

Abstract
Neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis and prion-based neurodegeneration are associated with the accumulation of misfolded proteins, resulting in neuronal dysfunction and cell death. However, current treatments for these diseases predominantly address disease symptoms, rather than the underlying protein misfolding and cell death, and are not able to halt or reverse the degenerative process. Studies in cell culture, fruitfly, worm and mouse models of protein misfolding-based neurodegenerative diseases indicate that enhancing the protein-folding capacity of cells, via elevated expression of chaperone proteins, has therapeutic potential. Here, we review advances in strategies to harness the power of the natural cellular protein-folding machinery through pharmacological activation of heat shock transcription factor 1 — the master activator of chaperone protein gene expression — to treat neurodegenerative diseases.
Many neurodegenerative diseases are associated with the misfolding of specific — although structurally unrelated — proteins (TABLE 1) that share a common tendency to misfold and form aggregates, which may be enhanced by mutations. Interestingly, following their misfolding, these functionally unrelated proteins frequently adopt a highly stable β-sheet structure that is instrumental in their aggregation and toxicity1,2. Once the β-sheet structures are formed, misfolded proteins multimerize into intermediate-sized soluble oligomers, which are thought to promote oxidative stress, disrupt calcium homeo stasis, titrate chaperone proteins away from other essential cellular functions and engage in other processes that are disruptive to cellular health, thus generating considerable cellular toxicity in neurodegenerative diseases3. Misfolded protein oligomers proceed to aggregate, eventually forming insoluble, high-molecular-weight amyloid fibrils that are incorporated into inclusions4 (FIG. 1). These inclusions were historically thought to be the major source of cytotoxicity in neurodegenerative diseases. Although aggregates and inclusions are still considered to be causative in diseases such as Alzheimer’s disease, recent evidence suggests that in other neurodegenerative diseases — such as Huntington’s disease — larger aggregates may serve a cytoprotective function5. As such, the role and context of misfolded oligomers and aggregates will be an important consideration in the development of therapeutic interventions (FIG. 1).
Table 1.
Neurodegenerative diseases that are associated with protein misfolding
| Neurodegenerative diseases |
Aggregation- prone protein |
Neuronal population lost |
Disease symptoms |
Current therapies |
Refs |
|---|---|---|---|---|---|
| Polyglutamine disorders: Huntington’s disease, spinal and bulbar muscular atrophy, spinocerebellar ataxias and others |
Polyglutamine tracts within distinct pathogenic proteins |
Variable | Chorea, cognitive decline, depression and anxiety, dementia and ataxia |
Tetrabenazine, amantadine, remacemide, antipsychotics and antidepressants |
13 |
| Parkinson’s disease | α-synuclein | Substantia nigra pars compacta, ventral tegmental area, autonomic ganglia and neurons of the myenteric plexus |
Tremors, bradykinesia, postural instability, rigidity, dementia, depression, anxiety, gastrointestinal dysfunction and hallucinations |
Levodopa/ carbidopa, dopamine agonists and monoamine oxidase inhibitors |
7,8 |
| Alzheimer’s disease | Amyloid-β, hyperphosphory- lated tau |
Cortical and subcortical neurons, locus coeruleus and cholinergic neurons |
Learning and memory impairments, motor dysfunction, and irritability |
Acetylcholin esterase inhibitors, memantine and antipsychotics |
9,10 |
| Amyotrophic lateral sclerosis/ Lou Gehrig’s disease |
Superoxide dismutase, TAR DNA binding protein 43 and others |
Motor neurons of the cortex, brain stem and spinal cord |
Muscular atrophy and motor dysfunction |
Riluzole | 11 |
| Prion disorders: Creutzfeldt–Jakob disease, Gerstmann– Straussler syndrome, fatal familial insomnia and kuru |
Various forms of the prion protein such as scrapie (PrPSc) |
Variable | Spongiform encephalopathy, personality changes, depression and insomnia |
None | 12 |
Figure 1. Chaperone proteins and maintenance of protein homeostasis.

Misfolding of disease-causing proteins results in the disruption of protein homeostasis when misfolded monomers accumulate and begin to form intermediate soluble oligomers or fibrils, and eventually form mature insoluble aggregates. Chaperone proteins assist in the correct folding of proteins and prevent the formation of toxic oligomeric species. Increasing the expression of chaperone proteins enhances the ability of cells to maintain protein homeostasis even in the presence of aggregation-prone proteins. It is not yet clear whether increased expression of chaperone proteins will prevent the formation of mature aggregates and promote their degradation.
Coiled-coil domains, which are characterized by multiple intertwined α-helices that interact via specific hydrophobic and ionic associations, have also been proposed to accelerate the aggregation of polyglutamine expansion (polyQ) proteins such as the huntingtin protein in Huntington’s disease or ataxin 3 in Machado–Joseph disease6. Interestingly, these domains are not required for the aggregation of proteins such as amyloid-β or α-synuclein — associated with Alzheimer’s disease and Parkinson’s disease, respectively — which appear to require only the formation of β-sheets to promote aggregation6. Because coiled-coil interactions are essential for many physiologically important protein–protein interactions, it is hypothesized that the propensity for coiled-coil interactions in polyQ proteins may also contribute to their toxicity in polyQ-based diseases, as a result of these proteins engaging in novel and inappropriate protein–protein interactions6.
The current standard treatments for neurodegenerative diseases such as Alzheimer’s disease, Huntington’s disease and Parkinson’s disease are primarily aimed at providing relief from symptoms, and do not address the underlying degenerative processes (TABLE 1). For example, tremors associated with Parkinson’s disease can be suppressed by administration of l-DOPA, the synthetic precursor of dopamine, which augments signalling in compromised dopaminergic pathways7. However, chronic treatment with l-DOPA results in the development of adverse effects such as dyskinesia, and treatment becomes less effective with time because neurons continue to degenerate7,8. Symptomatic treatment of Alzheimer’s disease with acetylcholinesterase inhibitors or memantine, a selective NMDA (N-methyl-d-aspartate) receptor antagonist, has also shown some efficacy9,10. These therapies augment neurotransmission in cholinergic neurons, which represent a large population of neurons that are affected in Alzheimer’s disease, but fail to prevent neuronal degeneration9,10. Treatments used in other neurodegenerative diseases include anti-psychotics and neuroleptics in Huntington’s disease, and riluzole in amyotrophic lateral sclerosis (ALS), but there are currently no efficacious treatments for prion-based diseases11-13.
There is, therefore, an urgent need for novel therapeutic strategies that could halt or reverse the underlying disease process in neurodegenerative disorders, which could have a profound impact on their long-term clinical management. Indeed, various disease-modifying approaches specific to particular neurodegenerative diseases are currently being investigated. For example, for Alzheimer’s disease, several agents that inhibit enzymes involved in the production of amyloid-β — a key constituent of the aggregated plaques that are a hallmark of the disease — are currently in clinical trials. An attractive alternative — and potentially more widely applicable — therapeutic strategy is to ameliorate the protein misfolding that is a shared underlying characteristic of many neurodegenerative diseases. Studies in various models of neurodegenerative diseases indicate that enhancing cellular protein-folding capacity by elevating the expression of chaperone proteins could represent such a strategy. In this article, we first provide an overview of the role of chaperone proteins in protein homeostasis. We then describe recent advances indicating the potential of pharmacologically activating heat shock transcription factor 1 (HSF1) — the master activator of chaperone protein gene expression — to treat neurodegenerative diseases, and discuss the challenges in developing HSF1 activators as drugs.
Protein homeostasis via chaperone proteins
In normal cells, protein homeostasis is maintained by regulating the expression, folding, modification, translocation and, ultimately, degradation of proteins. To achieve this, cells use sophisticated mechanisms to ensure the proper execution of these processes in response to cellular stress. A crucial aspect of cellular protein homeostasis is the utilization of chaperone proteins, which stabilize protein structures, assist in correct folding and unfolding of proteins, and facilitate the assembly of multimeric protein complexes14. Chaperone proteins, including αB-crystallin, heat shock protein 27 (HSP27), HSP40, HSP70 and HSP90, as well as class I and class II chaperonins, function individually or as part of larger heterocomplexes to prevent protein misfolding and protein aggregation14.
Many neurodegenerative diseases are characterized by the accumulation of misfolded and aggregation-prone proteins, and studies indicate that chaperone proteins have an important role in cellular function and survival in these diseases. This has perhaps been most elegantly demonstrated in fruitfly and worm models of several neurodegenerative diseases, including Huntington’s disease, ataxia, ALS and Alzheimer’s disease15-25. Interestingly, chaperone proteins are also powerful antiapoptotic factors that can stabilize pro-survival proteins such as the kinase AKT and inhibit the function of proapoptotic proteins such as JUN-activated N-terminal kinase, the apoptosis regulator BAX, apoptosis-inducing factor and caspases26. As such, chaperone proteins can promote cell survival in neurodegenerative diseases by stabilizing and refolding misfolded proteins and by directly inhibiting apoptosis. Moreover, the inability to mount an adequate response to misfolded proteins may be a feature of ageing cells and neuronal cells in general27-33, as reports have shown that although proteins such as huntingtin are misfolded in all cell types, toxicity occurs almost exclusively in neurons13. This susceptibility to proteotoxic stress stems predominantly from an inability of neuronal cells to robustly respond to stress in general, as constitutive overexpression of chaperone proteins can protect neurons from proteotoxicity15,16,34-38.
Because chaperone proteins act both individually and as macromolecular heterocomplexes, elevated expression — via co-transfection experiments — of several chaperone proteins has been shown to have greater therapeutic effects in cell culture models of protein misfolding than overexpression of individual chaperone proteins17,34,36. The genetic overexpression of several chaperone proteins for disease treatment is impractical, but the identification of pharmacological activators that promote the coordinated and chronic enhancement of chaperone protein levels could be a powerful strategy for therapeutic intervention in neurodegenerative diseases. Furthermore, overexpression of chaperone proteins may be toxic in fruitfly cells39, and has been linked to oncogenic transformation40. As such, the greatest benefit of chaperone protein-based therapy would probably be achieved through modest but chronic elevation of chaperone protein levels.
HSF1: master regulator of chaperone expression
The cellular response to cytoplasmic proteotoxic stimuli — such as elevated temperature, oxidative stress, heavy metals, bacterial or viral infection, and so on — is primarily controlled in human cells via transcriptional activation by HSF1. HSF1 is a member of the vertebrate family of HSF proteins, which consists of four members and is conserved in its overall architecture from yeast to mammalian cells41. Microarray analyses in yeast, fruitfly and mammalian cells have shown that although HSF1 promotes the expression of genes encoding chaperone proteins in response to cellular stress, it also regulates the expression of genes involved in other aspects of cell survival, including protein degradation, ion transport, signal transduction, energy generation, carbohydrate metabolism, vesicular transport and cytoskeleton formation42-44.
Although HSF1 is the isoform that has been predominantly linked to chaperone protein-dependent amelioration of neurodegenerative diseases, other members of the HSF family may also prove to be useful therapeutic targets. Specifically, HSF2 has recently been associated with the expression of chaperone protein-encoding genes at febrile temperatures, in part via heteromultimerization with HSF1 (REFS 45,46). Furthermore, it has been proposed that HSF2, in concert with HSF1, promotes the expression of non-classical heat shock genes such as αB-crystallin47. Consistent with the notion that HSF2 also contributes to the mammalian heat shock response, deletion of HSF2 in a mouse model of polyQ disease reduced lifespan and accelerated the accumulation of ubiquitylated misfolded proteins47.
