

Abstract
Proteins endogenously secreted by human embryonic stem cells (hESCs) and those present in hESC culture medium are critical regulators of hESC self-renewal and differentiation. Current MS-based approaches for identifying secreted proteins rely predominantly on MS analysis of cell culture supernatants. Here we show that targeted proteomics of secretory pathway organelles is a powerful alternate approach for interrogating the cellular secretome. We have developed procedures to obtain subcellular fractions from mouse embryonic fibroblasts (MEFs) and hESCs that are enriched in secretory pathway organelles while ensuring retention of the secretory cargo. MS analysis of these fractions from hESCs cultured in MEF conditioned medium (MEF-CM) or MEFs exposed to hESC medium revealed 99 and 129 proteins putatively secreted by hESCs and MEFs, respectively. Of these, 53 and 62 proteins have been previously identified in cell culture supernatants of MEFs and hESCs, respectively, thus establishing the validity of our approach. Furthermore, 76 and 37 putatively secreted proteins identified in this study in MEFs and hESCs, respectively, have not been reported in previous MS analyses.
The identification of low abundance secreted proteins via MS analysis of cell culture supernatants typically necessitates the use of altered culture conditions such as serum-free medium. However, an altered medium formulation might directly influence the cellular secretome. Indeed, we observed significant differences between the abundances of several secreted proteins in subcellular fractions isolated from hESCs cultured in MEF-CM and those exposed to unconditioned hESC medium for 24 h. In contrast, targeted proteomics of secretory pathway organelles does not require the use of customized media. We expect that our approach will be particularly valuable in two contexts highly relevant to hESC biology: obtaining a temporal snapshot of proteins secreted in response to a differentiation trigger, and identifying proteins secreted by cells that are isolated from a heterogeneous population.
Human embryonic stem cells (hESCs)1 are pluripotent cells isolated from the inner cell mass of a pre-implantation blastocyst stage embryo (1). They have potential applications in regenerative medicine, are an attractive source of human cells for drug evaluation, and are useful models for understanding human development. The self-renewal or differentiation of hESCs is controlled by endogenous proteins secreted by hESCs and by exogenous factors present in cell culture medium (2, 3). For instance, hESCs are routinely cultured on feeder layers of mouse embryonic fibroblasts (MEFs) or on Matrigel-coated plates in mouse embryonic fibroblast–conditioned medium (MEF-CM). In these cases, cytokines secreted by MEFs and present in MEF-CM, together with cytokines and extracellular matrix (ECM) proteins secreted by hESCs, form a localized microenvironment that regulates hESC fate.
The comprehensive identification of proteins secreted by MEFs and hESCs—their cellular secretome—can help unravel the molecular mechanisms that regulate hESC fate. Yet the use of MS-based approaches for secretome analysis remains challenging. In general, secretome studies of various cell types have relied on MS analysis of cell culture supernatants (reviewed in Ref. 4). However, such an approach typically results in the identification of small numbers of extracellular proteins. This was indeed the case with MS analysis of conditioned medium (CM) from MEFs or other feeder cells that support the maintenance of undifferentiated hESCs (5–8). A low abundance of secreted proteins of interest and a high concentration of serum proteins in cell culture media significantly impede MS analysis. To overcome these limitations, Bendall et al. implemented an iterative-exclusion MS (IE-MS) strategy, in conjunction with the use of medium without serum or serum replacer, for the identification of proteins secreted by MEFs and hESCs (2). Using this approach, large numbers of previously unreported proteins secreted by MEFs and hESCs could be identified, showing that IE-MS is a powerful strategy for the identification of low abundance proteins. However, the use of medium without serum or serum replacer for secretomic analysis can be problematic. Specifically, the use of a “blank” or serum-free medium might alter cellular physiology and, consequently, the profile of secreted proteins. Indeed, we observe that hESCs are highly prone to apoptosis under such growth conditions. Moreover, an analysis of the cell culture supernatant is not specifically targeted toward endogenously secreted ECM proteins, which are also an important component of the cellular microenvironment. ECM proteins form a matrix that associates with the cell and might not be present in the cell culture supernatant. Moreover, many growth factors are known to be sequestered by ECM proteins and might not be released into the culture medium (9). Here we present a rigorous evaluation of an alternate strategy to interrogate the entire cellular secretome, including cytokines and ECM proteins. Notably, our approach does not require the use of customized media lacking serum and serum replacers, and it is compatible with cell culture systems utilizing media of unknown or poorly defined composition, such as CM from MEFs.
To identify the secretome of MEFs and hESCs, we carried out an MS analysis of their subcellular fractions that were enriched in secretory pathway organelles. The secretory pathway comprises the endoplasmic reticulum (ER), the Golgi apparatus, and the associated transport vesicles. Detailed MS analysis of these organelles identifies the secretory cargo (i.e. proteins destined to be secreted) in addition to the secretory pathway proteome (10). Indeed, we have previously identified several secreted proteins in hESCs as a result of contamination by the ER and Golgi (11) in our subcellular fractions. In light of these reports, we hypothesized that targeted proteomic analysis of the secretory pathway is a viable approach for comprehensive characterization of the cellular secretome. Accordingly, we developed protocols to isolate subcellular fractions enriched in the ER and Golgi compartments from MEFs and hESCs, and we subsequently carried out MS analysis on these samples. Several proteins secreted by MEFs and hESCs could be identified in this manner. Strikingly, the numbers of proteins identified were comparable to those obtained with the highly efficient IE-MS approach. Furthermore, we also show that short-term changes in medium composition affect the profile and quantitative levels of several proteins that transit through the secretory pathway, including secreted and membrane proteins. Taken together, our results validate the use of targeted secretory pathway proteomics as a powerful alternate approach to interrogate the cellular secretome.
