

Abstract
The thick ascending limb of the loop of Henle (THAL) reabsorbs ∼30% of the filtered NaCl in a process mediated by the apical Na-K-2Cl cotransporter NKCC2. Stimulation of β-adrenergic receptors in the THAL enhances NaCl reabsorption and increases intracellular cAMP. We found that intracellular cAMP stimulates NKCC2 trafficking to the apical membrane via protein kinase A (PKA). Several cAMP-specific phosphodiesterases (PDE) have been identified in rat THALs, and PDE4 decreases cAMP generated by β-adrenergic stimulation in other cells. However, it is not known whether β-adrenergic receptors activation stimulates NKCC2 trafficking. Thus we hypothesized that β-adrenergic receptor stimulation enhances THAL apical membrane NKCC2 expression via the PKA pathway and PDE4 blunts this effect. THAL suspensions were obtained from Sprague-Dawley rats, and surface NKCC2 expression was measured by surface biotinylation and Western blot. Incubation of THALs with the β-adrenergic receptor agonist isoproterenol at 0.5 and 1.0 μM increased surface NKCC2 by 17 ± 1 and 29 ± 5% respectively (P < 0.05). Preventing cAMP degradation with 3-isobutyl-methylxanthine (IBMX; a nonselective phosphodiesterase inhibitor) enhanced isoproterenol-stimulated surface NKCC2 expression to 51 ± 7% (P < 0.05 vs. isoproterenol). The β-adrenergic receptor antagonist propranolol or the PKA inhibitor H-89 completely blocked isoproterenol + IBMX-induced increase on surface NKCC2, while propranolol or H-89 alone had no effect. Selective inhibition of PDE4 with rolipram (20 μM) potentiated the effect of isoproterenol on surface NKCC2 and increased cAMP levels. We concluded that β-adrenergic receptor stimulation enhances surface NKCC2 expression in the THALs via PKA and PDE4 blunts this effect.
Keywords: isoproterenol, sodium reabsorption, rolipram, PDE4, hypertension
the thick ascending limb of the loop of Henle (THAL) reabsorbs ∼30% of the filtered NaCl in a process mediated by the apical Na-K-2Cl cotransporter NKCC2 (19, 24, 43, 47). To mediate NaCl reabsorption from the tubular lumen, NKCC2 must be present at the apical membrane of THALs. Several hormones such as arginine vasopressin (AVP), parathyroid hormone, glucagon, and β-adrenergic agonists stimulate THAL NaCl absorption by increasing cAMP (cyclic adenosine monophosphate) production (7, 17, 22, 24, 25, 31, 43, 47). We (3, 43) have previously shown that AVP stimulates NKCC2-dependent NaCl reabsorption by increasing trafficking of NKCC2 to the apical membrane in rat THALs via cAMP. However, it is not known whether other hormones or neurotransmitters that stimulate cAMP enhance surface NKCC2 in the THAL.
Enhanced renal nerve activity stimulates NaCl absorption by the THAL (7, 15, 24, 49). Various adrenergic receptors including β-1 and β-2 (58) are expressed in rat THALs. In isolated THALs, stimulation of β-adrenergic receptors enhances intracellular cAMP production by adenylyl cyclase (16) and stimulates Cl absorption (7, 41, 48). Chronic stimulation of β-adrenergic receptors enhances NKCC2 expression (56), and this is likely to contribute to long-term control of blood pressure. However, stimulation of β-adrenergic receptors enhance NaCl absorption within 20 to 30 min, suggesting an acute effect on NKCC2 activity. We (12) have reported that cAMP acutely stimulates NKCC2 trafficking via protein kinase A (PKA). However, it is not known whether stimulation of β-adrenergic receptors enhances apical surface expression of NKCC2 and whether this is mediated by cAMP and PKA.
β-Adrenergic receptor stimulation increases intracellular cAMP in THALs (7, 18). In most cells, cAMP is rapidly degraded by various cAMP-specific phosphodiesterases (PDEs) that modulate the magnitude of cAMP actions (3, 12, 44, 46). There are 11 different types of PDEs of which 8 are considered cAMP specific (29). PDEs differ on their structure, localization, cellular expression, inhibitor sensitivity (9, 10), and affinity for cAMP (9, 10). Some of them are expressed in the kidney (3, 9, 46, 59). In THALs, only PDE1 and PDE2 have been confirmed to be expressed (3, 9, 32). However, in collecting ducts, PDE-dependent cAMP activity is known to be contributed primarily by PDE4 (38, 57). In other cells, PDE4 is primarily responsible for decreasing cAMP generated by β-adrenergic stimulation (6, 60). Thus we hypothesized that β-adrenergic receptor stimulation enhances surface NKCC2 expression in rat THALs and this is blunted by PDE4.
MATERIALS AND METHODS
Suspensions of medullary THALs.
Male Sprague Dawley rats were anesthetized with ketamine (100 mg/kg bw ip) and xylazine (20 mg/kg bw ip). Medullary THAL suspensions were prepared according to the method described previously (3, 12, 23, 43). In brief, kidneys were perfused via the aorta with perfusion solution (in mM: 130 NaCl, 2.5 NaH2PO43−, 4.0 KCl, 1.2 MgSO4, 6 l-alanine, 1.0 disodium citrate, 5.5 glucose, 2.0 calcium lactate, and 10 HEPES, pH 7.4) containing 0.1% collagenase (Sigma, St. Louis, MO) and 100 U heparin. The outer medulla was dissected, minced, and digested in collagenase to obtain THAL suspensions.
Surface biotinylation of THAL suspensions.
Cell surface proteins were biotinylated as described in detail previously (3, 12, 23, 43). THAL suspensions were equilibrated at 37°C for 10 min followed by treatment with the corresponding drugs for 30 min and oxygenated every 5 min with 100% O2 with gentle mixing. When an inhibitor was used, it was added at the time of equilibration so that blockade was complete. Then, the suspensions were cooled rapidly to 4°C, washed twice with chilled perfusion solution, and centrifuged at 100 g for 2 min. THAL suspensions were incubated 30 min at 4°C in biotinylation solution (in mM: 10 HEPES, 130 NaCl, 2 MgSO4, 1 CaCl2, and 5.5 glucose, pH: 7.8–8.0) containing 0.9 mg/ml NHS-SS-biotin (Thermo Scientific) in a rocker. After biotinylation, tubules were washed three times at 4°C: once with perfusion solution and twice with perfusion solution containing 100 mM glycine to remove the excess of NHS-SS-biotin. THAL suspensions were centrifuged (100 g) and lysed in buffer containing the following (in mM): 150 NaCl, 50 HEPES, and 5 EDTA, plus 2% Triton X-100, 0.2% SDS, and protease inhibitors (10 μg/ml aprotinin, 5 μg/ml leupeptin, 4 mmol/l benzamidine, 5 μg/ml chymostatin, and 5 μg/ml pepstatin A; Sigma), pH 7.5.