Stress-dependent regulation of HSF1 is a multistep process that is controlled by intricate regulatory mechanisms41 (FIG. 2). Under basal conditions HSF1 exists largely as an inactive monomer in the cytoplasm, repressed in part through the activity of the chaperone proteins HSP90, HSP70, HSP40 and other co-chaperones48-54. HSP70 and HSP40 bind to the transcriptional activation domain of HSF1 and are thought to repress HSF1-dependent transactivation. HSP90, in conjunction with the co-chaperone p23 (also known as PTGES3), protein phosphatase 5 and cyclophilins, is thought to associate with the regulatory domain of HSF1 (FIG. 3) and repress both the multi merization of HSF1 and its ability to activate target gene expression. As HSP40 and HSP70 are also required for the correct assembly of HSP90 on some client proteins55-57, it is conceivable that the activity of these chaperones may impinge on HSF1 regulation via several indirect mechanisms. Under basal conditions HSF1 is also phosphorylated on Ser303 and Ser307 residues within its central regulatory domain, which contributes to its repression (FIG. 3). However, the specific mechanisms by which these phosphorylation events mediate HSF1 repression are presently not clear58-65.
Figure 2. HSF1 activation and attenuation cycle.

In the absence of cellular stress, heat shock transcription factor 1 (HSF1) exists as an inactive monomer in the cytoplasm. Its activity is repressed via the interaction of the chaperone proteins heat shock protein 90 (HSP90), HSP70 and HSP40 as well as its phosphorylation on Ser303 and Ser307 residues. In response to proteotoxic stress HSP90 is thought to bind to misfolded proteins and dissociate from HSF1, thereby allowing HSF1 to form homotrimers and become localized to the nucleus to bind to heat shock elements in the promoters of stress-responsive genes. Disulphide bonding occurs between HSF1 monomers to stabilize trimer formation. HSP70 is known to associate with HSF1 even when it is bound to DNA, and it may continue to repress HSF1 until a secondary stimulus promotes its dissociation. Following DNA binding HSF1 is sumoylated — this is a repressive modification that is attenuated with prolonged heat stress. HSF1 is hyperphosphorylated on up to 12 serine residues during HSF1-dependent transactivation. HSF1-dependent transactivation is repressed via a negative feedback loop, in which HSP70 and HSP40 re-associate with the HSF1 transactivation domain, and HSF1 becomes dissociated from DNA following acetylation of Lys80 in its DNA binding domain. It remains unclear whether the disulphide-linked HSF1 trimers are dissociated into cytoplasmic monomers or whether they are degraded.
Figure 3. HSF1 regulation by post-translational modifications.

In response to cellular stress, heat shock transcription factor 1 (HSF1) is hyperphosphorylated on up to 12 serine residues; this occurs in parallel with HSF1-dependent transactivation. Three sites of phosphorylation involved in HSF1 activation that have been studied in detail are shown. HSF1 activity is also repressed by constitutive phosphorylation of Ser303 and Ser307, and by stress-responsive sumoylation of Lys298 (represented on the figure as ‘Su’). The binding of HSF1 to DNA is inhibited by the acetylation of Lys80 (represented on the figure as ‘Ac’). AD, activation domain; DBD, DNA binding domain; LZ1, leucine zipper domain 1.
In response to proteotoxic stress HSF1 is thought to dissociate from HSP90 and other co-chaperones; this allows it to homotrimerize, accumulate in the nucleus and bind to cis-acting heat shock elements (consisting of inverted NGAAN repeats) in the promoters of stress-responsive genes, including those encoding chaperone proteins66-68. HSF1 trimerization is dependent on three amino-terminal leucine zipper domains directly adjacent to the DNA binding domain that form an extended coiled-coil domain69,70, and is stabilized via intermolecular disulphide bonding between HSF1 monomers71. Indeed, treatment of cells with reducing agents significantly reduces HSF1 homotrimerization and stress-dependent gene activation in response to most — but not all —stimuli54,72,73. A fourth leucine zipper domain, located in the carboxy-terminal region of HSF1, represses HSF1 activity74. This may occur via an intramolecular interaction with the N-terminal coiled-coil domain, thereby preventing interaction with other HSF1 monomers, although there is no direct evidence for this74. Although HSP90 and other co-chaperones are thought to dissociate from HSF1 in response to cellular stress, evidence suggests that HSP70 and HSP40 remain associated with HSF1 even while it is bound to promoter DNA48.
In parallel with DNA binding, robust stress-responsive HSF1 phosphorylation is observed. Although the specific requirement for these phosphorylation events in HSF1-dependent gene activation is currently unclear, 12 phosphorylated serine residues have been identified; surprisingly, however, the mutation of individual serine residues does not abrogate HSF1-dependent transactivation75. Although calcium/calmodulin-dependent protein kinase II and Polo-like kinase 1 have been shown to phosphorylate Ser230 and Ser419, respectively76,77, the protein kinases that catalyse the remaining phosphorylation events have not yet been identified. Concomitant with DNA binding, HSF1 is also transiently sumoylated on Lys298 (FIG. 3): this modification represses HSF1-dependent transactivation but is attenuated following prolonged exposure to thermal stress61. It has been clearly established that HSF1 sumoylation is dependent on the phosphorylation of Ser303 and contributes to HSF1 repression. However, the precise mechanism by which Lys298 sumoylation mediates HSF1 repression remains to be determined.
Even with chronic proteotoxic heat stress, HSF1-dependent transactivation of target genes is a transient response that is regulated at multiple levels62. First, HSF1 is negatively regulated via a feedback loop, as the newly synthesized chaperone proteins HSP70 and HSP40 bind to and inhibit the HSF1 transactivation domain, and HSP90 re-associates with the regulatory domain52,53. Subsequently, the residence time of HSF1 on promoter heat shock element sites is reduced by the acetylation of several lysine residues, in particular Lys80, located within the DNA binding domain78 (FIG. 3). Little is known about the fate of HSF1 after its dissociation from DNA. For example, it remains unclear whether HSF1 hyperphosphorylation is reversed by protein phosphatases after the completion of the HSF1 activation cycle. Similarly, the fate of the disulphide-linked HSF1 monomers is not known. Are the disulphide bonds reduced after completion of the HSF1 activation cycle, allowing the monomers to return to an inactive state in the cytoplasm, or is the HSF1 homotrimer degraded following its dissociation from DNA? Although recent evidence has suggested that HSF1 may be a substrate for chaperone-mediated autophagy, relatively little is known about HSF1 degradation79.
The multiplicity of mechanisms that influence the activation and repression of HSF1 indicates that the expression of HSF1 target genes is a highly regulated event. Therefore, obtaining a thorough understanding of the mechanisms by which HSF1 is regulated will broaden our knowledge of stress responses and expand the spectrum of opportunities for the identification of targets for therapeutic intervention in neurodegenerative diseases associated with protein misfolding.
Pharmacological activators of HSF1
Pharmacological activation of HSF1 and transcriptional activation of genes encoding chaperone proteins have been achieved through molecules that either cause proteotoxic stress or inhibit the chaperone proteins that limit HSF1 activity. Molecules that activate HSF1 by promoting protein misfolding or proteotoxic stress, such as the proteasome inhibitor MG132 or the proline analogue azetidine-2-carboxylic acid, are unlikely to be efficacious in the chronic treatment of neurodegenerative diseases as these molecules ultimately promote cellular dysfunction and lead to cell death. Therefore, in this Review we focus solely on pharmacological agents that promote HSF1 activity and could provide potential opportunities for the development of drugs for neurodegenerative diseases (TABLE 2).
Table 2.
Pharmacological activators of HSF1
| Compound | Structure | Mechanism of action | Toxicity | Refs |
|---|---|---|---|---|
| Geldanamycin |
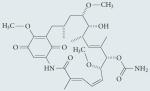
|
Amino-terminal HSP90 inhibitor |
Significant hepatotoxicity, leading to the development of analogues 17-AAG (currently in Phase I/II trials) and 17-DMAG (discontinued after Phase I development owing to toxicity) |
80,174-176 |
| Gedunin |

|
Carboxy-terminal HSP90 inhibitor |
Not well characterized in humans or mammalian animal models; gedunin and analogues exhibit toxicity in in vitro models of cell viability |
177,178 |
| Celastrol |
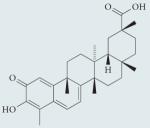
|
Not well characterized; hypothesized to act via C-terminal HSP90 inhibition, interruption of HSP90-CDC37 interaction, proteasome inhibition, thiol oxidation and topoisomerase inhibition |
Not well characterized in humans or mammalian animal models; significant toxicity in in vitro models of cell viability |
107-109 |
| Arimoclomol |
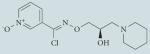
|
Not well characterized; hypothesized to increase duration of HSF1 binding to heat shock element and increase plasma membrane fluidity |
Well tolerated in Phase I trials; currently in Phase II/III clinical trials for amyotrophic lateral sclerosis |
120-122, 167,179 |
| HSF1A |

|
Not well characterized; hypothesized to modulate activity of TRIC or other protein targets |
Not tested in humans or mammalian animal models |
72 |
| Riluzole |
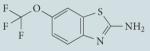
|
Not well characterized; hypothesized to have antiglutamatergic activity, and to increase steady-state levels of HSF1 |
Approved by the FDA for clinical use in amyotrophic lateral sclerosis; mild gastrointestinal toxicity |
180 |
| Geranylgeranylacetone |
|
Not well characterized; hypothesized to inhibit HSP70 substrate binding and redox activity |
Limited toxicity in humans |
181-183 |
17-AAG, 17-allylamino-17-demethoxy-geldanamycin; 17-DMAG, 17-dimethylaminoethylamino-17-demethoxygeldanamycin; CDC37, co-chaperone of HSP90; FDA, US Food and Drug Administration; HSF1, heat shock transcription factor 1; HSP90, heat shock protein 90; TRIC, T complex protein 1 ring complex.
HSP90 inhibitors
The chaperone protein HSP90 is one of the most abundant proteins in the cell, and is required for the folding and maintenance of many cellular proteins — referred to as client proteins — including steroid hormone receptors, protein kinases and transcription factors80. Under normal growth conditions, HSP90 is also thought to bind to the regulatory domain of HSF1 and repress both HSF1 multimerization and transactivation49,50,52,54. Accordingly, pharmacological inhibition of HSP90 promotes the activation of HSF1 and leads to increased expression of chaperone proteins54. Well-characterized HSP90 inhibitors such as the benzoquinone ansamycin antibiotic geldanamycin, as well as its derivatives 17-allylamino-17-demethoxygeldanamycin (17-AAG) and 17-dimethylaminoethyl-amino-17-demethoxygeldanamycin (17-DMAG), bind to the N-terminal ATP-binding pocket of HSP90, thereby inhibiting its ATPase activity. This promotes the degradation of its array of client proteins. As HSP90 is required for the stability and function of many essential cellular proteins, high levels of HSP90 inhibitors are associated with substantial cytotoxicity. However, although treatment with low levels of HSP90 inhibitors is tolerable for non-tumorigenic cells, tumorigenic cells are highly sensitive to HSP90 inhibitors, possibly owing to an increased demand for the pro-proliferative and pro-survival factors that are HSP90 client proteins.
Geldanamycin (TABLE 2) and its derivatives have proven to be efficacious in promoting HSF1-dependent chaperone protein expression, thereby ameliorating protein aggregation and cytotoxicity in experimental models of protein-misfolding diseases, including Alzheimer’s disease80-84, Parkinson’s disease18,85,86 and Huntington’s disease87-90. One of the most pronounced examples of the efficacy of geldanamycin in the treatment of a protein-misfolding disease was observed by Bonini and colleagues18 in a fruitfly model expressing mutant α-synuclein proteins in dopaminergic neurons. Exposure to as little as 3 μg per ml of geldanamycin protected dopaminergic neurons from α-synuclein-induced cell death18,19. Interestingly, elevations in HSP70 expression were not detected with geldanamycin concentrations that rescued neuronal cell viability, suggesting that very small increases in HSP70 expression may be sufficient to provide protection from α-synuclein-induced cytotoxicity.