MATERIALS AND METHODS
Cell Culture
MEFs were isolated from E13.5 pregnant CD-1 mice embryos (Charles River, Wilmington, MA) and cultured in MEF medium comprising DMEM-High Glucose (Invitrogen, Carlsbad, CA), 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), 1% penicillin-streptomycin (Invitrogen), and 1% l-glutamine (Invitrogen). H1 and H9 hESCs (WiCell, Madison, WI) were cultured on MEF feeders as described elsewhere (12). hESCs were passaged upon confluence, every 5 to 7 days. Feeder free culture of hESCs was carried out on Growth Factor Reduced MatrigelTM (BD Biosciences, Bedford, MA) in the presence of MEF-CM. For passaging, confluent colonies were scored manually, and subcolonies were lifted off the plate using Collagenase IV (Invitrogen). MEF-CM was prepared using previously published protocols (13), using passage-3 MEFs.
Flow Cytometry
H1 and H9 colonies were dissociated using Trypsin/EDTA (Sigma) and fixed in 4% paraformaldehyde (Fisher Scientific, Fair Lawn, NJ). Cells were permeabilized in saponin buffer containing 1 mg/ml saponin (Sigma-Aldrich) and 1% BSA in PBS. Cells were incubated with rabbit anti-human OCT4 antibody (Cell Signaling, Danvers, MA) at 1:200 dilution and mouse anti-human SSEA4 antibody (Millipore, Billerica, MA) at 1:1000 dilution for 1 h at room temperature and subsequently with goat anti-rabbit ALEXA-633 (Invitrogen) and goat anti-mouse ALEXA-488 (Invitrogen) antibodies at 1:500 dilution for 1 h at 4 °C. Cells were analyzed using a BD FacsAria flow cytometer.
Isolation of Subcellular Fractions and Whole Cell Lysate
Mitomycin C-treated MEFs were exposed to hESC medium, as per the protocol for making MEF-CM, prior to harvesting the cells. hESC medium comprised DMEM/F12 (Invitrogen), 20% knockout serum replacement (Invitrogen), 1 mm l-glutamine (Invitrogen), 0.1 mm β-mercaptoethanol (Sigma-Aldrich), 1% non-essential amino acids (Invitrogen), and 4 ng/ml basic fibroblast growth factor (Sigma-Aldrich). MEFs were exposed to fresh hESC medium every day for 4 days to allow the cells to adapt to the medium. MEFs were then washed with Dulbecco's PBS without Ca2+ and Mg2+ (DPBS) (Sigma-Aldrich) and incubated in hypotonic PBS (25% DPBS in water) for 20 min at 4 °C. The sheet of MEF cells that subsequently detached from the flask was collected and vigorously vortexed to dislodge individual cells from the ECM. Cells were centrifuged at 300g for 5 min at 4 °C, and the supernatant was discarded. The cell pellet was resuspended in sucrose buffer comprising 250 mm sucrose (Fisher), 25 mm potassium chloride (Fisher), 5 mm magnesium chloride (Fisher), 10 mm triethanolamine (Sigma-Aldrich), 10 mm acetic acid (Fisher), and cØmplete mini® protease inhibitor mixture tablets (Roche, Indianapolis, IN) with the pH adjusted to 7.6 using triethanolamine and/or acetic acid. The cells were lysed by being passed through a 25-gauge needle twice and centrifuged at 3000g for 10 min at 4 °C to remove nuclei (14). The supernatant was incubated with 50 μl anti-Tom22 magnetic microbeads (Miltenyi Biotec, Auburn, CA) at 4 °C for 1 h. The magnetic microbeads loaded with mitochondria were cleared using a DynaMag-15 magnet (Invitrogen) via incubation at 4 °C for 5 h. The cleared lysate was centrifuged at 15,000g for 30 min at 4 °C. The pellet was enriched for ER and Golgi (14) and was solubilized in 8 m urea (Sigma) and 50 mm ammonium bicarbonate (VWR, West Chester, PA) and stored at −80 °C until further use.
H1 and H9 cells were grown until confluence and washed with DPBS. For experiments with unconditioned medium, H1 and H9 cells were exposed to unconditioned hESC medium for 1 day, and cultures were washed with DPBS to remove dead cells. Cultures were incubated in DPBS with 1 mm EDTA (ACROS, Geel, Belgium) to dissociate colonies into single cells. The suspension was centrifuged at 300g for 5 min at 4 °C. The cell pellet was resuspended in sucrose buffer, and cells were lysed by being passed through a 25-gauge needle twice. The lysate was centrifuged at 3000g for 10 min at 4 °C to remove nuclei. The supernatant was incubated with anti-TOM22 magnetic microbeads (Miltenyi Biotec) at 4 °C for 1 h. Magnetic microbeads were cleared via incubation with a DynaMag-15 magnet at 4 °C for 5 h. The cleared lysate was centrifuged at 15,000g for 30 min at 4 °C, and the pellet was solubilized in 8 m urea with 50 mm ammonium bicarbonate. Samples were stored at −80 °C until further use. To obtain the whole cell lysate, H9 cells were directly lysed in 8 m urea with 50 mm ammonium bicarbonate and passed vigorously through a 22-gauge needle until the lysate was no longer viscous. The lysate was stored at −80 °C until further use.
Protein Digestion
Approximately 100 μg of total protein was digested using filtered-aided sample preparation, a protocol adapted from that of Mann and coworkers (15). Vivacon 30k molecular weight cutoff filters (Sartorius Stedim Biotech, Goettingen, Germany) were used as reaction vessels; samples were reduced with dithiothreitol, alkylated with iodoacetamide, and digested overnight with trypsin. The digestion was quenched by the addition of 1% formic acid solution, and the peptides were collected via centrifugation.
