

Abstract
There is an immediate need for improved methods to systematically and precisely quantify large sets of peptides in complex biological samples. To date protein quantification in biological samples has been routinely performed on triple quadrupole instruments operated in selected reaction monitoring mode (SRM), and two major challenges remain. Firstly, the number of peptides to be included in one survey experiment needs to be increased to routinely reach several hundreds, and secondly, the degree of selectivity should be improved so as to reliably discriminate the targeted analytes from background interferences. High resolution and accurate mass (HR/AM) analysis on the recently developed Q-Exactive mass spectrometer can potentially address these issues. This instrument presents a unique configuration: it is constituted of an orbitrap mass analyzer equipped with a quadrupole mass filter as the front-end for precursor ion mass selection. This configuration enables new quantitative methods based on HR/AM measurements, including targeted analysis in MS mode (single ion monitoring) and in MS/MS mode (parallel reaction monitoring). The ability of the quadrupole to select a restricted m/z range allows one to overcome the dynamic range limitations associated with trapping devices, and the MS/MS mode provides an additional stage of selectivity. When applied to targeted protein quantification in urine samples and benchmarked with the reference SRM technique, the quadrupole-orbitrap instrument exhibits similar or better performance in terms of selectivity, dynamic range, and sensitivity. This high performance is further enhanced by leveraging the multiplexing capability of the instrument to design novel acquisition methods and apply them to large targeted proteomic studies for the first time, as demonstrated on 770 tryptic yeast peptides analyzed in one 60-min experiment. The increased quality of quadrupole-orbitrap data has the potential to improve existing protein quantification methods in complex samples and address the pressing demand of systems biology or biomarker evaluation studies.
Shotgun proteomics has emerged over the past decade as the most effective method for the qualitative study of complex proteomes (i.e., the identification of the protein content), as illustrated by a wealth of publications (1, 2). In this approach, after enzymatic digestion of the proteins, the generated peptides are analyzed by means of liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS)1 in a data dependent mode. However, the complexity of the digested proteomes under investigation and the wide range of protein abundances limit the reproducibility and the sensitivity of this stochastic approach (3), which is critical if one aims at the systematic quantification of the proteins. Thus, alternative MS approaches have emerged for the systematic quantitative study of complex proteomes, the MS-based targeted proteomics (4). In this hypothesis-driven approach, only specific subsets of analytes (a few targeted peptides used as surrogates for the proteins of interest) are selectively measured in predefined m/z ranges and retention time windows, which overcomes the bias toward most abundant compounds commonly observed with shotgun proteomics. When applied to complex biological samples—for example, bodily fluids such as urine or plasma—targeted proteomics requires high performance instruments allowing measurements of a wide dynamic range (many orders of magnitude), with high sensitivity in order to detect peptides in the low amol range and sufficient selectivity to cope with massive biochemical background (5). Selected reaction monitoring (SRM) on triple quadrupole (6) or triple quadrupole-linear ion trap mass spectrometers (7) has emerged as a means to conduct such analyses (8). Initially applied in the MS analysis of small molecules (9, 10), SRM has gradually emerged as the reference quantitative technique for analyzing proteins (or peptides) in biological samples. When coupled with the isotope dilution strategy (11, 12), this very effective technique allows the precise quantification of proteins (13–18). However, despite the increased selectivity provided by the two-stage mass filtering of SRM (at the precursor and fragment ion levels), the low resolution of mass selection does not allow the systematic removal of interferences (19, 20). Moreover, in proteomics, the biochemical background has a composition similar to that of the analytes of interest, which remains a major hurdle limiting the sensitivity of assays, especially in a bodily fluid matrix. High resolution/accurate mass (HR/AM) analysis represents a promising alternative approach that might more efficiently distinguish the compounds of interest from interferences in targeted proteomics. Such analyses can be conducted on orbitrap-based mass spectrometers because of their high sensitivity and high mass accuracy capabilities (21). The introduction of the benchtop standalone orbitrap mass spectrometer (Exactive) (22) further strengthened the attractiveness of the approach, especially in the field of small molecule analysis (23, 24). However, as quantification using trapping devices intrinsically suffers from a limited dynamic range because of the overall ion capacity, the complexity of biological samples remains very challenging even with the HR/AM approach (25). Targeted protein analysis with triple quadrupole mass spectrometers keeps on showing significant superiority for such samples.2 The recently developed quadrupole-orbitrap mass spectrometer (Q-Exactive) can potentially address this issue.3 It is constituted of an orbitrap mass analyzer equipped with a quadrupole mass filter as the front-end for precursor ion mass selection (26, 27). This configuration combines advantages of triple quadrupole instruments for mass filtering and orbitrap-based mass spectrometers for HR/AM measurement. The ability of the instrument to select a restricted m/z range or (sequentially) a small number of precursor ions offers new opportunities for quantification in complex samples by selectively enriching low abundant components. The resulting data, acquired in the so-called single ion monitoring (SIM) mode, fully benefit from the trapping capability while keeping a high acquisition rate as a result of the fast switching time between targeted precursor ions of the quadrupole. Although this mode of data acquisition is possible with a configuration combining a linear ion trap with the orbitrap (as in the LTQ-Orbitrap mass spectrometer), its effectiveness is far more limited in this case. The quadrupole-orbitrap configuration presents significant benefits by selectively isolating a narrow population of precursor ions. Other features of the instrument include its multiplexed trapping capability (26) using either the C-trap or the higher energy collisional dissociation (HCD) cell (28, 29), which opens new avenues in the design of innovative acquisition methods for quantification studies. For the first time, a panel of acquisition methods is designed and applied to targeted quantification at the MS and MS/MS levels. In the latter case, the simultaneous monitoring of multiple MS/MS fragmentation channels, also called parallel reaction monitoring4 (PRM), is particularly promising for quantifying large sets of peptides with increased selectivity.
EXPERIMENTAL PROCEDURES
Sample Preparation
Yeast Digest Mixture
Yeast lysate was prepared as described elsewhere (30). The cell lysate was solubilized in 0.1 m ammonium bicarbonate buffer containing 8 m urea (Sigma, St. Louis, MO) at a final concentration of 6 mg/ml. The proteins were reduced with dithiothreitol (Sigma) at a final concentration of 12 mm for 30 min at 37 °C and alkylated with iodoacetamide (Sigma) at a final concentration of 40 mm for 1 h at 25 °C in the dark. The sample was diluted with 0.1 m ammonium bicarbonate buffer to a final concentration of 1 m urea, sequencing grade porcin trypsin (Promega, Madison, WI) was added to a final enzyme:substrate ratio of 1:100, and the sample was incubated for 16 h at 37 °C. The peptide mixture was cleaned using Sep-Pak tC18 cartridges (Waters, Milford, MA) and eluted with 60% acetonitrile (Sigma). The resulting peptide sample was lyophilized on a vacuum centrifuge and then resolubilized in 0.1% formic acid (Sigma) to a final concentration of 1 μg/μl. For large-scale parallel reaction monitoring analysis, yeast digest samples were diluted to a final concentration of 0.2 μg/μl.
Isotopolog Peptides
Eight synthetic stable isotope-labeled peptides (AQUA peptides) based on an LVALVR sequence with different combinations of 15N- and 13C-labeled amino acids were provided by Thermo Fisher Scientific (Ulm, Germany). For analysis in the absence of chemical background, they were prepared at different concentrations (0.01, 0.03, 0.09, 0.27, 0.81, 2.4, 7.3, and 21.9 fmol/μl) in water (Table I). For analysis in the presence of chemical background, the solution of the eight isotopolog peptides in water was spiked in 1 μg/μl of yeast digest mixture at a ratio of 1:1 (v/v). For the absolute sensitivity test, the peptide LVALVR was spiked at different concentrations (0.9, 0.3, 0.1, and 0.033 amol/μl) into aqueous solutions containing 20 fmol/μl of the peptide LVALVR.
