

Abstract
A complete understanding of the biological functions of large signaling peptides (>4 kDa) requires comprehensive characterization of their amino acid sequences and post-translational modifications, which presents significant analytical challenges. In the past decade, there has been great success with mass spectrometry-based de novo sequencing of small neuropeptides. However, these approaches are less applicable to larger neuropeptides because of the inefficient fragmentation of peptides larger than 4 kDa and their lower endogenous abundance. The conventional proteomics approach focuses on large-scale determination of protein identities via database searching, lacking the ability for in-depth elucidation of individual amino acid residues. Here, we present a multifaceted MS approach for identification and characterization of large crustacean hyperglycemic hormone (CHH)-family neuropeptides, a class of peptide hormones that play central roles in the regulation of many important physiological processes of crustaceans. Six crustacean CHH-family neuropeptides (8–9.5 kDa), including two novel peptides with extensive disulfide linkages and PTMs, were fully sequenced without reference to genomic databases. High-definition de novo sequencing was achieved by a combination of bottom-up, off-line top-down, and on-line top-down tandem MS methods. Statistical evaluation indicated that these methods provided complementary information for sequence interpretation and increased the local identification confidence of each amino acid. Further investigations by MALDI imaging MS mapped the spatial distribution and colocalization patterns of various CHH-family neuropeptides in the neuroendocrine organs, revealing that two CHH-subfamilies are involved in distinct signaling pathways.
Neuropeptides and hormones comprise a diverse class of signaling molecules involved in numerous essential physiological processes, including analgesia, reward, food intake, learning and memory (1). Disorders of the neurosecretory and neuroendocrine systems influence many pathological processes. For example, obesity results from failure of energy homeostasis in association with endocrine alterations (2, 3). Previous work from our lab used crustaceans as model organisms found that multiple neuropeptides were implicated in control of food intake, including RFamides, tachykinin related peptides, RYamides, and pyrokinins (4–6).
Crustacean hyperglycemic hormone (CHH)1 family neuropeptides play a central role in energy homeostasis of crustaceans (7–17). Hyperglycemic response of the CHHs was first reported after injection of crude eyestalk extract in crustaceans. Based on their preprohormone organization, the CHH family can be grouped into two sub-families: subfamily-I containing CHH, and subfamily-II containing molt-inhibiting hormone (MIH) and mandibular organ-inhibiting hormone (MOIH). The preprohormones of the subfamily-I have a CHH precursor related peptide (CPRP) that is cleaved off during processing; and preprohormones of the subfamily-II lack the CPRP (9). Uncovering their physiological functions will provide new insights into neuroendocrine regulation of energy homeostasis.
Characterization of CHH-family neuropeptides is challenging. They are comprised of more than 70 amino acids and often contain multiple post-translational modifications (PTMs) and complex disulfide bridge connections (7). In addition, physiological concentrations of these peptide hormones are typically below picomolar level, and most crustacean species do not have available genome and proteome databases to assist MS-based sequencing.
MS-based neuropeptidomics provides a powerful tool for rapid discovery and analysis of a large number of endogenous peptides from the brain and the central nervous system. Our group and others have greatly expanded the peptidomes of many model organisms (3, 18–33). For example, we have discovered more than 200 neuropeptides with several neuropeptide families consisting of as many as 20–40 members in a simple crustacean model system (5, 6, 25–31, 34). However, a majority of these neuropeptides are small peptides with 5–15 amino acid residues long, leaving a gap of identifying larger signaling peptides from organisms without sequenced genome. The observed lack of larger size peptide hormones can be attributed to the lack of effective de novo sequencing strategies for neuropeptides larger than 4 kDa, which are inherently more difficult to fragment using conventional techniques (34–37). Although classical proteomics studies examine larger proteins, these tools are limited to identification based on database searching with one or more peptides matching without complete amino acid sequence coverage (36, 38).
Large populations of neuropeptides from 4–10 kDa exist in the nervous systems of both vertebrates and invertebrates (9, 39, 40). Understanding their functional roles requires sufficient molecular knowledge and a unique analytical approach. Therefore, developing effective and reliable methods for de novo sequencing of large neuropeptides at the individual amino acid residue level is an urgent gap to fill in neurobiology. In this study, we present a multifaceted MS strategy aimed at high-definition de novo sequencing and comprehensive characterization of the CHH-family neuropeptides in crustacean central nervous system. The high-definition de novo sequencing was achieved by a combination of three methods: (1) enzymatic digestion and LC-tandem mass spectrometry (MS/MS) bottom-up analysis to generate detailed sequences of proteolytic peptides; (2) off-line LC fractionation and subsequent top-down MS/MS to obtain high-quality fragmentation maps of intact peptides; and (3) on-line LC coupled to top-down MS/MS to allow rapid sequence analysis of low abundance peptides. Combining the three methods overcomes the limitations of each, and thus offers complementary and high-confidence determination of amino acid residues. We report the complete sequence analysis of six CHH-family neuropeptides including the discovery of two novel peptides. With the accurate molecular information, MALDI imaging and ion mobility MS were conducted for the first time to explore their anatomical distribution and biochemical properties.
EXPERIMENTAL PROCEDURES
Materials and Chemicals
All chemical reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Optima grade formic acid, ACN, water, and methanol were purchased from Fisher Scientific (Pittsburgh, PA).
Animals, Tissue Dissection and Extraction
Blue crabs Callinectes sapidus and Jonah crabs Cancer borealis were shipped from the Fresh Lobster Company (Gloucester, MA), and then maintained in artificial seawater. The animals were anesthetized in ice, and the sinus glands (SGs) and pericardial organs (POs) were dissected and collected in acidified methanol. The tissue was then homogenized and extracted with acidified methanol. After centrifugation, supernatant fractions were combined and concentrated to dryness. The sample was re-suspended in 100 μl of water for further analysis (5). The detailed protocol is described in Supplementary Materials.
HPLC Fractionation
HPLC separations were performed with a Waters Alliance HPLC system (Milford, MA). The mobile phases included solution A (water containing 0.1% formic acid) and solution B (acetonitrile (ACN) containing 0.1% formic acid). Approximately 50 μl of extract was injected onto a Phenomenex Gemini C18 column (2.1 mm i.d., 150 mm length, 5 μm particle size; Torrance, CA). The separations consisted of a 120 min gradient of 5–95% solution B. The flow rate was 0.2 ml/min. Fractions were automatically collected every 2 min with a Rainin Dynamax FC-4 fraction collector, followed by lyophilized, re-suspended in 20 μl water, and stored in −80 °C.
MALDI-TOF/TOF Analysis
A model 4800 MALDI-TOF/TOF analyzer (Applied Biosystems, Framingham, MA) equipped with a 200 Hz, 355 nm Nd:YAG laser was used. Acquisitions were performed in positive ion reflectron mode. Instrument parameters were set using the 4000 Series Explorer software (Applied Biosystems). Mass spectra were obtained by averaging 1000 laser shots covering mass range m/z 500–4000 in reflectron mode and m/z 2000–10000 in linear mode. MS/MS was achieved by 1 kV CID. For sample analysis, 0.4 μl of sample was spotted on MALDI plate first and allowed to dry followed by the addition of 0.4 μl 2,5-dihydroxybenzoic acid (DHB) matrix (4).
Bottom-up MS on Nano-LC-ESI-QTOF
An aliquot of 3 μl peptide fraction was reduced and alkylated by dithiothreitol (DTT) and iodoacetamide (IAA), followed by digestion with trypsin, Glu-C and Lys-C (41) (see Supplemental materials for details). Nano-LC-ESI-QTOF MS/MS was performed using a Waters nanoAcquity UPLC system coupled to a QTOF Micro mass spectrometer (Waters, Milford, MA) as described previously (5). The MS/MS raw data were converted to peak list (.pkl) files using ProteinLynx software 2.4 (Waters) (5). Peptides were identified by searching against an NCBInr 20090726 protein database (9330197 sequences; 3196564765 residues) using the Mascot v2.1 search engine. Trypsin or Glu-C was selected as enzyme allowing up to two missed cleavages. Carboxylmethyl cysteine was specified as fixed modifications, and methionine oxidation and pyro-Glu as variable modifications. Precursor and MS/MS tolerances were set within 30 ppm and 0.6 Da for monoisotopic mass, respectively. Peptide charge states include 1+, 2+, and 3+ charged peptides.