Peptide Fractionation
Peptides were fractionated by means of an anion exchange protocol previously described by Wisniewski et al. (16). 200 μl pipette tips were filled with a solid phase anion exchange resin (3M, St. Paul, MN) to create StageTips for peptide fractionation (17). Britton & Robinson buffer pH 5.0 (Ricca Chemical Company, Arlington, TX) was titrated with either HCl or NaOH to different pH readings (3.0, 4.0, 5.0, 6.0, 8.0, and 11.0) to create a total of six different buffers. StageTips were conditioned with methanol followed by 1 m NaOH, followed by pH 11 buffer. Peptides were loaded in pH 11 buffer and subsequently eluted with each of the aforementioned buffers in descending order via centrifugation. Fractions were collected and evaporated under vacuum before being reconstituted in mobile phase A for LC-MS/MS analysis.
LC-MS/MS
An Eksigent 1D+ nano-LC system (Eksigent, Dublin, CA) utilizing a cHiPLC-Nanoflex system for reversed-phase separation of peptides was employed with a trap and elute configuration. The dimensions of the nano cHiPLC analytical column were 75 μm inner diameter × 15 cm, and the nano cHiPLC trapping column measured 200 μm inner diameter × 0.5 mm. Both columns were packed with ChromXP C18-CL (3 μm, 120 Å) by the manufacturer (Eksigent).
LC solvents were purchased from Burdick and Jackson (Muskegon, MI). Mobile phase A contained 98% water, 2% acetonitrile, and 0.2% formic acid, and mobile phase B consisted of 2% water, 98% acetonitrile, and 0.2% formic acid. Peptides were loaded on the trap column at 2 μl/min before switching in-line with the analytical column at a flow rate of 350 nL/min. The gradient was initiated at 2% B and adjusted to 7% B over the first 5 min. The gradient was then slowly ramped to 40% B over the next 211 min before being adjusted to 95% B over 2 min. The gradient was held at 95% B for 8 min before being adjusted to 2% B over the course of 2 min and was held at 2% B for an additional 10 min for re-equilibration.
Mass measurements were made using an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) using parameters optimized in a previous study (18). A broadband scan using 60,000 resolving power at 400 m/z was collected in profile mode by the Orbitrap mass analyzer, followed by eight data-dependent MS/MS scan events collected in centroid mode in the LTQ using collision-induced dissociation. Charge state screening was enabled, and unassigned, 1+, and ≥4+ charge states were rejected from MS/MS analysis. Dynamic exclusion was also enabled and set to 180 s with a repeat count of 1, a repeat duration of 0 s, and an exclusion list size as large as 500 ions. Automatic gain control settings were 8 × 103 ions for the LTQ and 1 × 106 ions for the Orbitrap.
For analysis of the whole cell lysate, 50 μg of protein was loaded onto a gel and separated at a constant 200 V before staining with BioSafe Coomassie stain (BioRad, Hercules, CA). The gel lane was cut into 10 sections which were reduced with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin. Digested peptides were separated using an Eksigent 1D+nano-LC system (Eksigent, Dublin, CA) with a vented column configuration (19). A 15 cm PicoFrit column (New Objective, Woburn, MA) and a 5 cm IntegraFrit trap column (New Objective) were packed in-house with Magic C18AQ stationary phase (5 μm particles, 200 Å pore size) (Microm Biosources, Auburn, CA). Data were acquired with a 7T LTQ-FT-ICR Ultra (Thermo Fisher Scientific, San Jose, CA) mass spectrometer using 100,000FWHM resolving power at 400 m/z. Up to eight data dependent MS/MS scan events were triggered for every precursor scan.
MS Data Analysis
RAW data files generated from LC-MS/MS analyses were analyzed using MASCOT Distiller version 2.4.2.0 (Matrix Science, Boston, MA) to create peak lists and subsequently searched using the MASCOT search engine version 2.3.2 (Matrix Science) (20). The parameters used for searching included the variable modifications asparagine and glutamine deamidation, as well as methionine oxidation. Cysteine carbamidomethylation was set as a fixed modification. Trypsin was selected as the protease, and a maximum of two missed cleavages were allowed. Search tolerances were set to ±5 ppm and ±0.6 Da for the precursor ions and product ions, respectively. The SwissProt human database containing 20,255 target sequences (downloaded January 28, 2012) was appended with reverse sequences for use as the target/decoy database. Search results were imported into ProteoIQ version 2.3.05 (NuSep, Athens, GA) to create protein lists filtered using a 1% false discovery rate. Normalized spectral abundance factor (NSAF) values were manually calculated using the raw spectral count data exported from ProteoIQ using the following formula:
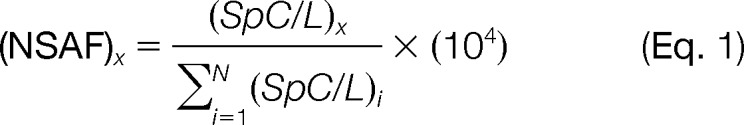 |
L = number of amino acids.
SpC = total number of MS/MS spectra that identify protein x.
Search results were imported into ProteoIQ version 2.3.05 to create protein lists filtered using a 1% false discovery rate to perform spectral counting normalization and relative quantification across hESC samples. All data reported are from three technical replicates for single biological replicates corresponding to MEFs, H1 and H9 hESCs cultured in MEF-CM, and H1 and H9 hESCs exposed to unconditioned hESC medium for 24 h.
Gene Ontology Annotation and Estimating Enrichment of Secretory Pathway Organelles in Subcellular Fractions
Gene Ontology (GO) enrichment analysis of identified proteins was carried out using DAVID (21, 22). Briefly, the DAVID algorithm maps the list of proteins to their GO annotations and outputs a statistical measure of over-representation of various GO terms (22). Lists of proteins annotated to the secretory pathway organelles—namely, the endoplasmic reticulum (GO:0005783), endoplasmic reticulum–Golgi intermediate compartment (GO:0005793), ER to Golgi transport vesicle (GO:0030134), Golgi apparatus (GO:0005794), secretory granule (GO:0030141), transport vesicle (GO:0030133), and ribosome (GO:0005840)—were obtained using the Princeton GO Term Mapper (go.princeton.edu/cgi-bin/GOTermMapper) and repeats were eliminated. Lists of proteins annotated as extracellular, comprising ECM (GO:0031012), extracellular region (GO:0005576), extracellular region part (GO:0044421), extracellular space (GO:0005615), and proteinaceous ECM (GO:0005578), were also obtained using the Princeton GO Term Mapper, and repeats were eliminated. These lists were further curated via comparison with the UniProt database. Proteins annotated as “Secreted” in the UniProt database were retained and denoted as “Secretory Cargo.”