Several coumarin antibiotics, most notably novobiocin, are thought to bind to the C-terminal region of HSP90, resulting in the inhibition of HSP90 activity91. Although most molecules in this class bind to HSP90 with weak affinity, the novobiocin analogue A4 had neuroprotective effects at concentrations 70-fold lower than novobiocin92. Administration of the A4 analogue protected cultured neuronal cells from amyloid-β-induced cytotoxicity at concentrations as low as 5 nM93. Interestingly, at these concentrations HSP70 levels only increased by approximately 50%, further supporting the notion that relatively small increases in chaperone protein levels can have potent therapeutic effects on protein aggregation and cell viability.
Several additional compounds, including (−)-epigallo-catechin-3-gallate, gedunin, AEG3482 and ITZ-1, also bind to the C-terminal ATP-binding pocket of HSP90, inhibit HSP90 activity94,95 and promote the activation of HSF1-dependent chaperone protein expression. Interestingly, AEG3482 and ITZ-1 promoted the activation of HSF1 without leading to significant degradation of other HSP90 client proteins. It remains to be determined whether this deviation from the activity of N- and C-terminal HSP90 inhibitors stems from experimental variability (that is, cell type) or whether these molecules inhibit HSP90 without affecting the stability of HSP90 client proteins.
Given the potentially hundreds of HSP90 client proteins involved in cellular growth, proliferation and signalling, it is not surprising that many HSP90 inhibitors have strong toxic side effects such as defective T cell activation96. Consequently, the feasibility of using HSP90 inhibitors for the treatment of neurodegenerative diseases remains unclear. Thirteen HSP90 inhibitors are currently in clinical trials, primarily as cancer therapeutics80.
Celastrol
Celastrol is a quinone methide triterpene isolated from Tripterygium wilfordii (thunder god vine) root extracts (TABLE 2). It has well-established antioxidant and anti-inflammatory properties, and has been shown to be a potent activator of HSF1 and chaperone protein expression97. Although the mechanism by which celastrol promotes HSF1 activation is unclear, various hypotheses have been proposed. Recent studies suggest that celastrol binds to the C-terminal domain of HSP90 and, similarly to novobiocin and gedunin, inhibits the chaperone activity of HSP90, promotes client protein degradation and promotes the activation of HSF1 (REF. 98). More specifically, celastrol is thought to inhibit the interaction between HSP90 and its co-chaperone CDC37 (REF. 99). In addition to its ability to inhibit HSP90, celastrol has been shown to inhibit the proteasome, which is required for the degradation of damaged and misfolded proteins100.
Celastrol-dependent proteasome inhibition could result in the accumulation of misfolded proteins, which could lead to the induction of HSF1 activity. Celastrol has also been shown to covalently react with nucleophilic thiol groups of cysteine residues73. As such, it is possible that celastrol-mediated thiol oxidation alone could result in the damage and misfolding of various cellular proteins or the oxidation of cysteine residues in HSF1 (REF. 71). Consistent with this hypothesis, co-administration of celastrol with dithiothreitol blocked celastrol-dependent activation of HSF1 as well as chaperone protein expression in both HeLa and yeast cells73. Although the ability of celastrol to promote chaperone protein expression has proven to be efficacious in reducing protein aggregation and cytotoxicity in models of ALS101, Alzheimer’s disease102, Huntington’s disease103,104 and Parkinson’s disease105,106, the therapeutic potential of celastrol will require further evaluation owing to its inherent cytotoxicity107-109. Because celastrol-mediated cytotoxicity is likely to be associated with its propensity to promote thiol oxidation, co-administration of celastrol with antioxidants might be valuable in therapeutic applications.
Geranylgeranylacetone
Several anti-ulcer drugs — including geranylgeranylacetone (GGA) (TABLE 2), carbenoxolone, zinc l-carnosine and rebamipide — have been identified as activators of chaperone protein expression110. However, so far only the acyclic isoprenoid GGA has been studied in detail. Treatment with GGA promotes the activation of HSF1 and the expression of chaperone proteins in mammalian tissues, leading to cytoprotection from various stresses111. Pretreatment of neuronal precursor cells expressing a polyQ derivative of the androgen receptor — a model for spinal and bulbar muscular atrophy — with GGA promoted the expression of chaperone proteins, reduced protein aggregation and reduced polyQ protein-dependent cell death112. Moreover, administration of GGA to mice expressing a mutant androgen receptor protein promoted chaperone protein expression, improved motor neuron function and expanded lifespan. GGA is thought to promote HSF1 activation by binding to the substrate binding domain of HSP70 (REF. 113). However, as other HSP70 inhibitors do not promote HSF1 activity114, it is possible that GGA might also promote HSF1 activity through an HSP70-independent mechanism. Interestingly, GGA was also shown to induce the expression of thioredoxin and cause apoptosis in human promyelocytic leukaemia (HL-60) cells, suggesting that it might activate HSF1 through a potential redox mechanism115,116. GGA is also a potent activator of the unfolded protein response in the endoplasmic reticulum117.
HSF1A
Studies from our laboratory72 have identified the benzyl pyrazole derivative HSF1A (TABLE 2) as an activator of HSF1-dependent chaperone protein expression in both mammalian and fruitfly cells as well as in adult fruitflies. Although the specific mechanisms by which HSF1A promotes HSF1 expression remain unclear, HSF1A interacts with the T complex protein 1 ring complex (TRIC; also known as CCT) — a cytosolic chaperonin complex — and other cellular proteins. Further studies showed that pretreatment with HSF1A alleviated both the aggregation and the cytotoxicity of polyQ proteins expressed in neuronal precursor cells as well in a fruitfly model of polyQ-dependent toxicity. HSF1A was unable to ameliorate polyQ-dependent toxicity in flies lacking a functional Hsf1 allele, supporting the notion that HSF1A ameliorates cytotoxicity by stimulating the obligatory HSF1 target. Interestingly, similarly to examples noted above, HSF1A-mediated protection from polyQ-induced cytotoxicity was observed at concentrations that promoted only modest increases in HSP70 levels. Furthermore, unlike the HSF1 activation observed in response to celastrol or heat shock exposure, HSF1A-dependent expression of chaperone proteins was not diminished by dithiothreitol co-administration, suggesting that HSF1A may not mediate HSF1 activation through redox-sensitive mechanisms.
The possibility that HSF1A binds to TRIC directly and potentially modulates TRIC activity is of interest. TRIC has been shown to bind to the first 17 amino acids of human huntingtin (which stabilize downstream coiled-coil domains that are important for disease progression6) and prevent its aggregation118,119. As such, HSF1A might ameliorate polyQ-induced cytotoxicity via HSF1-dependent and -independent mechanisms. Alternatively, TRIC could be a modulator of HSF1 activity. It will be important to carry out further research to characterize the effects of HSF1A on TRIC activity, and to examine whether HSF1A can also ameliorate cytotoxicity in models of Parkinson’s disease and Alzheimer’s disease, in which disease-causing proteins lack the coiled-coil structure of many polyQ proteins.
Hydroxylamine derivatives
Several hydroxylamine derivatives, including bimoclomol and its derivative arimoclomol (also known as BRX-220) (TABLE 2), have been identified as co-inducers of the heat shock response120,121. Although these compounds alone do not promote HSF1-dependent chaperone protein expression, their activity prolongs stress-induced activation of HSF1 (REF. 120). The specific mechanism by which the hydroxylamine derivatives amplify HSF1 activation remains unclear. Early reports suggested that bimoclomol directly bound to HSF1, thereby increasing the duration that HSF1 remains bound to the promoter heat shock element120. Interestingly, this outcome is consistent with the phenotypes observed with reduced HSF1 acetylation78. However, the effects of bimoclomol on HSF1 acetylation have not been tested.
Subsequent studies have also suggested that molecules in this class can modulate the fluidity of the plasma membrane, thereby stimulating HSF1 activation122. Despite their inability to induce HSF1 activity on their own, these molecules may be promising therapeutics for several neurodegenerative diseases. Experiments have shown that bimoclomol amplifies the expression of chaperone proteins and has cytoprotective effects in disease models of diabetes mellitus, cardiovascular disease and renal failure123. The bimoclomol derivative arimoclomol is also being tested for efficacy in the treatment of ALS. Treatment of SOD1G93A mice — a model for familial ALS — with arimoclomol alleviated the disease symptoms and prolonged the lifespan of the mice even when it was administered after the onset of symptoms124,125. A Phase II/III clinical trial with arimoclomol is currently underway in patients with superoxide dismutase 1 (SOD1)-positive ALS126 (ClinicalTrials.gov identifier: NCT00706147).
Riluzole
Riluzole is currently the only approved pharmacological agent for the treatment of ALS, but it only extends survival by 3-5 months. The mechanisms by which riluzole alleviates symptoms of ALS are currently unclear. Although riluzole is thought to antagonize glutamatergic signalling, some other anti-glutamate agents have been ineffective in the treatment of ALS127. Recent evidence has suggested that riluzole also stimulates the expression of chaperone proteins by increasing the steady-state levels of HSF1 (REF. 79). This may occur by selectively inhibiting chaperone-mediated autophagy-based HSF1 degradation. It is important to note that relatively little is known about the mechanisms underlying HSF1 degradation. Nevertheless, these and other data further support the notion that increased expression of chaperone proteins might be beneficial in ALS, and warrant further investigations into HSF1 turnover mechanisms.
NSAIDS
The non-steroidal anti-inflammatory drugs (NSAIDs) sodium salicylate and indomethacin robustly induce the binding of HSF1 to DNA in the absence of cellular stress in mammalian, fruitfly and yeast cells, but they do not strongly promote the transcriptional activation of HSF1 target genes128-130. However, similarly to the hydroxylamine derivatives, NSAIDs synergize with exposure to temperatures that are suboptimal for heat shock to promote the expression of HSF1 target genes129. Other NSAIDs — including sulindac, nabumetone and phenylbutazone — appear to be more robust activators of HSF1 and promote the expression of HSP70 even in the absence of cellular stress131. Although the specific mechanism by which these compounds activate HSF1 is unclear, experiments in cultured fruitfly S2 cells have shown that salicylate treatment resulted in a pronounced reduction in cellular ATP levels130. As the chaperone proteins that repress HSF1 activity are ATPases, reduced levels of cellular ATP might reduce the activity of these chaperones as well as their ability to repress HSF1 activity.
In addition, at high concentrations NSAIDs have been proposed to function as protein kinase inhibitors, and may promote the activation of HSF1 through inhibition of kinases that phosphorylate and repress HSF1 activity132,133. Despite their inability to strongly induce HSP70 expression, recent studies have shown that treatment with either sodium salicylate or indomethacin can reduce polyQ protein aggregation and cytotoxicity in mammalian cell cultures134. Because extremely high concentrations of NSAIDs are required to promote the binding of HSF1 to DNA, and given the pleiotropic nature of NSAIDs, it is difficult to ascertain the contribution of HSF1 to this phenomenon. The antiinflammatory compound cyclopentenone prostaglandin 2-cyclopenten-1-one is also a potent inducer of HSF1 activity135,136.
Additional compounds
Additional HSF1 co-inducers that have been identified include chloro-oximine137 and pyrimido(5,4-e)(1,2,4)triazine-5,7(1H,6H)-dione derivatives138, as well as gedunin and sappanone A derivatives139. Exposure of cultured mammalian cells to these molecules did not promote HSF1 activation, but it increased the heat shock response when the cells were exposed to subthreshold elevations in temperature. In addition, these compounds protected cells from cytotoxicity, further supporting the notion that maximal induction of HSF1 and chaperone protein expression may not be required for therapeutic efficacy. As gedunin was previously identified as an HSP90 inhibitor, it is possible that these derivatives also promote HSF1 activation via the inhibition of HSP90.