Table I. List of isotopolog peptides prepared in various amounts.
Isotopically labeled amino acids are in italic.
| m/z [M+2H]2+ | Peptide | Amount (fmol) |
|---|---|---|
| 335.73 | LVALVR | 0.01 |
| 337.74 | LVALVR | 0.03 |
| 340.74 | LVALVR | 0.09 |
| 343.74 | LVALVR | 0.27 |
| 347.25 | LVALVR | 0.81 |
| 349.26 | LVALVR | 2.4 |
| 352.26 | LVALVR | 7.3 |
| 355.77 | LVALVR | 21.9 |
Urine Sample Treatment
Urine pooled from de-identified human specimens was provided by Integrated Biobank of Luxembourg and treated as “not human subjects research” material for samples prepared in this study. Urine samples corresponding to 250 μg of protein, estimated via quantitative protein assays (pyrogallol assay, Sigma), were precipitated with 100% stock solutions of acetonitrile at a ratio of 1:9 (v/v). Samples were incubated at room temperature overnight. After precipitation, urine samples were centrifuged at 14,000 g for 30 min at 4 °C. The pellet was washed twice with acetonitrile, air dried, and resuspended with 250 μl of 8 m urea and 0.1 m ammonium bicarbonate. The samples were reduced with 20 mm dithiothreitol in 50 mm ammonium bicarbonate at 37 °C and 800 rpm for 30 min and then alkylated with 80 mm iodoacetamide in 50 mm ammonium bicarbonate at 37 °C and 800 rpm for 30 min. Volume samples were adjusted at 2 m urea with 100 mm ammonium bicarbonate. Then, urine samples were digested with trypsin, which cleaves at the C terminus of lysine and arginine residues, using a ratio of 1:20 (w/w). The trypsin digestion was carried out at 37 °C overnight. Sep-Pak tC18 cartridges were used to clean and desalt the samples after the protein digestion. The peptides were eluted using 1 ml of 50% acetonitrile and 0.1% formic acid and then dried. The dried samples were stored at −20 °C until the next preparation steps were to be carried out.
Dilution Series of Stable Isotope-labeled Peptides in Urine Samples
Urine samples were resolubilized in 0.1% formic acid at a final concentration of 1 μg/μl and supplemented with three individual yeast proteins (Carboxypeptidase Y, Enolase 1, and Alcohol deshydrogenase 1; Sigma) at a final concentration of 1 ng/μl. Synthetic stable isotope-labeled peptides (AQUA peptides) with C-terminal 15N- and 13C-labeled arginine and lysine residues were provided by Thermo Fisher Scientific (Ulm, Germany) and were spiked at different concentrations (0.002, 0.006, 0.018, 0.054, 0.162, 0.486, 1.458, 4.374, 13.122, and 39.366 fmol/μl) into urine samples (for a list of the 28 peptides, see supplementary data 1).
Liquid Chromatography and MS
LC Separation
All peptide separations were carried out using an Ultimate 3000 RSLCnano system (Dionex/Thermo Fisher Scientific). For each analysis, the sample was loaded into a trap column (Acclaim PepMap, 2 cm × 75 μm inner diameter, C18, 3 μm, 100 A) (Dionex, Sunnyvale, CA) at 5 μl/min with aqueous solution containing 0.05% (v/v) trifluoroacetic acid and 1% acetonitrile. After 3 min, the trap column was set on-line with an analytical column (Acclaim PepMap RSLC, 15 cm × 75 μm inner diameter, C18, 2 μm, 100 A) (Dionex). Peptide elution was performed by applying a mixture of solvents A and B. Solvent A was HPLC grade water with 0.1% (v/v) formic acid, and solvent B was HPLC grade acetonitrile with 0.1% (v/v) formic acid. Separations were performed by applying either a linear gradient of 2% to 35% solvent B at 300 nL/min over 48 min followed by a washing step (5 min at 90% solvent B) and an equilibration step (10 min at 2% solvent B) or a stepwise gradient of 17% solvent B over 5 min followed by a washing step (4 min at 90% solvent B) and an equilibration step (10 min at 2% solvent B). One microliter of each sample was injected.
Analyses on Quadrupole-Orbitrap Instrument
SIM and PRM analyses were performed using a Q-Exactive mass spectrometer (Thermo Scientific, Bremen, Germany). A dynamic nano-electrospray source housing was utilized with uncoated SilicaTips (12 cm length, 360 μm outer diameter, 20 μm inner diameter, 10 μm tip inner diameter). For ionization, 1500 V of liquid junction voltage and a 250 °C capillary temperature were used.
For the analysis of the eight isotopolog peptides, LC separation was performed by applying a stepwise gradient (absence of chemical background) or a linear gradient (presence of chemical background). The mixture of eight isotopolog peptides was analyzed using an acquisition method combining two scan events corresponding to two SIM methods. The first SIM method employed an m/z 300–1000 mass selection, an orbitrap resolution of 70,000 (at m/z 200), target automatic gain control (AGC) values of 1 × 106, and maximum fill times of 250 ms. This method is referred to herein as the full scan method. The second SIM method employed a multiplexing degree of 8 (eight-plex method), an individual isolation of predefined target ions with a 2-Th window (supplementary data 1), a resolution of 70,000, target AGC values of 5 × 105, and maximum individual fill times of 250 ms. For the absolute sensitivity test in SIM mode, only target ions corresponding to peptides LVALVR and LVALVR were multiplexed using target AGC values of 3 × 106 and maximum fill times of 3 s. For the absolute sensitivity test in PRM mode, the acquisition method was modified by substituting the multiplexed SIM method with a sequential PRM method. This PRM method employed an isolation of target ions corresponding to peptides LVALVR and LVALVR with a 2-Th window, a resolution of 70,000 (at m/z 200), target AGC values of 3 × 106, and maximum fill times of 3 s. Fragmentation was performed with a normalized collision energy of 25, and MS/MS scans were acquired with a starting mass of m/z 100.
For the analysis of the dilution series of 28 stable isotope-labeled peptides in urine samples, LC separation was performed with the linear gradient. The acquisition method in SIM mode combined a full scan method with a time-scheduled multiplexed SIM method targeting the 28 pairs of isotopically labeled peptides/endogenous peptides in ±2-min retention time windows. The doubly charged ions within each pair of peptides (for a list of target ions, see supplementary data 1) were multiplexed using 2-Th individual isolation windows, target AGC values of 1 × 106, and maximum individual fill times of 100 ms. MS scans were acquired at a resolution of 70,000 (or 140,000 for specific experiments). For analysis in PRM mode, the time-scheduled multiplexed SIM method was substituted by a time-scheduled duplex PRM method using a resolution of 35,000 (at m/z 200) and fixed fill times of 10, 50, and 100 ms for target doubly charged ions (for list of target ions, see supplementary data 1). Fragmentation was performed with a normalized collision energy of 25, and MS/MS scans were acquired with a starting mass of m/z 100.
Large-scale PRM analyses of yeast digest samples were conducted using the linear gradient. The acquisition method combined a full scan method with a time-scheduled multiplexed PRM method. The PRM method employed a multiplexing degree of 4 to target in 1.5- to 2.5-min retention time windows with 770 tryptic yeast peptides (corresponding to 436 proteins) grouped in subsets of 4 peptides according to their chromatographic elution order. The selection of target precursor ions was performed based on the resources of peptide MS/MS data from SRM-Atlas (31) (lists of target peptides, target precursor ions, and selected fragment ions are provided in supplementary data 2). PRM acquisition was performed using a resolution of 35,000, individual isolation windows of 2 Th, target AGC values of 1 × 106, and equal individual fill times within each subset with maximum values of 30 ms. Fragmentation was performed with a normalized collision energy of 25, and MS/MS scans were acquired with a starting mass of m/z 100.
