perturb cellular redox homeostasis and result in oxidative damage, which contributes to stress injury. To counterbalance the ROS production, plants subjected to low temperatures induce and activate scavenging systems. For example, catalases and superoxide dismutases are induced by cold stress in rice and A. thaliana (59, 60). SOD acts as a first line of defense against ROS, dismutating superoxide to H2O2 and hence decreasing the risk of hydroxyl radical formation from superoxide via the metal-catalyzed Haber-Weiss-type reaction (61). The differential expression of SOD and CAT in response to cold stress in plantain was also apparent in this study (Figs. 7B, 7D, Table II). Cu/Zn SOD was up-regulated at all 3 time points, whereas Mn SOD was down-regulated at C6h, and recovered at C24h and R24h (Table II). Western blot analysis confirmed the Cu/Zn SOD change in proteomic data. Despite the fact that up-regulated Cu/Zn SOD in response to the old stress was found in both banana and plantain (Fig. 7A), the total activities of SOD (including Cu/Zn SOD, Fe SOD, and Mn SOD activities) increased by 36% in plantain, whereas decreased by 27% in cold-sensitive banana at C24h treatment (Fig. 7D), suggesting that plantain may more effectively reduce the production of H2O2 than banana. Furthermore, the results indicate that Cu/Zn SOD is a major SOD isoform responsible for the total SOD activities in plantain, whereas Mn SOD and/or Fe SOD appear to play more important roles in the total SOD activities for banana under cold stress. CAT catalyzes the decomposition of H2O2 to water and oxygen in the CAT pathway, which is mainly localized in peroxisomes. The abundance of two CAT isoforms was affected by the cold treatment in plantain: catalase isozyme 2 was up-regulated compared with the control, whereas levels of Catalase isozyme A decreased. Because the total plant CAT activity increased by 31% following cold treatment, CAT isozyme 2 is likely to play a dominant role in response to cold stress. In contrast, both Western blot analysis and activity assays showed a reduction in CAT in banana, suggesting that plantain can more effectively reduce H2O2 levels in response to cold shock. Recently, it was reported that banana has higher levels of malondialdehyde (MDA), H2O2 and superoxide radicals (O2●−), whereas higher activities of ascorbate peroxidase (APX), peroxidase (POD), SOD, and 2,2-Diphenyl-1-picrylhydrazyl (DPPH●) scavenging capability are present in plantain (19). Similar results were reported for cold-tolerant versus cold-sensitive rice (Oryza sativa L.) (59). Moreover, the application of certain chemicals (e.g. low concentration H2O2, Ca2+, salicylic acid, brassinolide, or methyl jasmonate) can induce the activities of antioxidant enzymes and the resulting higher ROS scavenging ability has been reported to result in increased cold tolerance in banana seedlings (23–26).
Taking together, the iTRAQ data, Western blot analyses and enzyme activity results lead us to conclude that more effective ROS scavenging through the CAT pathway may at least in part explain the greater tolerance of plantain to cold stress than banana. It should be noted that several key proteins, including ascorbate peroxidase, glutathione S-transferase DHAR2 and several types of peroxidase were found to be significantly down-regulated in plantain response to cold stress. This suggests that the glutathione-ascorbate cycle may not play a major role as an antioxidant protection system in plantain response to cold stress.
Proteins Involved in Lipid Peroxidation and Cell Wall
The ROS produced during cold exposure can also cause lipid peroxidation, resulting in ion leakage. The level of unsaturation of lipids is particularly critical in the response to cold stress, because unsaturated lipids yield to a more fluid membrane, prevent the formation of expansion-induced lysis and therefore, are very important for chloroplast maintenance. Formation of oxidized polyenoic fatty acids (PUFAs), called oxylipins, is one of the main reactions in lipid alteration (62). The metabolism of PUFAs via the LOX-catalyzed step and the subsequent reactions are collectively named the LOX pathway. All four proteins (Allene oxide synthase and three Lipoxygenase iosforms) involved in LOX pathway were down-regulated in response to cold stress in plantain and enabled to return to normal at R24h point (Fig. 7B, Table II). Furthermore, both immune-blot and activity assays on LOX expression confirmed our proteomics data. On contrary, the LOX expression and activity were up-regulated in cold-sensitive banana under the same cold stress (Figs. 7A, 7B, 7D), which can facilitate the formation of saturated lipids. These results suggest the impact of cold stress on lipid peroxidation in cold-sensitive banana, which may ultimately affect cell membrane permeability. This finding may also at least in part to explain the previous observations that the low-temperature treatment was found for significantly increased MDA content and enhanced cell membrane permeability rates in cold-sensitive banana, but not in cold-tolerant plantain (19). MDA is a harmful lipid peroxidation product, which indicated that the plasma membranes in banana suffered more damages than in plantain under cold stress (19).