For measurement of surface NKCC2, 80 μg of biotinylated THAL proteins from each experimental sample were incubated in two rounds with streptavidin-coated agarose beads. Surface proteins were extracted from the beads by boiling in 50 μl SDS-loading buffer containing 50 μM dl-dithiothreitol and 5% β-mercaptoethanol as described in detail in our previous studies (3, 4, 12, 23, 43). For total NKCC2 measurement, a one-tenth fraction of the supernatant (8 μg of total protein), containing intracellular nonbiotinylated proteins, was loaded in the same gels with surface fractions recovered from the beads. NKCC2 was detected by Western blot. Optical densities from surface and intracellular NKCC2 bands were used to calculate total NKCC2 and the ratio of surface to total NKCC2. Given the small percentage of NKCC2 at the surface (3–5%; Ref. 43), changes in the surface fraction do not result in detectable changes in the intracellular fraction. Day-to-day variation in surface biotinylation experiments in vehicle-treated samples (controls) was 8.1% across data sets. Thus the variability of surface NKCC2 in control THALs was acceptable.
Western blot analysis.
THAL lysates were resolved by SDS polyacrylamide gel electrophoresis in 6.5% gels and transferred to Hy-bond PVDF membranes (GE Health Care). The PVDF membrane was incubated for 60 min in blocking buffer containing 50 mM Tris, 150 mM NaCl, 3% BSA, and 0.1% Tween-20. Then, 1:1,000 dilution of anti-NKCC2 raised in chicken was incubated overnight at 4°C in blocking buffer. This antibody was raised against a synthetic peptide corresponding to amino acids 33–55 of rat NKCC2 (3, 43). Next, the membrane was incubated with anti chicken secondary antibody (1:10,000 conjugated with horseradish peroxidase; Thermo Fisher Scientific; Ref. 43). Bands were developed with ECL solution (GE Health Care). Surface NKCC2 and total NKCC2 were calculated by densitometry.
To assure that the surface fraction does not contain intracellular protein, we measured GAPDH in the surface and intracellular fraction in each experiment. For GAPDH immunoblotting, PVDF membranes were blocked with a mixture of 2.5% BSA and 2.5% milk for 60 min. We used primary monoclonal anti-GAPDH (Chemicon) at 1:60,000 for 120 min at room temperature in 3% BSA. Finally, anti-mouse secondary antibody conjugated to horseradish peroxidase (GE Health Care) was used at a 1:12,000 for 60 min at room temperature in 3% BSA.
To determine which PDE4 isoforms are expressed in THALs, we used 15–25 μg of total THAL lysate, resolved them by SDS-PAGE (7% gels), and transferred to Hy-bond PVDF membranes. PVDF membranes were incubated for 60 min in blocking buffer containing 50 mM Tris, 150 mM NaCl, 3% BSA, and 0.1% Tween-20. Then, the membranes were incubated with previously characterized isotype-specific pan-antibodies (Fabgennix): PDE4A (PD4-112AP), PDE4B (PD4-231), PDE4C (PD4-301), or PDE4D (PD4-401). For all antibodies except PDE4C, we used 1:1,000 for primary and secondary antibodies. For PDE4C, we used 1:2,000 primary antibody and 1:5,000 for the secondary antibody.
Intracellular cAMP measurements.
Intracellular cAMP was determined by RIA after sample acetylation (Biomedical Technologies) as described earlier (3). In brief, THAL suspensions were aliquoted and equilibrated in physiological perfusion solution (mentioned above) at 37°C for 10 min. Then, vehicle, isoproterenol, or rolipram was added to the bath and incubated for 10 min. The reaction was stopped by addition of 1.5 ml of chilled perfusion solution containing the general PDE inhibitor 3-isobutyl-1-methylxanthine (IBMX; 1 mM). To prevent endogenous nitric oxide (NO) production and decrease the variability of baseline cAMP, the NO synthase inhibitor Nω-l-arginine methyl ester was included in the solution. THALs were then washed by centrifugation (160 g) at 4°C, lysed in chilled solution containing 50% methanol/distilled water, and stored at −80°C for 1 h. Lysates were spun at 16,000 g, and the supernatant was recovered and stored at −80°C. The pellet was used for determination of total protein (in duplicate) by colorimetric commercial assay (Thermo Fisher Scientific). Samples were dried overnight on a Savant centrifuge dryer, the dried pellet was reconstituted in 240 μl of assay buffer, and cAMP was determined by RIA. Internal standards and recoveries were run with each experiment. Intracellular cAMP content was expressed as femtomoles per micrograms of protein.
Statistics.
Results are expressed as means ± SE. Differences between means were evaluated with one-way ANOVA, using Bonferroni adjustment for multiple comparisons. P < 0.05 was considered significant.
RESULTS
Effect of β-adrenergic receptor stimulation on surface NKCC2 expression in rat THALs.
First, we studied whether β-adrenergic receptor stimulation with the agonist isoproterenol (Iso) enhances surface NKCC2 in THALs. Rat THAL suspensions obtained from the same animal were divided into four aliquots and treated with vehicle (control) or isoproterenol to reach a final concentration of 0.2, 0.5, and 1.0 μM. After treatment (30 min), we measured surface and total NKCC2 in THALs by surface biotinylation as described in materials and methods. Data were normalized to surface NKCC2 in controls that was set to 100%. We found that isoproterenol at 0.2 μM did not affect surface NKCC2 (102 ± 3%), whereas at 0.5 and 1.0 μM it enhanced surface NKCC2 to 117 ± 1 and 129 ± 5%, respectively (Fig. 1; n = 6). We also measured total NKCC2 expression and found no effect of Iso on total NKCC2 levels (control: 100, 0.2 μM Iso: 108 ± 3%, 0.5 μM Iso: 115 ± 13%, and 1 μM Iso: 109 ± 15%). GAPDH was not detected in the surface fraction while observed in the total/intracellular fractions. These data indicate that β-adrenergic receptor stimulation increases steady-state surface NKCC2 in THALs.
Fig. 1.