The SIM experiments described in the study were performed with a high target AGC value setting (≥ 5 × 105) in order to increase fill times, which can induce overfilling effects (i.e., saturation, lower mass accuracy) if the target AGC value is actually reached. However, most often, the maximum fill times are reached at lower AGC values, which prevents the observation of this drawback unless high abundance components are selected together with target analytes within the m/z isolation window.
Analyses on Triple Quadrupole Instrument
SRM analyses of the dilution series of 28 stable isotope-labeled peptides in urine samples were performed using a TSQ Vantage extended mass range triple quadrupole mass spectrometer (Thermo Scientific, San Jose, CA) with identical nano-electrospray and chromatographic settings. The selectivity for both Q1 and Q3 was set to 0.7 Da (full width at half-maximum). The collision gas pressure of Q2 was set at 1.5 mTorr argon. For each peptide, the selection of the two monitored transitions and the optimization of their collision energy required preliminary experiments that were performed as described elsewhere (32). The optimized time-scheduled SRM method targeted the 28 pairs of isotopically labeled peptides/endogenous peptides in ±2-min retention time windows by monitoring two transitions for each peptide within a cycle time of 2.5 s.
Data Processing
Data analysis was performed using Xcalibur (version 2.2; Thermo Fisher Scientific) and/or Pinpoint (version 1.2; Thermo Fisher Scientific). Ion chromatograms were extracted with a mass tolerance of 5 to 10 ppm for SIM data and 10 to 20 ppm for PRM data.
Quantification Based on Isotope Dilution Strategy
The area under the curve (AUC) of each target peptide ion (SIM analysis), target transition (SRM analysis), and selected fragment ion (PRM analysis) (a list of selected transitions is presented in supplementary data 1) was calculated for each sampling point based on the co-elution profiles of differently labeled peptides. The AUCs of each individual SRM or PRM transition were then summed to obtain AUCs at the peptide level.
For the analysis of the eight isotopolog peptides, the peptide AUCs were directly used to establish the corresponding dilution curves. For the analyses of the dilution series of the 28 stable isotope-labeled peptides in urine samples, the peptide AUCs were employed to calculate heavy/light peptide AUC ratios. These peptide area ratios were then used to establish the dilution curves of each peptide after applying a correction accounting for slight dilution factors induced by the sample preparation. For each technique (SRM, SIM, and PRM), a linear regression analysis was performed on the dilution curves of peptides to evaluate their linearity range. Various approaches for determining the linearity range (and limit of quantification (LOQ)) can be used. In this study, the only requirement was the use of a common set of metrics to obtain a consistent comparison of the performances of the different techniques. Here, the linearity range of measurement of each peptide was defined as the range of spiked peptide amounts for which the relative difference between deduced values of peptide area ratios from regression analysis and experimentally determined ones was lower than 25%. An additional criterion was established for the lower limit of the linearity range, referred as the LOQ. The peptide area ratio at the LOQ should be at least three times the area ratio obtained for matrix blank (no spiked peptide). The lowest sampling point of the dilution series satisfying all the evaluation criteria for a given peptide was assigned as its LOQ without further extrapolation. The regression analysis was also employed to estimate the amount of endogenous peptides in urine samples. All dilution curves, along with corresponding regression analysis, linearity range evaluation, and determined amount of endogenous peptide, are included in supplementary data 1.
Quantification Based on Label-free Strategy
All raw files obtained from large-scale PRM analyses of yeast digest samples were processed using Pinpoint (version 1.2; Thermo Fisher Scientific). The AUCs of all PRM transitions selected from resources of peptide MS/MS data from SRM-Atlas were computed by the software. The composite MS/MS spectrum of each targeted peptide was reconstructed from the AUCs of the five to eight transitions selected for the peptide. The identity of targeted peptides was verified by the correlation between the composite MS/MS spectrum and the MS/MS spectral library entry, assessed by the calculation of a Bonferonni corrected p value of correlation (33, 34). The peptide that gave a p value less than 0.1 was considered to be confidently confirmed by the library match. The AUCs and coefficients of variation (CVs) of peptides were computed by the software for quantification and reproducibility assessment. The computed AUCs and CVs for each targeted peptide/selected transition, along with calculated p values, are provided in supplementary data 2. The full set of experimental composite MS/MS spectra and reference composite MS/MS spectra is provided in supplementary data 3, and the raw data are available from the Tranche proteome repository (www.ProteomeCommons.org) with the following hash code: kx+aUJP8xwZJC+lRW0PBJecszQ0VCw5Al4d24nH40k6ZkJ9tE5eOXsf5bTUiQ40QEyETdZpHEmjXW6PpfV7TwotKNzkAAAAAAAACww== (passphrase: sebastien.gallien).
RESULTS AND DISCUSSION
The new quadrupole-orbitrap mass spectrometer presents unique capabilities in terms of sensitivity and speed, resulting from improvements in the ion transmission interface and orbitrap data processing that facilitate qualitative experiments (26, 27).
In addition, these features can be exploited for quantitative proteomic studies, as illustrated in Fig. 1A for targeted analysis workflows. In these hypothesis-driven workflows, only a predefined set of peptides, representing the proteins of interest in the context of a specific biological question, is systematically analyzed. The systematic analysis of these peptides, in contrast to the data-dependent analysis characterizing shotgun proteomics, is based on the design of specific acquisition methods including peptide attributes and instrument parameters. For SRM analysis on a triple quadrupole instrument, collecting all information required for method development can be a tedious process.
Fig. 1.

Targeted proteomic workflow employing the quadrupole-orbitrap mass spectrometer. A, From the proteins of interest defined in the context of a biological question, a set of peptides used as surrogates is selected and specifically analyzed. The targeted analysis of the peptides requires a minimal set of instrument control parameters (precursor ion m/z value and chromatographic retention time window) and can be conducted using two main operation modes of the quadrupole-orbitrap instrument: the selected ion monitoring (SIM) mode, relying on single-stage mass spectrometry, and the parallel reaction monitoring (PRM) mode, relying on tandem mass spectrometry. B, In SIM mode, a limited m/z range including the precursor ions of interest is isolated by the quadrupole (1), transmitted and accumulated in the C-trap (2), and finally transferred and analyzed in the orbitrap mass analyzer (3). The actual quantification of endogenous peptides is performed on the peak areas of corresponding precursor ions. C, In PRM mode, a limited m/z range including the precursor ions of interest is isolated by the quadrupole (1), transmitted via the C-trap to the HCD cell where corresponding fragment ions are generated and accumulated (2). Fragment ions are then transferred back into the C-trap (3) and eventually injected and analyzed in the orbitrap mass analyzer (4). The actual quantification of endogenous peptides is performed on the peak areas of corresponding selected fragment ions.
For quadrupole-orbitrap analysis, two main modes of operation, which are discussed extensively below, were designed for targeted quantification: the SIM mode and the PRM mode, relying on single-stage and tandem mass analysis, respectively. Both operation modes require a minimal set of instrument control parameters limited to the precursor ion m/z value and the predicted chromatographic retention time window.
The actual quantification of endogenous peptides is performed on the peak areas of corresponding precursor ions and selected fragment ions for SIM and PRM analyses, respectively. When an isotope dilution strategy is applied, the extraction of ion traces is carried out on both endogenous and isotopically labeled peptides. This is the typical workflow for targeted proteomic experiments that was used throughout this study.