Amelioration of protein-folding cytotoxicity by HSF1-inducing compounds
A common feature of many HSF1-activating molecules discussed here is their ability to ameliorate the cytotoxicity of misfolded proteins at concentrations that result in only mild increases in the abundance of chaperone proteins. Does this suggest that only small increases in chaperone protein expression are required for the amelioration of proteotoxicity induced by protein misfolding, or is it possible that the expression of other HSF1 target genes, in addition to chaperone proteins, influences toxicity? Data show that following exposure to heat shock, genes encoding chaperone proteins — in particular HSP70 — are substantially upregulated in response to HSF1 activation44. However, recent evidence shows that HSF1 also promotes the expression of the transcription factor NFATC2 (nuclear factor of activated T cells cytoplasmic 2), the scaffold protein PDZ domain-containing protein 3, transmembrane protease serine 3 and other genes that contribute to the amelioration of polyQ aggregation and polyQ-induced cyto toxicity140. Additional expression studies using small-molecule activators at both low and high concentrations, combined with genetic analyses, are needed to identify the HSF1 target genes that are expressed in response to minimal levels of HSF1 activation and contribute to therapeutic efficacy, especially in neuronal cells.
Indirect mechanisms to promote HSF1 activation
HSF1 activity is regulated through various complex mechanisms, including post-translational modifications, intramolecular coiled-coil interactions as well as interactions with macromolecular chaperone complexes and other proteins41. The pharmacological modulation of these regulatory mechanisms to promote HSF1 activation is a promising avenue for further exploration.
HSF1 phosphorylation
Data from mass spectrometry analyses indicate that HSF1 is phosphorylated on at least 12 serine residues75 and that phosphorylation of Ser121, Ser303, Ser307, Ser320 and Ser363 is associated with repression of HSF1 activity60,63-65 (FIG. 3). However, only the substitution of Ser303 or Ser307 with alanine has been reported to result in an increase in HSP70 expression141, suggesting that these specific phosphorylation events could be targeted for pharmacological activation of HSF1. In support of this hypothesis, expression of an HSF1 mutant (with an S303G mutation) in HeLa cells resulted in significantly reduced polyQ protein aggregation compared with wild-type HSF1 (REF. 142).
Several protein kinases have been linked to the phosphorylation of HSF1 at Ser303 and Ser307, although the specific kinases that phosphorylate these residues remain unknown. Early in vitro experiments suggested that Ser303 was phosphorylated via glycogen synthase kinase 3 (GSK3) and that Ser307 was phosphorylated via extracellular signal-regulated kinase 1 or extracellular signal-regulated kinase 2 (REF. 59). However, recent data from our laboratory suggest that although GSK3 does repress HSF1 activity, it appears to do so independently of Ser303 phosphorylation141. Rather, these and other data suggest that Ser303 may be phosphorylated by a mitogen-activated protein kinase62,63,141. The identification of the kinase (or kinases) that phosphorylate Ser303 and Ser307 in vivo could be important for developing agents to target neurodegenerative diseases via HSF1 activation.
Although the precise mechanisms by which GSK3 represses HSF1 activity are not fully understood, GSK3 inhibition by either lithium or SB-216763 results in increased binding of HSF1 to DNA and modest activation of HSP70 expression in the absence of additional stress141,143-145. Inhibition of GSK3 activity could therefore serve as a useful therapeutic approach in the treatment of neurodegenerative diseases. Indeed, treatment of cells with GSK3 inhibitors has been shown to be protective against polyQ-induced cell death58. In addition, GSK3 inhibition promotes the activation of autophagy, which is known to be efficacious in the treatment of neurodegenerative diseases58.
HSF1 sumoylation
Sumoylation of HSF1 at Lys298 occurs via the activity of the E1 small ubiquitin-related modifier (SUMO)-activating enzymes SAE1 and SAE2 as well as the E2 SUMO-conjugating enzyme UBC9. Sumoylation represses HSF1-dependent target gene transactivation146 (FIG. 3). In some instances protein sumoylation may occur solely via the activity of the E1 SUMO-activating enzymes and the E2 SUMO-conjugating enzyme, without requiring activity of the E3 SUMO-ligating enzyme147. It is currently unknown whether HSF1 sumoylation requires an E3 enzyme, although recent data have shown that large oligomers of the chaperone protein HSP27 bind to UBC9 and are required for HSF1 sumoylation148. As such, it is possible that HSP27 itself acts as the E3 enzyme in HSF1 sumoylation.
Ginkgolic and anacardic acid have recently been identified as pharmacological inhibitors of the sumoylation machinery149. Both small molecules inhibit protein sumoylation in vitro and in vivo, without affecting protein ubiquitylation, by directly binding to the E1 enzyme and inhibiting the formation of the E1–SUMO intermediate, thereby inhibiting the attachment of SUMO to target proteins. It remains to be determined whether treatment with ginkgolic acid or anacardic acid results in HSF1 activation. However, owing to the ubiquitous nature of the E1 and E2 enzymes, it is likely that pharmacological modulation of their activity would have pleiotropic effects. Future research efforts could focus on the identification and inhibition of the E3 enzymes that may be required for HSF1 sumoylation.
HSF1 acetylation
The HSF1 activation cycle is a dynamic process that is regulated by intra- and inter-molecular interactions as well as post-translational modifications (FIG. 2). Stress-induced binding of HSF1 to DNA is reversed by acetylation of HSF1 at Lys80, which is mediated by histone acetyltransferase p300 and cyclic AMP-responsive element-binding protein. Conversely, DNA binding is prolonged by NAD-dependent deacetylase sirtuin 1 (SIRT1)-mediated deacetylation of HSF178. Treatment of mammalian cells with the SIRT1 agonist resveratrol resulted in decreased acetylation of HSF1 at Lys80 and prolonged stress-dependent binding of HSF1 to DNA. Although resveratrol-dependent activation of SIRT1 is likely to affect many cellular processes, it is important to note that resveratrol treatment has been shown to rescue polyQ-dependent neuronal dysfunction in Caenorhabditis elegans as well as in mammalian neurons150. In addition, the efficacy of resveratrol has been explored in models of Alzheimer’s disease151-153, Parkinson’s disease154,155 and ALS151. The regulation of HSF1 acetylases and deacetylases may therefore provide crucial intervention points for the modulation of chaperone protein expression.
Direct activation of HSF1
Achieving HSF1 activation through the inhibition of negative regulators such as chaperones, protein kinases and acetyltransferases has therapeutic potential in neurodegenerative diseases. However, the inhibition of these negative regulators is likely to have pleiotropic effects on many cellular targets and functions, which could complicate disease treatment. As such, further investigation into the identification of pharmacological HSF1 activators could be directed towards the identification of molecules that directly bind and activate HSF1.The interconversion of HSF1 monomers to homotrimers is an early and crucial step in the HSF1 activation pathway, and so small molecules that directly interact with the monomeric HSF1 transcription factor to promote its homotrimerization could be highly specific HSF1 activators.
Direct activators of HSF1 have not yet been reported, so we propose three potential approaches to directly promote HSF1 activation (FIG. 4). First, we envision a small molecule that would recognize and disrupt the protein interface between HSF1 and HSP90, without directly inhibiting either protein. Because the interaction between HSP90 and HSF1 is thought to be relatively weak54, it is likely that this interaction could be antagonized with relatively low-affinity molecules that compete for this interface. Unfortunately, only limited information exists regarding the HSF1–HSP90 interaction. Further structural information regarding the sites encompassing the HSP90–HSF1 interaction as well as the binding affinity of HSP90 for HSF1 could facilitate the development of such molecules. Elucidation of the HSP90–HSF1 interaction site could enable the development of an in vitro screen (using recombinant HSP90 and a small peptide encompassing the region of HSF1 that interacts with HSP90) to search for small-molecule inhibitors of the HSP90-HSF1 interaction. We envision a similarly designed secondary screen, using the in vitro interaction between HSP90 and another client protein (for example, the glucocorticoid receptor), to exclude molecules that directly inhibit HSP90.
Figure 4. Proposed mechanisms to promote the direct activation of HSF1.

We propose three potential approaches to promote the direct activation of heat shock transcription factor 1 (HSF1). a | First, a small molecule specifically designed to bind the interface between heat shock protein 90 (HSP90) and HSF1 would prevent the interaction between the two proteins and allow for HSF1 trimerization and target gene expression. b | Second, HSF1 could be activated using a small molecule designed to sequester the carboxy-terminal coiled-coil domain that is thought to repress HSF1 function, allowing for HSF1 trimerization. c | Last, a multivalent molecule specifically designed to bind to HSF1 monomers could bring these monomers into close proximity and increase the rate of HSF1 trimerization. AD, activation domain; DBD, DNA binding domain; LZ1, leucine zipper domain 1.
A second approach to directly activate HSF1 may be to unleash the intramolecular repressive interaction between the C-terminal coiled-coil domain and the N-terminal multimerization domain of HSF1. However, this intramolecular interaction is currently only speculative and direct physical evidence for this interaction has not yet been reported74. A crystal structure of the full-length HSF1 protein would significantly aid in this regard. Nevertheless, it is possible to envisage small molecules that could inhibit such an interaction. This might be effectively achieved via the use of protein or RNA aptamers that could sequester the C-terminal repressive coiled-coil domain and thereby free the N-terminal multimerization domain for homotrimerization.
As a proof of concept, Lis and colleagues156 have recently shown that RNA aptamers can be used to bind and sequester the DNA binding domain of HSF1 and, as a result, effectively inhibit HSF1 activation. Although these aptamers inhibit rather than activate HSF1 function, aptamer technology could be developed to promote HSF1 activation. It should be noted that mutations in the fourth leucine zipper domain result in constitutive HSF1 trimerization and constitutive DNA binding, but may only modestly increase the expression of chaperone proteins. However, as previously noted, the use of pharmacological activators of HSF1 has shown that high-level expression of chaperone proteins is not required for therapeutic efficacy18,72,93,121. Rather, small and chronic increases in levels of chaperone proteins are highly effective in the amelioration of toxicity induced by protein misfolding. Therefore, future investigations into pharmacological activators of chaperone protein expression could be directed to identify modest but chronic activators of HSF1.
A third approach to directly activate HSF1 could involve designing a multivalent molecule that binds to three independent HSF1 monomers, bringing them into close proximity such that the probability of trimer formation is substantially enhanced. Without a crystal structure of HSF1 it is difficult to predict where such a molecule would interact with the HSF1 monomer. Furthermore, it is only speculative that bringing multiple HSF1 monomers into closer proximity would promote spontaneous homotrimerization. However, as HSF1 is thought to exist in an equilibrium between the monomeric and trimeric state157 — even in the absence of cellular stress — simply increasing the local concentration of HSF1 monomers could stimulate trimer formation.
Future perspectives
The identification of disease-modifying therapies for neurodegenerative diseases has been challenging, yet the market for novel treatments is enormous158. Although many molecules that activate HSF1 have been identified, the majority are thought to act by causing cellular stress, and a direct pharmacological activator of HSF1 has not yet been reported. The discovery of such a class of molecules would be a major accomplishment for the field. We envision the ideal activator of HSF1 as a small molecule that directly binds to HSF1 and promotes its transition from a monomer to a homotrimer. As the ideal HSF1 activator would not cause proteotoxic stress, we propose that such a molecule would stimulate modest but chronic expression of chaperone proteins, resulting in the amelioration of neurodegenerative disease phenotypes. Below, we outline major hurdles from the discovery to the clinical application of HSF1 activators.