To quantify the fractional abundance of a particular subcellular organelle in the whole cell lysate or subcellular fractions isolated using our protocols, we used the method previously described by Gilchrist et al. (10). Briefly, for each sample analyzed, we obtained a list of all nonredundant peptides identified in MS analysis and the corresponding spectral count data. The fractional abundance of peptides associated with a particular organelle i (Fi) in a given sample was estimated as follows: peptides that map to proteins annotated to the specific organelle i were identified, and this set of peptides was denoted by Si. Subsequently, Fi was estimated as
 |
Fi was used as a metric to quantify the fractional abundance of organelle i in a given sample. Note that Fi accounts for the abundance of all identified peptides associated with a particular organelle. Subsequently, the fold-enrichment of organelle i in subcellular fractions was calculated as
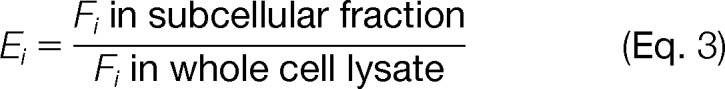 |
The two-tailed Fisher's exact test was used to calculate the p value associated with Ei. A fold-enrichment value greater than 1 indicates that the organelle is enriched in the subcellular fraction isolated. Conversely, a value of Ei less than 1 shows depletion of the organelle during subcellular fractionation.
RESULTS AND DISCUSSION
Isolation of Subcellular Fractions Enriched in Secretory Pathway Organelles from hESCs and MEFs
The secretory pathway comprises the ER, the Golgi apparatus, and the associated transport vesicles that transport proteins from ER to Golgi and finally from Golgi to the plasma membrane and the extracellular space. Protocols for the extraction of smooth ER, rough ER, cis-Golgi, trans-Golgi, and the transport vesicles have been worked out (10, 14). However, these protocols involve extensive ultracentrifugation steps that might potentially rupture the ER and Golgi membranes and lead to loss of the secretory cargo. Therefore, we focused on isolating the Golgi and ER organelles without ultracentrifugation steps, using our modified protocol. Note that the lysate obtained after pelleting of the nucleus, as in our procedure, is a widely used source for ER and Golgi fractionation (14). We isolated subcellular fractions putatively enriched in secretory pathway organelles from H1 and H9 hESCs and MEFs and subsequently conducted MS analysis on these fractions. The complete MS dataset is available in supplemental Tables S1–S3.
The DAVID software has been commonly used for determining organelle enrichment in subcellular fractions (23–25). GO annotation analysis using DAVID shows a statistically significant enrichment of ER proteins in these subcellular fractions (Fig. 1). For instance, the frequency of proteins annotated to the ER is 2.3-fold higher in the list of proteins identified in our samples from H1 hESCs than the corresponding frequency of ER-annotated proteins among all human proteins (p value < 10−42 using three different statistical tests). Similar enrichment is seen in H9 hESCs and MEFs. Enrichment for other parts of the secretory pathway, such as the Golgi, ER–Golgi intermediate compartment, cytoplasmic vesicle, nuclear envelope–ER network, and ribosomes, was also observed. Proteins with GO annotation for the cytoplasm were most abundant because ER and Golgi are a part of the cytoplasm of the cell. Additionally, some enrichment for plasma membrane proteins was seen. This is expected because plasma membrane proteins are synthesized in the ER.
Fig. 1.

Subcellular fractionation of secretory pathway organelles was carried out from hESCs and MEFs. Distribution of proteins annotated to the secretory pathway organelles (endoplasmic reticulum, GO:0005783, endoplasmic reticulum-Golgi intermediate compartment, GO:0005793; ER to Golgi transport vesicle, GO:0030134; Golgi apparatus, GO:0005794; secretory granule, GO:0030141; transport vesicle, GO:0030133; and ribosome, GO:0005840) are shown for (A) H1 hESCs, (B) H9 hESCs, and (C) MEFs. GO enrichment analysis for identified proteins was carried out using DAVID, and statistically significant enrichment for secretory pathway organelles was obtained for (D) H1 hESCs, (E) H9 hESCs, and (F) MEFs. G, fractional abundance of various organelles in the whole cell lysate and subcellular fractions putatively enriched in secretory pathway organelles, obtained from H9 hESCs. “Secretory cargo” denotes proteins that are annotated as secreted in the Uniprot database. “Extracellular” refers to proteins annotated as extracellular (GO:0031012, GO:0005576, GO:0044421, GO:0005615, and GO:0005578). Secretory pathway organelles and mitochondria are considered a part of the cytoplasm, and many plasma membrane and nuclear proteins are also annotated as cytoplasmic. Therefore, proteins that were already accounted for in these organelles were not included in the category “remaining cytoplasm.”
A limitation of the DAVID analysis is that the number of proteins annotated to an organelle—and not the abundance of these proteins—is used for calculating enrichment. Therefore, to further confirm enrichment of secretory pathway organelles, we adapted a method previously described by Gilchrist et al. (10). We estimated the fraction of various organelles in the whole cell lysate and in the subcellular fraction from H9 cells (Fig. 1G). As expected, we observed that proteins associated with the secretory pathway, such as the ER, Golgi, and plasma membrane, were overrepresented in our subcellular fraction relative to whole cell lysate (p value < 6 × 10−310). This is highly consistent with the DAVID analysis. A significant increase in the fractional abundance of the secretory cargo was observed in the subcellular fraction (p value < 4 × 10−82) as a result of the enrichment of secretory pathway organelles. In contrast, the fractional abundance of peptides associated with mitochondria, nuclei, and cytoplasm was lower in the subcellular fraction relative to whole cell lysate (p value < 6 × 10−133). Interestingly, despite the use of a negative selection step to reduce mitochondria from the subcellular fractions, we observed a very modest decrease in the fractional abundance of mitochondrial proteins. In this context, it should be noted that complete elimination of mitochondrial contamination without rupturing of the ER, and concomitant loss of lumenal content, might not be possible because mitochondria are physically associated with the ER through mitochondria-associated ER membranes (26). The list of nonredundant peptides and corresponding spectral counts for whole cell lysate and subcellular fractions from H9 hESCs are given in supplemental Table S4; detailed analysis for enrichment or depletion of specific organelles in subcellular fractions relative to whole cell lysate can be found in supplemental Table S5.