Effect of the β-adrenergic receptor agonist isoproterenol on surface NKCC2 in rat thick ascending limbs (THALs). A: representative Western blot for the Na-K-2Cl cotransporter NKCC2 and GAPDH measured from surface and intracellular protein after 0.2, 0.5, and 1 μM Iso treatment for 30 min. We loaded 80 μg THAL proteins for surface NKCC2 measurement, and for intracellular fraction we loaded 8 μg proteins. GAPDH is not detectable in the surface fraction. B: average data show that isoproterenol-stimulated surface NKCC2 expression is in a concentration response pattern. *P < 0.05 vs. control; n = 6.
Effect of phosphodiesterase inhibition on β-adrenergic receptor-stimulated surface NKCC2 expression in rat THALs.
β-Adrenergic receptor stimulation increases cAMP production in THALs (18). However, cAMP produced by β-adrenergic receptor stimulation is quickly degraded by phosphodiesterases, which may decrease the effect of isoproterenol. To test this, we used the nonspecific phosphodiesterase inhibitor IBMX (0.5 mM). THAL suspensions were divided into four aliquots and treated with vehicle, IBMX, 1 μM Iso, or IBMX + 1 μM Iso. IBMX alone did not change surface NKCC2, whereas it potentiated the stimulatory effect of Isoproterenol (control: 100%, IBMX: 109 ± 5%, Iso: 129 ± 6%, and IBMX + Iso: 151 ± 8%; Fig. 2). Inhibition of PDEs with IBMX enhanced the effect of isoproterenol on surface NKCC2 by ∼75% (P < 0.02 vs. Iso). No significant change in total NKCC2 was observed in this group of experiments (control: 100%, IBMX: 100 ± 2%, Iso: 94 ± 3%, and IBMX + Iso: 95 ± 2%). GAPDH was not present in the surface fraction. These data indicate that the stimulatory effect of isoproterenol on surface NKCC2 expression is blunted by the action of phosphodiesterases.
Fig. 2.

Effect of nonspecific phosphodiesterase (PDE) inhibition on β-adrenergic receptor-stimulated surface NKCC2 in rat THALs. A: representative Western blot for surface NKCC2 expression. B: average data show that phosphodiesterase inhibition with 3-isobutyl-methylxanthine (IBMX) further enhanced the stimulatory effect of isoproterenol (Iso) on surface NKCC2. *P < 0.05 vs. control; #P < 0.025 vs. Iso alone; n = 12.
Effect of β-adrenergic receptor antagonism on isoproterenol-stimulated surface NKCC2 expression in rat THALs.
Because PDE inhibition with IBMX enhanced the stimulatory effect of isoproterenol, we used this combination in subsequent blockade experiments. To test whether the effect of isoproterenol is solely mediated by β-adrenergic receptor stimulation, we used the β-adrenergic receptor antagonist propranolol (Pro, 10 μM). THAL suspensions were separated into four aliquots and treated with vehicle, Pro, IBMX + Iso (1 μM), and Pro + IBMX + Iso. Propranolol did not affect surface NKCC2, but it completely blocked the stimulatory effect of IBMX + Iso on surface NKCC2 (control: 100%, Pro: 107 ± 3%, IBMX+ Iso: 144 ± 8%, and Pro + IBMX + Iso: 104 ± 4%; P < 0.05 vs. IBMX + Iso; Fig. 3). No significant changes in total NKCC2 were observed in any group (control: 100%, Pro: 93 ± 4%, IBMX + Iso: 103 ± 10%, and Pro + IBMX + Iso: 92 ± 6%). GAPDH was not detected in the surface fraction. These data indicate that isoproterenol stimulates NKCC2 trafficking by acting on β-adrenergic receptors.
Fig. 3.

Effect of β-adrenergic receptor antagonist propranolol (Pro) on Iso-stimulated surface NKCC2 in rat THALs. A: representative Western blot of surface NKCC2. B: cumulative data shows that the β-adrenergic antagonist Pro blocked surface NKCC2 expression stimulated by the β-adrenergic agonist Iso. *P < 0.05 vs. control; #P < 0.025 vs. IBMX + Iso; n = 6.
Effect of PDE4 inhibition on isoproterenol-stimulated surface NKCC2 expression in rat THALs.
During the dose-response effect to isoproterenol, we observed that 0.2 μM did not increase surface NKCC2 (Fig. 1). Therefore, we used this lower concentration to test whether selective PDE4 inhibition with rolipram enhances the effect of β-adrenergic stimulation on surface NKCC2 expression in THALs. THAL suspensions were divided into four aliquots and treated with either vehicle, 20 μM rolipram, 0.2 μM Iso, or 20 μM rolipram + 0.2 μM Iso for 30 min. We found that 0.2 μM Iso enhanced surface NKCC2 only when PDE4 was inhibited by rolipram (control: 100%, 0.2 μM Iso: 100 ± 5%, 20 μM rolipram: 106 ± 6%, and rolipram + Iso: 126 ± 8%; P < 0.05 vs. Iso; Fig. 4, A and B). No significant changes were observed in total NKCC2 expression (control: 100%, 20 μM rolipram: 105 ± 2%, 0.2 μM Iso: 99 ± 5%, and rolipram + Iso: 102 ± 3%). GAPDH was not observed in the surface fraction. These data indicate that β-adrenergic receptor-mediated stimulation of NKCC2 trafficking is blunted by the action of PDE4.
Fig. 4.

PDE4 inhibition with rolipram (Roli; 20 μM) potentiates the effect of Iso on surface NKCC2 and cAMP in rat THALs. A: surface NKCC2 expression in rat THAL suspensions treated with 0.2 μM Iso and Iso (0.2 μM) + rolipram (20 μM); representative Western blot of NKCC2. B: cumulative data showing the effect of 20 μM rolipram on a nonstimulatory concentration of Iso (0.2 μM) on surface NKCC2. When rolipram and Iso were added together, they increased surface NKCC2 expression (n = 6, *P < 0.05 vs. control). C: effect of 20 μM rolipram on a nonstimulatory concentration of Iso (0.2 μM) on intracellular cAMP in THALs. Iso (0.2 μM) increased cAMP in the presence of 20 μM rolipram, a PDE4 inhibitor. *P < 0.05 vs. control; #P < 0.025 vs. Iso; n = 8 in each cases.
Effect of PDE4 inhibition on β-adrenergic receptor-mediated stimulation of cAMP levels in rat THALs.
