

Summary
Natural products are often large, synthetically intractable molecules, yet frequently offer surprising inroads into previously unexplored chemical space for enzyme inhibitors. Argifin is a cyclic pentapeptide that was originally isolated as a fungal natural product. It competitively inhibits family 18 chitinases by mimicking the chitooligosaccharide substrate of these enzymes. Interestingly, argifin is a nanomolar inhibitor of the bacterial-type subfamily of fungal chitinases that possess an extensive chitin-binding groove, but does not inhibit the much smaller, plant-type enzymes from the same family that are involved in fungal cell division and are thought to be potential drug targets. Here we show that a small, highly efficient, argifin-derived nine-atom fragment is a micromolar inhibitor of the plant-type chitinase ChiA1 from the opportunistic pathogen Aspergillus fumigatus. Evaluation of the binding mode with the first crystal structure of an A. fumigatus plant-type chitinase reveals that the compound binds the catalytic machinery in the same manner as observed for argifin with the bacterial-type chitinases. The structure of the complex was used to guide synthesis of derivatives to explore a pocket near the catalytic machinery. This work provides synthetically tractable plant-type family 18 chitinase inhibitors from the repurposing of a natural product.
Introduction
Compounds isolated from natural sources, natural products, provide a wealth of bioactives that in some cases are directly used as drugs and/or lead to the development of potent inhibitors useful for characterization of enzymes of interest and the design of future therapeutic drugs. Argifin is a natural product that was first isolated from Gliocladium fungal cultures grown from a soil sample collected in Micronesia (Omura et al., 2000). The structure of argifin was shown to be an unusual arginine-containing cyclopentapeptide (Fig. 1A) (Arai et al., 2000). Initial studies of argifin found that it showed inhibition of a family 18 chitinase from Lucilia cuprina in a dose-dependent manner as well as inhibiting chitinases from Streptomyces griseus and Vibrio alginolyticus within the micromolar range (Omura et al., 2000).
Figure 1. Chemical structures and inhibitory properties.

A) Chemical structure of the cyclopentapeptide argifin with its dimethylguanylurea moiety highlighted in red.
B) Steady-state initial velocities measured at different substrate concentrations for AfChiA1. Reaction mixtures contained 200 nM of AfChiA1, 0.1 mg/ml BSA and 50 – 800 μM of the 4MU-NAG3 in McIlvaine buffer (100 mM citric acid, 200 mM sodium phosphate [pH 5.5]) to a final volume of 50 μl. Production of 4-methylumbelliferone was linear with time for the incubation period used (70 min at 37°C), and less than 10% of available substrate was hydrolyzed. The Km was measured to be 300 ± 27 μM with a kcat of 3.0 s−1.
C) Lineweaver-Burk plot showing a mixed mode of inhibition for the dimethylguanylurea fragment against AfChiA1. Mode of inhibition assays were set up as described previously using 50 μM – 1 mM 4MU-NAG3, and 60 μM – 6 mM inhibitor.
D) Lineweaver-Burk plot showing a non-competitive mode of inhibition for the phenyl derivative 6 using the same reaction parameters.
Chitinases are enzymes that cleave the β-(1,4) glycosidic bond of chitin, a structural component in insect exoskeletons and fungal cell walls. The CAZy glycoside hydrolase family 18 (GH18), as defined by amino acid sequence similarity (Cantarel et al., 2009), contains a large number of chitinases expressed in archaea, prokaryotes and eukaryotes. It is subdivided into two subfamilies, bacterial-type and plant-type family 18 chitinases, based on sequence similarity, active site construction, corresponding preferred activity (exo-chitinases vs. endo-chitinases, respectively) and occurrence in different organisms: plants generally express plant-type GH18 enzymes, bacteria generally use bacterial-type GH18 chitinases, while members of both subfamilies have been found in fungi and vertebrates. It has been hypothesised that bacterial-type chitinases in fungi and bacteria are used to process chitin as a carbohydrate source, while the fungal plant-type chitinases are involved in cell wall remodelling and maintenance (Cantarel et al., 2009; Griffith, 1991; Jaques et al., 2003; Takaya et al., 1998).