Taken in total, our analysis conclusively shows that subcellular fractions isolated using our fractionation procedure are enriched in secretory pathway organelles. Further, this enrichment is consistent across H1 and H9 hESCs and MEFs; peptides associated with the secretory pathway organelles and extracellular proteins constituted a significant fraction (40% to 45%) of all our fractionated samples (Fig. 1 and supplemental Fig. S1; see supplemental Table S6 for a list of nonredundant peptides and corresponding spectral counts used for the calculation of numbers in supplemental Fig. S1). Further enrichment of secretory pathway organelles may be obtained through optimization of the fractionation procedure. However, it is important to note that our central goal was to interrogate the secretory cargo through enrichment of the secretory pathway organelles. To that end, the extent of enrichment provided by our procedure is sufficient and enables us to elucidate the secretome of MEFs and hESCs (vide infra).
The Secretome of MEFs
We conducted MS analysis on subcellular fractions that were enriched in secretory pathway components, isolated from MEFs, to identify secreted proteins that are present in MEF-CM. MEFs were exposed to hESC medium, as specified in the protocol for producing MEF-CM, prior to the isolation of subcellular fractions. 129 proteins that are known to be extracellular were identified through MS analysis. These proteins are putatively secreted by MEFs into the hESC medium during the process of generating MEF-CM, and they are listed in Fig. 2 (also see supplemental Table S7). 76 of these proteins have not been identified in previous MS analyses of MEF-CM. Fig. 2 also shows plots of the relative abundances of these proteins (their NSAF values) observed during MS analysis. The NSAF values correspond to the relative abundance of proteins (27–30) within the secretory pathway organelles when the cells were harvested. The NSAF value for a particular protein depends on the rate of protein secretion, as well as its residence time in the secretory pathway. For proteins with identical residence times in the secretory pathway, differences in NSAF values indicate differences in rates of protein secretion.
Fig. 2.

The secretome of MEFs in hESC medium was identified through MS analysis of the secretory pathway organelles. These are listed in descending order of their NSAF values, which are shown in the plot. Proteins that were not previously identified in MEFs are highlighted in green. Error bars denote standard deviation from triplicate measurements.
The Secretome of hESCs
We further conducted MS analysis on subcellular fractions that were enriched in secretory pathway components, isolated from H1 and H9 hESCs, to identify proteins endogenously secreted by hESCs. Subcellular fractions were isolated from hESCs cultured in MEF-CM, as the culture of hESCs in serum or serum-replacer free medium was not necessary in our protocol. MS analysis revealed several cytokines and ECM proteins that were known to be extracellular. These proteins are putatively secreted by hESCs and constitute the hESC microenvironment. Fig. 3 lists 99 such proteins that were identified in both H1 and H9 hESCs, along with plots of their NSAF values from each cell line (also see supplemental Table S8). Of these, 37 proteins have not been previously identified by MS analysis, as indicated in Fig. 3. Among these, the endogenous secretion of growth and differentiation factor-3 (GDF3) by H9 hESCs, identified via ELISA, has been previously reported (3).
Fig. 3.

The secretome of H1 and H9 hESCs in MEF-conditioned medium was identified through MS analysis of the secretory pathway organelles. Only those proteins that were identified in both cell lines are listed. The NSAF values obtained in both H1 and H9 hESCs are plotted. Proteins that were not previously identified in hESCs are highlighted in green. Error bars denote standard deviation from triplicate measurements.
Our analysis of both MEFs and hESCs revealed several putatively secreted proteins that were previously unreported. However, it is important to note that we found 53 and 62 proteins secreted by MEFs and hESCs, respectively, that have been identified in other MS analyses (2, 11). These findings strongly support the validity of secretomic analysis through targeted proteomics of the secretory pathway organelles.
HESC Proteome Changes upon Exposure to Unconditioned Medium
Endogenous factors secreted by hESCs play an important role in regulating hESC fate (2, 3). However, it is likely that the profile of secreted proteins in hESCs is dependent on the media conditions. Indeed, hESCs exposed to unconditioned medium (i.e. hESC medium used for generating MEF-CM) are reported to rapidly lose transcripts for Lefty and Nodal (31). Yet secretome studies on cell culture supernatants necessitate the use of altered media such as unconditioned serum-free or unconditioned serum-replacer-free media. To investigate the effect of medium alteration on hESCs, we carried out MS analysis on hESCs exposed to unconditioned medium for 24 h.
Exposure of H1 and H9 hESCs to unconditioned serum replacer-free hESC medium for 24 h caused extensive cell death. Therefore, we restricted our analysis to H1 and H9 hESCs exposed to unconditioned hESC medium for 24 h. Vast cell death was also seen in this case, but a population of viable cells could be recovered. These cells continued to express the pluripotency markers OCT4 and SSEA4 (supplemental Fig. S2). Subcellular fractions enriched in secretory pathway organelles were isolated from these cells and subjected to an MS analysis. Statistically significant enrichment for the ER was obtained (Figs. 4A, 4B). The proportion of secretory pathway organelle proteins was also similar to that obtained for H1 and H9 hESCs cultured in MEF-CM, as previously described (Fig. 1, Fig. 4C). This shows that our procedure is feasible even in the presence of extensive cell debris.