Off-line Top-down MS on ESI-LTQ-FTICR
A 0.5 μl of peptide fraction was reduced by incubation in 2.5 mm DTT for 1 h at 37 °C and desalted by C18 ZipTip and resuspended in 10 μl of 50% ACN containing 2% formic acid. The sample was analyzed using a 7T linear trap quadrupole (LTQ)/Fourier transform ion cyclotron resonance (FTICR) (LTQ-FT Ultra) hybrid mass spectrometer (Thermo Scientific Inc., Bremen, Germany) equipped with an automated chip-based nano-ESI source (Triversa NanoMate; Advion BioSciences, Ithaca, NY) as described previously (42). For CID and ECD fragmentation, individual charge states of peptide molecular ions were first isolated and then dissociated using 22–28% of normalized collision energy for CID or 4% electron energy for ECD with a 60 ms duration with no delay. Typically, 1000 transients were averaged to ensure high quality MS/MS spectra (42). All FTICR spectra were processed with Xtract Software (Xcalibur 2.0.5, Thermo Scientific Inc., Bremen, Germany) using a S/N threshold of 1.5 and fit factor of 40% and validated manually. The resulting mass lists were further assigned using the in-house developed “Ion Assignment” software. The assigned ions were manually validated to ensure the quality of assignments (42).
On-line Top-down MS on Nano-LC-ESI-LTQ-Orbitrap Elite
A 1 μl of crude tissue extract was reduced by incubation in 2.5 mm DTT for 1 h at 37 °C and desalted by C18 ZipTip and resuspended in 10 μl of water containing 0.2% formic acid. On-line top-down MS was carried out on an Ultimate 3000 RSLCnano system coupled to an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). A 0.5 μl of peptide sample was injected onto a 2 cm, 150 μm i.d. PLRP-S (dp 5 μm, pore size 1000Å) trap column. A 10 cm, 75 μm i.d. PLRP-S column was used for separation. The gradient was delivered at 300 nL/min starting at 5% B (95% acetonitrile and 0.2% formic acid) and rose to 10% B at 7 min, 50% B at 50 min, and 85% B at 58 min. The mass spectrometer was operated in the data-dependent mode to switch automatically between full-MS (scan 1), higher-energy collision dissociation (HCD)-MS2 (scan 2), and electron transfer dissociation (ETD)-MS2 (scan 3). The isolation width was set at 10 Da (36). The data processing method is the same as off-line top-down method described above.
MALDI Imaging
Immediately following dissection, the eyestalk was embedded in gelatin (100 mg/ml aqueous) and snap-frozen. Sectioning into 12 μm slices at −25 °C was performed on a cryostat (Microtom HM505E, Waldorf, Germany), and the slices were thaw-mounted onto a glass slide (Bruker Daltonics). An airbrush was used to spray coat the tissues with DHB. The airbrush was held perpendicular to the glass slide at a distance of 35 cm. Five coats of matrix were applied by spraying each sample for 30 s with 1 min dry time between each application (6).
Mass spectrometric analyses were performed in the linear, positive mode at +20 kV accelerating potential on a time-of-flight mass spectrometer (Bruker Autoflex III TOF-TOF; Bruker Daltonics, Bremen, Germany), which was equipped with a Smartbeam laser capable of operating at a repetition rate of 200 Hz with optimized delayed extraction time and laser beam size was set to medium. Laser energy was optimized for signal-to-noise in each preparation. Using Bruker Protein Standard 1 (Bruker Daltonics, Bremen, Germany), a linear external calibration was applied to the instrument before data collection. Mass spectral data sets were acquired over a whole eyestalk section using FlexImaging software (Bruker Daltonics, Bremen, Germany) in the mass range of m/z 3000 to 10,000, with a raster step size of 50 μm and 500 laser shots per spectrum. After data acquisition, molecular images were reconstituted using the FlexImaging software. Data was normalized using FlexImaging software, and each m/z signal plotted ± 10 mass units (6).
RESULTS
Establishment and Validation of the High-definition De Novo Sequencing Strategy
Our MS strategy for identification of CHH-family neuropeptides from the crustacean nervous system (Fig. 1) involves three steps: peptide candidate scanning, in silico homology searching, and de novo sequencing. Although the cDNA sequence of C. sapidus Cas−SG−CHH (Cas C. sapidus, SG sinus gland) preprohormone has been obtained by PCR-based cloning strategy, its amino acid sequence has not been identified by MS or Edman degradation (16, 17). Here, the multifaceted strategy is established and validated by performing MS-based sequencing of the Cas−SG−CHH peptide.
Fig. 1.
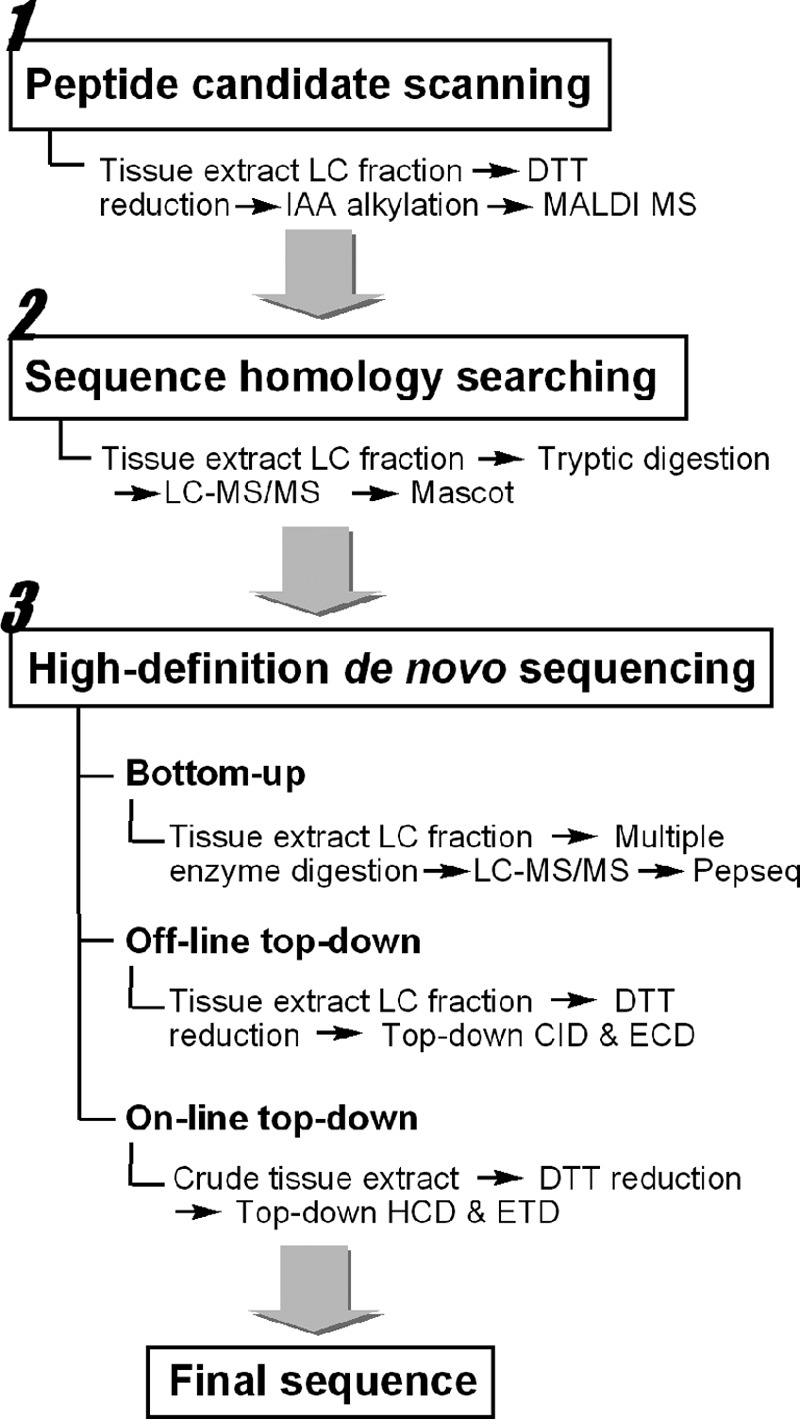
Workflow of the multifaceted strategy for identification and characterization of the CHH-family neuropeptides.
Peptide Candidate Scanning
Two unique features of the CHH-family neuropeptides, a molecular weight (MW) ranging from 8 to 10 kDa and the presence of three disulfide bonds (12), were used as screening criteria for candidate identification. The sinus gland organs were pooled from ten animals, homogenized, lyophilized and subjected to reversed phase (RP)-HPLC fractionation (Fig. 2A), followed by off-line direct infusion on an ESI-LTQ-FTICR mass spectrometer. Multiply charged CHH-family peptide ions were detected in the high-resolution spectrum of Fraction #17 (Fig. 2B), and the accurate MW of this large peptide was determined as 8472.948 Da.