The fungal cell wall protects the cell from the environment; it is a dynamic structure that is continually modified by enzymes to facilitate growth (Latge, 2001). All fungal cell walls contain chitin as a major component that is broken down by chitinases during cell wall remodelling. Disrupting this process is expected to result in a decrease of fungal viability and/or virulence, making plant-type fungal chitinases potential targets for drugs against fungal pathogens. Unfortunately, while there are numerous inhibitor families targeting bacterial-type GH18 enzymes (Rao et al., 2005; Schuttelkopf et al., 2006; Vaaje-Kolstad et al., 2004) most of them perform poorly against these fungal plant-type family 18 chitinases. An exception is the natural product allosamidin isolated from Streptomyces sp., that acts on both plant-type and bacterial-type family 18 chitinases (Sakuda et al., 1987), competitively inhibit the plant-type chitinase CTS1 from Saccharomyces cerevisiae (ScCTS1, Ki = 0.61 μM (Hurtado-Guerrero and van Aalten, 2007)) and hevamine (Ki = 3.1 μM (Vaaje-Kolstad et al., 2004)). Unfortunately allosamidin is a substrate analogue with poor drug-like properties (high molecular weight, glycosidic bonds and a cLogP of −5.2) as well as complicated and costly synthesis (Blattner et al., 1996; Kassab and Ganem, 1999). Interestingly, a fragment identified from the large natural product argifin, dimethylguanylurea (Fig. 1A), has recently been identified as the minimal fragment necessary and sufficient for competitive inhibition of the bacterial-type chitinases (Andersen et al., 2008). As the catalytic machinery is conserved across all GH18 family members, it can be hypothesized that the fragment molecule would also inhibit the plant-type family 18 enzymes and provide a template for the much needed development of potent, specific inhibitors for this class of chitinases.
Here, we report the inhibitory activity of both argifin-derived and newly synthesized dimethylguanylurea fragments against the Aspergillus fumigatus plant-type chitinase ChiA1 (AfChiA1) as well as the crystal structure of this enzyme in complex with the most potent of these inhibitors. A. fumigatus is a saprotrophic fungus that is widespread in nature and involved in carbon and nitrogen recycling (Chazalet et al., 1998). Although the spores are not harmful to healthy individuals, A. fumigatus is an opportunistic pathogen that can colonize the lungs of immunocompromised individuals, frequently resulting in life-threatening invasive aspergillosis (Brakhage and Langfelder, 2002; Singh and Paterson, 2005). The deconstruction of the natural product, argifin, and the characterization of its fragments as inhibitors of AfChiA1, demonstrates the importance of combining natural product investigation with chemical deconvolution and establishes a new family of efficient and synthetically attractive leads for the development of inhibitors against medically relevant GH18 chitinases.
Results and Discussion
An argifin fragment inhibits fungal chitinases involved in cell division
Argifin (Fig. 1A) is a natural product that inhibits bacterial-type family 18 chitinases in the low nanomolar range by mimicking enzyme-substrate interactions (Houston et al., 2002; Rao et al., 2005). We recently reported a chemical dissection of argifin, revealing that the dimethylguanylurea fragment (compound 1 in Table I, Fig. 1A), still competitively inhibits the bacterial-type chitinase B1 from A. fumigatus (AfChiB1) (Andersen et al., 2008). This encouraged us to investigate inhibition of the plant-type fungal chitinases that are thought to be involved in cell wall remodelling during cell division (Selvaggini et al., 2004). We cloned the previously uncharacterized chitinase A1 from A. fumigatus (AfChiA1, 38 % catalytic core sequence identity to the plant-type chitinase CTS1 from S. cerevisiae, ScCTS1) and purified the protein from a Pichia expression system. The enzyme hydrolyses 4MU-NAG3 with a Km of 300 ± 27 μM and a kcat of 3.0 s−1 (Fig. 1B). Strikingly, the large allosamidin pseudotrisaccharide, generally a potent GH18 inhibitor, inhibits AfChiA1 with an IC50 of only 127 ± 14 μM (Table I). Similarly, the natural product argifin (Fig. 1A) does not inhibit AfChiA1 (IC50 > 3.8 mM). However, the nine-atom fragment 1 (Fig. 1A), derived from argifin, has an IC50 of 79 ± 8 μM against the enzyme (Table I), compared to 500 ± 20 μM previously reported for AfChiB1 (Andersen et al., 2008). Mode of inhibition studies suggest that 1 is not a purely competitive inhibitor of AfChiA1 (Fig. 1C). In terms of ligand efficiency, i.e. the contribution to the free energy of binding per inhibitor atom, compound 1 achieves an excellent value of −0.93 kcal·mol−1·atom−1, while allosamidin exhibits only moderate efficiency at −0.19 kcal·mol−1·atom−1.
Table I.
Structures and enzyme inhibition of the argifin-derived fragments.
