

Abstract
A detailed account of the enantioselective total synthesis of (−)-lasonolide A is described. Our initial route to the top tetrahydropyran ring synthesis involved an Evans asymmetric alkylation as the key step. In itially, we rely upon a diastereoselective alkylation of an α-alkoxyacetimide derivative containing an α’-stereogenic center. In this context, we investigated asymmetric alkylation of α-alkoxy acetimide derivatives containing an adjacent stereocenter. While alkylation proceeded in good yield, the lack of diastereoselectivity prompted us to explore alternative routes. Our subsequent successful synthetic strategies involved highly diastereoselective cycloaddition routes to both tetrahydropyran rings of lasonolide A. The top tetrahydropyran ring was constructed stereoselectively by an intramolecular 1,3-dipolar cycloaddition reaction. The overall process constructed a bicyclic isoxazoline which was later unraveled to a functionalized tetrahydropyran ring as well as a quaternary stereocenter present in the molecule. The lower tetrahydropyran ring was assembled by Jacobsen’s catalytic asymmetric hetero-Diels-Alder reaction as the key step. The synthesis also featured a Lewis acid catalyzed opening of epoxide to form substituted ether stereoselectively.
Keywords: total synthesis, lasonolide A, macrocycle, antitumor agent, cycloaddition
Introduction
Lasonolide A (1), a structurally unique 20-membered macrolide, was isolated from the Caribbean marine sponge, Forcepia sp., by McConnell and co-workers in 1994.[1] Lasonolide A exhibited potent cytotoxic activity against proliferation of A-549 human lung carcinoma and P388 murine leukemia cells. It has shown cell adhesion in the EL-4.IL-2 cell line that detects signal transduction agents.[1] Thus far, lasonolide A’s biological mechanism of action remained unknown due to its scarce natural abundance. The proposed structure of lasonolide A was initially determined through extensive NMR studies by McConnell and co-workers.[1] During total synthesis and biological studies of lasonolide A, Lee and co-workers, revised the geometry of two double bonds and absolute stereochemistry of lasonolide.[2] The most prominent structural features of lasonolide A are a 20-membered macrolactone which contains two highly substituted tetrahydropyran units bearing a total of eight stereogenic centers and five disubstituted and trisubstituted double bonds as part of the macrolide backbone.
The novel structural features and promising antitumor activities of lasonolide A prompted significant interest in synthesis and biological studies. Since the first total synthesis by Lee and co-workers,[2] a number of other total syntheses[3-5] and synthetic studies on both tetrahydropyran rings[6] have been reported in the literature. A number of diverse strategies and methodologies have been developed toward the synthesis of lasonolide A, especially for building the two highly substituted tetrahydropyran rings. Recently, we reported a convergent and enantioselective synthesis of (−)-lasonolide A.[5] Herein, we report the details of our synthetic efforts that have led to the convergent total synthesis of (−)-lasonolide A. The synthesis involved a Lewis acid catalyzed hetero Diels-Alder reaction to construct the lower tetrahydropyran ring and an intramolecular 1,3-dipolar cycloaddition reaction to assemble the upper tetrahydropyran ring. Other key reactions include a Lewis acid-catalyzed opening of epoxide to form a substituted ether stereoselectively, an efficient cross metathesis reaction to construct the functionalized olefins and an intramolecular Horner-Emmons reaction to form the macrolactone.
Results and Discussion
Preliminary strategy for two core tetrahydropyrans
Our initial retrosynthetic analysis of lasonolide A is outlined in Figure 1. Strategic disconnection of lasonolide at C25-C26 resulted a phosphonium salt 2 and the Horner-Emmons substrate 3 for the 20-membered macrolide core of lasonolide A. Further disconnection of phosphonoacetate 3 provided functionalized tetrahydropyran rings 4, 6 and tin-derivative 5. We planned to carry out a Stille coupling of 5 and 6 followed by a Julia-Kocienski olefination to construct the Horner-Emmons precursor 3. Functionalized tetrahydropyran ring 4 was planned to be derived from reduction of isoxazoline 7. This isoxazoline derivative would be derived from acid 8 through an intramolecular [3+2] nitrile oxide cycloaddition reaction. Both double bonds in tetrahydropyran ring 6 would be installed from precursor 9 by using Wittig reaction. The tetrahydropyran ring in 9 can be constructed by a Lewis acid-catalyzed hetero Diels-Alder reaction of silyloxy diene 10 and an appropriately protected aldehyde 11.