Fig. 2.

CHH-family neuropeptide candidate scanning in C. sapidus sinus gland. A, Reversed phase HPLC fractionation of C. sapidus sinus gland tissue extract showing the presence of putative CPRP, CHH, and MIH peptide peaks. B, ESI-LTQ-FTICR spectra of LC Fraction #17. C, MALDI-TOF/TOF mass spectra of original, intact Cas−SG−CHH and D, DTT-reduced and IAA-treated LC Fraction #17 showing multiple alkylated CHH peptide forms.
To screen for disulfide bonds, the peptide candidate was treated with DTT and IAA respectively to reduce the disulfide bonds and then alkylate the free thiol groups followed by analysis via MALDI-MS. MALDI can tolerate higher levels of salt and mostly produces singly charged ions facilitating mass comparison. Fig. 2C and 2D show the MALDI mass spectra of the original and derivatized peptides, respectively. The ions at m/z 8728, 8777, and 8824 correspond to the reduced peptides alkylated with attachment of 4, 5, and 6 carbamidomethyl groups, respectively. Incomplete alkylation might be caused by the large peptide size. These results suggest the presence of three disulfide bonds in the peptide Cas−SG−CHH.
In Silico Sequence Homology Searching
Sequence homology searching can aid in de novo sequencing of small neuropeptides. Here, we designed a bottom-up method to extend the utility of the protein database for sequencing of large neuropeptides. The candidate peptide was digested by trypsin and analyzed by LC-MS/MS, followed by Mascot searching against NCBI database. The first two hits were the CHH preprohormones of C. sapidus (52% sequence match) and Portunus trituberculatus (29% sequence match). The goal of our method is to analyze neuropeptides from species without proteome and genome databases. So, we assumed that there were no such knowledge for this target peptide Cas−SG−CHH, and the homologous CHH preprohormone from P. trituberculatus (supplemental Fig. S1) was used as a reference sequence for de novo sequencing. CHH family members share the characteristic feature of six cysteines located in the identical or similar positions, i.e. C7, C23/C24, C26/C27, C39/C40, C43/C44, C52/C53. Therefore, the sequence AA64-AA139 of P. trituberculatus CHH preprohormone (supplemental Fig. S1) was mined as a reference peptide for the following de novo sequencing.
Bottom-up De Novo Sequencing
The tryptic fragments of Cas−SG−CHH were analyzed by LC-QTOF-MS/MS and MALDI-TOF/TOF-MS/MS. Fig. 3A is the MALDI-TOF/TOF mass spectrum of tryptic peptides of the Fraction #17. Fig. 3B displays a representative MS/MS spectrum of the tryptic peptide. Using the preprohormone sequence of P. trituberculatus CHH as a reference, the MS/MS spectra were carefully analyzed by software PepSeq (6) to assign fragment ions and determine peptide sequences. supplemental Table S1 lists all the sequenced tryptic peptides arising from Cas−SG−CHH. By this bottom-up sequencing method, 81% of the sequence was determined.
Fig. 3.

Bottom-up MS strategy for the analysis of Cas−SG−CHH. MALDI-TOF/TOF MS (A) and MS/MS (B) spectra of tryptic digest of LC Fraction #17 from C. sapidus sinus gland tissue extract.
Off-line Top-down De Novo Sequencing
To determine the rest of the sequence, the intact Cas−SG−CHH was fragmented with top-down MS using CID on an ESI-ultra-high resolution (UHR)-QTOF maXis mass spectrometer via direct infusion (Fig. 4A). Two sets of b55-b65 and y7-y15 ions, generated by cleavage of amide bonds close to the peptide C terminus, were detected with intense signals. However, only a few fragment ions from middle region of the peptide chain were observed. Although only 14% of b and y ions were assigned, a sequence tag 56LLIMDNFEEY65 (in this study, the isobaric I/L residues were assigned using homologous sequences) was confidently determined from the intense C-terminal fragmentation (Fig. 4A). Similarly, this intact peptide was fragmented on a different instrument, an ESI-LTQ-FTICR mass spectrometer, with CID and ECD, resulting in poor fragmentation as well (data not shown). A possible explanation is that fragmentation of this peptide is hindered by its tertiary structure resulting from three disulfide bonds which crosslink the residues between Cys7-Cys52. Therefore, denaturation of peptides by breaking disulfide bonds with DTT and IAA could facilitate peptide fragmentation. However, the large peptide size causes incomplete alkylation as shown previously (Fig. 2D).
Fig. 4.

Top-down MS strategy for the analysis of Cas−SG−CHH. A, CID spectrum of intact Cas−SG−CHH acquired on ESI-UHR-QTOF maXis. B, ECD spectrum of DTT-reduced Cas−SG−CHH acquired on ESI-LTQ-FTICR. C, HCD spectrum of DTT-reduced Cas−SG−CHH acquired on ESI-LTQ-Orbitrap Elite.
Here, we adopted a different strategy for peptide denaturation, in which the peptide was treated with DTT in urea solution for complete reduction of disulfide bonds, and then stored in 50% ACN containing 2% formic acid. The acidic environment prevented the disulfide bonds from reforming although the free thiol groups were not blocked by protective groups. After storing the sample either at 5 °C for 5 h or −20 °C for 4 days, no disulfide bond formation was observed (data not shown). The subsequent fragmentation of the reduced peptide on the LTQ-FTICR with CID and ECD produced 42% of b- and y-type ions and 39% of c- and z.- type ions, respectively. Fig. 4B is the ECD spectrum, and Fig. 5A shows the fragmentation map from ECD and CID. As a result, 75% of the primary sequence and the location of two PTMs, N-terminal pyroGlu and C-terminal amidation, were identified by this off-line top-down strategy.
Fig. 5.

Peptide fragmentation maps and sequence coverage. A, Fragmentation map of Cas−SG−CHH. B, Fragmentation map of Cab−SG−MIH. C, Sequence coverage map of Cas−SG−CHH. Green square, unidentified; blue square, identified.
On-line Top-down De Novo Sequencing
Top-down LC-MS/MS was carried out on an LTQ-Orbitrap Elite mass spectrometer. Here, the crude tissue extract, treated with DTT, was directly analyzed with top-down LC-MS/MS under data-dependent mode switching between ETD and HCD. Fig. 4C shows the HCD spectrum, and supplemental Fig. S2 shows the fragmentation map of ETD and HCD for the Cas−SG−CHH peptide.
Sequence Assembling
Fig. 5C summarizes the sequence coverage identified by the three de novo sequencing methods (bottom-up, off-line top-down and on-line top-down). These methods are complementary and maximize the sequence identification. The identified sequence percentages are 81%, 75%, and 50%, respectively. Only two residues AA32 and AA35 were not identified, which can be further determined by so-called Dipeptide Mass Deduction and Peptide Mass Deduction.
Manual De Novo Sequencing
Dipeptide Mass Deduction: The mass of AA31+AA32 can be calculated by c32 minus c30 (3826.720 - 3556.569 = 270.151 Da), only matching to dipeptide RN (Dipeptide mass Table in ProteinProspector, http://prospector.ucsf.edu/prospector/html/misc/dipep.htm). So, AA32 is an asparagine because AA31 was already determined as an arginine (Fig. 5C). Peptide Mass Deduction: The residue AA35 can be determined by peptide MW minus the rest of the sequence (8478.878 - 8379.802 = 99.076 Da, mass value of a valine residue). Finally, the complete sequence was determined.
In summary, this multifaceted MS strategy offers a systematic approach to elucidate amino acid sequences of the CHH-family neuropeptides, which is applicable to characterization of large intact peptide hormones with or without known cDNA sequences.
CHH-family Neuropeptidome in C. sapidus and C. borealis
With the strategies described above, we have discovered and identified six CHH-family neuropeptides and two modified isoforms from C. sapidus and C. borealis (Table I). For each peptide, the MWs of both intact and “reduced” peptides were measured to improve the identification confidence (“reduced” refers to the peptides after DTT reduction), in which the theoretical and experimental MWs match within 17.5 ppm. The sequence alignments among homologous species are shown in Fig. 6. This high-level confidence for complete sequence coverage arises from a combination of the three de novo sequencing methods.