Our proteomics data in plantain showed that many cell wall-degrading enzymes (63) including heparanase-like protein 2, probable beta-d-xylosidase 6, beta-galactosidase, beta-hexosaminidase (β-Hex) subunit B2, beta-mannosidase 2, were down-regulated in response to cold stress. Western blot has confirmed our proteomics discovery for one of the above enzyme: β-Hex in plantain. In contrast, β-Hex was found with steadily up-regulated through C6h, C24h, and R24h treatment in banana by Western blot analysis (Table II and Fig. 7A). This result suggests that cold stress might affect biochemical metabolisms in cell wall.
Other Cold Responsive Proteins
It was reported that one of the common responses in plants to cold stress is the accumulation of hydrophilic proteins (64). Some of these proteins have been named as COR (cold responsive), RAB (responsive to abscisic acid), etc. We have identified a COR protein (COR47) in plantain, which was up-regulated by 3.5-fold, 1.8-fold, and 5.6-fold at C6h and C24h cold stress and R24h recovery, respectively (Supplemental Table S2). RAB18 and COR47 (two dehydrins) were ectopically expressed as pairs in transgenic Arabidopsis, this resulted in plants with reduced ion leakage during freezing test when compared with the wild-type plants (65), suggesting the COR47 in plantain plays an important role in functioning as self-protection to cope with cold stress.
In conclusion, our results show that molecular mechanisms for the higher cold resistance found in plantain can be associated with increased antioxidation with adapted ROS scavenging capability, reduced ROS production and decreased lipid peroxidation. These results illustrate the value of new sequencing technologies in enabling research in traditionally nonmodel crops, and suggest strategies that may form foundation for developing cold-tolerant banana varieties through a better understanding of antioxidation and cellular redox systems. Our proteomic data also support the general observation that impacts posed by cold stress to plant cells appear systematic and on variable aspects, and plant responses to cold stress may include physical (e.g. membrane rigidification), chemical (e.g. ROS production) and biochemical (e.g. metabolic disequilibrium) adaptations.
Supplementary Material
Acknowledgments
We thank Mr. Robert Sherwood for technical support and Professors Jian Hua, Li Li, and Dr. Ted Thannhauser for critical reading of this manuscript and helpful discussion.
Footnotes
* This work was supported by National High-tech R&D Program (863 Program) No. 2011AA10020602, National Natural Science Foundation of China No. U1131004, Guangdong Science and Technology plan project No. 2011A020201006, and Dean funding project of Guangdong Academy of Agricultural Sciences No. 201113.
 This article contains supplemental Tables S1 to S3 and Figs S1 to S5.
This article contains supplemental Tables S1 to S3 and Figs S1 to S5.
1 The abbreviations used are:
- iTRAQ
- Isobaric Tags for Relative and Absolute Quantitation
- ROS
- reactive oxygen species
- FA
- formic acid
- SCX
- strong cation exchange
- SPE
- solid phase extraction
- TCEP
- Tris-(2-carboxyethyl) phosphine
- MMTS
- methyl methanethiosulfonate
- hpRP
- high pH reverse phase
- DDA
- data-dependent acquisition
- HCD
- high energy collision dissociation
- FDR
- false discovery rate
- SOD
- superoxide dismutase
- CAT
- catalase
- LOX
- lipoxygenase
- GGAT
- glutamate glyoxylate aminotransferase
- SGAT
- serine glyoxylate aminotransferase
- SHMT
- serine hydroxymethyltransferase
- β-Hex
- β-hexosaminidase
- PVPP
- polyvinyl polypyrrolidone
- RSD
- relative standard deviation
- GO
- Gene Ontology
- GDC
- glycine decarboxylase
- MDA
- malondialdehyde
- PUFAs
- polyenoic fatty acids
- COR
- cold responsive.