Figure 1.
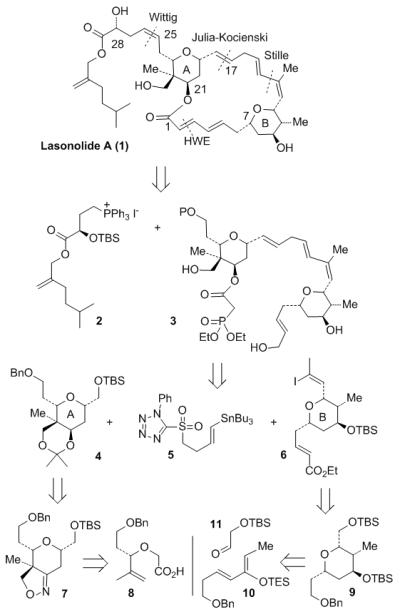
Initial retrosynthetic analysis of lasonolide A (1).
Initial synthetic route to the top tetrahydropyran ring A
The synthesis of A ring began with addition of isopropenylmagnesium bromide to aldehyde 12 to form racemic allylic alcohol rac-13 in 94% yield as shown in Scheme 1. Optically active alcohol (−)-13 was prepared from kinetic resolution of racemic mixture rac-13 by means of Sharpless asymmetric epoxidation.[7] Alkylation of alcohol (−)-13 with bromoacetic acid and NaH provided acid 8 in poor yield. However, using KH as a base gave excellent yield. The carboxylic acid 8 was converted to its mixed pivalic anhydride and the resulting anhydride was treated with different lithio oxazolidinones to give the corresponding N-acyl-oxazolidinones (14-17) in 60% yield (2 steps).
Scheme 1.
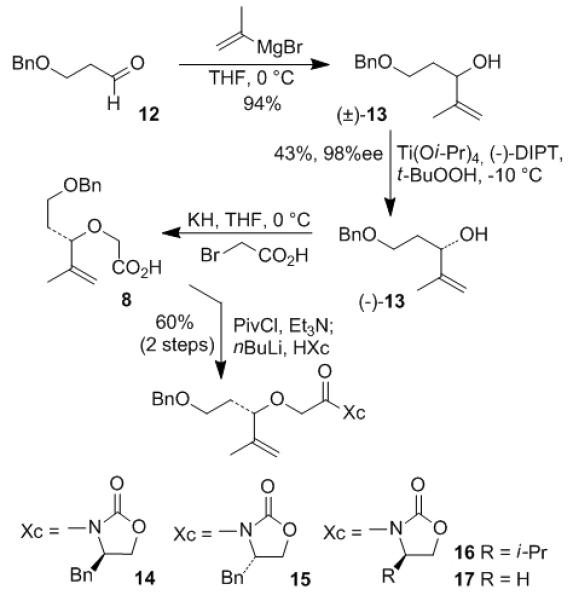
Preparation of oxazolidinoes (14-17).