Table I. CHH-family neuropeptides identified in this study.
| Species | Peptide name | Calcd. MWa | Exptl. MWa | Error/ppm | PTMb | Organ |
|---|---|---|---|---|---|---|
| C. sapidus | Cas−SG−CHH | 8472.800/8478.847 | 8472.948/8478.878 | 17.5/3.7 | pQ,C-NH2 | SG |
| Cas−SG−MIH | 9065.364/9071.435 | 9065.391/9071.424 | 3.0/1.2 | − | SG | |
| Cas−PO−CHH | 8395.644/8401.691 | 8395.643/− | 0.1/− | pQ,3Mox | PO | |
| C. borealis | Cab−SG−CHH-I | 8539.876/8545.923 | 8539.892/8545.934 | 1.9/1.3 | pQ,C-NH2 | SG |
| Cab−SG−CHH-II | 8556.903/8562.950 | 8556.913/8562.958 | 1.2/0.9 | C-NH2 | SG | |
| Cab−SG−MIH | 8932.189/8938.235 | 8932.302/8938.230 | 12.7/0.6 | C-NH2 | SG | |
| Cab−SG−MOIH | 9252.258/9258.305 | 9252.346/9258.278 | 9.5/2.9 | − | SG |
a MW of intact peptide/MW of “reduced” peptide. The observed m/z values and charge states of intact peptides are listed in Table S2.
b pQ, pyro-Glu; C-NH2, C-terminal amidation; Mox, methionine oxidation.
Fig. 6.

Sequence alignment of CHH-family neuropeptides. Cas, Callinectes sapidus; Cab, Cancer borealis; Cap, Cancer pagurus.
Identification of Two Novel CHH-family Neuropeptides in C. borealis
The Novel MIH
Although C. borealis is a widely used animal model in neurobiological studies, there is no available genomic database, making peptide identification in this model organism challenging (25, 27). With the multifaceted MS strategy described here, we found one novel MIH in the sinus gland. The accurate MWs of intact and “reduced” forms were determined as 8932.302 and 8938.230 Da, respectively. From the 6-Da mass difference, we inferred that this peptide contains three disulfide bonds and thus placing this peptide as a putative candidate for the CHH-family neuropeptides. The homology search using bottom-up proteomic method resulted in a hit of the MIH from a homologous species, Cancer pagurus (43), with 89% of matched peptide coverage (supplemental Fig. S3). In the subsequent bottom-up de novo sequencing, multiple proteases (trypsin, Glu-C and Lys-C) were employed to generate proteolytic peptides from different cleavage site. Followed by LC-MS/MS analysis, computer-assisted sequencing resulted in identification of proteolytic peptides listed in Table II. In addition, these results indicated that a Mascot search caused false positive identification of the tryptic peptide 68QWVGILGAGRE78 (supplemental Fig. S3). By overlapping these proteolytic peptides and referring the homologous sequence of Cancer pagurus Cap−SG−MIH, we identified the sequence AA1-AA59 of the new Cab−SG−MIH in C. borealis. The sequence AA60-AA77 remained unknown.
Table II. Proteolytic peptides of Cab−SG−MIH identified by bottom-up de novo sequencing.
| Obsd. m/z | Charge state | Calcd. MW | Sequencea | Proteolytic peptide | Mascot ion scoreb | Protease |
|---|---|---|---|---|---|---|
| 828.49 | 2 | 1654.82 | RVINDDCPNLIGNR | MIH [1–14] | 65 | Trypsin |
| 750.40 | 2 | 1498.71 | VINDDCPNLIGNR | MIH [2–14] | 70 | Trypsin |
| 1010.11 | 2 | 2017.99 | VINDDCPNLIGNRDLYK | MIH [2–18] | 76 | Trypsin |
| 928.50 | 2 | 1854.84 | KVEWICEDCSNIFR | MIH [19–32] | 90 | Trypsin |
| 864.48 | 2 | 1726.74 | VEWICEDCSNIFR | MIH [20–32] | 53 | Trypsin |
| 512.30 | 2 | 1022.46 | NTGMATLCR | MIH [33–41] | 50 | Trypsin |
| 1200.16 | 2 | 2398.05 | KNCFFNEDFLWCVYATER | MIH [42–59] | 79 | Trypsin |
| 1136.11 | 2 | 2269.96 | NCFFNEDFLWCVYATER | MIH [43–59] | 95 | Trypsin |
| 590.96 | 2 | 1769.85 | RVINDDCPNLIGNRD | MIH [1–15] | 49 | Glu-C |
a Fixed carbamidomethyl is on cysteine.
b Matching to homologous sequence.
Previous work on sequencing of the C. sapidus Cas−SG−CHH showed that CID fragmentation of intact (nonreduced) CHH peptide scanning resulted in ample cleavage of C-terminal amide bonds, facilitating identification of the C-terminal residues. Similarly, we fragmented this putative Cab−SG−MIH peptide on ESI-LTQ-FTICR using CID, and observed one set of b ions with intense signals arising from sequential cleavage of the C-terminal amide bonds. Accordingly, AA68-AA77was determined as 68EWVGILGAGS77 with a C-terminal amidation. The remaining task was to identify AA60-AA67.
In the homologous Cap−SG−MIH (supplemental Fig. S3), AA67 is an arginine and thus AA60-AA67 forms a tryptic peptide during digestion. So, it is possible that in the putative Cab−SG−MIH the residue AA60-AA67 also form a tryptic peptide. Based on this hypothesis, the MW of the tryptic peptide can be calculated by Peptide Mass Deduction, i.e. the measured Cab−SG−MIH peptide MW minus the calculated remaining sequence mass (8938.230 - 8003.781 = 934.449 Da). By carefully searching the bottom-up de novo sequencing data, one tryptic peptide was found matching the MW 934.449 Da (Fig. 7B). Analysis of the MS/MS spectrum using PepSeq determined its sequence as 60TAEMSQLR67 (Fig. 7C). With the addition of this fragment, the complete sequence of this novel Cab−SG−MIH was determined. To confirm it, off-line and on-line top-down MS/MS were carried out. In the resulting ECD spectrum (Fig. 7D), observation of a series of z. ions confirms the sequence 63MSQL66 which was determined by the bottom-up tryptic peptide in Fig. 7C. The fragmentation map resulting from ECD and CID (Fig. 5B) shows 58% of identified sequence coverage, and that of ETD and HCD gives 50% of sequence coverage (supplemental Fig. S4). Fig. 7E illustrates the sequencing process step by step. This approach offers a confident and accurate sequence analysis for the discovery of novel large neuropeptides without genomic information.
Fig. 7.

De novo sequencing of novel Cab−SG−MIH. A, CID spectrum of intact Cab−SG−MIH (m/z 1278.055, charge state +7). B, C, MS and MS/MS spectrum of tryptic peptide TAEMSQLR from Cab−SG−MIH. D, Zoom-in ECD spectrum of DTT-reduced Cab−SG−MIH. E, Sequence assembling of Cab−SG−MIH.
The Novel MOIH
In C. borealis sinus gland, we found one putative Cab−SG−MOIH whose MWs of intact and “reduced” forms were observed as 9252.346 and 9258.278 Da, respectively. Once again, 6-Da mass difference confirmed the candidate identity. A homology search hit the Cancer pagurus Cap−SG−MOIH (supplemental Fig. S5) with 89% of matched sequence coverage (44). With the bottom-up de novo sequencing method using multiple proteases for digestion, twelve proteolytic peptides corresponding to 97% sequence coverage (Table III) were identified to generate a putative sequence RRINNDCQNFIGNRAMYEKVDWICKDCANIFRQDGLLNNCRSNCFYNTEFLWCIDATENTRXXEQLEQWAAILGAGWN (X stands for unknown amino acid residue). The mass of the unknown residues AA62-AA63 was calculated by Peptide Mass Deduction: measured MW of the intact peptide minus theoretical mass of the rest of residues 9258.278 - 8993.151 = 265.127 Da, which matches the only dipeptide HK in Dipeptide mass table of ProteinProspector. Compared with the homologous Cap−SG−MOIH sequence, the sequence order should be HK rather than KH. Using an off-line top-down method, this putative Cab−SG−MOIH could not be selected for fragmentation because of its low abundance and suppressed ionization (data not shown). The on-line top-down method successfully fragmented this putative Cab−SG−MOIH using ETD and HCD, which produced a fragmentation map with 52% of sequence coverage (supplemental Fig. S6).