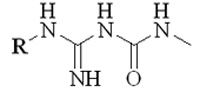
| ||
|---|---|---|
| R | IC50 (μM) | |
| 1 | CH3 | 79 ± 8 |
| 2 | CH3CH2 | 310 ± 7 |
| 3 | CH3(CH2)2 | 62 ± 1 |
| 4 | CH3(CH2)3 | 38 ± 7 |
| 5 |
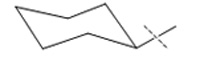
|
42 ± 1.3 |
| 6 | Ph | 112 ± 1 |
| 7 | PhCH2 | 299 ± 4 |
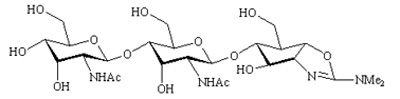
allosamidin |
127 ± 14 | |
| argifin | > 3.8 mM | |
AfChiA1 possesses a shallow active site that is efficiently occupied by the argifin fragment
To further investigate the mode of inhibition of the argifin-derived fragment 1, and provide a platform for rational optimisation, we determined the crystal structure of AfChiA1, both in its apo-form and in complex with 1 (Table II). Data for the apo-structure were collected to 2.0 Å resolution, solved by molecular replacement and refined to an Rfree of 0.249 with good stereochemistry. AfChiA1 has the (β/α)8 barrel fold common to all family 18 glycoside hydrolases (Fig. 1A, (Davies and Henrissat, 1995)). Most of the loops connecting the helices and strands are relatively short; exceptions are the β2-α2 loop, which contains three minimal β-strands (labelled β2′, β2″ and β2′″) that, together with the C-terminal end of the extended β2 itself, form a small four-stranded mixed β-sheet, and the β6-α6 loop, which includes the short α5′ helix. Helix α8 is interrupted by a minimal β-sheet comprising strands β9 and β10 (Fig. 1A). Comparison with existing structures of GH18 family member proteins shows that AfChiA1 is most similar to S. cerevisiae CTS1 (PDB id 2UY2). The agreement between the two structures is good for the core (β/α)8 barrel (overall RMSD=1.15 Å for 265 Cα atoms), whereas some conformational differences exist in the connecting loops, many of which harbour insertions/deletions.
Table II. Summary of data collection and structure refinement statistics.
Values for the highest resolution shell are given in brackets.
| apo-AfChiA1 | AfChiA1-1 | AfChiA1-6 | |
|---|---|---|---|
| Resolution (Å) | 20.00–2.00 (2.05–2.00) | 25.01 – 2.3 | 30.00 – 2.35 |
| Unit cell |
a=76.7 Å, b=126.1 Å, c=213.4Å |
a, b=100 Å, c=111.2 Å | a, b=100 Å, c=111.2 Å |
| # Unique reflection | 69723 | 55123 | 51912 |
| Redundancy | 4.1 (4.0) | 4.1 (3.1) | 3.0 (3.2) |
| R merge | 0.099 (0.438) | 0.109 (0.58) | 0.114 (0.406) |
| I/σ(I) | 10.8 (2.6) | 12.4 (5.4) | 17.3 (5.2) |
| Completeness (%) | 100.0 (99.8) | 99.9 (98) | 98.5 (94.1) |
| R, Rfree | 0.201 (0.241) | 0.241 (0.285) | 0.259 (0.298) |
| <B> protein (Å2) | 14.0 | 26.1 | 16.9 |
| <B> ligand (Å2) | n/a | 33.1 | 24.4 |
| RMSD from ideal geometry | |||
| Bond lengths (Å) | 0.015 | 0.0218 | 0.0202 |
| Bond angles (°) | 1.38 | 1.912 | 1.837 |
The AfChiA1 active site lies at the C-terminal opening of the β-barrel (Fig. 2A). It is thus defined by the (mostly) very short βα-loops, which gives it the overall open, and apparently featureless, architecture also seen in ScCTS1 (Hurtado-Guerrero and van Aalten, 2007) and common to plant-type family 18 chitinases (Davies and Henrissat, 1995). However, in the AfChiA1 active site the bulky Tyr125, replacing a serine in ScCTS1, appears to partially block access to the catalytic site. The ligand complexes on the other hand reveal Tyr125 to be conformationally flexible and show it pointing away from the active site, suggesting that its presence would not have a significant effect on substrate/ligand specificity.
Figure 2. Structural analysis of AfChiA1 inhibitor complexes.

A) Comparison of the AfChiB1-argifin complex structure (left, PDB id 1W9V) and the complex of AfChiA1 with 1 (right). The proteins are shown in cartoon representation (α-helices in red, β-strands in blue) with the ligands drawn as sticks (green). The extra α/β domain present in the bacterial-type fungal chitinases, but lacking in the plant-type fungal chitinases, is highlighted in yellow.
B) Surface view of the structures in panel A. The AfChiB1 and AfChiA1 surfaces are shaded by sequence identity amongst A. fumigatus bacterial-type and plant-type GH18 chitinases, respectively (white=non-conserved, dark blue=completely conserved). Ligands are shown as in panel A. The extra α/β domain present in the bacterial-type fungal chitinases, but lacking in the plant-type fungal chitinases, is highlighted in yellow.
C) The active site of AfChiA1 in complex with 1/6 and AfChiB1 in complex with argifin. Side chains for important residues are shown as grey sticks and labeled. Ligands are shown as purple sticks, a water molecule is represented as an orange ball. Likely hydrogen bonds are indicated by green dashed lines. Electron density is shown contoured to 2.5 σ as a cyan mesh for AfChiA1 ligands.