Screening direct HSF1 activators
An immediate limitation in the identification of the ideal HSF1 activator is the lack of a high-quality screen. Current screens using HSF1 reporter genes as readouts of HSF1 activation are susceptible to the identification of molecules that cause proteotoxic stress or inhibit the function of HSP90. As such, novel screens are required that are not sensitive to proteotoxic molecules and HSP90 inhibitors. Our laboratory has developed a pharmacological screen using humanized yeast to identify small molecules that can activate human HSF1 in yeast72. Although this screen is not sensitive to molecules that cause proteotoxic stress or directly inhibit HSP90, it does have the limitation of not discriminating between direct and indirect activators of HSF1. We suggest that future research could be directed towards the development of screens to specifically identify direct activators of HSF1.
Timing of pharmacological intervention
The status of protein misfolding throughout the course of the disease will have to be considered in the development of therapeutics that enhance protein-folding capacity via HSF1 activation (FIG. 5). The progressive nature of neurodegeneration complicates treatment, as a substantial amount of neuronal damage is often present at the time of clinical diagnosis, making the prospect of prophylactic treatment very appealing159-161. Early intervention is crucial, as pharmacological activation of HSF1 will ideally slow neuronal degeneration and delay disease onset, and may completely arrest disease progression if the recently discovered disaggregase activity found in mammalian cells is elevated in an HSF1-dependent manner162. It will be important to consider other treatments such as autophagy inducers that degrade mature protein aggregates, which could be more effective in late stages of the disease. Moreover, the clinical use of HSF1 therapeutics would be complicated by difficulties in the identification of individuals who are susceptible to late-onset and/or idiopathic forms of neurodegeneration, thus emphasizing the need for increased investigation into the identification of biomarkers that are reliable early predictors of disease158,160,161,163.
Figure 5. Importance of early intervention in neurodegenerative diseases.

The progressive nature of neurodegenerative diseases complicates treatment because neuronal damage and protein misfolding are already present at the stage of symptom onset (blue line). Early intervention with heat shock transcription factor 1 (HSF1) activators that promote chaperone protein expression will ameliorate protein misfolding, slow disease progression and delay the onset of symptoms (green line). In an ideal situation, HSF1 activators could be combined with currently available symptomatic treatments at the stage of symptom onset to further enhance disease management. By contrast, HSF1-based therapeutic strategies are unlikely to be as effective at a late stage owing to the severity of neuronal loss and protein-misfolding pathology that is unlikely to be reversed (pink line). In late-stage neurodegenerative disease, alternative or combination therapies — such as inducers of autophagy coupled with chaperone protein-based therapies — may be more effective.
As HSF1 activators would probably be used as a prophylactic treatment, it is important to consider the potential benefits of coupling HSF1 activators with currently available symptomatic treatments. This type of combination therapy would address the underlying protein-misfolding pathology and provide symptomatic relief. If HSF1 activators delay the onset of disease, the use of current treatments when symptoms manifest might substantially enhance the long-term clinical management of neurodegenerative diseases.
Blood–brain barrier
A major bottleneck in the development of therapeutics for central nervous system disorders is the identification of molecules that have the ability to cross the blood–brain barrier158,164,165. Size, lipophilicity and other physicochemical properties of small molecules generally determine their ability to cross the blood–brain barrier (for a detailed review of the ideal characteristics for penetrance, see REF. 166). Activators of HSF1 — such as arimoclomol and riluzole — have been shown to penetrate the blood–brain barrier, yet penetrance of direct HSF1 activators is difficult to predict as such a class of molecules does not exist167,168. Effective surrogate screening technologies for blood–brain barrier penetrance are not widely available; such a model could serve as a powerful secondary screen for direct HSF1 activators and substantially streamline their development164. Another useful approach in addressing the limitations of the blood–brain barrier is the use of molecular libraries of compounds that are known to cross the blood–brain barrier, such as the CNS-Set available from ChemBridge. Using such libraries in novel screens that identify HSF1 activators would help to bypass the limitations of the blood–brain barrier in the development of therapeutics for neuro degenerative diseases.
Potential limitations of HSF1 activators
HSF1 activation may have negative effects in some situations that may need to be addressed for its development as a therapeutic strategy for neurodegenerative diseases. Lindquist and colleagues40 have shown that HSF1 significantly contributes to tumour development in the p53R172H mouse model of carcinogenesis. Potential oncogenic side effects will therefore have to be considered in the development of HSF1 therapies, especially in populations harbouring genetic susceptibility to various cancers. Furthermore, it will be important to consider certain disease states in which the expression of cytosolic chaperone proteins will be ineffective. Extracellular amyloid-β aggregates present in Alzheimer’s disease are particularly neurotoxic and inaccessible to chaperone proteins169,170. Situations such as this may be more amenable to chemical chaperone-based therapies171, whereas diseases that are thought to be caused by intracellular oligomers, such as polyQ disorders or Parkinson’s disease, may be more effectively treated with HSF1 activators.
Studies in mouse models have provided some evidence for the differential efficacy of HSF1-activating molecules in the treatment of various neurodegenerative diseases. For example, treatment of the SOD1G93A mouse model of ALS with arimoclomol resulted in significant amelioration of disease phenotypes with little to no loss in efficacy125. However, in separate studies treatment of the R6/2 mouse model of Huntington’s disease with an HSP90 inhibitor also resulted in the amelioration of disease phenotypes but these benefits waned with disease progression172.
Aged cells appear to have a high threshold for HSF1 activation41,173, and this must be taken into consideration in the development of therapies based on HSF1 activation. Aged neurons have lower steady-state levels of HSF1 and SIRT1, a positive regulator of HSF1 activity; this is likely to contribute to diminished stress responsiveness78. The therapeutic implications of this phenomenon remain unclear, and a greater understanding is needed of why aged cells have an impaired chaperone expression response. If it is not possible to effectively activate HSF1 in aged neurons, treatment strategies targeting this pathway may be less effective. By contrast, aged neurons may respond to pharmacological intervention that lowers the threshold of HSF1 activation and is independent of proteotoxic stress.
In summary, pharmacological activation of HSF1 is a promising avenue for therapeutic intervention in neurodegenerative diseases. A concerted effort to understand HSF1 biology and the pharmacological activation of chaperone protein expression will ultimately reveal whether this strategy will be successful.
Acknowledgements
We thank T. Nevitt for her critical comments on the manuscript. This work was supported in part by the US National Institutes of Health (NIH) National Research Service Award Postdoctoral Fellowship GM076954 (to D.W.N.) and the NIH grant R01-GM059911 (to D.J.T.). A.M.J. is a trainee of the Duke University Pharmacological Sciences Training Program.
Glossary
- Dyskinesia
A condition in which voluntary movement is lost and an increase in chorea-like involuntary movement is observed.
- Leucine zipper
A structural motif that stabilizes inter- or intramolecular protein–protein interactions via hydrophobic and charged interactions across coiled-coils and is commonly found in oligomerization domains.
- Sumoylation
A post-translational modification that is indicated by the addition of a small ubiquitin-like modifier (SUMO) moiety that can affect protein stability, localization and activity.
- Residence time
The duration of time that heat shock transcription factor 1 is bound to heat shock elements in the promoter region of target genes such as those encoding chaperone proteins.
- Chaperone-mediated autophagy
A process by which cytosolic proteins are selectively degraded through interaction with heat shock cognate protein 70, which facilitates direct translocation into lysosomes for proteolysis.
- Unfolded protein response
A conserved physiological response involving endoplasmic reticulum (ER)-initiated signaltransduction events, induced by accumulation of unfolded proteins in the lumen of the ER.
- SOD1G93A mice
Transgenic mice expressing the G93A mutant form of human superoxide dismutase 1 (SOD1) that causes familial amyotrophic lateral sclerosis (ALS), which are commonly used as a model for ALS.
- RNA aptamer
A specifically designed oligonucleotide with a secondary structure that elicits high affinity for a desired target.
- p53R172H mouse model
A mouse model expressing a mutated form of the tumour suppressor protein p53, R172H, which results in increased oncogenesis.
- R6/2 mouse model
A widely used transgenic mouse model – expressing exon 1 of the human huntingtin gene containing 150 CAG repeats – that rapidly develops Huntington’s disease-like symptoms.
Footnotes
Competing interests statement
The authors declare competing financial interests: see Web version for details.
References
- 1.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nature Med. 2004;10:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 2.Zhang QC, et al. A compact β model of huntingtin toxicity. J. Biol. Chem. 2011;286:8188–8196. doi: 10.1074/jbc.M110.192013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nature Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 4.Muchowski PJ. Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron. 2002;35:9–12. doi: 10.1016/s0896-6273(02)00761-4. [DOI] [PubMed] [Google Scholar]
- 5.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 6.Fiumara F, Fioriti L, Kandel ER, Hendrickson WA. Essential role of coiled coils for aggregation and activity of Q/N-rich prions and polyQ proteins. Cell. 2010;143:1121–1135. doi: 10.1016/j.cell.2010.11.042. This study demonstrated that coiled-coil motifs in polyQ proteins contribute to the aggregation and cytotoxicity of these proteins.
- 7.Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nature Rev. Neurosci. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jankovic J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatr. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- 9.Buschert V, Bokde ALW, Hampel H. Cognitive intervention in Alzheimer disease. Nature Rev. Neurol. 2010;6:508–517. doi: 10.1038/nrneurol.2010.113. [DOI] [PubMed] [Google Scholar]
- 10.Carter MD, Simms GA, Weaver DF. The development of new therapeutics for Alzheimer’s disease. Clin. Pharmacol. Ther. 2010;88:475–486. doi: 10.1038/clpt.2010.165. [DOI] [PubMed] [Google Scholar]
- 11.Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 12.Verity NC, Mallucci GR. Rescuing neurons in prion disease. Biochem. J. 2010;433:19–29. doi: 10.1042/BJ20101323. [DOI] [PubMed] [Google Scholar]
- 13.Walker FO. Huntington’s disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 14.Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 15.Chai Y, Koppenhafer SL, Bonini NM, Paulson HL. Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J. Neurosci. 1999;19:10338–10347. doi: 10.1523/JNEUROSCI.19-23-10338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warrick JM, et al. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nature Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 17.Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum. Mol. Genet. 2000;9:2811–2820. doi: 10.1093/hmg/9.19.2811. This study showed that HSP40 and HSP70 synergize to ameliorate the cytotoxicity of polyQ proteins in fruitfly disease models by modulating the solubility of these proteins.
- 18.Auluck PK, Bonini NM. Pharmacological prevention of Parkinson disease in Drosophila. Nature Med. 2002;8:1185–1186. doi: 10.1038/nm1102-1185. This paper showed that pharmacological activation of HSF1 via the HSP90 inhibitor geldanamycin can ameliorate disease phenotypes in a fruitfly model of Parkinson’s disease.
- 19.Auluck P, Meulener M, Bonini N. Mechanisms of suppression of α-synuclein neurotoxicity by geldanamycin in Drosophila. J. Biol. Chem. 2005;280:2873–2878. doi: 10.1074/jbc.M412106200. [DOI] [PubMed] [Google Scholar]
- 20.Alavez S, Vantipalli MC, Zucker DJ, Klang IM, Lithgow GJ. Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature. 2011;472:226–229. doi: 10.1038/nature09873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl Acad. Sci. USA. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. This study describes a widespread failure in protein folding that occurs in early adulthood and coincides with reduced activation of HSF1 and chaperone protein expression in C. elegans.
- 22.Fonte V, et al. Interaction of intracellular β amyloid peptide with chaperone proteins. Proc. Natl Acad. Sci. USA. 2002;99:9439–9444. doi: 10.1073/pnas.152313999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satyal SH, et al. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA. 2000;97:5750–5755. doi: 10.1073/pnas.100107297. This study shows that the expression of polyQ proteins in C. elegans disrupts general protein folding, causes aggregation of otherwise soluble proteins and constitutively promotes the activation of HSF1 and chaperone proteins.