REFERENCES
- 1. Zhou M. Q., Shen C., Wu L. H., Tang K. X., Lin J. (2011) CBF-dependent signaling pathway: a key responder to low temperature stress in plants. Crit. Rev. Biotechnol. 31, 186–192 [DOI] [PubMed] [Google Scholar]
- 2. Sanghera G. S., Wani S. H., Hussain W., Singh N. B. (2011) Engineering cold stress tolerance in crop plants. Curr. Genomics 12, 30–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chinnusamy V., Zhu J., Zhu J. K. (2007) Cold stress regulation of gene expression in plants. Trends Plant Sci. 12, 444–451 [DOI] [PubMed] [Google Scholar]
- 4. Chen W., Provart N. J., Glazebrook J., Katagiri F., Chang H.-S., Eulgem T., Mauch F., Luan S., Zou G., Whitham S. A., Budworth P. R., Tao Y., Xie Z., Chen X., Lam S., Kreps J. A., Harper J. F., Si-Ammour A., Mauch-Mani B., Heinlein M., Kobayashi K., Hohn T., Dangl J. L., Wang X., Zhu T. (2002) Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell Online 14, 559–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fowler S., Thomashow M. F. (2002) Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell 14, 1675–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Greenup A. G., Sasani S., Oliver S. N., Walford S. A., Millar A. A., Trevaskis B. (2011) Transcriptome analysis of the vernalization response in barley (Hordeum vulgare) seedlings. PLoS One 6, e17900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winfield M. O., Lu C., Wilson I. D., Coghill J. A., Edwards K. J. (2010) Plant responses to cold: Transcriptome analysis of wheat. Plant Biotechnol. J. 8, 749–771 [DOI] [PubMed] [Google Scholar]
- 8. Yan S. P., Zhang Q. Y., Tang Z. C., Su W. A., Sun W. N. (2006) Comparative proteomic analysis provides new insights into chilling stress responses in rice. Mol. Cell. Proteomics 5, 484–496 [DOI] [PubMed] [Google Scholar]
- 9. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 10. Pradet-Balade B., Boulmé F., Beug H., Müllner E. W., Garcia-Sanz J. A. (2001) Translation control: bridging the gap between genomics and proteomics? Trends Biochem. Sci 26, 225–229 [DOI] [PubMed] [Google Scholar]
- 11. Bartel D. P. (2009) MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo H., Ingolia N. T., Weissman J. S., Bartel D. P. (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466, 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu H. H., Tian X., Li Y. J., Wu C. A., Zheng C. C. (2008) Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14, 836–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lv D. K., Bai X., Li Y., Ding X. D., Ge Y., Cai H., Ji W., Wu N., Zhu Y. M. (2010) Profiling of cold-stress-responsive miRNAs in rice by microarrays. Gene 459, 39–47 [DOI] [PubMed] [Google Scholar]
- 15. Kosová K., Vitámvas P., Prášil I. T., Renaut J. (2011) Plant proteome changes under abiotic stress–contribution of proteomics studies to understanding plant stress response. J. Proteomics 74, 1301–1322 [DOI] [PubMed] [Google Scholar]
- 16. Simmonds N. W., Weatherup S. T. C. (1990) Numerical taxonomy of the wild bananas (Musa). New Phytologist 115, 567–571 [DOI] [PubMed] [Google Scholar]
- 17. Davey M. W., Graham N. S., Vanholme B., Swennen R., May S. T., Keulemans J. (2009) Heterologous oligonucleotide microarrays for transcriptomics in a non-model species; a proof-of-concept study of drought stress in Musa. BMC Genomics 10, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carpentier S. C., Vertommen A., Swennen R., Witters E., Fortes C., Souza M. T., Jr., Panis B. (2010) Sugar-mediated acclimation: the importance of sucrose metabolism in meristems. J. Proteome Res. 9, 5038–5046 [DOI] [PubMed] [Google Scholar]
- 19. Zhang Q., Zhang J., Chow W., Sun L., Chen J., Chen Y., Peng C. (2011) The influence of low temperature on photosynthesis and antioxidant enzymes in sensitive banana and tolerant plantain (Musa sp.) cultivars. Photosynthetica 49, 201–208 [Google Scholar]
- 20. Lü Q. F., Feng F., Zhang X. Z. (2000) Cold tolerance of banana in relation to cell structure in leaf tissue. J. Zhanjiang Ocean Univ. 20, 48–51 (In Chinese) [Google Scholar]
- 21. Ganry J. (1973) Étude du développement du système foliaire du bananier en fonction de la température. Fruits 28, 499–516 [Google Scholar]