Main Modes of Operation for Targeted Analysis (SIM and PRM Modes)
The implementation of a quadrupole mass filter in the new configuration of the mass spectrometer enables the design of various acquisition methods for targeted analysis based on two main modes of operation, namely, SIM mode and the PRM mode. The basic operating procedures of these modes are described in Figs. 1B and 1C. SIM mode consists of the isolation of a limited m/z range including the precursor ions of interest by the quadrupole, followed by the accumulation of transmitted ions in the C-trap and their eventual transfer and analysis in the orbitrap mass analyzer (Fig. 1B). In PRM mode, the isolation of the targeted precursor ions is similarly performed, but they are transferred via the C-trap to the HCD cell, where they undergo fragmentation as soon as they are introduced. The generated fragment ions are thus accumulated in the HCD cell before being transferred back into the C-trap and eventually injected and analyzed in the orbitrap mass analyzer (Fig. 1C). In these acquisition modes, the ability of the quadrupole to select a restricted m/z range offers new opportunities for quantification in complex samples by selectively enriching low abundant components (i.e., increasing the ratio of analytes of interest/matrix). It also allows the C-trap to fill for longer times and thus enables an increased signal-to-noise ratio of targeted ions measured in the orbitrap. Over the acquisition period, the fill time for a given m/z range is calculated according to a previous MS scan, the desired number of charges, and a maximal preset value.
Design of Acquisition Methods for Targeted Quantification Experiments
The aim of a targeted analysis is to quantify many peptides during one LC separation. To monitor the full set of targeted analytes, several acquisition methods were designed in SIM mode. In all experiments, the LC retention time was used as a constraint to schedule monitoring of the peptides of interest during a time window corresponding to their expected retention time, which is equivalent to time-scheduled SRM (35). However, in practice, several peptides of the predefined set could co-elute and thus be monitored within the same elution time windows. In such a case, the SIM method is designed to perform the sequential isolations of individual target ions using narrow windows (typically 2 Th) and their accumulation in C-trap, followed by their transfer and their final detection in the orbitrap (Fig. 2A, left panel). This method is applicable as long as the number of co-eluting peptides is relatively small. With many co-eluting peptides, the sequential acquisition process would significantly increase the cycle time and thus compromise the precise quantification because of the limited number of data points collected across the elution profiles. In this scheme, assuming fill times shorter than the MS transient length, the cycle time is defined by the number of peptides and the orbitrap transient length, which can be as short as 64 ms. To overcome this issue, two additional acquisition methods were explored that rely on the simultaneous measurement of several peptides of interest in a single orbitrap scan. A first option consists in applying the full acquisition process to different peptides simultaneously. In this case, all the different precursor ions are isolated in one single window, accumulated in the C-trap, and eventually transferred and detected in the orbitrap (Fig. 2B, left panel). Nevertheless, if one wishes to keep the wide dynamic range provided by SIM mode, the isolation window cannot be dramatically wide, which occurs if the precursor ions cover a wide m/z range. This method is thus particularly indicated for, but limited to, the isotope dilution strategy, which implies the analysis of pairs of isotopically labeled/nonlabeled peptides with close m/z values. A more versatile and elegant approach to dealing with the analysis of high numbers of peptides exploits the multiplexing capabilities of this instrument, and more specifically of the C-trap in this instance. In such multiplexed experiments, the targeted peptides are mass analyzed concomitantly by applying multiple isolation cycles using narrow windows (typically 2 Th) to accumulate and store the selected precursor ions in the C-trap. They are then transferred and detected in one single scan in the orbitrap (Fig. 2C, left panel). The current acquisition program allows one to conduct up to 10-plex experiments (i.e., 10 isolation cycles/orbitrap scan) covering discontinuously the full m/z range, with a required time to switch from the isolation of one m/z range to the following one being negligible (ms range). In contrast to the sequential and simultaneous methods that use fill times typically shorter than the MS transient length, with the multiplexed method the summation of the fill times is prone to exceed the transient length, especially if low abundant compounds are targeted. However, this method remains very pertinent for providing a shorter overall cycle time than the sequential method while keeping the advantages of SIM mode, and it is preferred in the experiments described in this work. As described below, these methods can also be used in conjunction with the PRM mode (Fig. 2, right panel). Practical examples of large-scale PRM experiments, along with appropriate time management settings, are discussed in the section “Exploiting Multiplexing Capabilities for Directed Discovery Experiments.”
Fig. 2.

Acquisition methods for targeted quantification experiments. A, Sequential SIM method (left panel). Target ions are individually and sequentially isolated by the quadrupole in narrow windows, accumulated in the C-trap, and eventually transferred in the orbitrap to be detected. B, Simultaneous SIM method (left panel). All target ions are isolated by the quadrupole in one single window and then accumulated in the C-trap and detected in the orbitrap simultaneously. C, Multiplexed SIM method (left panel). Target ions are sequentially isolated in multiple windows by the quadrupole and accumulutated in the C-trap, where they are stored together before being finally detected in the orbitrap in one single scan. All these methods can be used in conjunction with the PRM modes (right panel). In this case, the HCD cell is used as the temporary accumulation/storage device for fragment ions.
Analytical Performances of the Quadrupole-Orbitrap Mass Spectrometer Operated in SIM Mode
To evaluate the performance of the instrument and, more specifically, its new quantification capabilities based on HR/AM measurements, an evaluation of the sensitivity and the dynamic range was conducted using two SIM methods. The first one used a broad band mass selection (m/z 300–1000, 700 Th) and was very similar to a full scan MS analysis, whereas the second was a multiplexed method using narrow windows (8 × 2 Th). Both methods operated in high resolution mode (70,000 at m/z 200, which corresponds to transient lengths of 256 ms), using a target AGC value of 1e6 for broad band mass selection and 5 × 105 for narrow band mass selection, and with maximum fill times of 250 ms. These methods were combined in a unique acquisition as two scan events and were applied to the analysis of a mixture of eight synthetic isotopolog peptides. These peptides had the same amino acid sequence (LVALVR) and different isotope labeling on individual amino acids based on 15N and 13C incorporation. They were prepared in various amounts between 10 amol and 21.9 fmol, with a 3-fold dilution factor for each point (Table I). With this mixture, an eight-points dilution series can be measured in one single LC/MS analysis, which constitutes a new effective method for assessing the performance of a mass spectrometer. The neat mixture was submitted to triplicate nano-LC/MS analyses using the previously described method. The AUCs were calculated based on the ion chromatograms of the corresponding doubly charged precursor ions and were used to establish the dilution curve. The two acquisition methods exhibited similar results. The full set of synthetic peptides was unambiguously detected, and the corresponding dilution curves clearly demonstrate that all the amounts lie in the linearity range of measurement (supplemental Fig. S1). An intrascan linearity range exceeding 3.5 orders of magnitude can thus be expected, as the instrument limits were not reached.
Although the range of abundance measured in these experiments corresponds to the expected amount of potential biomarkers in clinical samples, the analyses were here performed in the absence of the massive chemical background characterizing this type of sample. To better mimic this real situation, analysis was repeated on the mixture spiked in 500 ng of yeast digest. In this case, the benefits of using narrow isolation windows clearly arose. Relative to the wide mass selection (or full scan MS), the multiplexed SIM method provided a 10-fold increase in sensitivity, illustrated by a decrease in the LOQ from 405 amol to 45 amol (Fig. 3, supplementary data 1). As expected, for the narrow isolation windows of the multiplexed SIM method, the drastic background cleaning (Fig. 3A, bottom panel) induced a dramatic increase in fill times—up to 250 ms for the ions of lowest abundance, versus 0.3 ms for full scan at the apex of the elution profile. The high background isolated along with the targeted peptides in the full scan MS method (Fig. 3B, bottom panel) resulted in a lower ion capacity available for the compounds of interest and thus in a linearity range shrinking.
Fig. 3.

Comparison of the analytical performance of multiplexed SIM (A) and full scan MS (B) analyses. The dilution curves were obtained from one single LC/MS analysis with both acquisition methods on a set of isotopolog peptides (Table I) prepared in various amounts (between 5 amol and 10.9 fmol, with a 3-fold dilution factor for each point) in 500 ng of yeast digest. The linearity range of the dilution curves is displayed in the upper panel (i.e., 45 amol to 10.9 fmol for SIM analysis and 405 amol to 10.9 fmol for full scan analysis) (supplementary data 1). The corresponding mass spectra at the apex of the elution profile are shown in the bottom panel, along with corresponding zoomed image. Detected precursor ions of isotopologs are indicated by dark blue circles or light blue circles for measurements that lie in or out of the linear range, respectively.