- 24.Teixeira-Castro A, et al. Neuron-specific proteotoxicity of mutant ataxin-3 in C. elegans: rescue by the DAF-16 and HSF-1 pathways. Hum. Mol. Genet. 2011;20:2996–3009. doi: 10.1093/hmg/ddr203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, et al. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009;5:e1000350. doi: 10.1371/journal.pgen.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanneau D, de Thonel A, Maurel S, Didelot C, Garrido C. Apoptosis versus cell differentiation: role of heat shock proteins HSP90, HSP70 and HSP27. Prion. 2007;1:53–60. doi: 10.4161/pri.1.1.4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batulan Z, et al. High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J. Neurosci. 2003;23:5789–5798. doi: 10.1523/JNEUROSCI.23-13-05789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonelli MA, et al. Attenuated expression of 70-kDa heat shock protein in WI-38 human fibroblasts during aging in vitro. Exp. Cell Res. 1999;252:20–32. doi: 10.1006/excr.1999.4614. [DOI] [PubMed] [Google Scholar]
- 29.Gutsmann-Conrad A, Heydari AR, You S, Richardson A. The expression of heat shock protein 70 decreases with cellular senescence in vitro and in cells derived from young and old human subjects. Exp. Cell Res. 1998;241:404–413. doi: 10.1006/excr.1998.4069. [DOI] [PubMed] [Google Scholar]
- 30.Gutsmann-Conrad A, Pahlavani MA, Heydari AR, Richardson A. Expression of heat shock protein 70 decreases with age in hepatocytes and splenocytes from female rats. Mech. Ageing Dev. 1999;107:255–270. doi: 10.1016/s0047-6374(98)00132-8. [DOI] [PubMed] [Google Scholar]
- 31.Fargnoli J, Kunisada T, Fornace AJ, Schneider EL, Holbrook NJ. Decreased expression of heat shock protein 70 mRNA and protein after heat treatment in cells of aged rats. Proc. Natl Acad. Sci. USA. 1990;87:846–850. doi: 10.1073/pnas.87.2.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fawcett TW, Sylvester SL, Sarge KD, Morimoto RI, Holbrook NJ. Effects of neurohormonal stress and aging on the activation of mammalian heat shock factor 1. J. Biol. Chem. 1994;269:32272–32278. [PubMed] [Google Scholar]
- 33.Pahlavani MA, Harris MD, Moore SA, Weindruch R, Richardson A. The expression of heat shock protein 70 decreases with age in lymphocytes from rats and rhesus monkeys. Exp. Cell Res. 1995;218:310–318. doi: 10.1006/excr.1995.1160. [DOI] [PubMed] [Google Scholar]
- 34.Bailey CK, Andriola IFM, Kampinga HH, Merry DE. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2002;11:515–523. doi: 10.1093/hmg/11.5.515. [DOI] [PubMed] [Google Scholar]
- 35.Fujimoto M, et al. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J. Biol. Chem. 2005;280:34908–34916. doi: 10.1074/jbc.M506288200. This study demonstrates that the expression of a constitutively active HSF1 allele ameliorates pathogenic phenotypes in a mouse model of Huntington’s disease.
- 36.Muchowski PJ, et al. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl Acad. Sci. USA. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nature Struct. Mol. Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- 38.Wyttenbach A, et al. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington’s disease. Proc. Natl Acad. Sci. USA. 2000;97:2898–2903. doi: 10.1073/pnas.97.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feder JH, Rossi JM, Solomon J, Solomon N, Lindquist S. The consequences of expressing hsp70 in Drosophila cells at normal temperatures. Genes Dev. 1992;6:1402–1413. doi: 10.1101/gad.6.8.1402. [DOI] [PubMed] [Google Scholar]
- 40.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nature Rev. Mol. Cell Biol. 2010;11:545–555. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonsalves SE, Moses AM, Razak Z, Robert F, Westwood JT. Whole-genome analysis reveals that active heat shock factor binding sites are mostly associated with non-heat shock genes in Drosophila melanogaster. PLoS ONE. 2011;6:e15934. doi: 10.1371/journal.pone.0015934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hahn J-S, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trinklein ND, Murray JI, Hartman SJ, Botstein D, Myers RM. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol. Biol. Cell. 2004;15:1254–1261. doi: 10.1091/mbc.E03-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostling P, Björk JK, Roos-Mattjus P, Mezger V, Sistonen L. Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J. Biol. Chem. 2007;282:7077–7086. doi: 10.1074/jbc.M607556200. [DOI] [PubMed] [Google Scholar]
- 46.Sandqvist A, et al. Heterotrimerization of heat-shock factors 1 and 2 provides a transcriptional switch in response to distinct stimuli. Mol. Biol. Cell. 2009;20:1340–1347. doi: 10.1091/mbc.E08-08-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shinkawa T, et al. Heat shock factor 2 is required for maintaining proteostasis against febrile range thermal stress and polyglutamine aggregation. Mol. Biol. Cell. 2011;22:3571–3583. doi: 10.1091/mbc.E11-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abravaya K, Myers MP, Murphy SP, Morimoto RI. The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev. 1992;6:1153–1164. doi: 10.1101/gad.6.7.1153. [DOI] [PubMed] [Google Scholar]
- 49.Ali A, Bharadwaj S, O’Carroll R, Ovsenek N. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol. Cell Biol. 1998;18:4949–4960. doi: 10.1128/mcb.18.9.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bharadwaj S, Ali A, Ovsenek N. Multiple components of the HSP90 chaperone complex function in regulation of heat shock factor 1 in vivo. Mol. Cell Biol. 1999;19:8033–8041. doi: 10.1128/mcb.19.12.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conde R, Xavier J, McLoughlin C, Chinkers M, Ovsenek N. Protein phosphatase 5 is a negative modulator of heat shock factor 1. J. Biol. Chem. 2005;280:28989–28996. doi: 10.1074/jbc.M503594200. [DOI] [PubMed] [Google Scholar]
- 52.Guo Y, et al. Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J. Biol. Chem. 2001;276:45791–45799. doi: 10.1074/jbc.M105931200. [DOI] [PubMed] [Google Scholar]
- 53.Shi Y, Mosser DD, Morimoto RI. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998;12:654–666. doi: 10.1101/gad.12.5.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 55.Arlander SJH, et al. Chaperoning checkpoint kinase 1 (Chk1), an Hsp90 client, with purified chaperones. J. Biol. Chem. 2006;281:2989–2998. doi: 10.1074/jbc.M508687200. [DOI] [PubMed] [Google Scholar]
- 56.Hernández MP, Chadli A, Toft DO. HSP40 binding is the first step in the HSP90 chaperoning pathway for the progesterone receptor. J. Biol. Chem. 2002;277:11873–11881. doi: 10.1074/jbc.M111445200. [DOI] [PubMed] [Google Scholar]
- 57.King FW, Wawrzynow A, Höhfeld J, Zylicz M. Co-chaperones Bag-1, Hop and Hsp40 regulate Hsc70 and Hsp90 interactions with wild-type or mutant p53. EMBO J. 2001;20:6297–6305. doi: 10.1093/emboj/20.22.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carmichael J, Sugars KL, Bao YP, Rubinsztein DC. Glycogen synthase kinase-3β inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J. Biol. Chem. 2002;277:33791–33798. doi: 10.1074/jbc.M204861200. [DOI] [PubMed] [Google Scholar]
- 59.Chu B, Soncin F, Price BD, Stevenson MA, Calderwood SK. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J. Biol. Chem. 1996;271:30847–30857. doi: 10.1074/jbc.271.48.30847. [DOI] [PubMed] [Google Scholar]
- 60.Chu B, Zhong R, Soncin F, Stevenson MA, Calderwood SK. Transcriptional activity of heat shock factor 1 at 37 °C is repressed through phosphorylation on two distinct serine residues by glycogen synthase kinase 3 and protein kinases Cα and Cζ. J. Biol. Chem. 1998;273:18640–18646. doi: 10.1074/jbc.273.29.18640. [DOI] [PubMed] [Google Scholar]
- 61.Hietakangas V, et al. Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol. Cell Biol. 2003;23:2953–2968. doi: 10.1128/MCB.23.8.2953-2968.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kline MP, Morimoto RI. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol. Cell Biol. 1997;17:2107–2115. doi: 10.1128/mcb.17.4.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knauf U, Newton EM, Kyriakis J, Kingston RE. Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev. 1996;10:2782–2793. doi: 10.1101/gad.10.21.2782. [DOI] [PubMed] [Google Scholar]
- 64.Murshid A, et al. Protein kinase A binds and activates heat shock factor 1. PLoS ONE. 2010;5:e13830. doi: 10.1371/journal.pone.0013830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang X, et al. Phosphorylation of HSF1 by MAPK-activated protein kinase 2 on serine 121, inhibits transcriptional activity and promotes HSP90 binding. J. Biol. Chem. 2006;281:782–791. doi: 10.1074/jbc.M505822200. [DOI] [PubMed] [Google Scholar]
- 66.Pelham HR. A regulatory upstream promoter element in the Drosophila Hsp 70 heat-shock gene. Cell. 1982;30:517–528. doi: 10.1016/0092-8674(82)90249-5. [DOI] [PubMed] [Google Scholar]
- 67.Pelham HR, Bienz M. A synthetic heat-shock promoter element confers heat-inducibility on the herpes simplex virus thymidine kinase gene. EMBO J. 1982;1:1473–1477. doi: 10.1002/j.1460-2075.1982.tb01340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perisic O, Xiao H, Lis JT. Stable binding of Drosophila heat shock factor to head-to-head and tail-to-tail repeats of a conserved 5 bp recognition unit. Cell. 1989;59:797–806. doi: 10.1016/0092-8674(89)90603-x. [DOI] [PubMed] [Google Scholar]
- 69.Clos J, et al. Molecular cloning and expression of a hexameric Drosophila heat shock factor subject to negative regulation. Cell. 1990;63:1085–1097. doi: 10.1016/0092-8674(90)90511-c. [DOI] [PubMed] [Google Scholar]
- 70.Sorger PK, Nelson HC. Trimerization of a yeast transcriptional activator via a coiled-coil motif. Cell. 1989;59:807–813. doi: 10.1016/0092-8674(89)90604-1. [DOI] [PubMed] [Google Scholar]
- 71.Ahn S-G, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17:516–528. doi: 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neef DW, Turski ML, Thiele DJ. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010;8:e1000291. doi: 10.1371/journal.pbio.1000291. In this study the authors generated a humanized HSF1-based yeast screen to identify HSF1A, a novel pharmacological activator of HSF1 that is efficacious in ameliorating polyQ protein-associated protein aggregation and cytotoxicity in cell culture and fruitfly disease models.
- 73.Trott A, et al. Activation of heat shock and antioxidant responses by the natural product celastrol: transcriptional signatures of a thiol-targeted molecule. Mol. Biol. Cell. 2008;19:1104–1112. doi: 10.1091/mbc.E07-10-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rabindran SK, Haroun RI, Clos J, Wisniewski J, Wu C. Regulation of heat shock factor trimer formation: role of a conserved leucine zipper. Science. 1993;259:230–234. doi: 10.1126/science.8421783. [DOI] [PubMed] [Google Scholar]
- 75.Guettouche T, Boellmann F, Lane WS, Voellmy R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005;6:4. doi: 10.1186/1471-2091-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Holmberg CI, et al. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J. 2001;20:3800–3810. doi: 10.1093/emboj/20.14.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim S-A, Yoon J-H, Lee S-H, Ahn S-G. Polo-like kinase 1 phosphorylates heat shock transcription factor 1 and mediates its nuclear translocation during heat stress. J. Biol. Chem. 2005;280:12653–12657. doi: 10.1074/jbc.M411908200. [DOI] [PubMed] [Google Scholar]
- 78.Westerheide SD, Anckar J, Stevens SM, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. This study demonstrated that the DNA binding activity of HSF1 is inhibited by acetylation within the DNA binding domain, and HSF1 is maintained in a deacetylated state via SIRT1.