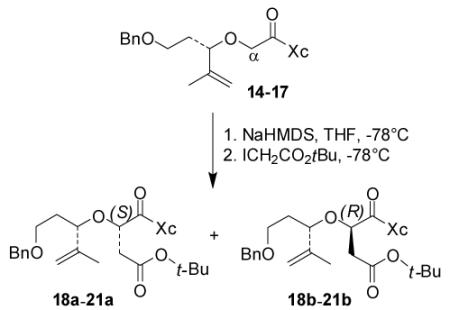
We planned to use the auxiliaries in 14-17 for introduction of an α-stereocenter by diastereoselective alkylation reaction developed by Evans.[8] After our extensive investigation, we chose tert-butyl iodoacetate as the alkylating agent. Sodium bis(trimethylsilyl)amide turned out to be superior to its lithium or potassium counterparts as the base. While asymmetric alkylation proceeded with good to excellent yield, the observed diastereoselectivity was far from satisfactory for our synthesis. Influence of substrate structure on the selectivity was shown in Table 1. In entry 1, alkylation of acyl (R)-benzyloxazolidinone 14 provided the expected diastereomer 18a as the minor product in 20% yield. The undesired 18b was the major product in 44% yield. The (S)-configuration of the newly generated α-stereocenter in 18a was determined by conversion of 18a to isoxazoline 7 following similar reactions as shown in Scheme 2. The presence of α-alkoxy group as well as the α’-stereogenic center may be responsible for the poor observed diastereoselectivity in this asymmetric alkylation reaction.[9] We hoped that changing the configuration of the auxiliary from R to S may provide the desired alkylation product as the major product. Unfortunately, alkylation of 15 gave the similar result as in 14. As shown in entry 2, asymmetric alkylation of 15 afforded desired isomer 19a in 24% yield and undesired isomer 19b in 40% isolated yield. By replacing the (R)-benzyl group with (R)-isopropyl group in the auxiliary (entry 3), the products ratio in the alkylation of 16 was improved to 1:1.1 (20a : 20b). For comparison, we carried out alkylation with an achiral oxazolidinone 17 (entry 4). The alkylation was selective (21a : 21b; 1:3.1), however, the minor isomer 21a was our desired isomer. The stereochemistry of compounds 19a/b-21a/b was confirmed after reductive removal of oxazolidinone with LiBH4 followed by comparison with the reduction of 18a/b. Changing the protecting group from benzyl to TIPS did not improve the selectivity. Treatment of 16 with NaHMDS at −78 °C followed by addition of tert-butyl iodoacetate at the same temperature afforded the desired product 20a in 40% yield along with its diastereomer 20b in 44% yield (entry 3). The diastereomers were separated and we elected to carry out subsequent steps with 20a at this point.
Table 1.
Alkylation of acyl oxazolidinones.
| Entry | Starting Material |
Desired Product (yield) |
Undesired Product (yield) |
Ratio a:b |
|---|---|---|---|---|
| 1 | 14 | 18a(20%) | 18b(44%) | 1:2.2 |
| 2 | 15 | 19a(24%) | 19b(40%) | 1:1.6 |
| 3 | 16 | 20a(40%) | 20b(44%) | 1:1.1 |
| 4 | 17 | 21a(18%) | 21b(57%) | 1:3.1 |
Scheme 2.
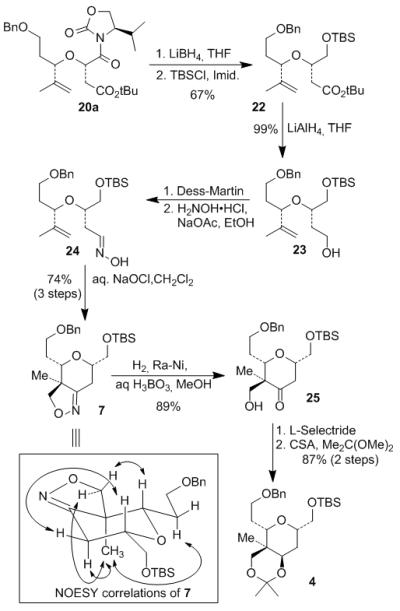
Preparation of top tetrahydropyran 4.
As outlined in Scheme 2, selective reduction of N-acyl-oxazolidinone 20a using LiBH4 afforded hydroxy ester, which was protected with TBSCl and imidazole to provide TBS-ether 22. The tert-butyl ester was reduced to the corresponding alcohol 23 by LiAlH4 in near quantitative yield. Oxidation of 23 with Dess-Martin periodinane[10] followed by treatment of the resulting aldehyde with hydroxylamine and sodium acetate in ethanol furnished the oxime 24. Exposure of 24 to sodium hypochlorite led to facile intramolecular 1,3-dipolar cycloaddition via the nitrile oxide to afford isoxazoline 7 as a single diastereomer.[11] The tetrahydropyran ring as well as the quaternary stereocenter at C22 were constructed efficiently in the cycloaddition process. Raney-nickel catalyzed hydrogenolysis of isoxazoline 7 provided β-hydroxy ketone 25 in 89% yield.[12] L-Selectride reduction of the ketone gave the corresponding alcohol with excellent diastereoselectivity (9:1 dr, axial/equatorial). The resulting diol was protected as acetonide 4 in the following step. The stereochemical outcome of the key cycloaddition process was confirmed by NOESY correlation of 7 as shown.