To push the limits of the instrument and to determine its absolute sensitivity and dynamic range, an additional experiment was performed. Here, a series of samples were prepared by spiking the peptide LVALVR (m/z 343.745, [M+2H]2+) in tiny concentrations (0.9 amol/μl, 0.3 amol/μl, 0.1 amol/μl, 0.033 amol/μl) into aqueous solutions containing 20 fmol/μl of the peptide LVALVR (m/z 355.773, [M+2H]2+). The two peptides were targeted in multiplexed SIM analyses of the mixtures with a very high fill time for the low abundance peptide (up to 3 s). It turned out that this peptide could be detected with as little as 0.035 amol injected. The intensities at the apexes of the ion chromatograms of the two peptides displayed in supplemental Fig. S2 were 29.4 and 21,500,000 (arbitrary units) for injected amounts of 0.035 amol and 20 fmol, respectively. An intrascan dynamic range exceeding 5 orders of magnitude can thus be estimated. It is noteworthy that the ratio of the precursor ion intensities (730,000) is relatively similar to the ratio of the corresponding peptide amounts (600,000). These exceptional results are not representative of the performance routinely achieved with complex biological samples, but they demonstrate the exceptional intrinsic sensitivity and dynamic range of the instrument. They clearly result here from the low chemical background, which is reflected in a significant increase in the signal-to-noise ratio of targeted ions, as well as the 3-s fill time applied to the accumulation of the low abundance ion. Such high fill times are not achievable in full scan MS analysis, even with this type of sample, without overfilling trapping devices.
Application of SIM Analysis to Protein Quantification in Urine Samples
To provide a relevant estimation of the performance of the quadrupole-orbitrap mass spectrometer with biological samples, a study aiming at the targeted quantification of proteins in urine sample was designed. In this study, quantification was performed using as internal standards 28 highly purified isotopically labeled peptides representing seven endogenous proteins and three exogenous yeast proteins spiked in the matrix. Dilution series of internal standards were prepared to obtain isotopically labeled peptides at a concentration ranging between 2 amol/μl and 40 fmol/μl in 1 μg/μl of proteins from urine digest (nine sampling points and one matrix blank point).
The samples were analyzed in triplicate on the quadrupole-orbitrap instrument, which was operated in time-scheduled multiplexed SIM method targeting the 28 pairs of heavy and light peptides. More precisely, the doubly charged precursor ions of each pair of heavy/light peptides were targeted in the multiplexed SIM method at a resolution of 70,000 (at m/z 200), and a full scan event was also included in order to monitor the full profile as a quality control. A preset target AGC value of 1 × 106 and a maximum fill time of 100 ms for each target ion were used in order to detect compounds of very low abundance. In the course of the analysis, up to ten pairs of heavy/light peptides were monitored in overlapping retention time windows. The heavy/light AUC ratios were calculated based on the ion chromatograms of each pair of doubly charged precursor ions and were used to establish the dilution curves.
For benchmarking the SIM method with the reference proteomic quantitative technique, the analyses were also performed in triplicate on a triple quadrupole mass spectrometer operated in time-scheduled SRM mode. In that case, for each peptide, two transitions (pairs of precursor/fragment ions) were selected on the basis of their intensity and their purity in the matrix and monitored continuously at their optimal collision energy, which required preliminary experiments and thus made the method's development less straightforward. The dilution curves were established for each pair of targeted peptides from the heavy/light AUC ratios of the traces recorded for the two transitions.
From the dilution curves obtained on both instruments, the LOQ and the linearity range of measurements of each isotopically labeled peptide were determined (supplementary data 1). A typical example is illustrated with the quantification of endogenous human transferrin in urine (Fig. 4). The dilution curves of the three isotopically labeled peptide surrogates for this protein are reported in Figs. 4A (SIM analysis) and 4B (SRM analysis). They indicate better sensitivity with SIM analysis, illustrated by lower LOQs, for two peptides of three (DGAGDVAFVK and EGYYGYTGAFR), whereas the last one is analyzed with more sensitivity with the SRM technique (SASDLTWDNLK). It is noteworthy that the amounts of endogenous peptides were determined from the dilution curves with high consistency (less than 10% difference between the techniques) (Fig. 4C). Comparable results were obtained on both instruments, and this was also the overall trend observed at the level of the study. Within the linearity range of measurements, quantification results were obtained with low CVs (≤15%) for 94% and 97% of sampling points analyzed via SIM and SRM, respectively (supplementary data 1). Fig. 5A presents the number of peptides that could be quantified at the different sampling points of the dilution series. It attests to an overall similar distribution of the LOQs with both instruments. However, with SIM analysis on the quadrupole-orbitrap instrument, a higher number of peptides could be quantified at the lowest spiked-in amounts. The correlation plot presenting the comparison of determined amounts of endogenous peptides for SIM and SRM analyses (Fig. 5B) indicates an excellent consistency of quantification results (slope ≈ 0.94, r2 ≈ 0.96). A high LOQ was determined for some peptides, which can be explained by poor ionization and/or fragmentation efficiencies or by the presence of interferences.
Fig. 4.

Quantification of endogenous human transferrin in urine samples using SIM and SRM analysis. Three isotopically labeled peptides representing the protein were spiked in various amounts in urine samples to obtain their dilution series. The dilution curves were established for each pair of endogenous/isotopically labeled peptides from the AUC ratios calculated on predefined precursor ions (SIM) or transitions (SRM). A, Linearity range of the dilution curves obtained from SIM analysis. B, Linearity range of the dilution curves obtained from SRM analysis. C, A lower LOQ was determined for two peptides via SIM analysis (DGAGDVAFVK and EGYYGYTGAFR), whereas SRM analysis provided a lower LOQ for the third one (SASDLTWDNLK). The amounts of endogenous peptides were determined with less than 10% difference between the techniques.
Fig. 5.

Comparison of quantification results obtained with SIM and SRM analysis. A, Numbers of peptides that could be quantified with each technique at the different amounts of the dilution series. B, Correlation between the amounts of endogenous peptides determined via SIM analysis (y-axis) and SRM analysis (x-axis). Only determined amounts higher than corresponding LOQs are displayed and were used to calculate the linear regression.
In the latter case, the very high resolution capabilities of the Q-Exactive were evaluated to address the issue and increase the selectivity of measurements. This evaluation was conducted on the peptide SDLAVPSELALLK, which presents a relatively high LOQ (more than 4 fmol) in both SIM and SRM analysis. The SIM mass spectrum of the corresponding doubly charged precursor ion (m/z 682.40, [M+2H]2+) acquired at the determined LOQ definitely validates the presence of an interfering ion of similar mass (m/z 682.37, [M+3H]3+) (Fig. 6A). Doubling the resolution to 140,000 allowed the separation of the targeted ion from the interfering signal (Fig. 6B) and extended the linearity by 2.5 orders of magnitude, resulting in an LOQ of 20 amol (Fig. 6C, supplementary data 1). However, as doubling the resolution requires doubling the transient length, this very high resolution mode should not be systematically used when large sets of peptides are targeted, so as to avoid the disruption of cycle time.
Fig. 6.

Selectivity of measurements in SIM and PRM modes. The peptide SDLAVPSELALLK was spiked at a concentration of 4 fmol/μl in 1 μg/μl of urine digest. A, SIM mass spectrum acquired in the m/z range of 681.40–683.40 at a resolution of 70,000 (at m/z 200). The doubly charged ion (m/z 682.40, [M+2H]2+) of the peptide SDLAVPSELALLK (orange circle) is interfered with by ions of similar m/z (m/z 682.37, [M+3H]3+). B, SIM mass spectrum acquired at a resolution of 140,000 (at m/z 200). Doubling the resolution allowed us to distinguish the ion of interest (orange circle) from interferences. C, Linearity range of the dilution curves established by SIM (resolutions of 70,000 and 140,000) and PRM analyses from the dilution series of SDLAVPSELALLK (2 amol/μl to 40 fmol/μl) prepared in 1 μg/μl of urine digest. The LOQ of 20 amol observed with SIM analysis at a resolution of 140,000 and PRM analysis is a significant improvement over the LOQ observed with SIM analysis at a resolution of 70,000.