- 79.Yang J, Bridges K, Chen KY, Liu AY-C. Riluzole increases the amount of latent HSF1 for an amplified heat shock response and cytoprotection. PLoS ONE. 2008;3:e2864. doi: 10.1371/journal.pone.0002864. This work reported that riluzole, which is a treatment for ALS, promotes an increase in steady-state HSF1 levels potentially via the inhibition of chaperone-mediated autophagy.
- 80.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nature Rev. Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dickey CA, et al. HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB J. 2006;20:753–755. doi: 10.1096/fj.05-5343fje. [DOI] [PubMed] [Google Scholar]
- 82.Dickey CA, et al. Development of a high throughput drug screening assay for the detection of changes in tau levels — proof of concept with HSP90 inhibitors. Curr. Alzheimer Res. 2005;2:231–238. doi: 10.2174/1567205053585927. [DOI] [PubMed] [Google Scholar]
- 83.Dou F, et al. Chaperones increase association of tau protein with microtubules. Proc. Natl Acad. Sci. USA. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Petrucelli L, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- 85.Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN. Heat shock prevents α-synuclein-induced apoptosis in a yeast model of Parkinson’s disease. J. Mol. Biol. 2005;351:1081–1100. doi: 10.1016/j.jmb.2005.06.060. [DOI] [PubMed] [Google Scholar]
- 86.Shen H-Y, He J-C, Wang Y, Huang Q-Y, Chen J-F. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J. Biol. Chem. 2005;280:39962–39969. doi: 10.1074/jbc.M505524200. [DOI] [PubMed] [Google Scholar]
- 87.Agrawal N, et al. Identification of combinatorial drug regimens for treatment of Huntington’s disease using Drosophila. Proc. Natl Acad. Sci. USA. 2005;102:3777–3781. doi: 10.1073/pnas.0500055102. This study demonstrated that the HSP90 inhibitor geldanamycin and the histone deacetylase inhibitor suberoylanilide hydroxamic acid have combinatorial efficacy in ameliorating cytotoxicity in a fruitfly model of neurodegenerative disease.
- 88.Fujikake N, et al. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 2008;283:26188–26197. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hay DG, et al. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum. Mol. Genet. 2004;13:1389–1405. doi: 10.1093/hmg/ddh144. [DOI] [PubMed] [Google Scholar]
- 90.Sittler A, et al. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum. Mol. Genet. 2001;10:1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 91.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J. Biol. Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 92.Yu XM, et al. Hsp90 inhibitors identified from a library of novobiocin analogues. J. Am. Chem. Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 93.Ansar S, et al. A non-toxic Hsp90 inhibitor protects neurons from Aβ-induced toxicity. Bioorg. Med. Chem. Lett. 2007;17:1984–1990. doi: 10.1016/j.bmcl.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 94.Kimura H, et al. ITZ-1, a client-selective Hsp90 inhibitor, efficiently induces heat shock factor 1 activation. Chem. Biol. 2010;17:18–27. doi: 10.1016/j.chembiol.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 95.Salehi AH, et al. AEG3482 is an antiapoptotic compound that inhibits Jun kinase activity and cell death through induced expression of heat shock protein 70. Chem. Biol. 2006;13:213–223. doi: 10.1016/j.chembiol.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 96.Schnaider T, Somogyi J, Csermely P, Szamel M. The Hsp90-specific inhibitor, geldanamycin, blocks CD28-mediated activation of human T lymphocytes. Life Sci. 1998;63:949–954. doi: 10.1016/s0024-3205(98)00352-x. [DOI] [PubMed] [Google Scholar]
- 97.Westerheide S, et al. Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- 98.Hieronymus H, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 99.Zhang T, et al. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 2008;7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 100.Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–4765. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- 101.Kiaei M, et al. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neurodegener. Dis. 2005;2:246–254. doi: 10.1159/000090364. [DOI] [PubMed] [Google Scholar]
- 102.Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2001;25:1341–1357. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- 103.Wang J, Gines S, MacDonald ME, Gusella JF. Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci. 2005;6:1. doi: 10.1186/1471-2202-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang Y-Q, Sarge KD. Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J. Mol. Med. 2007;85:1421–1428. doi: 10.1007/s00109-007-0251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cleren C, Calingasan NY, Chen J, Beal MF. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem. 2005;94:995–1004. doi: 10.1111/j.1471-4159.2005.03253.x. [DOI] [PubMed] [Google Scholar]
- 106.Faust K, et al. Neuroprotective effects of compounds with antioxidant and anti-inflammatory properties in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2009;10:109. doi: 10.1186/1471-2202-10-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hansen J, Bross P. A cellular viability assay to monitor drug toxicity. Methods Mol. Biol. 2010;648:303–311. doi: 10.1007/978-1-60761-756-3_21. [DOI] [PubMed] [Google Scholar]
- 108.Kalmar B, Greensmith L. Activation of the heat shock response in a primary cellular model of motoneuron neurodegeneration — evidence for neuroprotective and neurotoxic effects. Cell. Mol. Biol. Lett. 2009;14:319–335. doi: 10.2478/s11658-009-0002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang S, Liu K, Wang X, He Q, Chen X. Toxic effects of celastrol on embryonic development of zebrafish (Danio rerio) Drug Chem. Toxicol. 2011;34:61–65. doi: 10.3109/01480545.2010.494664. [DOI] [PubMed] [Google Scholar]
- 110.Ohtsuka K, Kawashima D, Gu Y, Saito K. Inducers and co-inducers of molecular chaperones. Int. J. Hyperthermia. 2005;21:703–711. doi: 10.1080/02656730500384248. [DOI] [PubMed] [Google Scholar]
- 111.Hirakawa T, Rokutan K, Nikawa T, Kishi K. Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology. 1996;111:345–357. doi: 10.1053/gast.1996.v111.pm8690199. [DOI] [PubMed] [Google Scholar]
- 112.Katsuno M, et al. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc. Natl Acad. Sci. USA. 2005;102:16801–16806. doi: 10.1073/pnas.0506249102. This work demonstrated that pharmacological activation of HSF1 via geranylgeranylacetone promotes the activation of chaperone protein expression and ameliorates cytotoxicity in a mouse model of spinal and bulbar muscular atrophy.
- 113.Otaka M, et al. The induction mechanism of the molecular chaperone HSP70 in the gastric mucosa by geranylgeranylacetone (HSP-inducer) Biochem. Biophys. Res. Commun. 2007;353:399–404. doi: 10.1016/j.bbrc.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 114.Patury S, Miyata Y, Gestwicki JE. Pharmacological targeting of the Hsp70 chaperone. Curr. Top. Med. Chem. 2009;9:1337–1351. doi: 10.2174/156802609789895674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hirota K, et al. Geranylgeranylacetone enhances expression of thioredoxin and suppresses ethanol-induced cytotoxicity in cultured hepatocytes. Biochem. Biophys. Res. Commun. 2000;275:825–830. doi: 10.1006/bbrc.2000.3392. [DOI] [PubMed] [Google Scholar]
- 116.Okada S, et al. Geranylgeranylacetone induces apoptosis in HL-60 cells. Cell Struct. Funct. 1999;24:161–168. doi: 10.1247/csf.24.161. [DOI] [PubMed] [Google Scholar]
- 117.Endo S, et al. Geranylgeranylacetone, an inducer of the 70-kDa heat shock protein (HSP70), elicits unfolded protein response and coordinates cellular fate independently of HSP70. Mol. Pharmacol. 2007;72:1337–1348. doi: 10.1124/mol.107.039164. [DOI] [PubMed] [Google Scholar]
- 118.Tam S, Geller R, Spiess C, Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nature Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. This study shows that the TRIC cytosolic chaperone complex binds to the pathogenic huntingtin protein and reduces huntingtin-mediated cytotoxicity.
- 119.Tam S, et al. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nature Struct. Mol. Biol. 2009;16:1279–1285. doi: 10.1038/nsmb.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hargitai J, et al. Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem. Biophys. Res. Commun. 2003;307:689–695. doi: 10.1016/s0006-291x(03)01254-3. [DOI] [PubMed] [Google Scholar]
- 121.Vígh L, et al. Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nature Med. 1997;3:1150–1154. doi: 10.1038/nm1097-1150. [DOI] [PubMed] [Google Scholar]
- 122.Török Z, et al. Heat shock protein coinducers with no effect on protein denaturation specifically modulate the membrane lipid phase. Proc. Natl Acad. Sci. USA. 2003;100:3131–3136. doi: 10.1073/pnas.0438003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nánási PP, Jednákovits A. Multilateral in vivo and in vitro protective effects of the novel heat shock protein coinducer, bimoclomol: results of preclinical studies. Cardiovasc. Drug Rev. 2001;19:133–151. doi: 10.1111/j.1527-3466.2001.tb00060.x. [DOI] [PubMed] [Google Scholar]
- 124.Kalmar B, et al. Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1G93A mouse model of ALS. J. Neurochem. 2008;107:339–350. doi: 10.1111/j.1471-4159.2008.05595.x. [DOI] [PubMed] [Google Scholar]
- 125.Kieran D, et al. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nature Med. 2004;10:402–405. doi: 10.1038/nm1021. This study demonstrates that pharmacological activation of HSF1 via arimoclomol ameliorates pathogenic phenotypes and extends lifespan in a mouse model of ALS.