- 22. Israeli Y., Lahav E. (2000) Injuries to banana caused by adverse climate and weather, CAB International, UK [Google Scholar]
- 23. Kang G. Z., Tao J., Sun G. C., Wang Z. X. (2002) Physiological effects of H2O2 and Ca2+ on cold-stressed banana seedlings. Acta Hort. Sin. 29, 119–122 (In Chinese) [Google Scholar]
- 24. Kang G. Z., Wang C. H., Sun G. C., Wang Z. X. (2003) Salicylic acid changes activities of H2O2-metabolizing enzymes and increases the chilling tolerance of banana seedlings. Environ. Exp. Bot. 50, 9–15 [Google Scholar]
- 25. Liu D. B., Wei J. Y., Li S. P., Cui B. M., Peng M. (2008) Effects of brassinolid on chilling-resistance in banana seedlings. Bull. Bot. Res. 28, 195–198 (In Chinese) [Google Scholar]
- 26. Feng D., Xuan W. Y., Huang Z. S., Deng F. Y., Tan X. (2009) Effects of MeJA on cold tolerance of banana seedlings. J. Fruit Sci. 26, 390–393 (In Chinese) [Google Scholar]
- 27. Santos E., Remy S., Thiry E., Windelinckx S., Swennen R., Sági L. (2009) Characterization and isolation of a T-DNA tagged banana promoter active during in vitro culture and low temperature stress. BMC Plant Biol. 9, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feng D. R., Liu B., Li W. Y., He Y. M., Qi K. B., Wang H. B., Wang J. F. (2009) Over-expression of a cold-induced plasma membrane protein gene (MpRCI) from plantain enhances low temperature-resistance in transgenic tobacco. Environ. Exp. Bot. 65, 395–402 [Google Scholar]
- 29. Schulze W. X., Usadel B. (2010) Quantitation in mass-spectrometry-based proteomics. Annu. Rev. Plant Biol. 61, 491–516 [DOI] [PubMed] [Google Scholar]
- 30. Oeljeklaus S., Meyer H. E., Warscheid B. (2009) Advancements in plant proteomics using quantitative mass spectrometry. J. Proteomics 72, 545–554 [DOI] [PubMed] [Google Scholar]
- 31. Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P. (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2, 43–50 [DOI] [PubMed] [Google Scholar]
- 32. Washburn M. P., Wolters D., Yates J. R., 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 33. Ross P. L., Huang Y. L. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 34. Lopez-Casado G., Covey P. A., Bedinger P. A., Mueller L. A., Thannhauser T. W., Zhang S., Fei Z., Giovannoni J. J., Rose J. K. (2012) Enabling proteomic studies with RNA-Seq: The proteome of tomato pollen as a test case. Proteomics 12, 761–774 [DOI] [PubMed] [Google Scholar]
- 35. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 36. Yang Y., Qiang X., Owsiany K., Zhang S., Thannhauser T. W., Li L. (2011) Evaluation of different multidimensional LC-MS/MS pipelines for isobaric tags for relative and absolute quantitation (iTRAQ)-based proteomic analysis of potato tubers in response to cold storage. J. Proteome Res. 10, 4647–4660 [DOI] [PubMed] [Google Scholar]
- 37. Wang Y., Yang F., Gritsenko M. A., Wang Y., Clauss T., Liu T., Shen Y., Monroe M. E., Lopez-Ferrer D., Reno T., Moore R. J., Klemke R. L., Camp D. G., 2nd, Smith R. D. (2011) Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elias J. E., Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 39. Conesa A., Götz S. (2008) Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Götz S., Garcia-Gomez J. M., Terol J., Williams T. D., Nagaraj S. H., Nueda M. J., Robles M., Talón M., Dopazo J., Conesa A. (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Giannopolitis C. N., Ries S. K. (1977) Superoxide dismutases: I. Occurrence in higher plants. Plant Physiol. 59, 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aebi H. (1984) Catalase in vitro. Methods Enzymol. 105, 121–126 [DOI] [PubMed] [Google Scholar]
- 43. Lara I., Mio R. M., Fuentes T., Sayez G., Graell J., Lopez M. L. (2003) Biosynthesis of volatile aroma compounds in pear fruit stored under long-term controlled-atmosphere conditions. Postharvest Biol. Tec. 29, 29–39 [Google Scholar]
- 44. Allen D. J., Ort D. R. (2001) Impacts of chilling temperatures on photosynthesis in warm-climate plants. Trends Plant Sci. 6, 36–42 [DOI] [PubMed] [Google Scholar]
- 45. Gan C. S., Chong P. K., Pham T. K., Wright P. C. (2007) Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ). J. Proteome Res. 6, 821–827 [DOI] [PubMed] [Google Scholar]