Fig. 4.

The proteome of hESCs changes in unconditioned medium. HESCs were exposed to unconditioned medium for 1 day, and secretory pathway organelles were isolated for MS analysis. GO enrichment analysis using DAVID shows statistically significant enrichment of the secretory pathway organelles from (A) H1 hESCs and (B) H9 hESCs. Distribution of proteins annotated to the secretory pathway organelles (endoplasmic reticulum, GO:0005783; endoplasmic reticulum-Golgi intermediate compartment, GO:0005793; ER to Golgi transport vesicle, GO:0030134; Golgi apparatus, GO:0005794; secretory granule, GO:0030141; transport vesicle, GO:0030133; and ribosome, GO:0005840) shown for (C) H1 hESCs and (D) H9 hESCs. The protein NSAF values obtained from these cells were compared with those obtained from H1 and H9 hESCs grown in MEF-CM. Statistically significant differences in NSAF values of secretory proteins are shown for (E) H9 hESCs and (F) H1 hESCs.
We compared the NSAF values of proteins identified in these samples to those obtained from H1 and H9 hESCs cultured in MEF-CM. This allowed us to quantitatively assess changes in protein abundances in the secretory pathway upon exposure of hESCs to unconditioned medium for 24 h. Statistically significant changes seen are listed in supplemental Table S9. Pertinently, we also observed significant changes in relative abundances of multiple secreted proteins in both H9 and H1 hESCs. These are shown in Figs. 4E and 4F. Interestingly, VASORIN was identified only in hESCs exposed to unconditioned medium, and not in hESCs cultured in MEF-CM. These findings underscore a key problem associated with studying the secretome of cells under altered media conditions: the medium composition may directly influence the profile of secreted proteins. In contrast, targeted proteomics of the secretory pathway obviates the need for altering media conditions.
Both HESCs and MEFs Express Regulators of TGF-β, FGF, IGF, and Wnt Signaling Pathways
As discussed earlier, MS analysis of subcellular fractions enriched in secretory pathway organelles revealed several endogenous factors and ECM proteins putatively secreted by hESCs, as well as putative components of MEF-CM. Collectively, these factors constitute the hESC microenvironment that regulates hESC fate. Furthermore, our analysis also identified several membrane proteins expressed by hESCs. This is because membrane proteins are also processed in the ER. Notably, we identified components of the transforming and growth factor-β (TGF-β) pathway, the fibroblast growth factor (FGF) pathway, the Wnt pathway, and the insulin-like growth factor (IGF) pathway from both MEFs and hESCs (Fig. 5). These pathways have been implicated previously in hESC self-renewal and/or differentiation (32–37).
Fig. 5.

Identification of signaling pathway proteins in hESCs and MEFs. Cytokines and growth factor receptors identified in hESCs and cytokines identified in MEFs that are associated with the TGF-β pathway, Wnt pathway, FGF pathway, and Insulin/IGF pathway are listed. Also shown are proteases and protease inhibitors identified in hESCs and MEFs.
We observed both positive and negative regulators of the TGF-β and Wnt pathway in the hESC microenvironment. Ligands in the TGF-β superfamily can be broadly classified into two branches: the Activin/Nodal branch that signals through the Smad2/3 pathway, and the BMP branch that signals through the Smad1/5/8 pathway (38). We found expression of receptors associated with both branches in hESCs. Self-renewal of hESCs is associated with the presence of Activin/Nodal signaling and suppression of BMP activity (34, 37). Consistent with this paradigm, we identified BMP inhibitors in both hESCs and MEFs. Follistatin-related protein 1 (FSTL1) was identified in MEFs, and FSTL1 and GDF3 were identified in hESCs. We also identified TGF-β1 in MEFs; TGF-β1 and GDF3 can act as agonists of the Activin/Nodal pathway. Intriguingly, we identified Nodal modulator proteins (NOMO1–3) in hESCs that might negatively regulate Activin/Nodal signaling. Negative regulators of Wnt signaling were identified in both MEFs and hESCs. These include secreted frizzled-related proteins (SFRP1, -2) and Dickkopf-related protein 3, identified in hESCs and MEFs, respectively. This is consistent with recent studies that suggest a role for Wnt signaling in hESC differentiation but not hESC self-renewal (33). Additionally, various proteases and protease inhibitors were also identified in MEFs and hESCs. Thus, taken together, these data reveal a picture wherein positive and negative regulators of multiple signaling pathways, expressed by both MEFs and hESCs, potentially control hESC self-renewal in MEF-CM.
CONCLUSIONS
It is increasingly becoming evident that autocrine and paracrine factors play an important role in the self-renewal and differentiation of hESCs. Identification of these endogenously secreted factors can help elucidate the molecular mechanisms that govern hESC fate and aid in the development of efficient protocols for generating desired differentiated cell types. MS analyses of cell culture supernatants have enabled the identification of several factors secreted by MEFs and hESCs. However, this approach has an important limitation: practical considerations often necessitate the use of serum or serum-replacer free medium. This in turn might lead to a significant observer effect wherein the experimental design influences the object of study; that is, changing the medium composition for MS analysis might alter the secreted protein profile being investigated. Here we show that targeted proteomics of the secretory pathway organelles is a powerful alternate approach for interrogating the cellular secretome. Most important, this strategy does not require the use of serum-free or other customized medium formulation and therefore eliminates the observer effect.
We hypothesized that proteomic analysis of secretory pathway organelles would allow identification of secreted proteins that constitute the cargo in the secretory pathway. To investigate this hypothesis, we first developed a subcellular fractionation procedure to obtain fractions enriched in secretory pathway components. Subsequently, we showed that MS analysis of these fractions indeed enabled us to elucidate the secretome of MEFs and hESCs. The significant overlap between proteins identified in this study and those previously identified strongly supports the validity of our approach. Notably, this targeted secretory pathway proteomics approach can further be combined with sophisticated MS strategies, such as the repeated analysis of the same protein sample using increasingly stringent exclusion lists, to further improve coverage of the secretome. Also, the subcellular fractionation procedures described herein may be optimized further to improve the enrichment of secretory pathway components, possibly leading to additional improvements in secretome coverage. However, the extensive use of ultracentrifugation steps and hypertonic sucrose density gradients, as are commonly used for isolating pure organelles, might potentially compromise the integrity of the ER and Golgi organelles. This might result in the loss of the secretory cargo. Therefore, the implications of such improvements should be carefully evaluated.