The exposed AfChiA1 active site contrasts with AfChiB1 (Rao et al., 2005) and other bacterial-type chitinases, which harbour larger βα-loops (including one incorporating an entire α/β domain) that form a deep active site groove (Fig. 2B), lined by several solvent-exposed aromatic residues, including Trp137 (cf. Fig. 2C). It is these differences in active site construction that explain the different affinities for argifin by AfChiA1 and AfChiB1 – the AfChiB1-argifin complex structure shows that the inhibitor is engulfed by the wall of the active site groove, whereas these walls are non-existent in AfChiA1 (Fig. 2B).Nonetheless, the catalytic machinery itself is relatively well-conserved among all GH18 chitinases and AfChiA1 is no exception, sporting hallmark residues of the GH18 family, including the solvent-exposed tryptophan (Trp312) that forms the bottom of the −1 sugar-binding subsite, the catalytic DxE motif (Asp172 and Glu174) and conserved tyrosines Tyr232 and Tyr34 (Fig. 2C).
The structure of AfChiA1 in complex with 1 was refined against 2.3 Å synchrotron diffraction data, revealing clear density for the inhibitor (Table II; Fig. 2C). The ligand is bound in the active site in a position and orientation similar to the equivalent fragment in the AfChiB1-argifin complex (Andersen et al., 2008) (Fig. 2B,C) where 1 forms a bidentate hydrogen bond with the side chain of Glu174 (the catalytic acid) and also donates a single hydrogen bond to the Asp172 carboxylate. Additionally the inhibitor accepts a weak hydrogen bond from Tyr232 and stacks with the Trp312 side chain. Surprisingly, clear density for a second ligand molecule can be observed sitting in a shallow groove just outside the catalytic pocket (Fig. 2C). The groove is lined by the side chains of Trp312 and Gln37 on one side and the backbone atoms of Ala124 and Tyr125 on the other side, while its floor is contributed by the phenyl moiety of Phe60. The ligand molecule forms one direct hydrogen bond with the protein, from the backbone amide of Ala124 to a (deprotonated) ligand backbone nitrogen. The ligand backbone also donates a hydrogen bond to a water molecule, which is bound in the groove through two hydrogen bonds to the backbone carbonyls of Gly122 and Tyr125. It is possible that the presence of this second inhibitor binding site explains the observed mixed inhibition pattern (Fig. 1C).
Chemical elaboration of the argifin fragment identifies additional binding pockets
As stated earlier, compound 1 is a highly efficient (-0.93 kcal·mol−1·atom−1) plant-type chitinase inhibitor, compared to the widely studied, but less synthetically tractable, natural product pseudotrisaccharide inhibitor allosamidin (-0.19 kcal·mol−1·atom−1) (Hopkins et al., 2004). Furthermore, the guanylurea moiety would be a suitable core for high-throughput synthesis of a library of fragments, similar to what has been achieved recently for alpha-glucosidases, starting from a valienamine scaffold (Lysek et al., 2006).
To explore this, a number of analogues of 1 were synthesised using a three step procedure, via conversion of the appropriate amine to the N-,N′-bis(tert-butoxycarbonyl)guanidine derivative (Feichtinger, 1998), followed by treatment with methylamine to generate the desired guanylurea (Miel, 1998) (Table I). Compounds 2-7 were designed based on the binding mode of 1 in the catalytic pocket, which suggests that only the methyl group at the guanyl end of 1 is available for derivatization (Fig. 2C). Only moderately sized moieties were explored, given the relatively featureless surface beyond the immediate surroundings of the catalytic pocket and the knowledge that the full argifin molecule itself does not inhibit AfChiA1. A number of alkyl and ring extensions were explored (Table I), and found to inhibit AfChiA1 with IC50 values between 38 μM and 310 μM, compared to 79 μM for the starting compound. Although some of the derivatives appeared to give modest improvements in inhibition (i.e. up to 2-fold), this is not considered to be significant given the general accuracy of IC50 measurements. Further examinations into the effect from substitutions of 1 on mode of inhibition and binding, were focused on compound 6. As shown in Fig. 1C, the mode of inhibition is non-competitive (Fig. 1D). This compound was also a 10-fold stronger inhibitor of AfChiB1 (IC50 = 210 ± 40) compared to parent compound 1.
A structure of AfChiA1 in complex with 6 was refined against 2.4 Å synchrotron diffraction data (Table II). The complex shows that 6 adopts a virtually identical binding mode to the parent compound 1 (Fig. 2C, maximum atomic positional shift = 0.4 Å), with an identical hydrogen bonding pattern. The phenyl ring does not make any specific interactions with the protein and correspondingly is poorly ordered (Fig. 2C). This lack of interactions in combination with the increased disorder of 6 compared to 1 explains the reduced IC50 observed for 6. In contrast to the AfChiA1-1 complex, there is no evidence for a secondary binding site, compatible with the notion that the phenyl ring of a ligand molecule bound in the secondary site would clash with the ligand occupying the catalytic pocket (Fig. 2).