Initial approach to bottom tetrahydropyran
We initially planned to construct the Z,E-conjugated double bonds at C12-C15 via a palladium-mediated Stille coupling[13] between E-vinylstannane 5 and Z-trisubstituted alkenyl iodide 6. Preparation of fragment 5 is shown in Scheme 3. Because direct hydrostannation of the terminal alkyne 26 exhibited low regioselectivity, we used catalytic hydrostannation of 1-bromo-1-alkyne to obtain the regio- and stereodefined E-vinylstannane.[14] Thus, 3-butyn-1-ol 26 was converted to acetylenic sulfone 28 via Mitsunobu substitution[15] and selective molybdate oxidation.[16] Bromination of 28 with NBS in the presence of silver acetate provided acetylenic bromide,[17] which was converted to vinylstannane 5 as an E/Z mixture (10:1) by treatment with tributyltin hydride and catalytic amounts of Pd(PPh3)4 and PPh3.
Scheme 3.
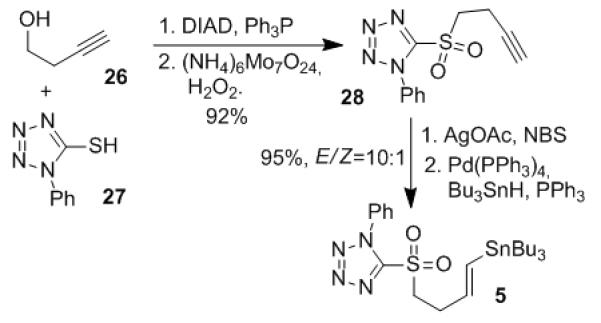
Preparation of vinylstannane 5.
Synthesis of bottom tetrahydropyran started from preparation of nucleophilic diene 10 as outlined in Scheme 4. Protection of alcohol 26 with BnBr and NaH gave the benzyl ether in quantitative yield. Deprotonation of the alkyne with nBuLi followed by addition of propionaldehyde afforded the propargylic alcohol 29 in 86% yield. Reduction of the triple bond with LiAlH4 in THF provided the E-allylic alcohol in 93% yield. Following PCC oxidation produced the α, β-unsaturated ketone 30. The enone was converted to the Diels-Alder precursor dienol silyl ether 10 by treatment with Et3N and TESOTf. The chiral tridentate Schiff base chromium (III) complex (1S, 2R)-31 developed by Jacobsen[18] was used as catalyst (10 mol%) in the asymmetric hetero-Diels-Alder reaction between diene 10 and (tert-butyldimethylsilyloxy)acetaldehyde 11. The resulting dihydropyran silyl enol ether was treated with TBAF/AcOH in the same pot to remove the TES group and give the corresponding ketone 32 in 71% yield and 94% ee. Reduction of the ketone with Dibal-H gave axial alcohol 33 and equatorial alcohol 34 as a 1:2 separable mixture in 96% combined yield. The other reducing agents we tried, including L-Selectride, NaBH4, LiAlH4, Red-Al, LiEt3BH, SmI2 and BH3, afforded the undesired equatorial hydroxy pyran 34 as the predominant product. Therefore, we recycled 34 back to 33 using Swern oxidation followed by reduction and the overall conversion yield from 32 to 33 was 53% after one cycle.
Scheme 4.
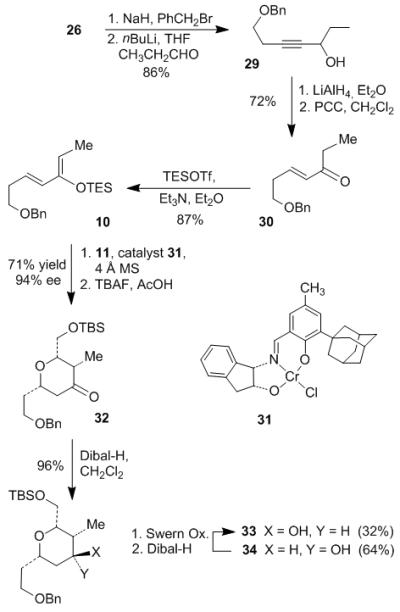
Preparation of bottom tetrahydropyran 33.