Table III. Proteolytic peptides of Cab−SG−MOIH identified by bottom-up de novo sequencing.
| Obsd. m/z | Charge state | Calcd. MW | Sequencea | Proteolytic peptide | Mascot ion scoreb | Protease |
|---|---|---|---|---|---|---|
| 810.94 | 2 | 1619.76 | RINNDCQNFIGNR | MOIH [2–14] | −c | Trypsin |
| 732.88 | 2 | 1463.66 | INNDCQNFIGNR | MOIH [3–14] | 84 | Trypsin |
| 566.30 | 3 | 1695.79 | VDWICKDCANIFR | MOIH [20–32] | 70 | Trypsin |
| 448.23 | 2 | 894.40 | DCANIFR | MOIH [26–32] | 36 | Trypsin |
| 656.00 | 3 | 1964.89 | DCANIFRQDGLLNNCR | MOIH [26–41] | 68 | Trypsin |
| 545.29 | 2 | 1088.50 | QDGLLNNCR | MOIH [33–41] | 49 | Trypsin |
| 1271.10 | 2 | 2540.07 | SNCFYNTEFLWCIDATENTR | MOIH [42–61] | 103 | Trypsin |
| 843.46 | 2 | 1684.82 | EQLEQWAAILGAGWN | MOIH [64–78] | −c | Trypsin |
| 757.65 | 3 | 2270.04 | RRINNDCQNFIGNRAMYE | MOIH [1–18] | 45 | Glu-C |
| 763.00 | 3 | 2286.04 | RRINNDCQNFIGNRAMoxYE | MOIH [1–18] | 36 | Glu-C |
| 956.08 | 3 | 2865.27 | CANIFRKDGLLNNCRSNCFYNTE | MOIH [27–49] | −c | Glu-C |
| 593.84 | 2 | 1185.59 | QWAAILGAGWN | MOIH [68–78] | −c | Glu-C |
a Fixed carbamidomethyl is on cysteine; Mox, methionine oxidation.
b Matching to homologous sequence.
c Not matched in Mascot search.
MS-based Distribution Mapping and Conformation Analysis Reveal Biological Significance
Distribution Mapping by MALDI Imaging
The functional roles that various compounds play in an organism are closely related to their locations. The X-organ/sinus gland complex located in eyestalks represents a major neurodocrine structure in decapod crustaceans (10). Previous studies using immunohistochemical techniques for peptide profiling indicate that the subfamily-I peptides (CHH) rarely overlap with the subfamily-II peptides (MIH and MOIH) (9, 14, 15, 45). However, the immunohistochemical method suffers from antibody cross-reaction problem as the epitope peptides share a high degree of sequence homology (see Fig. 6) (14). To overcome this limitation, we demonstrated the first use of MALDI MS imaging technique to map the endogenous CHH-family peptides in crustacean neurosecretory structures.
Fig. 8A shows direct tissue MALDI MS analysis of Jonah crab C. borealis sinus gland, which contains Cab−SG−CPRP dimer (5), Cab−SG−CHH-I, Cab−SG−CHH-II, and Cab−SG−MOIH (MWs and PTMs are listed in Table I). The MALDI imaging results of the entire eyestalk reveal that all these peptides are located in the sinus gland (Fig. 8B). Interestingly, the zoom-in images (Figs. 8C–8G) of the sinus gland illustrate that Cab−SG−CPRP and subfamily-I peptides, Cab−SG−CHH-I and Cab−SG−CHH-II are co-localized; and subfamily-II peptides, Cab−SG−MIH and Cab−SG−MOIH are also co-localized. Moreover, the Cab−SG−CPRP, Cab−SG−CHH-I and Cab−SG−CHH-II have almost no overlap with Cab−SG−MIH and Cab−SG−MOIH, exhibiting distinct distribution patterns for the two CHH subfamily peptides. Furthermore, a similar distribution pattern was observed from MALDI imaging of the blue crab C. sapidus sinus gland, which shows that the Cas−SG−MIH and Cas−SG−CHH are differentially distributed, and Cas−SG−CPRP and Cas−SG−CHH are co-localized (data not shown). These results are consistent with previous studies using immunohistochemical methods.
Fig. 8.

Spatial distribution mapping of the CHH-family neuropeptides in C. borealis sinus gland. A, MALDI mass spectrum of direct tissue analysis of C. borealis sinus gland. B, MALDI-MSI image of Cab−SG−CHH-I on C. borealis eyestalk. Zoom-in MALDI images of Cab−SG−CHH-I (C), Cab−SG−CHH-II (D), Cab−SG−MIH (E), Cab−SG−MOIH (F), and Cab−SG−CPRP (G) on the sinus gland of C. borealis eyestalk.
Conformation Analysis by Ion Mobility MS
The N-terminal glutamine of neuropeptides can be modified by cyclization of the glutamine via condensation of the α-amino group with the side-chain carboxyl group (pyro-Glu modification). Modified peptides show variation of half-life and biological activity related to conformational changes (46). The Cab−SG−CHH-I and Cab−SG−CHH-II in Fig. 8A are the modified and unmodified isoforms, respectively. The release sites of the two isoforms are co-localized in the sinus gland (Fig. 8C and 8D), suggesting that they exhibit similar tissue-specific distribution patterns. Thus, it is interesting to investigate the potential peptide conformational change induced by this modification. Supplemental Fig. S7 shows the calibrated collision cross section (CCS) (47) of the Cab−SG−CHH-I and Cab−SG−CHH-II with 40 Å2 of CCS difference measured by ion mobility spectrometry, suggesting that the N-terminal pyro-Glu modification may cause intrinsic shape or conformational changes of neuropeptides.
Statistical Analysis Evaluates Performance Characteristics of the Three De Novo Sequencing Strategies
Complementary Characteristics
Implementation of three de novo sequencing strategies provides more confident and effective sequence elucidation of large peptides. Here, we use Cas−SG−CHH, Cas−SG−MIH, Cab−SG−CHH and Cab−SG−MIH as model peptides to evaluate three figures of merit: number of identified residues (Fig. 9A), number of residues in sequence tags (Fig. 9B), and number of cleavage sites (Fig. 9C). These results indicate that the three strategies provide complementary information. Combined use of these strategies leads to 60%, 100%, and 36% boosts in the values of the three evaluated aspects, respectively (Supplemental Fig. S8).
Fig. 9.

Statistical evaluation of bottom-up, off-line top-down and on-line top-down de novo sequencing strategies. Contributions of the three de novo sequencing methods to number of identified residues (A), number of residues in sequence tags (B), and number of cleavage sites (C).
Local Identification Confidence
The combination of off-line and on-line top-down methods can also improve the local identification confidence (48) for each amino acid residue (Supplemental Fig. S9A). The confidence scores of each residue of Cas−SG−CHH are illustrated in Supplemental Fig. S9B. Supplemental Fig. S9C summarizes the average scores of off-line top-down, on-line top-down and a combination for the four model peptides, Cas−SG−CHH, Cas−SG−MIH, Cab−SG−CHH and Cab−SG−MIH. All the confidence scores are increased when two methods are combined. In addition, Supplemental Fig. S10 plots the correlation between length of peptide fragment ions and fragmentation techniques (ECD, ETD, CID, and HCD), which suggests that use of multiple fragmentation techniques produces fragment ions with broader mass ranges in top-down fragmentation of large peptides. More detailed discussion of the statistical evaluation of these complementary techniques can be found in Supplementary Materials.
DISCUSSION
Advantages of the High-definition De Novo Sequencing Approach
We have described a multifaceted strategy for identification and characterization of the CHH-family neuropeptides. High-definition de novo sequencing not only offers complementary sequencing information but also improves confidence for elucidation of individual amino acid residues. Specifically, the bottom-up de novo sequencing method, employing multiple protease digestion and LC-MS/MS technique, enables deep amino acid sequencing by analysis of the overlapped proteolytic peptides (37, 49). However, complications arise from incomplete characterization of alternative splice forms, labile PTMs and truncated isoforms, leading to misidentification of the native intact peptide isoforms (35, 50, 51). Combining bottom-up and top-down methods can overcome this issue. Off-line and on-line top-down de novo sequencing methods combined use of multiple fragmentation techniques including CID, ECD, HCD and ETD (52), offering comprehensive cleavage of intact large peptide molecules while retaining site-specific PTMs. This allows us to obtain the precise molecular details of these important peptide hormones.