To assure that the effect of rolipram at 20 μM was due to enhanced cAMP levels caused by PDE4 inhibition, we measured intracellular cAMP by RIA. THAL suspensions were divided into four aliquots and treated with either vehicle, 20 μM rolipram, 0.2 μM Iso or 20 μM rolipram + 0.2 μM Iso. We observed that cAMP was significantly enhanced by isoproterenol during inhibition of PDE4 (control: 39 ± 3 fmol/μg protein, 20 μM rolipram: 73 ± 9 fmol/μg protein, 0.2 μM Iso: 40 ± 6 fmol/μg protein, and 20 μM rolipram + 0.2 μM Iso: 116 ± 22 fmol/μg protein; P < 0.05 vs. rolipram alone; n = 5; Fig. 4C). Rolipram alone tended to increase cAMP, but this was not statistically significant.
Effect of PKA inhibitor H-89 on β-adrenergic receptor-stimulated surface NKCC2 expression in rat THALs.
cAMP stimulates apical exocytosis of NKCC2 in rat THALs via the PKA pathway (12). Thus we tested whether β-adrenergic receptor stimulated surface NKCC2 is mediated by PKA. We used the PKA inhibitor H-89 (10 μM). THAL suspensions were separated into four aliquots and then treated with vehicle, H-89, 1 μM IMBX + Iso, and 1 μM H-89 + IBMX + Iso. H-89 alone did not affect surface NKCC2 but completely blocked the stimulatory effect of IBMX + Iso on surface NKCC2 (control: 100%, H-89: 106 ± 16%, IBMX + Iso: 154 ± 10%, and H-89 + IBMX + Iso: 113 ± 13%; P < 0.05 vs. IBMX + Iso; Fig. 5). Total NKCC2 was not different between control and treatments (control: 100%, H-89: 95 ± 7%, IBMX + Iso: 111 ± 7%, and H-89 + IBMX + Iso: 103 ± 7%). GAPDH was not detected in the surface fraction. These data indicate that β-adrenergic receptor stimulation enhances surface NKCC2 expression via PKA.
Fig. 5.

Effect of protein kinase A (PKA) inhibition on β-adrenergic receptor-stimulated surface NKCC2 in rat THALs. A: representative Western blot of surface NKCC2 in THALs treated with Iso/IBMX or Iso/IBMX plus H-89. B: cumulative data showing that H-89 (10 μM) completely blocked surface NKCC2 expression stimulated by Iso (1 μM). *P < 0.05 vs. control; #P < 0.025 vs. IBMX + Iso; n = 6.
Western blot analysis of PDE4 isoforms present in rat THALs.
Pharmacological blockade of PDE4 by rolipram indicates that PDE4 is present in THALs, yet the expression of PDE4 has not been studied in THALs. There are four PDE4 families: PDE4A/B/C/D, each with many splice variants. Using antibodies directed to conserved regions in the different PDE4 families, we studied which isoforms were detected in THALs by Western blot. Splice variants for each family were identified based on their different molecular mass. A pan-PDE4A antibody recognized three predominant bands at ∼ 76, 102, and 109 kDa corresponding to isoforms PDE4A1 (76 kDa), PDEAX (102 kDa), and PDE4A5 (109 kDa; Fig. 6) (20, 34). Two of these bands were also present in brain homogenate (76 and 102 kDa). A PDE4B pan antibody detected one predominant band at ∼85 kDa and two less dense bands at 60 and 68 kDa. They might correspond to PDE4B4 (85 kDa; Ref. 52), PDE4B5 (58–60 kDa), and a short form of PDE4B2 (68 kDa; Fig. 6) (13, 52). A single band at 86 kDa was predominant in brain homogenate. The PDE4C pan antibody detected a predominant band at ∼ 64 kDa and less dense bands at 80 and 94 kDa. The 64-kDa band may correspond to a short PDE4C variant (Δ54; Ref. 42) and is similar to that reported in lung and other tissues (61), while the 80-kDa band may correspond to PDE4C2 (Fig. 6) (26, 30, 42, 45). The PDE4C antibody reacted strongly with recombinant GST-PDE4C (75 kDa). Finally, a PDE4D pan-antibody detected bands at ∼68, 78, 90, and 100 kDa corresponding to PDE4D2/6 (68 kDa), PDE4D1 (78 kDa), PDE4D3/8/9 (90 kDa), and PDE4D5/7 (102 kDa; Fig. 6) (51, 57). In brain homogenate, three bands were observed at 72, 78, and 102 kDa. These data indicate that multiple PDE4 isoforms and variants are expressed in THALs.
Fig. 6.
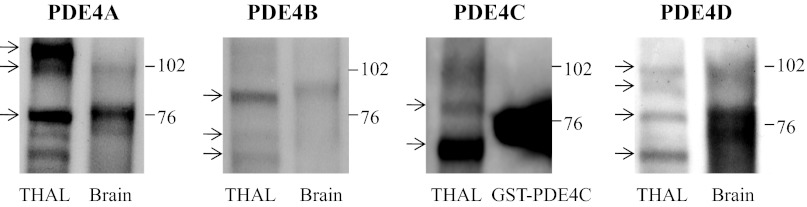
Expression of various PDE4 isoforms in rat medullary THALs. Antibodies against conserved but distinct domains in PDE4A/B/C/D were used to detect PDE4 variants in THAL lysates. Whole brain homogenate was used as positive control since most of the PDE4 isoforms are expressed in that tissue. GST-PDE4C (SignalChem) was used as positive control for PDE4C. As detailed in the results section, PDE4A/B/C/D and some of their variants were detected in THAL lysates. Arrows indicates splice variants.
DISCUSSION
β-Adrenergic receptor stimulation enhances NaCl reabsorption by the THAL. However, the molecular mechanisms by which this occurs remain poorly explored. In the current study, we have addressed the role of β-adrenergic stimulation on trafficking of NKCC2 to the cell surface and the signaling pathway involved. We found that stimulation of β-adrenergic receptors enhanced cAMP and surface NKCC2 levels and that PKA was required for this stimulatory effect. We also found that inhibition of PDE4 enhanced the stimulatory effect of β-adrenergic receptors on intracellular cAMP and NKCC2 trafficking to the surface in THALs. Taken together, these data indicate that β-adrenergic receptor stimulation enhances NKCC2 trafficking in THALs via cAMP and that PDE4 blunts this effect.