As depicted in Scheme 5, protection of the secondary alcohol 33 with TBSOTf and following debenzylation under Pd/H2 gave primary alcohol 35 in 97% yield. Spectral data of alcohol 35 was identical with that of the same intermediate in Lee’s synthesis.[2] This confirmed the stereochemical outcome of our asymmetric hetero-Diels-Alder reaction. Alcohol 35 was oxidized to the corresponding aldehyde under Swern condition and then treated with (carbethoxymethylene)triphenylphosphorane to furnish the E-unsaturated ester 36 in 84% yield over 2 steps. Selective removal of the primary TBS protecting group in 36 using CSA in MeOH gave the corresponding primary alcohol, which was subsequently oxidized to its corresponding aldehyde 37. The Stork-Zhao Wittig reaction[19] converted the aldehyde to the trisubstituted vinyl iodide by condensation with α-iodoethylidene triphenylphosphorane. This protocol typically provides the Z-vinyl iodide as the major product and has been utilized in a number of total syntheses.[20] However, in our case, the Wittig reaction between aldehyde 37 and α-iodoethylidene triphenylphosphorane produced a mixture of Z (6) and E (38) isomers with 1:2.6 ratio. The desired Z-vinyl iodide 6 was obtained as a minor isomer in 12% yield. The Z/E stereochemistry in 6 and 38 was determined by comparison of the chemical shifts of the vinyl proton. The shift of Ha in 6 is shown to be 5.60 ppm and the shift of Ha in 38 is shown to be 6.19 ppm. Because the vinyl hydrogen cis to iodine should appear in the lower field than the hydrogen trans to iodine,[21] we concluded that 6 is the Z isomer and 38 is the E isomer. The stereochemistry was also confirmed by the NOESY correlation analysis of the two isomers. There was interaction between the protons on the methyl group and the vinyl proton Ha in 6 and this proved their cis relationship. On the contrary, no such correlation was found in 38.
Scheme 5.
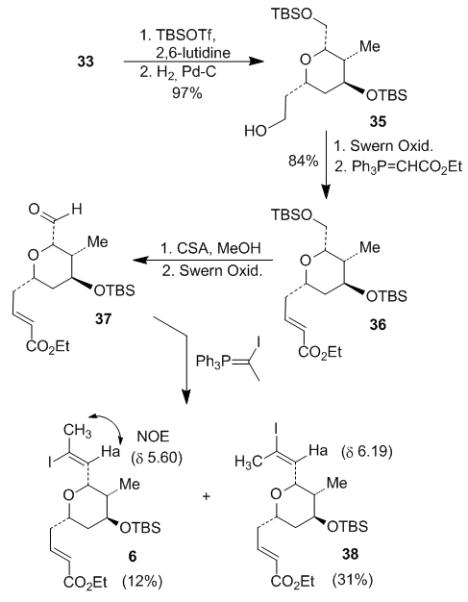
Preparation of vinyl iodide 6.
Though fragments 4 and 6 could be obtained in optically pure form, the yield and selectivity for the crucial auxiliary-directed alkylation in fragment 4 and installing Z-vinyl iodide in fragment 6 was far from satisfactory for our total synthesis. Our attempts to convert the undesired isomer to the desired one were fruitless. It appeared necessary for us to modify our synthetic strategy in order to improve selectivity and efficiency for both tetrahydropyrans of lasonolide A.
New strategy for two core tetrahydropyrans
The second generation retrosynthetic analysis is illustrated in Figure 2. Disconnection of lasonolide A (1) at C25-C26 would give side chain fragment phosphonium salt 2 and the 20-membered macrolide core 39 containing tetrahydropyrans A and B. Further disassembly of macrolactone 39 would lead to sulfone 40 and aldehyde 41. Construction of the macrocycle can be achieved by Julia-Kocienski[22] coupling between 40 and 41 at C14-C15 and subsequent intramolecular Horner–Wadsworth–Emmons (HWE) reaction[23] at C2-C3. The E-double bond in fragment 40 was planned to be installed by a cross metathesis process[24] and the 6-membered ring A would be prepared through an intramolecular [3+2] 1,3-dipolar cycloaddition from ether 42. The E-olefin in fragment 41 can also be connected by cross metathesis and the Z-double bond would be set up through HWE reaction with Ando’s modification[25] from the same precursor 9 used in our previous approach.
Figure 2.