MS/MS Capabilities and Performances of Parallel Reaction Monitoring Mode
The MS/MS capabilities of the quadrupole-orbitrap instrument using the HCD cell can be exploited not only to fragment peptides in the data dependent acquisition of shotgun proteomic experiments or confirm the identity of targeted peptides in SIM analysis (through data dependent triggering), but also to systematically fragment targeted peptides for quantification purposes (Fig. 1). In the parallel reaction monitoring mode, the quantification process is similar to SRM; that is, it relies on measuring peak areas of transition (pairs of precursor/fragment ions) traces. However, in contrast to SRM experiments, here all transitions from a common precursor ion are monitored in parallel, and their selection is performed post-acquisition. A priori knowledge of MS/MS fragmentation patterns is not required in order to design the acquisition method, making the method development more straightforward. In addition, the high resolution of fragment ion measurement combined with the MS1 stage narrow filter provides additional selectivity. The different acquisition methods extensively described for the SIM mode can also be used with the PRM mode (Fig. 2).
To assess the sensitivity of the PRM mode relative to that of the SIM mode, an absolute sensitivity test measuring tiny amounts of the isotopically labeled peptide LVALVR (“Analytical Performance of the Quadrupole-Orbitrap Mass Spectrometer Operated in SIM Mode”) was adapted. Here the peptides LVALVR (m/z 343.745, [M+2H]2+) and LVALVR (m/z 355.773, [M+2H]2+) were targeted in a sequential analysis of the mixture with a very high fill time for the low abundant peptide (up to 3 s), and the ion chromatograms of the fragment y4+ (the most intense fragment ion) were extracted for both peptides. Although slightly higher than the limit of detection determined with SIM mode (3-fold increase), which can be explained by the dilution of the precursor ion signal into several fragment ions, a minimal amount of peptide as low as 0.1 amol could be detected with the MS/MS mode (supplemental Fig. S3). A very satisfying correlation was also observed between the ratio of the fragment ion intensities and the ratio of the corresponding peptide amounts (195,000 versus 200,000).
The hypothesized improved selectivity of the PRM mode was verified by a subset of pairs of heavy/light peptides targeted in the urine study (“Application of SIM Analysis to Protein Quantification in Urine Samples”) by analyzing the same samples on the quadrupole-orbitrap instrument operated in time-scheduled duplex PRM mode using a resolution of 35,000 (at m/z 200). In this acquisition method, for each targeted pair of analytes, first the endogenous peptide (“light”) was isolated in a 2-Th window and transferred to the HCD cell to undergo dissociation. This was followed by the accumulation and storage of its fragment ions; then the process was repeated on the isotopically labeled counterpart (“heavy”). Finally, fragment ions of both peptides, which had been intermediately stored together in the HCD cell, were injected and analyzed in one single scan in the orbitrap mass analyzer (Fig. 2C). Within each pair of targeted peptides, fragment ion chromatograms were extracted from the same MS/MS spectra (supplemental Fig. S4) and used to establish the dilution curves. Although quantification could be done on many fragment ions, only the traces of the two transitions monitored in SRM analysis were extracted in the PRM quantification process for a direct comparison between both techniques. The pairs of targeted peptides in the experiment were selected on the basis of the analytical performance of their monitoring with SIM and SRM techniques in order to evaluate the benefits of targeted quantification with high resolution PRM in various situations. The most interesting case corresponded to the peptide SDLAVPSELALLK interfered in SIM analysis at a resolution of 70,000 in the urine study and for which a high LOQ (more than 4 fmol) was determined in both SIM and SRM analysis. Similar to the impact of doubling the resolution of the SIM measurement from 70,000 to 140,000, the PRM mode provided increased selectivity and allowed the determination of an LOQ of 20 amol for the isotopically labeled peptide, as illustrated by the dilution curve displayed in Fig. 6C. In addition, unlike SIM analysis at a resolution of 140,000, the increase in selectivity resulting from the PRM mode does not compromise the cycle time, as the HCD fragmentation time is negligible in the overall acquisition process. Other examples were the peptides LLLTSAPSLATSPAFR, EGYYGYTGAFR, VSTLPAITLK, and NVNDVIAPAVFK, which previously exhibited LOQs of 50, 20, 50, and 490 amol for SIM analyses and 160, 490, 160, and 160 amol for SRM analyses. Quantification using PRM analysis provided a constant LOQ of 20 amol for all four peptides, as illustrated by the dilution curves shown in supplemental Fig. S5 and supplementary data 1, which matched or outperformed the most effective technique included in the comparison (i.e., SIM or SRM). Although evaluated on a limited set of peptides, parallel reaction monitoring turned out to be a very promising quantification approach on the quadrupole-orbitrap instrument to significantly increase the selectivity of the measurement without noticeably compromising the sensitivity performance. In addition, this method has the advantage of monitoring in parallel multiple transitions without a priori knowledge, which is critical for assessing the presence of interferences.
Exploiting Multiplexing Capabilities for Directed Discovery Experiments
Although initially focused on the absolute quantification of a relatively small number of peptides, the objective of targeted proteomics has extended over the past few years toward the quantification of very large sets of peptides (i.e., the detection of relative changes between samples). This type of experiment, defined as a directed discovery experiment, can, for instance, establish the detection of biomarker candidates in bodily fluids. In SRM analyses, despite the latest developments in acquisition techniques (33, 36), scaling up the number of targeted analytes often implies decreasing the number of transitions monitored for each peptide to maintain acceptable cycle times (8), which can limit the overall specificity of measurements. In this context, PRM analysis on the quadrupole-orbitrap instrument was evaluated because of its ability to record the signal of the full set of transitions without any direct impact on cycle times. The critical parameters driving such a large-scale time-scheduled PRM experiment are the number of peptides monitored at a given retention time, the fill time for each peptide, and the transient length used for orbitrap acquisition, as briefly mentioned in the section “Main Modes of Operation for Targeted Analysis (SIM and PRM Modes).”
Assuming that in directed discovery experiments scale comes first to the detriment of sensitivity, the maximum fill times were set lower than the transient length. Using a sequential acquisition method, the cycle time at a given point in time is thus expressed as follows: Cycle_time = Number_of_peptides × Transient_length.
In practice, a maximal cycle time is adjusted to allow the collection of enough data points to describe properly the elution profile of targeted peptides. As a consequence, the number of peptides that can be monitored is directly dependent on the transient length. Relaxing resolution, which decreases transient length, is thus an effective way to increase the number of peptides to be analyzed, but it can affect the selectivity of measurements. Typical sets of parameters used in large-scale PRM analysis using a sequential acquisition method are displayed in Table II under the designation “multiplexing degree 1.” Relaxing the resolution from 35,000 to 17,500 (at m/z 200), together with a maximum fill time of 60 instead of 120 ms, results in a 2-fold increase in the number of peptides analyzed (15 to 30) within the cycle time (2 s). As mentioned, the use of a multiplexed acquisition method can overcome this practical limit of the sequential acquisition method by analyzing several peptides in the same orbitrap scan. In this case, the maximal number of peptides that can be analyzed within the cycle time is expressed as
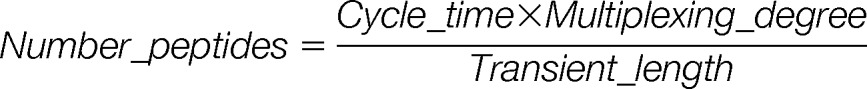 |
assuming the adjustment
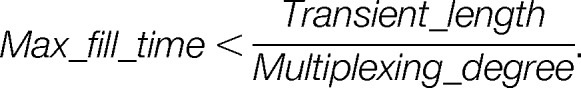 |
Table II. Typical sets of parameters used in large scale parallel reaction monitoring analysis.