In the present study, we demonstrated that acute stimulation of β-adrenergic receptors in THALs increases NKCC2 trafficking to the plasma membrane. We also found that this occurs within a time frame (20–30 min) where total NKCC2 expression did not change. We (4) previously showed that trafficking of NKCC2 to the cell surface is essential to acutely stimulate NaCl absorption by the THAL, independently of changes on total NKCC2 expression. Thus we think this mechanism provides a way to quickly enhance salt reabsorption during heightened norepinephrine/epinephrine release by renal nerves. In addition to the acute effect on NKCC2, a previous report by Sonalker et al. (55) showed that chronic infusion of norepinephrine (NE) increases the total expression of NKCC2 in rats. Thus our data and that of others suggest that NKCC2 stimulation in the THAL is an essential component of catecholamine-induced antinatriuresis.
Isoproterenol stimulates both, β1- and β2-adrenergic receptors, which are expressed in the THAL (18, 39). Previous evidence indicates that β1-receptors are the main contributors to adrenergic stimulation of cAMP in THALs (18). While we have used isoproterenol to stimulate β-receptors, α-adrenergic receptors are also expressed in the THAL (39, 40). However, α2-receptor activation inhibits NaCl transport in the THAL (48) by increasing NO production (48, 49), which directly inhibits NKCC2 trafficking and activity. In light of these observations, it is unlikely that α-adrenergic receptors play a role in isoproterenol-induced stimulation of NKCC2 trafficking. To confirm the role of β-adrenergic receptors on NKCC2 trafficking, we used the antagonist propranolol. We observed that blockade of β-adrenergic receptors with propranolol in THALs completely prevented the stimulation induced by isoproterenol on surface NKCC2 expression even in the presence of the nonspecific phosphodiesterase inhibitor IBMX. Propranolol or IBMX alone did not affect surface NKCC2 (Figs. 2 and 3), suggesting that β-adrenergic receptors are inactive in THAL suspensions in the absence of agonists. This is most likely due to washout of endogenous catecholamines during the preparation of THAL suspensions. These data confirm the stimulatory role of β-adrenergic receptors on NKCC2 trafficking in the THAL. The effect of isoproterenol on surface NKCC2 is comparable to the effect of higher concentrations of NE on net Cl reabsorption. Plato (48) reported that NE causes a biphasic response in THAL Cl absorption. At lower concentrations (0.01–0.1 μM), NE decreased Cl transport by activating α2-receptors, whereas at 10 μM NE increased net Cl transport by 20%. In agreement with our data, Baum (8) found that isoproterenol (1 μM) stimulated Cl reabsorption in rat medullary THALs by 20–25%. These data suggest that maximal β-adrenergic receptor stimulation is required to stimulate NKCC2 surface expression.
It has long been recognized that stimulation of β-adrenergic receptors enhances intracellular cAMP in THALs. However, cAMP levels are regulated by stimulated production and also by degradation via phosphodiesterases. We found that the nonspecific phosphodiesterase inhibitor IBMX enhanced the stimulatory effect of isoproterenol on surface NKCC2. These data suggest that cAMP-phosphodiesterases blunt the action of β-adrenergic receptors on NKCC2 trafficking. We (12) previously found that general phosphodiesterase inhibition enhanced the effect of vasopressin on NKCC2 trafficking to a maximum of 52%. Because vasopressin also enhances NKCC2 activity via cAMP, it is possible that cAMP phosphodiesterases are part of a negative feedback regulatory mechanism that limits the stimulation of NKCC2 and NaCl transport in the THAL.
Most cells express multiple phosphodiesterases. The specific subset of phosphodiesterases expressed in THALs is not known. To our knowledge, only the expression of PDE1 and PDE2 has been reported at the protein level in THALs (3, 9, 32). However, PDE4 is expressed in most cells and is known to blunt β-adrenergic signaling in other cells (1, 6, 11, 60). Thus we focused on PDE4. In the present study, we used the PDE4 inhibitor rolipram (28) to differentiate the effects of PDE4 from other cAMP-phosphodiesterases that are inhibited by IBMX. We tested whether rolipram (20 μM) potentiated the effect of isoproterenol on surface NKCC2. From our initial concentration-response curve, we selected 0.2 μM isoproterenol, the highest concentration that did not stimulate surface NKCC2. At this concentration, isoproterenol only enhanced surface NKCC2 in the presence of 20 μM rolipram. Taken together, these data indicate that PDE4 blunts the stimulatory effect of β-adrenergic receptor stimulation on NKCC2 trafficking in THALs. Studies (1, 6, 11, 60) in other cells also point to a role of PDE4 on limiting β-adrenergic receptor signaling. To examine the role of PDE4 in β-adrenergic receptor-stimulated intracellular cAMP, we measured cAMP levels in THALs. As expected, a nonstimulatory concentration of isoproterenol or rolipram did not increase cAMP. However, a combination of both drugs significantly increased intracellular cAMP. These observations indicate that mild stimulation of β-adrenergic receptors with isoproterenol enhances cAMP production, which is immediately degraded by PDE4. Surface NKCC2 was also enhanced by PDE4 inhibition under these conditions. Thus our data indicate that β-adrenergic stimulation produces cAMP that enhances NKCC2 trafficking, whereas PDE4 decreases the magnitude of this stimulatory response by immediately degrading cAMP.
There are 4 genes encoding PDE4 A/B/C/D generating >13 PDE4 isoforms that result from alternative splicing (27). We used antibodies recognizing conserved but distinct regions of the four PDE4 families to explore their expression in THALs. Given the large number of splice variants and the tissue specificity of each variant there is some controversy on the molecular mass for each PDE4. In general, all PDE4 isoforms run at a higher molecular mass than predicted by their sequence (20, 30, 37, 50, 51, 52) in SDS-PAGE. However, the splice variants for each family exhibit different molecular mass allowing their identification. Using this approach, we report that at least twelve PDE4 variants of the four families (PDE4A/B/C/D) are detected in THAL lysates. This is not surprising as most cell types express PDE4A/B/C/D and multiple variants (20, 30, 37, 50, 51, 52). The PDE4 isoforms PDE4D and to a lesser extent PDE4B seem to be the main contributors limiting β-adrenergic signaling in other cells (5, 60). PDE4D seems to be responsible for inhibiting vasopressin action in the collecting duct (57). The relative contribution of the twelve different PDE4 isoforms in THAL physiology is unknown. In the future, it could be important to determine the specific PDE4 isoform that blunts β-adrenergic signaling and vasopressin signaling in the THAL.