Off-line Top-down Strategy
One challenge for top-down MS/MS of large peptides and proteins is the limited capability to detect low-abundance fragment ions. To overcome this problem, our off-line top-down experiment was performed on a platform established by Ge and coworkers (42), in which a 7T LTQ-FTICR is coupled with a chip-based nano-ESI, enabling femtomole level of sample consumption and consistent acquisition of spectrum. Thousands of MS/MS scans were averaged to generate a high-quality top-down MS/MS spectrum which facilitated the detection of numerous low-abundance fragment ions. For example, the ECD spectrum in Fig. 4B resulted from averaging 1000 scans with enhanced S/N ratio, which allows us to detect the low-abundance ions and obtain a 75% of sequence coverage (Figs. 5A and 5C).
On-line Top-down Strategy
Another challenge of top-down MS/MS is the incompatibility of long duty cycle of FT transient with fast LC-MS time scale in which analytes of interest are (co)eluted in a short time window (36). Here, we carried out the on-line top-down experiment on a platform constructed by Kelleher and co-workers (36), in which a newly developed ultrahigh resolution LTQ-Orbitrap Elite FTMS system (53) was coupled with nano-LC for high throughput LC-MS/MS analysis. The optimized ion transfer optics facilitates high speed top-down MS/MS and fast switching between CID, HCD, and ETD, enabling sensitive and rapid identification of the eluted peptide ions. For instance, the Cab−SG−MOIH was co-eluted with Cab−SG−CHH from LC fractionation. The major component Cab−SG−CHH suppressed the detection of Cab−SG−MOIH and thus the off-line top-down fragmentation of Cab−SG−MOIH cannot be performed. In contrast, using on-line top-down system, HCD and ETD spectra were collected with one scan resulting in a 52% of sequence coverage (supplemental Fig. S6).
Sample Preparation Strategy
In addition, we employed a simple strategy to dramatically improve the fragmentation efficiency of large peptides containing extensive disulfide bonds. The peptides are reduced by DTT and stored in acidic solvent followed by MS analysis, instead of IAA alkylation which usually causes incomplete reaction because of large peptide sizes and thus decreases the target ion intensities. Compared with our previous study (34) using IAA alkylation on CHH, this method resulted in ∼3 fold increase of observed fragment ions and identified sequence coverage (comparison of fragmentation maps shown in supplemental Fig. S11). Furthermore, this method can potentially be applied to top-down proteomics to enhance fragmentation efficiency and sequence coverage.
Homology Search Strategy
In this study, the CHH-family peptides share high degree of sequence homology (Fig. 6). Therefore, a homology search using tryptic peptides in Mascot (54) ensures finding the desired homologous preprohormone. Alternatively, other algorithms with homology search function, such as PLGS (55) and PEAKS (48), have been examined to obtain similar results. For utility of the on-line top-down LC-MS/MS data (fragmentation of entire large peptides), ProSightPC (36) enables searching against both a manually curated peptide database and a database of homologous preprohormones. In addition, manual homology search using NCBI Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi) with the sequence tag LLIMDNFEEY (Fig. 4A) yielded the same preprohormones as Mascot.
Applicability of the Multifaceted Strategies
Although in this work we employed multiple mass spectrometers to demonstrate the complementary nature of various instruments and applicability of the strategies, the high-definition sequencing of large peptides can be accomplished using two or three instruments, depending on the goals of the study. For example, a combined use of a MALDI-based mass spectrometer and high-resolution nano-ESI-tandem MS instrument with multiple fragmentation techniques would enable both high sequence coverage of peptides and spatial mapping of the peptides of interest. The former instrument can be used for fast peptide candidate screening and MALDI imaging; and the latter MS-platform can be employed for bottom-up, off-line top-down and on-line top-down experiments, in which the nano-ESI source is coupled with either off-line infusion device or on-line LC system.
Functional Aspects of the Identified CHH-family Neuropeptides
Previous studies in crustacean endocrinology highlighted the central role of the CHH-family neuropeptides in signaling system of energy homeostasis, and proposed a model that employs neurotransmitters to control secretion and release of CHHs, followed by triggering the downstream energy metabolism by the second messengers (7, 8). However, the precise molecular mechanisms underlying the interactions between neurotransmitters and CHHs at the cellular and network levels remain elusive. This is, in part, because of a lack of analytical capabilities to identify and characterize these low abundance endogenous signaling molecules in a complex microenvironment.
Our current large neuropeptidome analysis by use of high-definition de novo sequencing allowed precise characterization of six CHH-family peptides with PTMs, including the novel MIH and MOIH neuropeptides. Their detailed molecular information will contribute to further functional and physiological studies exploring the mechanisms modulating the animal's metabolism, osmoregulation, molting and reproduction (9). One of the applications is to study their regulatory roles on energy homeostasis, with a goal to establish a simplified neuroendocrine model of energy regulation using crustaceans (8). A critical element of studying the complex interactions between multiple molecular players is the ability to map their spatial distributions and colocalization patterns. Toward this end, we employed MALDI imaging mass spectrometry and ion mobility MS to analyze the spatial distribution and molecular conformation of several CHH-family neuropeptides.
Generally, each CHH-family peptide plays distinct functional role. For example, CHHs regulate the blood glucose metabolism (16); MIHs suppress the synthesis of ecdysteroids delaying molting(12); and MOIHs inhibit the synthesis of methyl farnesoate and thus control somatic and gonadal growth (43). Nevertheless, many previous studies have suggested that these large peptides are multifunctional hormones and often display overlapping biological activities (8, 9). For instance, the osmoregulatory function of CHH is related with crustacean molt cycles (56). Thus, visualization of the CHH-family peptide hormone release sites may provide insights into the neurosecretion and signal transduction pathways as well as complex hormonal integration of these processes. Many efforts have been directed to the use of immunohistochemical techniques for distribution analysis of the CHH-family peptides in the neurosecretory system (9, 14, 15, 45). In general, the immunoreactivities of subfamily-I peptides rarely overlap with those of subfamily-II peptides, but co-localization of individual isoforms among subfamily-II peptides has been observed. Our MALDI imaging results of both C. sapidus and C. borealis sinus glands (Fig. 8) provide the first direct biochemical evidence to confirm this distribution pattern. This MS-based imaging method provides unambiguous identification and simultaneous measurement and mapping of multiple CHH-family peptides including intact CHH and CPRP as well as MIH and MOIH using a single eyestalk tissue section. This new method overcomes several limitations of traditional immunohistochemical techniques such as cross-reactivity and limited throughput by offering accurate and simultaneous mapping of numerous endogenous CHH-family peptide isoforms in a single experiment. Nonetheless, the MALDI MS imaging technique does not rival the cellular spatial resolution offered by antibody-based staining techniques. Here, we used 50 × 50 μm2 pixels for MS image acquisition and visualization, enabling the generation of spatial distribution of various CHH-family neuropeptides within a ∼1 × 1 mm2 tissue area of this important neuroendocrine organ.
CONCLUSIONS
The overall work described here represents a new route to discovery and characterization of large neuropeptides. This multifaceted MS-based strategy involves a comprehensive and systematic implementation of peptide candidate scanning, in silico homology searching, de novo sequencing, distribution mapping, and conformation analysis. The accurate sequence, spatial distribution pattern, and conformational analysis of the CHH-family neuropeptides were elucidated with a combination of MS tools. This high-definition de novo sequencing strategy combines well-established bottom-up, on-line top-down and off-line top-down methods, offering complementary sequence information at the residue level. Because CHH-family peptides represent the typical molecular features of large neuropeptides, this multifaceted strategy is applicable to comprehensive characterization of large peptidomes in other biological systems.
With the knowledge of the precise molecular details of these CHH-family neuropeptides, future work will focus on studying their functional roles and modulation mechanism. Ongoing project based on a novel quantitative top-down MS method will enable monitoring of CHH-family peptide release and deciphering the regulatory pathways in energy homeostasis and feeding behavior.
Supplementary Material
Acknowledgments
We thank Bruker Daltonics for graciously loaning the Autoflex III MALDI TOF/TOF mass spectrometer. We are also grateful to the UW School of Pharmacy Analytical Instrumentation Center for access to UHR-TOF maXis, to Lisa Xu and Huseyin Guner for experimental assistance with LTQ-FTICR, and to Emma Doud for assistance in editing this manuscript.
Footnotes
* This work is supported in part by the National Institutes of Health (NIH) grant (R01DK071801 to LL) and the National Science Foundation grant (CHE-0967784 to LL). YG acknowledges support from the Wisconsin Partnership Fund for a Healthy Future and NIH R01HL096971. NLK thanks support from NIH grants R01 GM067193 and P30 DA018310 and the Searle Funds at the Chicago Community Trust (to Chicago Biomedical Consortium). LL acknowledges an H. I. Romnes Faculty Research Fellowship. CJ thanks an Oversea Training Fellowship and UW Vilas Conference Presentation Funds.