Concluding Remarks
Based on the dissection of the natural product argifin, an inhibitor of bacterial-type GH18 chitinases (Fig. 1A), we have characterised the activity of dimethylguanylurea (1) and its derivatives (2-7) against a plant-type chitinase, ChiA1 from the fungal pathogen Aspergillus fumigatus. The small compounds are found to be attractive starting points for AfChiA1 inhibitors, with IC50 values in the micromolar range. In fact, 1 is a better inhibitor of AfChiA1 (IC50 = 79 μM) than it is of the bacterial-type chitinase AfChiB1 (IC50 = 500 μM; (Andersen et al., 2008)), reversing the relative affinities observed for its parent compound, argifin. Moreover, 1 is already a better AfChiA1 inhibitor than the broad-spectrum (and generally low nanomolar) allosamidin inhibitor. This is particularly striking when expressed as ligand efficiency (-0.93 kcal·mol−1·atom−1 for 1 versus −0.19 kcal·mol−1·atom−1 for allosamidin), taking into account the almost five-fold larger mass of allosamidin. The first structures of AfChiA1 show that 1 itself, as well as its derivative 6, bind to the enzyme exclusively through interactions with the catalytic machinery conserved across GH18 hydrolases, explaining their ability to inhibit AfChiA1 and suggesting that they might represent a family of pan-GH18 chitinase inhibitors. The structures also reveal AfChiA1 to adopt the (minimal) TIM barrel fold typical of (plant-type) GH18 family members and shows that it is closely related to ScCTS1, the only other known fungal plant-type GH18 structure, sharing its open active site architecture, which explains the poor affinity of this enzyme for argifin and other inhibitors of bacterial-type chitinases.
Based on its observed binding mode to AfChiA1, several simple derivatives of 1 were synthesised and tested against the enzyme. None of these derivatives showed significantly improved binding, which is not unexpected, given that the solvent-exposed AfChiA1 active site does not provide many functional groups to interact with. At the same time it is encouraging that most of the derivatives were also not significantly worse binders, suggesting that it could be energetically feasible to further build up the outwards-pointing side of 1 to eventually reach interactable groups on the enzyme surface. Such modifications could allow to take this scaffold further and design bulkier compounds that retain their affinity for plant-type GH18 proteins, but lose binding to the more restricted bacterial-type GH18 active site, thus yielding inhibitor specific to plant-type chitinases that would be of interest as chemical tools to dissect the precise biological roles of different chitinases.
Intriguingly the structure of AfChiA1 in complex with 1 reveals that in addition to the catalytic-site bound inhibitor, a second ligand molecule occupies a shallow groove close to the catalytic pocket. While this second ligand makes few obviously energetically favourable interactions with the enzyme, the clear electron density observed for it suggests that it does bind with reasonable affinity. It might thus be possible to exploit its observed binding mode as a template for the further elaboration of the scaffold, possibly by simply joining two molecules of 1 into a new inhibitor able to bind in both pockets.
Experimental Procedures
Synthesis of dimethylguanylurea and derivatives
Compounds 1-7 were synthesised by a three-step procedure as follows. Starting from the amine, R-NH2 (R as designated in Table I), the corresponding bis-tert- butoxycarbonylguanidine derivatives were generated by reaction with 1,3-di-tert-butoxycarbonyl-2-(trifluoromethylsulfonyl)guanidine, according to the method of Goodman (Feichtinger, 1998). Transformation to the desired guanylureas was then effected in two steps, by aminolysis with methylamine in tetrahydrofuran (Miel, 1998), followed by removal of the remaining tert-butoxycarbonyl group with trifluoroacetic acid. The final compounds were all isolated as the corresponding trifluoroacetate salts, with the exception of 7, which was converted to the free base by neutralisation with saturated aqueous sodium hydrogencarbonate. 1-7 were fully characterised by 1H NMR (JEOL Delta at 270 MHz, or Varian Mercury-VX at 400 MHz) 13C NMR (JEOL Delta at 68 MHz), and high resolution mass spectrometry (Bruker MicroTOF autospec electrospray ionisation instrument). Purities were verified by analytical RP-HPLC on a Dionex system with a Gemini 5μ C18 110A column, 150 × 4.6 mm (Phenomenex, UK).