A potential drawback of our approach is that secretory pathway organelle fractions might be contaminated with other organelles. This is problematic when an identified protein is annotated as being secreted as well as intracellular or membrane-bound. Because we do not directly assay the cell culture supernatant, such proteins are putatively secreted and need further validation. It must, however, be noted that this limitation exists with MS analysis of cell culture supernatants as well. Indeed, a large proportion of identified proteins are annotated as intracellular in this case as well (2), likely because of cell lysis during culture. Nevertheless, our strategy provides a starting point for investigating putative secreted factors that constitute the cellular microenvironment, without alterations to culture conditions. Additionally, this approach is not biased against cell-surface-associated ECM proteins and growth factors that may be captured by ECM proteins and not released into the cell culture supernatant. We expect that secretory pathway proteomics for identification of secreted proteins will be particularly useful in two contexts that are of immense relevance in hESC biology: the identification of proteins secreted by cells undergoing differentiation, and the identification of specific secretomes of various cell types present in a heterogeneous population. Our approach can be used to obtain a temporal snapshot of the secreted protein profile in response to a particular differentiation trigger. Additionally, the secretome of specific cells in a heterogeneous population may be interrogated by means of MS analysis of their secretory pathway organelles after isolating cells using techniques such as flow cytometry. Thus, targeted proteomics of the secretory pathway organelles is a powerful tool for investigating hESC biology.
Supplementary Material
Footnotes
* This work was supported by funding from the NSF (grant no. CBET-0966859).
 This article contains supplemental Tables S1 to S9 and Figures S1 and S2.
This article contains supplemental Tables S1 to S9 and Figures S1 and S2.
1 The abbreviations used are:
- CM
- conditioned medium
- ECM
- extracellular matrix
- ER
- endoplasmic reticulum
- FGF
- fibroblast growth factor
- GDF3
- growth and differentiation factor-3
- GO
- Gene Ontology
- HESCs
- human embryonic stem cells
- IE-MS
- iterative exclusion MS
- IGF
- insulin-like growth factor
- MEF
- mouse embryonic fibroblast
- NSAF
- normalized spectral abundance factor
- TGF-β
- transforming and growth factor-β.
REFERENCES
- 1. Thomson J. A., Itskovitz-Eldor J., Shapiro S. S., Waknitz M. A., Swiergiel J. J., Marshall V. S., Jones J. M. (1998) Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 [DOI] [PubMed] [Google Scholar]
- 2. Bendall S. C., Hughes C., Campbell J. L., Stewart M. H., Pittock P., Liu S., Bonneil E., Thibault P., Bhatia M., Lajoie G. A. (2009) An enhanced mass spectrometry approach reveals human embryonic stem cell growth factors in culture. Mol. Cell. Proteomics 8, 421–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peerani R., Rao B. M., Bauwens C., Yin T., Wood G. A., Nagy A., Kumacheva E., Zandstra P. W. (2007) Niche-mediated control of human embryonic stem cell self-renewal and differentiation. EMBO J. 26, 4744–4755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skalnikova H., Motlik J., Gadher S. J., Kovarova H. (2011) Mapping of the secretome of primary isolates of mammalian cells, stem cells and derived cell lines. Proteomics 11, 691–708 [DOI] [PubMed] [Google Scholar]
- 5. Chin A. C., Fong W. J., Goh L. T., Philp R., Oh S. K., Choo A. B. (2007) Identification of proteins from feeder conditioned medium that support human embryonic stem cells. J. Biotechnol. 130, 320–328 [DOI] [PubMed] [Google Scholar]
- 6. Lim J. W., Bodnar A. (2002) Proteome analysis of conditioned medium from mouse embryonic fibroblast feeder layers which support the growth of human embryonic stem cells. Proteomics 2, 1187–1203 [DOI] [PubMed] [Google Scholar]
- 7. Prowse A. B., McQuade L. R., Bryant K. J., Marcal H., Gray P. P. (2007) Identification of potential pluripotency determinants for human embryonic stem cells following proteomic analysis of human and mouse fibroblast conditioned media. J. Proteome Res. 6, 3796–3807 [DOI] [PubMed] [Google Scholar]
- 8. Prowse A. B., McQuade L. R., Bryant K. J., Van Dyk D. D., Tuch B. E., Gray P. P. (2005) A proteome analysis of conditioned media from human neonatal fibroblasts used in the maintenance of human embryonic stem cells. Proteomics 5, 978–989 [DOI] [PubMed] [Google Scholar]
- 9. ten Dijke P., Arthur H. M. (2007) Extracellular control of TGFbeta signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 8, 857–869 [DOI] [PubMed] [Google Scholar]
- 10. Gilchrist A., Au C. E., Hiding J., Bell A. W., Fernandez-Rodriguez J., Lesimple S., Nagaya H., Roy L., Gosline S. J., Hallett M., Paiement J., Kearney R. E., Nilsson T., Bergeron J. J. (2006) Quantitative proteomics analysis of the secretory pathway. Cell 127, 1265–1281 [DOI] [PubMed] [Google Scholar]
- 11. Sarkar P., Collier T. S., Randall S. M., Muddiman D. C., Rao B. M. (2012) The subcellular proteome of undifferentiated human embryonic stem cells. Proteomics 12, 421–430 [DOI] [PubMed] [Google Scholar]
- 12. Collier T. S., Sarkar P., Rao B., Muddiman D. C. (2010) Quantitative top-down proteomics of SILAC labeled human embryonic stem cells. J. Am. Soc. Mass Spectrom. 21, 879–889 [DOI] [PubMed] [Google Scholar]