We (12) previously found that cAMP enhances NKCC2 exocytosis in part by stimulating PKA. To begin exploring the signaling pathway by which β-adrenergic receptors stimulate NKCC2 trafficking we used H-89, a PKA inhibitor. We observed that H-89 completely prevented the stimulatory effect of β-adrenergic stimulation on surface NKCC2 expression. These data are in agreement with our previous report (12) showing that vasopressin stimulates surface NKCC2 by activating PKA. In addition to vasopressin and β-adrenergic stimulation, dopamine has been shown to stimulate NaCl transport in the THAL by PKA (2). How the same second messenger (cAMP) participates in different signaling pathways still requires further investigation. It is possible that cAMP is produced by the same pool of adenylyl cyclase following receptor stimulation, or that cAMP converges into PKA to stimulate trafficking, or that the various receptors stimulate different compartments of cAMP production and signaling. In most cells studied, compartmentalization provides an efficient mechanism to limit cAMP signal propagation, which in most cases includes a signaling complex with a unique phosphodiesterase and PKA anchoring (12).
Renal interstitial release of norepinephrine from renal sympathetic nerves is a potent source of catecholamines reaching the kidney. Renal nerve stimulation enhances Na and Cl absorption by the loop of Henle as indicated by in vivo micropuncture studies (15). Increased renal sympathetic nerve activity has long been associated with hypertension in humans and animals (14, 21, 35, 36, 54). In addition, animal models of salt sensitive hypertension like the Dahl rat exhibit higher renal sympathetic nerve activity (53) and abnormally enhanced NaCl absorption by the THAL (23, 33). Our observations provide a new mechanism by which catecholamines enhance NaCl absorption via stimulation of NKCC2 trafficking in the THAL. This mechanism may be enhanced in salt-sensitive hypertension.
In summary, we conclude that β-adrenergic receptor stimulation enhances NKCC2 trafficking to the apical membrane of THALs by increasing intracellular cAMP levels and subsequent activation of PKA. The stimulatory response is blunted by PDE4, which degrades cAMP. In this way, PDE4 provides a possible mechanism for restricting β-adrenergic signaling on NKCC2 trafficking in the THAL.
GRANTS
This work was supported in part by National Heart, Lung, and Blood Institute Grants R0-HL-080409 and P01-HL-090550 (subproject 2, to P. Ortiz).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.Z.H. and P.A.O. conception and design of research; M.Z.H. and P.S.C. performed experiments; M.Z.H. and P.A.O. analyzed data; M.Z.H. and P.A.O. interpreted results of experiments; M.Z.H. prepared figures; M.Z.H. drafted manuscript; M.Z.H., P.S.C., and P.A.O. edited and revised manuscript; M.Z.H., P.S.C., and P.A.O. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Becca Alviani for technical assistance.
REFERENCES
- 1.Afzal F, Aronsen JM, Moltzau LR, Sjaastad I, Levy FO, Skomedal T, Osnes JB, Qvigstad E. Differential regulation of beta2 -adrenoceptor-mediated inotropic and lusitropic response by PDE3 and PDE4 in failing and nonfailing rat cardiac ventricle. Br J Pharmacol 162: 54–71, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoki Y, Albrecht FE, Bergman KR, Jose PA. Stimulation of Na+-K+-2Cl− cotransport in rat medullary thick ascending limb by dopamine. Am J Physiol Regul Integr Comp Physiol 271: R1561–R1567, 1996 [DOI] [PubMed] [Google Scholar]
- 3.Ares GR, Caceres P, Alvarez-Leefmans FJ, Ortiz PA. cGMP decreases surface NKCC2 levels in the thick ascending limb: role of phosphodiesterase 2 (PDE2). Am J Physiol Renal Physiol 295: F877–F887, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ares GR, Ortiz PA. Constitutive endocytosis and recycling of NKCC2 in rat thick ascending limbs. Am J Physiol Renal Physiol 299: F1193–F1202, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baillie GS, Adams DR, Bhari N, Houslay TM, Vadrevu S, Meng D, Li X, Dunlop A, Milligan G, Bolger GB, Klussmann E, Houslay MD. Mapping binding sites for the PDE4D5 cAMP-specific phosphodiesterase to the N- and C-domains of beta-arrestin using spot-immobilized peptide arrays. Biochem J 404: 71–80, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD. beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci USA 100: 940–945, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Bailly C, Imbert-Teboul M, Roinel N, Amiel C. Isoproterenol increases Ca, Mg, and NaCl reabsorption in mouse thick ascending limb. Am J Physiol Renal Fluid Electrolyte Physiol 258: F1224–F1231, 1990 [DOI] [PubMed] [Google Scholar]
- 8.Baum M. Effect of catecholamines on rat medullary thick ascending limb chloride transport: interaction with angiotensin II. Am J Physiol Regul Integr Comp Physiol 298: R954–R958, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol Rev 75: 725–748, 1995 [DOI] [PubMed] [Google Scholar]
- 10.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58: 488–520, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Bruss MD, Richter W, Horner K, Jin SL, Conti M. Critical role of PDE4D in beta2-adrenoceptor-dependent cAMP signaling in mouse embryonic fibroblasts. J Biol Chem 283: 22430–22442, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caceres PS, Ares GR, Ortiz PA. cAMP stimulates apical exocytosis of the renal Na(+)-K(+)-2Cl(−) cotransporter NKCC2 in the thick ascending limb: role of protein kinase A. J Biol Chem 284: 24965–24971, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung YF, Kan Z, Garrett-Engele P, Gall I, Murdoch H, Baillie GS, Camargo LM, Johnson JM, Houslay MD, Castle JC. PDE4B5, a novel, super-short, brain-specific cAMP phosphodiesterase-4 variant whose isoform-specifying N-terminal region is identical to that of cAMP phosphodiesterase-4D6 (PDE4D6). J Pharmacol Exp Ther 322: 600–609, 2007 [DOI] [PubMed] [Google Scholar]