Cloning and purification
The coding sequence for residues Ser29-Leu335 of Aspergillus fumigatus chitinase A (AfChiA1; UniProt ID: Q873Y0) was cloned by PCR from an A. fumigatus cDNA library (kindly provided by Jean-Paul Latgé, Paris) using the forward primer 5′-GCGAATTCTCCAACCTTGCCATCTACTGGGGTC-3′ and the reverse primer 5′-CTGCGGCCGCTCAAAGAATATCCTTCATGTGGTCTGCATAGGG-3′, containing an EcoRI and a NotI restriction site, respectively. The PCR product was initially cloned into the PCR cloning vector pSC-B (Stratagene) and subcloned into the EcoRI and NotI sites of the Pichia pastoris expression vector pPICZαA (Invitrogen). Subsequently, to obtain a soluble and crystallisable protein, we re-added the codons for residues Leu336 and His337 by site directed mutagenesis using the mutagenesis oligonucleotide pair 5′-GACCACATGAAGGATATTCTTTTGCACTGAGCGGCCGCCAGCTTTCTAG-3′ and 5′-CTAGAAAGCTGGCGGCCGCTCAGTGCAAAAGAATATCCTTCATGTGGTC-3′. Site-directed mutagenesis was carried out following the QuikChange Site-Directed Mutagenesis protocol (Stratagene), using the KOD HotStart DNA polymerase (Novagene). All plasmids were verified by sequencing (DNA Sequencing Service, College of Life Sciences, University of Dundee; www.dnaseq.co.uk).
The plasmid was isolated from E. coli strain DH5α, linearised with SacII and used to transform Pichia pastoris strain X-33 using the LiCl method (Invitrogen) or using the Pichia EasyComp™ Transformation Kit (Invitrogen). Transformants were selected on YPD plates (1% (w/v) yeast extract, 2% (w/v) peptone, 2% (w/v) dextrose) containing 100 mg/ml of zeocin (Invitrogen). Batch cultures were performed in 100 ml volume of BMGY medium (1% (w/v) yeast extract, 2% (w/v) peptone, 100 mM potassium phosphate (pH 6.0), 1.34% (w/v) yeast nitrogen base and 1% (v/v) glycerol). 50 ml were used to inoculate 500 ml of BMGY medium overnight at 30° C and expression was induced by methanol 1% (v/v) for 72 h at room temperature in a shaking incubator (270 rpm). Yeast cells were harvested by centrifugation at 3480 g for 30 min. The supernatants containing soluble AfChiA1 were filtered to 0.2 micron, concentrated to 50 ml using a Vivaflow 200 cassette (10,000 MWCO, PES membrane; Vivascience) and dialyzed against water.
The samples were then loaded onto a 2 × 5 ml HiTrap Q FF column (Amersham Biosciences) that had been equilibrated with 10 column volumes of 25 mM Tris pH 7.5 on an AKTA purifier system. Following loading, the column was washed with 10 column volumes of 25 mM Tris pH 7.5. The protein was eluted with a salt gradient (0–500 mM NaCl) over 20 column volumes, collecting 2 ml fractions. The fractions containing the proteins were then pooled and concentrated to 5 ml. Subsequently, gel filtration was carried out using a Superdex 75 XK26/60 column in 25 mM Tris, 150 mM NaCl, pH 7.5. The concentrated AfChiA1 protein sample was used for both kinetic analysis and crystallization trials.
Crystallization and structure determination
The protein was concentrated to between 28 and 36 mg·ml−1 and crystallized by hanging drop vapour diffusion over a reservoir containing 0.42 – 0.58 M NaH2PO4 and 0.74 - 0.83 M K2HPO4; usable crystals grew over a few days. After cryoprotection by short immersion in 2.5 M Li2SO4, native data were collected at 100 K in house. To obtain complex structures, AfChiA1 crystals were soaked in the presence of inhibitor from a 10 mM stock. The inhibitor was added directly to the crystal growth drop at a final concentration of 1-5 mM. After soaking for 1-10 minutes and cryoprotection, data were collected at 100 K on beamline ID14-EH2 (wavelength 0.953 Å) at the European Synchrotron Radiation Facility (ESRF, Grenoble, France).
AfChiA1 forms either orthorhombic crystals (crystal form I, space group I212121, unit cell dimensions a=76.6 Å, b=125.8 Å, c=211.7 Å) with two molecules per asymmetric unit or trigonal crystals (crystal form II, space group P32, a=b=100.0 Å, c=111.2 Å) with three molecules per asymmetric unit. All data were processed and scaled using HKL software (Otwinowski et al., 2003) to resolutions of 2.0 Å (native), 2.3 Å (complex with 1), and 2.4 Å (complex with 6), Table II. The apo-data (crystal form I) were solved by molecular replacement with AMoRe (Navaza, 2001) using the S. cerevisiae CTS1 apo-structure (PDB id 2UY2, (Hurtado-Guerrero and van Aalten, 2007) as a search model yielding the expected two molecules per asymmetric unit, while the ligand complexes (crystal form II) were solved by molecular replacement with MolRep (Vagin and Teplyakov) using the refined AfChiA1 apo structure. Refinement of the AfChiA1 structures (Table II) proceeded through rounds of minimization with REFMAC5 (Vagin et al., 2004) and model building with Coot (Emsley and Cowtan, 2004). The R-factors for the complex structures are relatively high, most likely due to disorder of the third monomer. Because of this disorder as well as the structural similarity of the first two monomers for all structures (RMSD values between 0.15 Å and 0.35 Å for ca. 310 Cαs without NCS restraints) atoms, the further discussion will focus on the first monomer (i.e. chain A) only, unless noted otherwise. Ligand coordinates and topologies were generated with PRODRG for the complex structures (Schuttelkopf and van Aalten, 2004). In all AfChiA1 molecules built, Cys81 falls into the disallowed region of the Ramachandran plot, despite having been modelled in agreement with clear electron density. This residue participates in an intramolecular disulfide bridge, which might necessitate its strained backbone conformation.