| Parameters | Multiplexed MS/MS analysis (parallel reaction monitoring) | ||||||
|---|---|---|---|---|---|---|---|
| Multiplexing degree | 1 | 1 | 2 | 4 | 4 | 8 | 8 |
| Isolation window/peptide (Th) | 2 | 2 | 2 | 2 | 2 | 2 | 1 |
| Max injection time/peptide (ms) | 1–60 | 1–120 | 1–30 | 1–30 | 1–30 | 1–30 | 1–15 |
| Transient length (ms) @ m/z = 200 Th | 64 | 128 | 64 | 128 | 128 | 256 | 128 |
| Resolution | 17.5k | 35k | 17.5k | 35k | 35k | 70k | 35k |
| Overall isolation window (Th) | 2 | 2 | 4 | 8 | 8 | 16 | 8 |
| Preset cycle time | 2 | 2 | 2 | 2 | 3 | 3 | 3 |
| Nb peptides analyzed/cycle time | 31 | 15 | 62 | 62 | 92 | 92 | 184 |
Several combinations in the values of the multiplexing degree and the orbitrap resolution (transient length) can thus be made to increase the scale of the experiment (Table II), but in any case the maximal fill time becomes the limiting factor. Among the sets of parameters shown in Table II, an eight-plex acquisition method using an orbitrap resolution of 35,000 (at m/z 200) was suggested to monitor 184 peptides within the cycle time (3 s) but implied applying fill times lower than 15 ms, which can prevent proper analysis of low abundance peptides. The combinations also have an effect on the selectivity of PRM measurements, depending on one hand on the overall m/z isolation window resulting from the multiplexing degree and on the other hand on the resolution of the orbitrap.
Only an experimental study fully dedicated to the evaluation of the effect of different combinations of these instrumental parameters on sensitivity/selectivity/scale could really allow one to make recommendations about the most effective parameters for a given type of experiment. Here, to establish the baseline expectations, a basic multiplexed PRM acquisition method was prepared for the targeted analysis of 770 tryptic peptides from a yeast digest in one 60-min LC separation. This method used middle-range values of orbitrap resolution (35,000) and m/z isolation window (4 (multiplexing degree) × 2 Th (individual isolation window)) and a maximum fill time of 30 ms. A time-scheduled experiment was designed based on previously reported studies (33, 36). In the present experiment, a set of 770 tryptic peptides from yeast digest (representing 436 proteins) were analyzed using 1.5- to 2.5-min retention time windows. As a result, up to 60 peptides were monitored in overlapping retention time windows, which allowed the maintenance of a cycle time of less than 2 s in the course of the analysis (supplemental Fig. S6). The yeast digest sample (0.2 μg injected) was analyzed in duplicate, and quantification was performed on AUCs of the five to eight reference transitions reported in SRM-Atlas (31) for these peptides (lists of target peptides, target precursor ions, and selected fragment ions are provided in supplementary data 1). To confirm that the fragment ion chromatograms used for quantification corresponded to the targeted peptides, a composite MS/MS spectrum was reconstructed from the signals of individual transitions and compared with the reference composite MS/MS spectrum stored in SRM-Atlas for similarity assessment (33, 34). The processing process is illustrated in supplemental Fig. S7 for four multiplexed yeast peptides for which fragment ions were measured in the same MS/MS spectra. For the four peptides, a high correlation was obtained between experimental and reference composite MS/MS spectra (p value ≤ 0.1 and dot-product > 0.9), attesting to the correspondence between the signal and targeted peptides. At the level of the whole study, 605 peptides (≈ 80%) from yeast proteins expressed in a wide range of concentrations, as previously reported (37), were verified by the composite MS/MS spectra with a p value ≤ 0.1 (supplementary data 1). The full set of experimental and reference composite MS/MS spectra is provided in supplementary data 3. Among these peptides, very consistent quantification results were obtained, with over 95% of the peptides exhibiting coefficients of variation below 10% (supplementary data 2). Large-scale parallel reaction monitoring analysis on the quadrupole-orbitrap instrument thus turned out to be an effective approach for conducting directed discovery experiments. The systematic quantitative analysis of more than 1000 peptides while maintaining high specificity resulting from the monitoring of the full sets of transitions can hence realistically be considered with optimized instrumental parameters.
Conclusion and Outlook
A new quadrupole-orbitrap instrument was employed to conduct targeted proteomic quantification experiments. In order to carry out such experiments, new quantitative methods were explored to leverage the unique configuration of the instrument, exhibiting unique HR/AM capabilities. Two main modes were employed, including targeted analysis in SIM and PRM modes.
The SIM mode, benefiting from the ability of the quadrupole to isolate specific m/z ranges, can operate with significantly increased fill times. The resulting sensitivity/dynamic range performance dramatically exceeds that of the full scan MS mode for the analysis of both simple and complex samples. When benchmarked with the reference proteomic quantitative technique (i.e., SRM on the triple quadrupole instrument), the SIM technique appears to be a realistic quantification alternative for peptides in complex biological samples. It compares favorably with SRM by exhibiting LOQs in the low amol range.
The PRM mode provides an additional stage of selectivity without significantly compromising the sensitivity of measurements. It has the potential to improve the LOQs of peptides that suffer from interferences when measured in SIM and/or SRM modes, as illustrated by several examples in this study. The PRM mode offers additional performance for the precise quantification of peptides in complex samples such as bodily fluids. The effectiveness of PRM analysis is further enhanced by the multiplexing capabilities of the instrument, which allow one to increase the number of targeted analytes, as demonstrated by the directed discovery experiment applied to about 800 yeast peptides performed in one 60-min LC/MS experiment.
Quantitative experiments conducted on the quadrupole-orbitrap instrument generate data of increased quality that directly enhance analytical performance, especially when applied to complex biological samples. The experiments reported here on limited sets of peptides aimed to demonstrate proof of principle and to establish the baseline defining the expectations of targeted quantification using a trapping instrument. Additional refinements of the methods, including better control of some acquisition parameters, are likely to further expand the performance. The quality of data obtained on an HR/AM mass analyzer, together with a simplified experimental design requiring a very limited set of a priori parameters, opens new avenues in quantitative analysis in biological samples for which increased selectivity is required.
Supplementary Material
Footnotes
* This work was supported by a PEARL grant from the Fonds National de la Recherche Luxembourg (FNR).
 This article contains supplementary data 1 to 3 and supplemental Figs. S1 to S7.
This article contains supplementary data 1 to 3 and supplemental Figs. S1 to S7.
2 Gallien, S., Souady, J., and Domon, B., unpublished results.
3 Gallien, S., Duriez, E., Kim, Y. J., Domon, B., Hao, Z., Kellmann, M., Moehring, T., and Huhmer, A. (2011) Targeted protein quantification in urine samples using a new quadrupole-orbitrap mass spectrometer. Proceedings of the 59th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 5–9, 2011, abstract number 1900, American Society for Mass Spectrometry, Santa Fe, NM, USA.
4 The term of parallel reaction monitoring (PRM) is introduced by analogy to selected reaction monitoring (SRM). Whereas in SRM a single selected reaction (transition) is monitored for a peptide at a given point in time, in PRM virtually all reactions (fragments/transitions) from a common precursor ion are monitored in parallel.
1 The abbreviations used are:
- SRM
- selected reaction monitoring
- SIM
- single ion monitoring
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- LC
- liquid chromatography
- HCD
- higher energy collisional dissociation
- HR/AM
- high resolution/accurate mass
- LOQ
- limit of quantification
- AGC
- automatic gain control
- PRM
- parallel reaction monitoring
- AUC
- area under the curve
- CV
- coefficient of variation.