- 14.DiBona GF. The kidney in the pathogenesis of hypertension: the role of renal nerves. Am J Kidney Dis 5: A27–A31, 1985 [DOI] [PubMed] [Google Scholar]
- 15.DiBona GF, Sawin LL. Effect of renal nerve stimulation on NaCl and H2O transport in Henle's loop of the rat. Am J Physiol Renal Fluid Electrolyte Physiol 243: F576–F580, 1982 [DOI] [PubMed] [Google Scholar]
- 16.Dousa TP. Cyclic-3′,5′-nucleotide phosphodiesterase isozymes in cell biology and pathophysiology of the kidney. Kidney Int 55: 29–62, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Ecelbarger CA, Yu S, Lee AJ, Weinstein LS, Knepper MA. Decreased renal Na-K-2Cl cotransporter abundance in mice with heterozygous disruption of the Gsalpha gene. Am J Physiol Renal Physiol 277: F235–F244, 1999 [DOI] [PubMed] [Google Scholar]
- 18.Elalouf JM, Buhler JM, Tessiot C, Bellanger AC, Dublineau I, de Rouffignac C. Predominant expression of beta 1-adrenergic receptor in the thick ascending limb of rat kidney. Absolute mRNA quantitation by reverse transcription and polymerase chain reaction. J Clin Invest 91: 264–272, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eng B, Mukhopadhyay S, Vio CP, Pedraza PL, Hao S, Battula S, Sehgal PB, McGiff JC, Ferreri NR. Characterization of a long-term rat mTAL cell line. Am J Physiol Renal Physiol 293: F1413–F1422, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Farooqui SM, Zhang K, Makhay M, Jackson K, Farooqui SQ, Cherry JA, O'Donnell JM. Noradrenergic lesions differentially alter the expression of two subtypes of low Km cAMP-sensitive phosphodiesterase type 4 (PDE4A and PDE4B) in rat brain. Brain Res 867: 52–61, 2000 [DOI] [PubMed] [Google Scholar]
- 21.Grande MT, Pascual G, Riolobos AS, Clemente-Lorenzo M, Bardaji B, Barreiro L, Tornavaca O, Meseguer A, Lopez-Novoa JM. Increased oxidative stress, the renin-angiotensin system, and sympathetic overactivation induce hypertension in kidney androgen-regulated protein transgenic mice. Free Radic Biol Med 51: 1831–1841, 2011 [DOI] [PubMed] [Google Scholar]
- 22.Gunaratne R, Braucht DW, Rinschen MM, Chou CL, Hoffert JD, Pisitkun T, Knepper MA. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc Natl Acad Sci USA 107: 15653–15658, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haque MZ, Ares GR, Caceres PS, Ortiz PA. High salt differentially regulates surface NKCC2 expression in thick ascending limbs of dahl salt sensitive and salt resistant rats. Am J Physiol Renal Physiol 300: F1096–F1104, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hebert SC, Andreoli TE. Control of NaCl transport in the thick ascending limb. Am J Physiol Renal Fluid Electrolyte Physiol 246: F745–F756, 1984 [DOI] [PubMed] [Google Scholar]
- 25.Hebert SC, Friedman PA, Andreoli TE. Effects of antidiuretic hormone on cellular conductive pathways in mouse medullary thick ascending limbs of Henle: I. ADH increases transcellular conductance pathways. J Membr Biol 80: 201–219, 1984 [DOI] [PubMed] [Google Scholar]
- 26.Heimann E, Jones HA, Resjo S, Manganiello VC, Stenson L, Degerman E. Expression and regulation of cyclic nucleotide phosphodiesterases in human and rat pancreatic islets. PLos One 5: e14191, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today 10: 1503–1519, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Johnston LA, Erdogan S, Cheung YF, Sullivan M, Barber R, Lynch MJ, Baillie GS, Van HG, Adams DR, Huston E, Houslay MD. Expression, intracellular distribution and basis for lack of catalytic activity of the PDE4A7 isoform encoded by the human PDE4A cAMP-specific phosphodiesterase gene. Biochem J 380: 371–384, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ke H. Implications of PDE4 structure on inhibitor selectivity across PDE families. Int J Impot Res 16, Suppl 1: S24–S27, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Keravis T, Monneaux F, Yougbare I, Gazi L, Bourguignon JJ, Muller S, Lugnier C. Disease progression in MRL/lpr lupus-prone mice is reduced by NCS 613, a specific cyclic nucleotide phosphodiesterase type 4 (PDE4) inhibitor. PLos One 7: e28899, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim GH, Ecelbarger CA, Mitchell C, Packer RK, Wade JB, Knepper MA. Vasopressin increases Na-K-2Cl cotransporter expression in thick ascending limb of Henle's loop. Am J Physiol Renal Physiol 276: F96–F103, 1999 [DOI] [PubMed] [Google Scholar]
- 32.Kim JK, Jackson BA, Edwards RM, Dousa TP. Effect of potassium depletion on the vasopressin-sensitive cyclic AMP system in rat outer medullary tubules. J Lab Clin Med 99: 29–38, 1982 [PubMed] [Google Scholar]
- 33.Kirchner KA. Increased loop chloride uptake precedes hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 262: R263–R268, 1992 [DOI] [PubMed] [Google Scholar]
- 34.Kolosionek E, Savai R, Ghofrani HA, Weissmann N, Guenther A, Grimminger F, Seeger W, Banat GA, Schermuly RT, Pullamsetti SS. Expression and activity of phosphodiesterase isoforms during epithelial mesenchymal transition: the role of phosphodiesterase 4. Mol Biol Cell 20: 4751–4765, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kopp UC, Cicha MZ, Smith LA, Ruohonen S, Scheinin M, Fritz N, Hokfelt T. Dietary sodium modulates the interaction between efferent and afferent renal nerve activity by altering activation of alpha2-adrenoceptors on renal sensory nerves. Am J Physiol Regul Integr Comp Physiol 300: R298–R310, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lohmeier TE. The sympathetic nervous system and long-term blood pressure regulation. Am J Hypertens 14: 147S–154S, 2001 [DOI] [PubMed] [Google Scholar]