- 46. Redding A. M., Mukhopadhyay A., Joyner D. C., Hazen T. C., Keasling J. D. (2006) Study of nitrate stress in Desulfovibrio vulgaris Hildenborough using iTRAQ proteomics. Brief Funct. Genomic Proteomic 5, 133–143 [DOI] [PubMed] [Google Scholar]
- 47. Castagliola P. (1998) Approximation of the normal sample median distribution using symmetrical johnson SU distributions: application to quality control. Commun. Statistics 27, 289–301 [Google Scholar]
- 48. Mitra S. K., Walters B. T., Clouse S. D., Goshe M. B. (2009) An efficient organic solvent based extraction method for the proteomic analysis of Arabidopsis plasma membranes. J. Proteome Res. 8, 2752–2767 [DOI] [PubMed] [Google Scholar]
- 49. Chong P. K., Gan C. S., Pham T. K., Wright P. C. (2006) Isobaric tags for relative and absolute quantitation (iTRAQ) reproducibility: Implication of multiple injections. J. Proteome Res. 5, 1232–1240 [DOI] [PubMed] [Google Scholar]
- 50. Wall P. K., Leebens-Mack J., Chanderbali A. S., Barakat A., Wolcott E., Liang H., Landherr L., Tomsho L. P., Hu Y., Carlson J. E., Ma H., Schuster S. C., Soltis D. E., Soltis P. S., Altman N., dePamphilis C. W. (2009) Comparison of next generation sequencing technologies for transcriptome characterization. BMC Genomics 10, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Metzker M. L. (2010) Sequencing technologies - the next generation. Nat. Rev. Genet. 11, 31–46 [DOI] [PubMed] [Google Scholar]
- 52. Surget-Groba Y., Montoya-Burgos J. I. (2010) Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 20, 1432–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Balbuena T. S., Salas J. J., Martinez-Force E., Garcés R., Thelen J. J. (2011) Proteome analysis of cold acclimation in sunflower. J. Proteome Res. 10, 2330–2346 [DOI] [PubMed] [Google Scholar]
- 54. Ensminger I., Busch F., Huner N. P. A. (2006) Photostasis and cold acclimation: sensing low temperature through photosynthesis. Physiologia Plantarum 126, 28–44 [Google Scholar]
- 55. Zhang H., Han B., Wang T., Chen S., Li H., Zhang Y., Dai S. (2012) Mechanisms of plant salt response: insights from proteomics. J. Proteome Res. 11, 49–67 [DOI] [PubMed] [Google Scholar]
- 56. Taler D., Galperin M., Benjamin I., Cohen Y., Kenigsbuch D. (2004) Plant eR genes that encode photorespiratory enzymes confer resistance against disease. Plant Cell Online 16, 172–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Foyer C. H., Noctor G. (2005) Redox homeostasis and antioxidant signaling: a metabolic interface between stress perception and physiological responses. Plant Cell Online 17, 1866–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Verslues P., Kim Y. S., Zhu J. K. (2007) Altered ABA, proline and hydrogen peroxide in an Arabidopsis glutamate:glyoxylate aminotransferase mutant. Plant Mol. Biol. 64, 205–217 [DOI] [PubMed] [Google Scholar]
- 59. Guo Z., Ou W., Lu S., Zhong Q. (2006) Differential responses of antioxidative system to chilling and drought in four rice cultivars differing in sensitivity. Plant Physiol. Biochem. 44, 828–836 [DOI] [PubMed] [Google Scholar]
- 60. Goulas E., Schubert M., Kieselbach T., Kleczkowski L. A., Gardeström P., Schröder W., Hurry V. (2006) The chloroplast lumen and stromal proteomes of Arabidopsis thaliana show differential sensitivity to short- and long-term exposure to low temperature. Plant J. 47, 720–734 [DOI] [PubMed] [Google Scholar]
- 61. Apel K., Hirt H. (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 55, 373–399 [DOI] [PubMed] [Google Scholar]
- 62. Feussner I., Wasternack C. (2002) The lipoxygenase pathway. Annu. Rev. Plant Biol. 53, 275–297 [DOI] [PubMed] [Google Scholar]
- 63. Meli V. S., Ghosh S., Prabha T. N., Chakraborty N., Chakraborty S., Datta A. (2010) Enhancement of fruit shelf life by suppressing N-glycan processing enzymes. Proc. Natl. Acad. Sci. U.S.A. 107, 2413–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ruelland E., Vaultier M.-N., Zachowski A., Hurry V., Jean-Claude K., Michel D. (2009) Chapter 2, Cold Signalling and Cold Acclimation in Plants. Adv. Bot. Res. 35–150 [Google Scholar]
- 65. Puhakainen T., Hess M. W., Mäkelä P., Svensson J., Heino P., Palva E. T. (2004) Overexpression of multiple dehydrin genes enhances tolerance to freezing stress in Arabidopsis. Plant Mol. Biol. 54, 743–753 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.