Enzyme assays
Michaelis-Menten parameters of AfChiA1 were determined using 4-methylumbelliferyl β-D-N,N′,N′′-triacetylchitotrioside (4MU-NAG3). Standard reaction mixtures contained 200 nM of AfChiA1, 0.1 mg/ml BSA and 50 – 800 μM of the fluorogenic substrate in McIlvaine buffer (100 mM citric acid, 200 mM sodium phosphate [pH 5.5]) to a final volume of 50 μl. Reaction mixtures were incubated for 70 min at 37°C, after which the reaction was stopped with the addition of 25 μM of 3 M glycine-NaOH (pH 10.3). The fluorescence of the released 4-methyl-umbelliferone was quantified using a Flx 800 microtitreplate fluorescence reader (Bio-Tek Instruments Inc.) (excitation 366 nm, emission 445 nm). Experiments were performed in triplicate. Production of 4-methylumbelliferone was linear with time for the incubation period used, and less than 10% of available substrate was hydrolyzed. The IC50s of the argifin fragments against AfChiA1were determined using the same protocol but with a constant substrate concentration of 100 μM and 300 nM – 6 mM inhibitor. Similarly, the mode of inhibition assays were set up as previously described above using 50 μM – 1 mM 4MU-NAG3, and 60 μM – 6 mM inhibitor.
Significance
Natural products are often large, synthetically intractable molecules, yet frequently offer surprising inroads into previously unexplored chemical space for enzyme inhibitors. This work shows how, aided by detailed structural information, the large natural product chitinase inhibitor argifin can be dissected down to a tiny 9-atom active fragment that can then be used as a scaffold to develop micromolar inhibitors for a chitinase from the opportunistic pathogen Aspergillus fumigatus.
Acknowledgements
This work was supported by a Wellcome Trust Seeding Drug Discovery Award and an MRC Programme Grant. DvA is supported by a Wellcome Trust Senior Research Fellowship. We thank the European Synchrotron Radiation Facility, Grenoble, for the time at beamline ID14-EH3. The structure factors and models have been deposited in the Protein Data Bank (PDB ids 2xvp, 2xuc and 2xvn).
References
- Andersen OA, Nathubhai A, Dixon MJ, Eggleston IM, van Aalten DM. Structure-based dissection of the natural product cyclopentapeptide chitinase inhibitor argifin. Chem Biol. 2008;15:295–301. doi: 10.1016/j.chembiol.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai N, Shiomi K, Iwai Y, Omura S. Argifin, a new chitinase inhibitor, produced by Gliocladium sp. FTD-0668. II. Isolation, physico-chemical properties, and structure elucidation. J Antibiot (Tokyo) 2000;53:609–614. doi: 10.7164/antibiotics.53.609. [DOI] [PubMed] [Google Scholar]
- Blattner R, Furneaux RH, Lynch GP. Synthesis of allosamidin analogues. Carbohydr Res. 1996;294:29–39. doi: 10.1016/s0008-6215(96)90613-8. [DOI] [PubMed] [Google Scholar]
- Brakhage AA, Langfelder K. Menacing mold: the molecular biology of Aspergillus fumigatus. Annu Rev Microbiol. 2002;56:433–455. doi: 10.1146/annurev.micro.56.012302.160625. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazalet V, Debeaupuis JP, Sarfati J, Lortholary J, Ribaud P, Shah P, Cornet M, Vu Thien H, Gluckman E, Brucker G, et al. Molecular typing of environmental and patient isolates of Aspergillus fumigatus from various hospital settings. J Clin Microbiol. 1998;36:1494–1500. doi: 10.1128/jcm.36.6.1494-1500.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G, Henrissat B. Structures and mechanisms of glycosyl hydrolases. Structure. 1995;3:853–859. doi: 10.1016/S0969-2126(01)00220-9. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Feichtinger K, Zapf C, Sings HL, Goodman M. Diprotected triflylguanidines: A new class of guanidinylation reagents. The Journal of Organic Chemistry. 1998;63:3804–3805. [Google Scholar]
- Griffith DA, Danishefsky SJ. Total synthesis of allosamidin: an application of the sulfonamideoglycosylation of glycols. Journal American Chemical Society. 1991;113:5863–5864. [Google Scholar]
- Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- Houston DR, Shiomi K, Arai N, Omura S, Peter MG, Turberg A, Synstad B, Eijsink VG, van Aalten DM. High-resolution structures of a chitinase complexed with natural product cyclopentapeptide inhibitors: mimicry of carbohydrate substrate. Proc Natl Acad Sci U S A. 2002;99:9127–9132. doi: 10.1073/pnas.132060599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado-Guerrero R, van Aalten DM. Structure of Saccharomyces cerevisiae chitinase 1 and screening-based discovery of potent inhibitors. Chem Biol. 2007;14:589–599. doi: 10.1016/j.chembiol.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Jaques AK, Fukamizo T, Hall D, Barton RC, Escott GM, Parkinson T, Hitchcock CA, Adams DJ. Disruption of the gene encoding the ChiB1 chitinase of Aspergillus fumigatus and characterization of a recombinant gene product. Microbiology. 2003;149:2931–2939. doi: 10.1099/mic.0.26476-0. [DOI] [PubMed] [Google Scholar]
- Kassab DJ, Ganem B. An Enantioselective Synthesis of (-)-Allosamidin by Asymmetric Desymmetrization of a Highly Functionalized meso-Epoxide. J Org Chem. 1999;64:1782–1783. doi: 10.1021/jo990167e. [DOI] [PubMed] [Google Scholar]
- Latge JP. The pathobiology of Aspergillus fumigatus. Trends Microbiol. 2001;9:382–389. doi: 10.1016/s0966-842x(01)02104-7. [DOI] [PubMed] [Google Scholar]
- Lysek R, Schutz C, Favre S, O’Sullivan AC, Pillonel C, Krulle T, Jung PM, Clotet-Codina I, Este JA, Vogel P. Search for alpha-glucosidase inhibitors: new N-substituted valienamine and conduramine F-1 derivatives. Bioorg Med Chem. 2006;14:6255–6282. doi: 10.1016/j.bmc.2006.05.080. [DOI] [PubMed] [Google Scholar]
- Miel H, Rault S. Conversion of N, N′-bis(tert-butoxycarbonyl)guanidines to N, N′-bis(tert-butoxycarbonyl)amidino ureas. Tetrahedron Letters. 1998;39:1565–1568. [Google Scholar]
- Navaza J. Implementation of molecular replacement in AMoRe. Acta Crystallogr D Biol Crystallogr. 2001;57:1367–1372. doi: 10.1107/s0907444901012422. [DOI] [PubMed] [Google Scholar]
- Omura S, Arai N, Yamaguchi Y, Masuma R, Iwai Y, Namikoshi M, Turberg A, Kolbl H, Shiomi K. Argifin, a new chitinase inhibitor, produced by Gliocladium sp. FTD-0668. I. Taxonomy, fermentation, and biological activities. J Antibiot (Tokyo) 2000;53:603–608. doi: 10.7164/antibiotics.53.603. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Borek D, Majewski W, Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- Rao FV, Houston DR, Boot RG, Aerts JM, Hodkinson M, Adams DJ, Shiomi K, Omura S, van Aalten DM. Specificity and affinity of natural product cyclopentapeptide inhibitors against A. fumigatus, human, and bacterial chitinases. Chem Biol. 2005;12:65–76. doi: 10.1016/j.chembiol.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Sakuda S, Isogai A, Matsumoto S, Suzuki A. Search for microbial insect growth regulators. II. Allosamidin, a novel insect chitinase inhibitor. J Antibiot (Tokyo) 1987;40:296–300. doi: 10.7164/antibiotics.40.296. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf AW, Andersen OA, Rao FV, Allwood M, Lloyd C, Eggleston IM, van Aalten DM. Screening-based discovery and structural dissection of a novel family 18 chitinase inhibitor. J Biol Chem. 2006;281:27278–27285. doi: 10.1074/jbc.M604048200. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Selvaggini S, Munro CA, Paschoud S, Sanglard D, Gow NA. Independent regulation of chitin synthase and chitinase activity in Candida albicans and Saccharomyces cerevisiae. Microbiology. 2004;150:921–928. doi: 10.1099/mic.0.26661-0. [DOI] [PubMed] [Google Scholar]
- Singh N, Paterson DL. Aspergillus infections in transplant recipients. Clin Microbiol Rev. 2005;18:44–69. doi: 10.1128/CMR.18.1.44-69.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaya N, Yamazaki D, Horiuchi H, Ohta A, Takagi M. Cloning and characterization of a chitinase-encoding gene (chiA) from Aspergillus nidulans, disruption of which decreases germination frequency and hyphal growth. Biosci Biotechnol Biochem. 1998;62:60–65. doi: 10.1271/bbb.62.60. [DOI] [PubMed] [Google Scholar]
- Vaaje-Kolstad G, Vasella A, Peter MG, Netter C, Houston DR, Westereng B, Synstad B, Eijsink VG, van Aalten DM. Interactions of a family 18 chitinase with the designed inhibitor HM508 and its degradation product, chitobiono-delta-lactone. J Biol Chem. 2004;279:3612–3619. doi: 10.1074/jbc.M310057200. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]