REFERENCES
- 1. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 2. Domon B., Aebersold R. (2006) Mass spectrometry and protein analysis. Science 312, 212–217 [DOI] [PubMed] [Google Scholar]
- 3. Aebersold R. (2009) A stress test for mass spectrometry-based proteomics. Nat. Methods 6, 411–412 [DOI] [PubMed] [Google Scholar]
- 4. Domon B., Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 [DOI] [PubMed] [Google Scholar]
- 5. Anderson N. L., Anderson N. G. (2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 6. Yost R. A., Enke C. G. (1979) Triple quadrupole mass spectrometry for direct mixture analysis and structure elucidation. Anal. Chem. 51, 1251–1264 [DOI] [PubMed] [Google Scholar]
- 7. Hager J. W., Yves Le Blanc J. C. (2003) Product ion scanning using a Q-q-Q linear ion trap (Q TRAP) mass spectrometer. Rapid Commun. Mass Spectrom. 17, 1056–1064 [DOI] [PubMed] [Google Scholar]
- 8. Gallien S., Duriez E., Domon B. (2011) Selected reaction monitoring applied to proteomics. J. Mass Spectrom. 46, 298–312 [DOI] [PubMed] [Google Scholar]
- 9. Hoke S. H., Morand K. L., Greis K. D., Baker T. R., Harbol K. L., Dobson R. L. M. (2001) Transformations in pharmaceutical research and development, driven by innovations in multidimensional mass spectrometry-based technologies. Int. J. Mass Spectrom. 212, 135–196 [Google Scholar]
- 10. Kostiainen R., Kotiaho T., Kuuranne T., Auriola S. (2003) Liquid chromatography/atmospheric pressure ionization-mass spectrometry in drug metabolism studies. J. Mass Spectrom. 38, 357–372 [DOI] [PubMed] [Google Scholar]
- 11. Barr J. R., Maggio V. L., Patterson D. G., Jr., Cooper G. R., Henderson L. O., Turner W. E., Smith S. J., Hannon W. H., Needham L. L., Sampson E. J. (1996) Isotope dilution—mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin. Chem. 42, 1676–1682 [PubMed] [Google Scholar]
- 12. Desiderio D. M., Kai M. (1983) Preparation of stable isotope-incorporated peptide internal standards for field desorption mass spectrometry quantification of peptides in biologic tissue. Biomed. Mass Spectrom. 10, 471–479 [DOI] [PubMed] [Google Scholar]
- 13. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fortin T., Salvador A., Charrier J. P., Lenz C., Lacoux X., Morla A., Choquet-Kastylevsky G., Lemoine J. (2009) Clinical quantitation of prostate-specific antigen biomarker in the low nanogram/milliliter range by conventional bore liquid chromatography-tandem mass spectrometry (multiple reaction monitoring) coupling and correlation with ELISA tests. Mol. Cell. Proteomics 8, 1006–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Keshishian H., Addona T., Burgess M., Mani D. R., Shi X., Kuhn E., Sabatine M. S., Gerszten R. E., Carr S. A. (2009) Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 8, 2339–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuhn E., Addona T., Keshishian H., Burgess M., Mani D. R., Lee R. T., Sabatine M. S., Gerszten R. E., Carr S. A. (2009) Developing multiplexed assays for troponin I and interleukin-33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clin. Chem. 55, 1108–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuzyk M. A., Smith D., Yang J., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H. (2009) Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 8, 1860–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anderson L., Hunter C. L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]
- 19. Sherman J., McKay M. J., Ashman K., Molloy M. P. (2009) How specific is my SRM?: the issue of precursor and product ion redundancy. Proteomics 9, 1120–1123 [DOI] [PubMed] [Google Scholar]
- 20. Abbatiello S. E., Mani D. R., Keshishian H., Carr S. A. (2010) Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin. Chem. 56, 291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Olsen J. V., Schwartz J. C., Griep-Raming J., Nielsen M. L., Damoc E., Denisov E., Lange O., Remes P., Taylor D., Splendore M., Wouters E. R., Senko M., Makarov A., Mann M., Horning S. (2009) A dual pressure linear ion trap orbitrap instrument with very high sequencing speed. Mol. Cell. Proteomics 8, 2759–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Geiger T., Cox J., Mann M. (2010) Proteomics on an orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol. Cell. Proteomics 9, 2252–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seppala U., Dauly C., Robinson S., Hornshaw M., Larsen J. N., Ipsen H. (2011) Absolute quantification of allergens from complex mixtures: a new sensitive tool for standardization of allergen extracts for specific immunotherapy. J. Proteome Res. 10, 2113–2122 [DOI] [PubMed] [Google Scholar]
- 24. Wong R. L., Xin B., Olah T. (2011) Optimization of Exactive Orbitrap acquisition parameters for quantitative bioanalysis. Bioanalysis 3, 863–871 [DOI] [PubMed] [Google Scholar]
- 25. Kaufmann A., Widmer M., Maden K. (2010) Post-interface signal suppression, a phenomenon observed in a single-stage orbitrap mass spectrometer coupled to an electrospray interfaced liquid chromatograph. Rapid Commun. Mass Spectrom. 24, 2162–2170 [DOI] [PubMed] [Google Scholar]
- 26. Michalski A., Damoc E., Hauschild J. P., Lange O., Wieghaus A., Makarov A., Nagaraj N., Cox J., Mann M., Horning S. (2011) Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole orbitrap mass spectrometer. Mol. Cell. Proteomics 10, M111.011015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nagaraj N., Alexander Kulak N., Cox J., Neuhauser N., Mayr K., Hoerning O., Vorm O., Mann M. (2012) System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top orbitrap. Mol. Cell. Proteomics 11, M111.013722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Olsen J. V., Macek B., Lange O., Makarov A., Horning S., Mann M. (2007) Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 4, 709–712 [DOI] [PubMed] [Google Scholar]
- 29. McAlister G. C., Phanstiel D. H., Brumbaugh J., Westphall M. S., Coon J. J. (2011) Higher-energy collision-activated dissociation without a dedicated collision cell. Mol. Cell. Proteomics 10, O111.009456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Picotti P., Bodenmiller B., Mueller L. N., Domon B., Aebersold R. (2009) Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Picotti P., Lam H., Campbell D., Deutsch E. W., Mirzaei H., Ranish J., Domon B., Aebersold R. (2008) A database of mass spectrometric assays for the yeast proteome. Nat. Methods 5, 913–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim Y. J., Zaidi-Ainouch Z., Gallien S., Domon B. (2012) Mass spectrometry-based detection and quantification of plasma glycoproteins using selective reaction monitoring. Nat. Protoc. 7, 859–871 [DOI] [PubMed] [Google Scholar]
- 33. Kiyonami R., Schoen A., Prakash A., Peterman S., Zabrouskov V., Picotti P., Aebersold R., Huhmer A., Domon B. (2011) Increased selectivity, analytical precision, and throughput in targeted proteomics. Mol. Cell. Proteomics 10, M110.002931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prakash A., Tomazela D. M., Frewen B., Maclean B., Merrihew G., Peterman S., Maccoss M. J. (2009) Expediting the development of targeted SRM assays: using data from shotgun proteomics to automate method development. J. Proteome Res. 8, 2733–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stahl-Zeng J., Lange V., Ossola R., Eckhardt K., Krek W., Aebersold R., Domon B. (2007) High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol. Cell. Proteomics 6, 1809–1817 [DOI] [PubMed] [Google Scholar]
- 36. Gallien S., Peterman S., Kiyonami R., Souady J., Duriez E., Schoen A., Domon B. (2012) Highly multiplexed targeted proteomics using precise control of peptide retention time. Proteomics 12, 1122–1133 [DOI] [PubMed] [Google Scholar]
- 37. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.