 This article contains supplemental Figs. S1 to S12 and Tables S1 to S3.
This article contains supplemental Figs. S1 to S12 and Tables S1 to S3.
1 The abbreviations used are:
- CHH
- crustacean hyperglycemic hormone
- MIH
- molt-inhibiting hormone
- MOIH
- mandibular organ-inhibiting hormone
- CPRP
- CHH precursor related peptide
- PTM
- post-translational modification
- IM
- ion mobility
- IAA
- iodoacetamide
- FTICR
- Fourier transform ion cyclotron resonance
- UHR
- ultra-high resolution
- LTQ
- linear trap quadropole
- ECD
- electron capture dissociation
- ETD
- electron transfer dissociation
- HCD
- higher-energy collision dissociation
- DHB
- 2,5-dihydroxybenzoic acid
- CCS
- collision cross section
- SG
- sinus gland
- PO
- pericardial organ
- MS/MS
- tandem mass spectrometry.
REFERENCES
- 1. Li L., Sweedler J. V. (2008) Peptides in the brain: mass spectrometry-based measurement approaches and challenges. Annu. Rev. Anal. Chem. 1, 451–483 [DOI] [PubMed] [Google Scholar]
- 2. Morton G. J., Cummings D. E., Baskin D. G., Barsh G. S., Schwartz M. W. (2006) Central nervous system control of food intake and body weight. Nature 443, 289–295 [DOI] [PubMed] [Google Scholar]
- 3. Fricker L. D. (2007) Neuropeptidomics to study peptide processing in animal models of obesity. Endocrinology 148, 4185–4190 [DOI] [PubMed] [Google Scholar]
- 4. Chen R., Hui L., Cape S. S., Wang J., Li L. (2010) Comparative neuropeptidomic analysis of food intake via a multi-faceted mass spectrometric approach. ACS Chem. Neurosci. 1, 204–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hui L., Cunningham R., Zhang Z., Cao W., Jia C., Li L. (2011) Discovery and characterization of the Crustacean hyperglycemic hormone precursor related peptides (CPRP) and orcokinin neuropeptides in the sinus glands of the blue crab Callinectes sapidus using multiple tandem mass spectrometry techniques. J. Proteome Res. 10, 4219–4229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hui L., Zhang Y., Wang J., Cook A., Ye H., Nusbaum M. P., Li L. (2011) Discovery and functional study of a novel crustacean tachykinin neuropeptide. ACS Chem. Neurosci 2, 711–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chung J. S., Zmora N., Katayama H., Tsutsui N. (2010) Crustacean hyperglycemic hormone (CHH) neuropeptidesfamily: Functions, titer, and binding to target tissues. Gen. Comp. Endocrinol. 166, 447–454 [DOI] [PubMed] [Google Scholar]
- 8. Fanjul-Moles M. L. (2006) Biochemical and functional aspects of crustacean hyperglycemic hormone in decapod crustaceans: review and update. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 142, 390–400 [DOI] [PubMed] [Google Scholar]
- 9. Webster S. G., Keller R., Dircksen H. (2012) The CHH-superfamily of multifunctional peptide hormones controlling crustacean metabolism, osmoregulation, moulting, and reproduction. Gen. Comp. Endocrinol. 175, 217–233 [DOI] [PubMed] [Google Scholar]
- 10. Hopkins P. M. (2012) The eyes have it: A brief history of crustacean neuroendocrinology. Gen. Comp. Endocrinol. 175, 357–366 [DOI] [PubMed] [Google Scholar]
- 11. Mykles D. L., Adams M. E., Gäde G., Lange A. B., Marco H. G., Orchard I. (2010) Neuropeptide action in insects and crustaceans. Physiol. Biochem. Zool 83, 836–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakatsuji T., Lee C. Y., Watson R. D. (2009) Crustacean molt-inhibiting hormone: structure, function, and cellular mode of action. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 152, 139–148 [DOI] [PubMed] [Google Scholar]
- 13. Covi J. A., Chang E. S., Mykles D. L. (2009) Conserved role of cyclic nucleotides in the regulation of ecdysteroidogenesis by the crustacean molting gland. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 152, 470–477 [DOI] [PubMed] [Google Scholar]
- 14. Hsu Y. W., Messinger D. I., Chung J. S., Webster S. G., de la Iglesia H. O., Christie A. E. (2006) Members of the crustacean hyperglycemic hormone (CHH) peptide family are differentially distributed both between and within the neuroendocrine organs of Cancer crabs: implications for differential release and pleiotropic function. J. Exp. Biol. 209, 3241–3256 [DOI] [PubMed] [Google Scholar]
- 15. Chung J. S., Webster S. G. (2004) Expression and release patterns of neuropeptides during embryonic development and hatching of the green shore crab, Carcinus maenas. Development 131, 4751–4761 [DOI] [PubMed] [Google Scholar]
- 16. Chung J. S., Zmora N. (2008) Functional studies of crustacean hyperglycemic hormones (CHHs) of the blue crab, Callinectes sapidus - the expression and release of CHH in eyestalk and pericardial organ in response to environmental stress. FEBS J. 275, 693–704 [DOI] [PubMed] [Google Scholar]
- 17. Zheng J., Chen H. Y., Choi C. Y., Roer R. D., Watson R. D. (2010) Molecular cloning of a putative crustacean hyperglycemic hormone (CHH) isoform from extra-eyestalk tissue of the blue crab (Callinectes sapidus), and determination of temporal and spatial patterns of CHH gene expression. Gen. Comp. Endocrinol. 169, 174–181 [DOI] [PubMed] [Google Scholar]
- 18. Fricker L. D., Lim J., Pan H., Che F. Y. (2006) Peptidomics: identification and quantification of endogenous peptides in neuroendocrine tissues. Mass Spectrom. Rev. 25, 327–344 [DOI] [PubMed] [Google Scholar]
- 19. Zhang X., Petruzziello F., Zani F., Fouillen L., Andren P. E., Solinas G., Rainer G. (2012) High identification rates of endogenous neuropeptides from mouse brain. J. Proteome Res. 11, 2819–2827 [DOI] [PubMed] [Google Scholar]
- 20. Colgrave M. L., Xi L., Lehnert S. A., Flatscher-Bader T., Wadensten H., Nilsson A., Andren P. E., Wijffels G. (2011) Neuropeptide profiling of the bovine hypothalamus: thermal stabilization is an effective tool in inhibiting post-mortem degradation. Proteomics 11, 1264–1276 [DOI] [PubMed] [Google Scholar]
- 21. Petruzziello F., Fouillen L., Wadensten H., Kretz R., Andren P. E., Rainer G., Zhang X. (2012) Extensive characterization of Tupaia belangeri neuropeptidome using an integrated mass spectrometric approach. J. Proteome Res. 11, 886–896 [DOI] [PubMed] [Google Scholar]
- 22. Clynen E., Baggerman G., Huybrechts J., Vanden Bosch L., De Loof A., Schoofs L. (2003) Peptidomics of the locust corpora allata: identification of novel pyrokinins (-FXPRLamides). Peptides 24, 1493–1500 [DOI] [PubMed] [Google Scholar]
- 23. Lee J. E., Atkins N., Jr., Hatcher N. G., Zamdborg L., Gillette M. U., Sweedler J. V., Kelleher N. L. (2010) Endogenous peptide discovery of the rat circadian clock: a focused study of the suprachiasmatic nucleus by ultrahigh performance tandem mass spectrometry. Mol. Cell. Proteomics 9, 285–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hatcher N. G., Atkins N., Jr., Annangudi S. P., Forbes A. J., Kelleher N. L., Gillette M. U., Sweedler J. V. (2008) Mass spectrometry-based discovery of circadian peptides. Proc. Natl. Acad. Sci. U.S.A. 105, 12527–12532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma M., Wang J., Chen R., Li L. (2009) Expanding the Crustacean neuropeptidome using a multifaceted mass spectrometric approach. J. Proteome Res. 8, 2426–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma M., Bors E. K., Dickinson E. S., Kwiatkowski M. A., Sousa G. L., Henry R. P., Smith C. M., Towle D. W., Christie A. E., Li L. (2009) Characterization of the Carcinus maenas neuropeptidome by mass spectrometry and functional genomics. Gen. Comp. Endocrinol. 161, 320–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fu Q., Goy M. F., Li L. (2005) Identification of neuropeptides from the decapod crustacean sinus glands using nanoscale liquid chromatography tandem mass spectrometry. Biochem. Biophys. Res. Commun. 337, 765–778 [DOI] [PubMed] [Google Scholar]
- 28. Fu Q., Li L. (2005) De novo sequencing of neuropeptides using reductive isotopic methylation and investigation of ESI QTOF MS/MS fragmentation pattern of neuropeptides with N-terminal dimethylation. Anal. Chem. 77, 7783–7795 [DOI] [PubMed] [Google Scholar]