- 126.Lanka V, Wieland S, Barber J, Cudkowicz M. Arimoclomol: a potential therapy under development for ALS. Expert Opin. Investig. Drugs. 2009;18:1907–1918. doi: 10.1517/13543780903357486. [DOI] [PubMed] [Google Scholar]
- 127.Liu AYC, et al. Neuroprotective drug riluzole amplifies the heat shock factor 1 (HSF1)- and glutamate transporter 1 (GLT1)-dependent cytoprotective mechanisms for neuronal survival. J. Biol. Chem. 2011;286:2785–2794. doi: 10.1074/jbc.M110.158220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jurivich DA, Sistonen L, Kroes RA, Morimoto RI. Effect of sodium salicylate on the human heat shock response. Science. 1992;255:1243–1245. doi: 10.1126/science.1546322. [DOI] [PubMed] [Google Scholar]
- 129.Lee BS, Chen J, Angelidis C, Jurivich DA, Morimoto RI. Pharmacological modulation of heat shock factor 1 by antiinflammatory drugs results in protection against stress-induced cellular damage. Proc. Natl Acad. Sci. USA. 1995;92:7207–7211. doi: 10.1073/pnas.92.16.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Winegarden NA, Wong KS, Sopta M, Westwood JT. Sodium salicylate decreases intracellular ATP, induces both heat shock factor binding and chromosomal puffing, but does not induce hsp 70 gene transcription in Drosophila. J. Biol. Chem. 1996;271:26971–26980. doi: 10.1074/jbc.271.43.26971. [DOI] [PubMed] [Google Scholar]
- 131.Housby JN, et al. Non-steroidal anti-inflammatory drugs inhibit the expression of cytokines and induce HSP70 in human monocytes. Cytokine. 1999;11:347–358. doi: 10.1006/cyto.1998.0437. [DOI] [PubMed] [Google Scholar]
- 132.Palayoor ST, Youmell MY, Calderwood SK, Coleman CN, Price BD. Constitutive activation of IκB kinase α and NF-κB in prostate cancer cells is inhibited by ibuprofen. Oncogene. 1999;18:7389–7394. doi: 10.1038/sj.onc.1203160. [DOI] [PubMed] [Google Scholar]
- 133.Stevenson MA, Zhao MJ, Asea A, Coleman CN, Calderwood SK. Salicylic acid and aspirin inhibit the activity of RSK2 kinase and repress RSK2-dependent transcription of cyclic AMP response element binding protein- and NF-κ B-responsive genes. J. Immunol. 1999;163:5608–5616. [PubMed] [Google Scholar]
- 134.Ishihara K, Yamagishi N, Hatayama T. Suppression of heat- and polyglutamine-induced cytotoxicity by nonsteroidal anti-inflammatory drugs. Eur. J. Biochem. 2004;271:4552–4558. doi: 10.1111/j.1432-1033.2004.04419.x. [DOI] [PubMed] [Google Scholar]
- 135.Ianaro A, et al. Anti-inflammatory activity of 15-deoxy-δ12,14-PGJ2 and 2-cyclopenten-1-one: role of the heat shock response. Mol. Pharmacol. 2003;64:85–93. doi: 10.1124/mol.64.1.85. [DOI] [PubMed] [Google Scholar]
- 136.Rossi A, Elia G, Santoro MG. 2-cyclopenten-1-one, a new inducer of heat shock protein 70 with antiviral activity. J. Biol. Chem. 1996;271:32192–32196. doi: 10.1074/jbc.271.50.32192. [DOI] [PubMed] [Google Scholar]
- 137.Zhou Y, et al. Chloro-oxime derivatives as novel small molecule chaperone amplifiers. Bioorg. Med. Chem. Lett. 2009;19:3128–3135. doi: 10.1016/j.bmcl.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 138.Zhou Y, et al. Pyrimido[5,4-e][1,2,4]triazine-5,7 (1H,6H)-dione derivatives as novel small molecule chaperone amplifiers. Bioorg. Med. Chem. Lett. 2009;19:4303–4307. doi: 10.1016/j.bmcl.2009.05.073. [DOI] [PubMed] [Google Scholar]
- 139.Zhang B, et al. Identification of small-molecule HSF1 amplifiers by high content screening in protection of cells from stress induced injury. Biochem. Biophys. Res. Commun. 2009;390:925–930. doi: 10.1016/j.bbrc.2009.10.079. [DOI] [PubMed] [Google Scholar]
- 140.Hayashida N, et al. Heat shock factor 1 ameliorates proteotoxicity in cooperation with the transcription factor NFAT. EMBO J. 2010;29:3459–3469. doi: 10.1038/emboj.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Batista-Nascimento L, Neef DW, Liu PCC, Rodrigues-Pousada C, Thiele DJ. Deciphering human heat shock transcription factor 1 regulation via post-translational modification in yeast. PLoS ONE. 2011;6:e15976. doi: 10.1371/journal.pone.0015976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rimoldi M, Servadio A, Zimarino V. Analysis of heat shock transcription factor for suppression of polyglutamine toxicity. Brain Res. Bull. 2001;56:353–362. doi: 10.1016/s0361-9230(01)00602-5. This study shows that constitutively active HSF1, via loss of repressive phosphorylation events, prevents protein aggregation in cell culture models of polyglutamine disease.
- 143.Banerjee Mustafi S, Chakraborty PK, Raha S. Modulation of Akt and ERK1/2 pathways by resveratrol in chronic myelogenous leukemia (CML) cells results in the downregulation of Hsp70. PLoS ONE. 2010;5:e8719. doi: 10.1371/journal.pone.0008719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Khaleque MA, et al. Induction of heat shock proteins by heregulin β1 leads to protection from apoptosis and anchorage-independent growth. Oncogene. 2005;24:6564–6573. doi: 10.1038/sj.onc.1208798. [DOI] [PubMed] [Google Scholar]
- 145.Xavier I, et al. Glycogen synthase kinase 3β negatively regulates both DNA-binding and transcriptional activities of heat shock factor 1. J. Biol. Chem. 2000;275:29147–29152. doi: 10.1074/jbc.M002169200. [DOI] [PubMed] [Google Scholar]
- 146.Anckar J, et al. Inhibition of DNA binding by differential sumoylation of heat shock factors. Mol. Cell Biol. 2006;26:955–964. doi: 10.1128/MCB.26.3.955-964.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell. 2002;108:345–356. doi: 10.1016/s0092-8674(02)00630-x. [DOI] [PubMed] [Google Scholar]
- 148.Brunet Simioni M, et al. Heat shock protein 27 is involved in SUMO-2/3 modification of heat shock factor 1 and thereby modulates the transcription factor activity. Oncogene. 2009;28:3332–3344. doi: 10.1038/onc.2009.188. [DOI] [PubMed] [Google Scholar]
- 149.Fukuda I, et al. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009;16:133–140. doi: 10.1016/j.chembiol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 150.Parker JA, et al. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nature Genet. 2005;37:349–350. doi: 10.1038/ng1534. This study shows that activation of SIR-2 (the C. elegans homolog of SIRT1) via resveratrol rescues neuronal dysfunction in C. elegans and mouse models of polyQ disease.
- 151.Kim D, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ladiwala ARA, et al. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J. Biol. Chem. 2010;285:24228–24237. doi: 10.1074/jbc.M110.133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Marambaud P, Zhao H, Davies P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-β peptides. J. Biol. Chem. 2005;280:37377–37382. doi: 10.1074/jbc.M508246200. [DOI] [PubMed] [Google Scholar]
- 154.Lu K-T, et al. Neuroprotective effects of resveratrol on MPTP-induced neuron loss mediated by free radical scavenging. J. Agric. Food Chem. 2008;56:6910–6913. doi: 10.1021/jf8007212. [DOI] [PubMed] [Google Scholar]
- 155.Zhang F, et al. Resveratrol protects dopamine neurons against lipopolysaccharide-induced neurotoxicity through its anti-inflammatory actions. Mol. Pharmacol. 2010;78:466–477. doi: 10.1124/mol.110.064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Salamanca HH, Fuda N, Shi H, Lis JT. An RNA aptamer perturbs heat shock transcription factor activity in Drosophila melanogaster. Nucleic Acids Res. 2011;39:6729–6740. doi: 10.1093/nar/gkr206. This work describes an RNA aptamer that interacts with the DNA binding domain of HSF1 and inhibits its binding to promoter heat shock elements.
- 157.Liu PC, Thiele DJ. Modulation of human heat shock factor trimerization by the linker domain. J. Biol. Chem. 1999;274:17219–17225. doi: 10.1074/jbc.274.24.17219. [DOI] [PubMed] [Google Scholar]
- 158.Finkbeiner S. Bridging the Valley of Death of therapeutics for neurodegeneration. Nature Med. 2010;16:1227–1232. doi: 10.1038/nm.2222. [DOI] [PubMed] [Google Scholar]
- 159.Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature Rev. Drug Discov. 2010;9:237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- 160.Hampel H, et al. Biomarkers for Alzheimer’s disease: academic, industry and regulatory perspectives. Nature Rev. Drug Discov. 2010;9:560–574. doi: 10.1038/nrd3115. [DOI] [PubMed] [Google Scholar]
- 161.Schapira AHV. Challenges to the development of disease-modifying therapies in Parkinson’s disease. Eur. J. Neurol. 2011;18(Suppl. 1):16–21. doi: 10.1111/j.1468-1331.2010.03324.x. [DOI] [PubMed] [Google Scholar]
- 162.Murray AN, Solomon JP, Wang YJ, Balch WE, Kelly JW. Discovery and characterization of a mammalian amyloid disaggregation activity. Protein Sci. 2010;19:836–846. doi: 10.1002/pro.363. This work describes the discovery of a mammalian disaggregase with the ability to disaggregate β-amyloid aggregates.
- 163.Opar A. Hope builds for earlier detection of Alzheimer’s disease. Nature Rev. Drug Discov. 2010;9:579–581. doi: 10.1038/nrd3237. [DOI] [PubMed] [Google Scholar]
- 164.Nielsen PA, Andersson O, Hansen SH, Simonsen KB, Andersson G. Models for predicting blood–brain barrier permeation. Drug Discov. Today. 2011;16:472–475. doi: 10.1016/j.drudis.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 165.Pardridge WM. Alzheimer’s disease drug development and the problem of the blood-brain barrier. Alzheimers Dement. 2009;5:427–432. doi: 10.1016/j.jalz.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 167.Cudkowicz ME, et al. Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve. 2008;38:837–844. doi: 10.1002/mus.21059. [DOI] [PubMed] [Google Scholar]
- 168.Milane A, et al. Brain and plasma riluzole pharmacokinetics: effect of minocycline combination. J. Pharm. Pharm. Sci. 2009;12:209–217. doi: 10.18433/j36c78. [DOI] [PubMed] [Google Scholar]
- 169.Kumar S, et al. Extracellular phosphorylation of the amyloid β-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J. 2011;30:2255–2265. doi: 10.1038/emboj.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Williams TL, Serpell LC. Membrane and surface interactions of the Alzheimer’s Aβ peptide: insights into the mechanism of cytotoxicity. FEBS J. 2011;278:3905–3917. doi: 10.1111/j.1742-4658.2011.08228.x. [DOI] [PubMed] [Google Scholar]
- 171.Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426:905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 172.Labbadia J, et al. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J. Clin. Invest. 2011;121:3306–3319. doi: 10.1172/JCI57413. This study demonstrates that activation of HSF1-dependent chaperone protein expression via an HSP90 inhibitor transiently ameliorates disease phenotypes in a mouse model of polyQ-based disease as a result of decreased promoter acetylation.
- 173.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 174.Biamonte MA, et al. Heat shock protein 90: inhibitors in clinical trials. J. Med. Chem. 2010;53:3–17. doi: 10.1021/jm9004708. [DOI] [PubMed] [Google Scholar]
- 175.Lancet JE, et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia. 2010;24:699–705. doi: 10.1038/leu.2009.292. [DOI] [PubMed] [Google Scholar]
- 176.Nowakowski GS, et al. A phase I trial of twice-weekly 17-allylamino-demethoxy-geldanamycin in patients with advanced cancer. Clin. Cancer Res. 2006;12:6087–6093. doi: 10.1158/1078-0432.CCR-06-1015. [DOI] [PubMed] [Google Scholar]
- 177.Brandt GEL, Schmidt MD, Prisinzano TE, Blagg BSJ. Gedunin, a novel Hsp90 inhibitor: semisynthesis of derivatives and preliminary structure–activity relationships. J. Med. Chem. 2008;51:6495–6502. doi: 10.1021/jm8007486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kikuchi T, et al. Cytotoxic and apoptosis-inducing activities of limonoids from the seeds of Azadirachta indica (neem) J. Nat. Prod. 2011;74:866–870. doi: 10.1021/np100783k. [DOI] [PubMed] [Google Scholar]
- 179.Traynor BJ, et al. Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology. 2006;67:20–27. doi: 10.1212/01.wnl.0000223353.34006.54. [DOI] [PubMed] [Google Scholar]
- 180.Bensimon G, et al. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain. 2009;132:156–171. doi: 10.1093/brain/awn291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nanke Y, et al. Geranylgeranylacetone, a non-toxic inducer of heat shock protein, induces cell death in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Mod. Rheumatol. 2009;19:379–383. doi: 10.1007/s10165-009-0183-z. [DOI] [PubMed] [Google Scholar]
- 182.Nishida T, et al. Geranylgeranylacetone protects against acetaminophen-induced hepatotoxicity by inducing heat shock protein 70. Toxicology. 2006;219:187–196. doi: 10.1016/j.tox.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 183.Shirakabe H, et al. Clinical evaluation of teprenone, a mucosal protective agent, in the treatment of patients with gastric ulcers: a nationwide, multicenter clinical study. Clin. Ther. 1995;17:924–935. doi: 10.1016/0149-2918(95)80070-0. [DOI] [PubMed] [Google Scholar]