- 13. Xu C., Inokuma M. S., Denham J., Golds K., Kundu P., Gold J. D., Carpenter M. K. (2001) Feeder-free growth of undifferentiated human embryonic stem cells. Nat. Biotechnol. 19, 971–974 [DOI] [PubMed] [Google Scholar]
- 14. Graham J. M., Rickwood D. (1997) Subcellular Fractionation: A Practical Approach, IRL Press at Oxford University Press, Oxford, UK [Google Scholar]
- 15. Wisniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, U359–U360 [DOI] [PubMed] [Google Scholar]
- 16. Wisniewski J. R., Zougman A., Mann M. (2009) Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 8, 5674–5678 [DOI] [PubMed] [Google Scholar]
- 17. Ishihama Y., Rappsilber J., Mann M. (2006) Modular stop and go extraction tips with stacked disks for parallel and multidimensional peptide fractionation in proteomics. J. Proteome Res. 5, 988–994 [DOI] [PubMed] [Google Scholar]
- 18. Andrews G. L., Dean R. A., Hawkridge A. M., Muddiman D. C. (2011) Improving proteome coverage on a LTQ-Orbitrap using design of experiments. J. Am. Soc. Mass Spectrom. 22, 773–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andrews G. L., Shuford C. M., Burnett J. C., Hawkridge A. M., Muddiman D. C. (2009) Coupling of a vented column with splitless nanoRPLC-ESI-MS for the improved separation and detection of brain natriuretic peptide-32 and its proteolytic peptides. J. Chromatogr. B 877, 948–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perkins D. N., Pappin D. J. C., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 21. Huang da W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 23. An H. J., Gip P., Kim J., Wu S., Park K. W., McVaugh C. T., Schaffer D. V., Bertozzi C. R., Lebrilla C. B. (2012) Extensive determination of glycan heterogeneity reveals an unusual abundance of high mannose glycans in enriched plasma membranes of human embryonic stem cells. Mol. Cell. Proteomics 11, M111.010660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reinhardt T. A., Lippolis J. D., Nonnecke B. J., Sacco R. E. (2012) Bovine milk exosome proteome. J. Proteomics 75, 1486–1492 [DOI] [PubMed] [Google Scholar]
- 25. Wilson R., Norris E. L., Brachvogel B., Angelucci C., Zivkovic S., Gordon L., Bernardo B. C., Stermann J., Sekiguchi K., Gorman J. J., Bateman J. F. (2012) Changes in the chondrocyte and extracellular matrix proteome during post-natal mouse cartilage development. Mol. Cell. Proteomics 11, M111.014159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Csordas G., Renken C., Varnai P., Walter L., Weaver D., Buttle K. F., Balla T., Mannella C. A., Hajnoczky G. (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu H., Sadygov R. G., Yates J. R., 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 28. Zybailov B., Mosley A. L., Sardiu M. E., Coleman M. K., Florens L., Washburn M. P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 29. Sardiu M. E., Cai Y., Jin J., Swanson S. K., Conaway R. C., Conaway J. W., Florens L., Washburn M. P. (2008) Probabilistic assembly of human protein interaction networks from label-free quantitative proteomics. Proc. Natl. Acad. Sci. U.S.A. 105, 1454–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gokce E., Shuford C. M., Franck W. L., Dean R. A., Muddiman D. C. (2011) Evaluation of normalization methods on GeLC-MS/MS label-free spectral counting data to correct for variation during proteomic workflows. J. Am. Soc. Mass Spectrom. 22, 2199–2208 [DOI] [PubMed] [Google Scholar]
- 31. Besser D. (2004) Expression of nodal, lefty-a, and lefty-B in undifferentiated human embryonic stem cells requires activation of Smad2/3. J. Biol. Chem. 279, 45076–45084 [DOI] [PubMed] [Google Scholar]
- 32. Bendall S. C., Stewart M. H., Menendez P., George D., Vijayaragavan K., Werbowetski-Ogilvie T., Ramos-Mejia V., Rouleau A., Yang J., Bosse M., Lajoie G., Bhatia M. (2007) IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature 448, 1015–1021 [DOI] [PubMed] [Google Scholar]
- 33. Davidson K. C., Adams A. M., Goodson J. M., McDonald C. E., Potter J. C., Berndt J. D., Biechele T. L., Taylor R. J., Moon R. T. (2012) Wnt/beta-catenin signaling promotes differentiation, not self-renewal, of human embryonic stem cells and is repressed by Oct4. Proc. Natl. Acad. Sci. U.S.A. 109, 4485–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. James D., Levine A. J., Besser D., Hemmati-Brivanlou A. (2005) TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 132, 1273–1282 [DOI] [PubMed] [Google Scholar]
- 35. Sumi T., Tsuneyoshi N., Nakatsuji N., Suemori H. (2008) Defining early lineage specification of human embryonic stem cells by the orchestrated balance of canonical Wnt/beta-catenin, Activin/Nodal and BMP signaling. Development 135, 2969–2979 [DOI] [PubMed] [Google Scholar]
- 36. Wang L., Schulz T. C., Sherrer E. S., Dauphin D. S., Shin S., Nelson A. M., Ware C. B., Zhan M., Song C. Z., Chen X., Brimble S. N., McLean A., Galeano M. J., Uhl E. W., D'Amour K. A., Chesnut J. D., Rao M. S., Blau C. A., Robins A. J. (2007) Self-renewal of human embryonic stem cells requires insulin-like growth factor-1 receptor and ERBB2 receptor signaling. Blood 110, 4111–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu R. H., Peck R. M., Li D. S., Feng X., Ludwig T., Thomson J. A. (2005) Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat. Methods 2, 185–190 [DOI] [PubMed] [Google Scholar]
- 38. Shi Y., Massague J. (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.