- 29. Chen R., Jiang X., Conaway M. C., Mohtashemi I., Hui L., Viner R., Li L. (2010) Mass spectral analysis of neuropeptide expression and distribution in the nervous system of the lobster Homarus americanus. J. Proteome Res. 9, 818–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma M., Sturm R. M., Kutz-Naber K. K., Fu Q., Li L. (2009) Immunoaffinity-based mass spectrometric characterization of the FMRFamide-related peptide family in the pericardial organ of Cancer borealis. Biochem. Biophys. Res. Commun. 390, 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma M., Szabo T. M., Jia C., Marder E., Li L. (2009) Mass spectrometric characterization and physiological actions of novel crustacean C-type allatostatins. Peptides 30, 1660–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dowell J. A., Heyden W. V., Li L. (2006) Rat neuropeptidomics by LC-MS/MS and MALDI-FTMS: Enhanced dissection and extraction techniques coupled with 2D RP-RP HPLC. J. Proteome Res. 5, 3368–3375 [DOI] [PubMed] [Google Scholar]
- 33. Jiménez C. R., Spijker S., de Schipper S., Lodder J. C., Janse C. K., Geraerts W. P., van Minnen J., Syed N. I., Burlingame A. L., Smit A. B., Li K. (2006) Peptidomics of a single identified neuron reveals diversity of multiple neuropeptides with convergent actions on cellular excitability. J. Neurosci. 26, 518–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma M., Chen R., Ge Y., He H., Marshall A. G., Li L. (2009) Combining bottom-up and top-down mass spectrometric strategies for de novo sequencing of the crustacean hyperglycemic hormone from Cancer borealis. Anal. Chem. 81, 240–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Resemann A., Wunderlich D., Rothbauer U., Warscheid B., Leonhardt H., Fuchser J., Kuhlmann K., Suckau D. (2010) Top-down de novo protein sequencing of a 13.6 kDa camelid single heavy chain antibody by matrix-assisted laser desorption ionization-time-of-flight/time-of-flight mass spectrometry. Anal. Chem. 82, 3283–3292 [DOI] [PubMed] [Google Scholar]
- 36. Tran J. C., Zamdborg L., Ahlf D. R., Lee J. E., Catherman A. D., Durbin K. R., Tipton J. D., Vellaichamy A., Kellie J. F., Li M., Wu C., Sweet S. M., Early B. P., Siuti N., LeDuc R. D., Compton P. D., Thomas P. M., Kelleher N. L. (2011) Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 480, 254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen Z. W., Fuchs K., Sieghart W., Townsend R. R., Evers A. S. (2012) Deep amino acid sequencing of native brain GABAA receptors using high-resolution mass spectrometry. Mol Cell Proteomics 11, doi: 10.1074/mcp.M111.011445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fang L., Kaake R. M., Patel V. R., Yang Y., Baldi P., Huang L. (2012) Mapping the protein interaction network of the human COP9 signalosome (CSN) complex using a label-free QTAX strategy. Mol. Cell. Proteomics 11, 138–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mita M., Yoshikuni M., Ohno K., Shibata Y., Paul-Prasanth B., Pitchayawasin S., Isobe M., Nagahama Y. (2009) A relaxin-like peptide purified from radial nerves induces oocyte maturation and ovulation in the starfish, Asterina pectinifera. Proc. Natl. Acad. Sci. U.S.A. 106, 9507–9512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nässel D. R., Wegener C. (2011) A comparative review of short and long neuropeptide F signaling in invertebrates: Any similarities to vertebrate neuropeptide Y signaling? Peptides 32, 1335–1355 [DOI] [PubMed] [Google Scholar]
- 41. Swaney D. L., Wenger C. D., Coon J. J. (2010) Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J Proteome Res 9, 1323–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ge Y., Rybakova I. N., Xu Q., Moss R. L. (2009) Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proc. Natl. Acad. Sci. U.S.A. 106, 12658–12663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wainwright G., Webster S. G., Wilkinson M. C., Chung J. S., Rees H. H. (1996) Structure and significance of mandibular organ-inhibiting hormone in the crab, Cancer pagurus. Involvement in multihormonal regulation of growth and reproduction. J. Biol. Chem. 271, 12749–12754 [DOI] [PubMed] [Google Scholar]
- 44. Tang C., Lu W., Wainwright G., Webster S. G., Rees H. H., Turner P. C. (1999) Molecular characterization and expression of mandibular organ-inhibiting hormone, a recently discovered neuropeptide involved in the regulation of growth and reproduction in the crab Cancer pagurus. Biochem. J. 343, 355–360 [PMC free article] [PubMed] [Google Scholar]
- 45. Ollivaux C., Gallois D., Amiche M., Boscaméric M., Soyez D. (2009) Molecular and cellular specificity of post-translational aminoacyl isomerization in the crustacean hyperglycaemic hormone family. FEBS J 276, 4790–4802 [DOI] [PubMed] [Google Scholar]
- 46. Vitorino R., Alves R., Barros A., Caseiro A., Ferreira R., Lobo M. C., Bastos A., Duarte J., Carvalho D., Santos L. L., Amado F. L. (2010) Finding new posttranslational modifications in salivary proline-rich proteins. Proteomics 10, 3732–3742 [DOI] [PubMed] [Google Scholar]
- 47. Bush M. F., Hall Z., Giles K., Hoyes J., Robinson C. V., Ruotolo B. T. (2010) Collision cross sections of proteins and their complexes: a calibration framework and database for gas-phase structural biology. Anal. Chem. 82, 9557–9565 [DOI] [PubMed] [Google Scholar]
- 48. Zhang J., Xin L., Shan B., Chen W., Xie M., Yuen D., Zhang W., Zhang Z., Lajoie G. A., Ma B. (2012) PEAKS DB: De novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 11, 1–8, doi: 10.1074/mcp.M111.010587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bandeira N., Pham V., Pevzner P., Arnott D., Lill J. R. (2008) Automated de novo protein sequencing of monoclonal antibodies. Nat. Biotechnol. 26, 1336–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu S. L., Jardine I., Hancock W. S., Karger B. L. (2004) A new and sensitive on-line liquid chromatography/mass spectrometric approach for top-down protein analysis: the comprehensive analysis of human growth hormone in an E. coli lysate using a hybrid linear ion trap/Fourier transform ion cyclotron resonance mass spectrometer. Rapid Commun. Mass Spectrom. 18, 2201–2207 [DOI] [PubMed] [Google Scholar]
- 51. Eliuk S. M., Maltby D., Panning B., Burlingame A. L. (2010) High resolution electron transfer dissociation studies of unfractionated intact histones from murine embryonic stem cells using on-line capillary LC separation: determination of abundant histone isoforms and post-translational modifications. Mol. Cell. Proteomics 9, 824–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shen Y., Tolić N., Xie F., Zhao R., Purvine S. O., Schepmoes A. A., Moore R. J., Anderson G. A., Smith R. D. (2011) Effectiveness of CID, HCD, and ETD with FT MS/MS for degradomic-peptidomic analysis: comparison of peptide identification methods. J. Proteome Res. 10, 3929–3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Michalski A., Damoc E., Lange O., Denisov E., Nolting D., Mueller M., Viner R., Schwartz J., Remes P., Belford M., Dunyach J. J., Cox J., Horning S., Mann M., Makarov A. (In press) Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol. Cell. Proteomics 11, doi: 10.1074/mcp.O111.013698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 55. Silva J. C., Gorenstein M. V., Li G. Z., Vissers J. P., Geromanos S. J. (2006) Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics 5, 144–156 [DOI] [PubMed] [Google Scholar]
- 56. Chang E. S., Chang S. A., Beltz B. S., Kravitz E. A. (1999) Crustacean hyperglycemic hormone in the lobster nervous system: localization and release from cells in the subesophageal ganglion and thoracic second roots. J. Comp. Neurol. 414, 50–56 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.