- 37.Mackenzie KF, Topping EC, Bugaj-Gaweda B, Deng C, Cheung YF, Olsen AE, Stockard CR, High ML, Baillie GS, Grizzle WE, De VM, Houslay MD, Wang D, Bolger GB. Human PDE4A8, a novel brain-expressed PDE4 cAMP-specific phosphodiesterase that has undergone rapid evolutionary change. Biochem J 411: 361–369, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McSorley T, Stefan E, Henn V, Wiesner B, Baillie GS, Houslay MD, Rosenthal W, Klussmann E. Spatial organisation of AKAP18 and PDE4 isoforms in renal collecting duct principal cells. Eur J Cell Biol 85: 673–678, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Meister B, Dagerlind A, Nicholas AP, Hokfelt T. Patterns of messenger RNA expression for adrenergic receptor subtypes in the rat kidney. J Pharmacol Exp Ther 268: 1605–1611, 1994 [PubMed] [Google Scholar]
- 40.Mohuczy-Dominiak D, Garg LC. Alpha-2 adrenoceptors in medullary thick ascending limbs of the rabbit kidney. J Pharmacol Exp Therapeut 266: 279–287, 1993 [PubMed] [Google Scholar]
- 41.Morgunov NS, You YD, Hirsch DJ. Response of mouse proximal straight tubule and medullary thick ascending limb to beta-agonist. J Am Soc Nephrol 4: 1151–1158, 1993 [DOI] [PubMed] [Google Scholar]
- 42.Obernolte R, Ratzliff J, Baecker PA, Daniels DV, Zuppan P, Jarnagin K, Shelton ER. Multiple splice variants of phosphodiesterase PDE4C cloned from human lung and testis. Biochim Biophys Acta 1353: 287–297, 1997 [DOI] [PubMed] [Google Scholar]
- 43.Ortiz PA. cAMP increases surface expression of NKCC2 in rat thick ascending limbs: role of VAMP. Am J Physiol Renal Physiol 290: F608–F616, 2006 [DOI] [PubMed] [Google Scholar]
- 44.Ortiz PA, Garvin JL. NO Inhibits NaCl Absorption by Rat Thick Ascending Limb Through Activation of cGMP-Stimulated Phosphodiesterase. Hypertension 37: 467–471, 2001 [DOI] [PubMed] [Google Scholar]
- 45.Owens RJ, Lumb S, Rees-Milton K, Russell A, Baldock D, Lang V, Crabbe T, Ballesteros M, Perry MJ. Molecular cloning and expression of a human phosphodiesterase 4C. Cell Signal 9: 575–585, 1997 [DOI] [PubMed] [Google Scholar]
- 46.Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 298: 834–836, 2002 [DOI] [PubMed] [Google Scholar]
- 47.Plata C, Meade P, Hall A, Welch RC, Vazquez N, Hebert SC, Gamba G. Alternatively spliced isoform of apical Na+-K+-Cl− cotransporter gene encodes a furosemide-sensitive Na+-Cl− cotransporter. Am J Physiol Renal Physiol 280: F574–F582, 2001 [DOI] [PubMed] [Google Scholar]
- 48.Plato CF. Alpha-2 and beta-adrenergic receptors mediate NE's biphasic effects on rat thick ascending limb chloride flux. Am J Physiol Regul Integr Comp Physiol 281: R979–R986, 2001 [DOI] [PubMed] [Google Scholar]
- 49.Plato CF, Garvin JL. Alpha(2)-adrenergic-mediated tubular NO production inhibits thick ascending limb chloride absorption. Am J Physiol Renal Physiol 281: F679–F686, 2001 [DOI] [PubMed] [Google Scholar]
- 50.Pullamsetti SS, Banat GA, Schmall A, Szibor M, Pomagruk D, Hanze J, Kolosionek E, Wilhelm J, Braun T, Grimminger F, Seeger W, Schermuly RT, Savai R. Phosphodiesterase-4 promotes proliferation and angiogenesis of lung cancer by crosstalk with HIF. Oncogene 2012 Apr 23 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 51.Richter W, Jin SL, Conti M. Splice variants of the cyclic nucleotide phosphodiesterase PDE4D are differentially expressed and regulated in rat tissue. Biochem J 388: 803–811, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shepherd M, McSorley T, Olsen AE, Johnston LA, Thomson NC, Baillie GS, Houslay MD, Bolger GB. Molecular cloning and subcellular distribution of the novel PDE4B4 cAMP-specific phosphodiesterase isoform. Biochem J 370: 429–438, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sjoquist M, Lee SL, Hansell P. CNS-induced natriuresis, neurohypophyseal peptides and renal dopamine and noradrenaline excretion in prehypertensive salt-sensitive Dahl rats. Exp Physiol 90: 847–853, 2005 [DOI] [PubMed] [Google Scholar]
- 54.Somova LI, Channa ML, Khan MS. An experimental rat model of salt-sensitive hypertension; biochemical and morphological parameters and sympathetic nervous system. J S Afr Vet Assoc 70: 14–17, 1999 [DOI] [PubMed] [Google Scholar]
- 55.Sonalker PA, Tofovic SP, Bastacky SI, Jackson EK. Chronic noradrenaline increases renal expression of NHE-3, NBC-1, BSC-1 and aquaporin-2. Clin Exp Pharmacol Physiol 35: 594–600, 2008 [DOI] [PubMed] [Google Scholar]
- 56.Sonalker PA, Tofovic SP, Jackson EK. Increased expression of the sodium transporter BSC-1 in spontaneously hypertensive rats. J Pharmacol Exp Ther 311: 1052–1061, 2004 [DOI] [PubMed] [Google Scholar]
- 57.Stefan E, Wiesner B, Baillie GS, Mollajew R, Henn V, Lorenz D, Furkert J, Santamaria K, Nedvetsky P, Hundsrucker C, Beyermann M, Krause E, Pohl P, Gall I, MacIntyre AN, Bachmann S, Houslay MD, Rosenthal W, Klussmann E. Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J Am Soc Nephrol 18: 199–212, 2007 [DOI] [PubMed] [Google Scholar]
- 58.Struyker-Boudier HA, Vervoort-Peters LH, Rousch MJ, Smits JF, Thijssen HH. Beta-adrenoceptors in kidney tubules of spontaneously hypertensive and normotensive rats. Life Sci 38: 137–145, 1986 [DOI] [PubMed] [Google Scholar]
- 59.Tawar U, Kotlo K, Jain S, Shukla S, Setty S, Danziger RS. Renal phosphodiesterase 4B is activated in the Dahl salt-sensitive rat. Hypertension 51: 762–766, 2008 [DOI] [PubMed] [Google Scholar]
- 60.Xin W, Tran TM, Richter W, Clark RB, Rich TC. Roles of GRK and PDE4 activities in the regulation of beta2 adrenergic signaling. J Gen Physiol 131: 349–364, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yougbare I, Morin C, Senouvo FY, Sirois C, Albadine R, Lugnier C, Rousseau E. NCS 613, a potent and specific PDE4 inhibitor, displays anti-inflammatory effects on human lung tissues. Am J Physiol Lung Cell Mol Physiol 301: L441–L450, 2011 [DOI] [PubMed] [Google Scholar]
- 62.Zou AP, Cowley AW., Jr α2-Adrenergic receptor-mediated increase in NO production buffers renal medullary vasoconstriction. Am J Physiol Regul Integr Comp Physiol 279: R769–R777, 2000 [DOI] [PubMed] [Google Scholar]