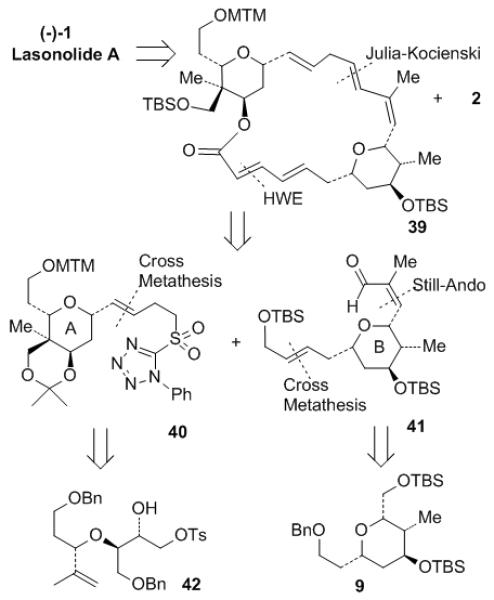
Modified retrosynthetic analysis of lasonolide A (1).
Second generation synthesis of top tetrahydropyran
Construction of top ring A started from the known epoxide 43[7] as shown in Scheme 6. Tosylation of alcohol 43 gave tosylate 44. The epoxide was regioselectively opened by alcohol (−)-13 in the presence of catalytic BF3•OEt2 to provide ether 42 according to the Hoffmann protocol.[26] Epoxidation of hydroxy tosylate 42 with K2CO3 afforded epoxide 45, which was heated with aqueous HClO4 in DMSO to give diol 46. Oxidative cleavage of diol 46 by NaIO4 followed by condensation of the corresponding aldehyde with nitromethane and KF afforded nitro alcohol 47 as a diastereomeric mixture. The mixture was converted into nitroalkene 48 with MsCl and Et3N. The resulting nitroalkene was reduced to oxime 49 by using Zn and AcOH. Intramolecular [3+2] cycloaddition of 49 as described in Scheme 2 afforded isoxazoline 50 as a single diastereomer.
Scheme 6.
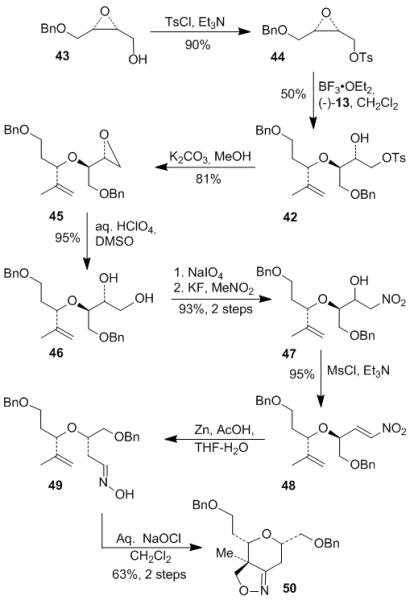
Preparation of isoxazoline 50 via [3+2] cycloaddition.
Raney-nickel catalyzed hydrogenolysis of isoxazoline 50 provided β-hydroxy ketone 51 as shown in Scheme 7. L-Selectride reduction of the ketone gave the corresponding axial alcohol as a single diastereomer. The resulting diol was protected as acetonide 52 in 87% yield in two steps. Differentiation of the two primary benzyl groups in 52 by palladium-catalyzed hydrogenolysis or hydrogen transfer was unsuccessful. Lipase catalyzed selective esterification was also fruitless. Thus, both benzyl groups were removed to provide the corresponding diol, which was then protected as bis-TBS-ether 53. Treatment of 53 with 1.2 equivalent TBAF provided desired alcohol 54 in 40% yield along with recovered 53 (32%) and the corresponding diol (24%) which could be converted back to 53. Alcohol 54 was obtained in 62% yield after one recycle. Dess-Martin oxidation of 54 followed by Wittig reaction afforded the terminal olefin, which was treated with TBAF to give alcohol 55. Cross metathesis between olefin 55 and sulfone 57 in the presence of Grubbs II catalyst[27] provided trans-olefin 58 in 81% yield (E/Z > 8:1). Due to the poor solubility of the homo-dimer of 57 in CH2Cl2, the cross metathesis required diluted reaction solution (0.02 M) and high catalyst loading (35% mol). Concentrated solution only gave incomplete reaction. Protection of alcohol 58 with benzoyl peroxide and Me2S provided MTM ether 40 in 88% yield.[28] Sulfone 57 was obtained from alcohol 56 via Mitsunobu substitution and selective molybdate oxidation.
Scheme 7.
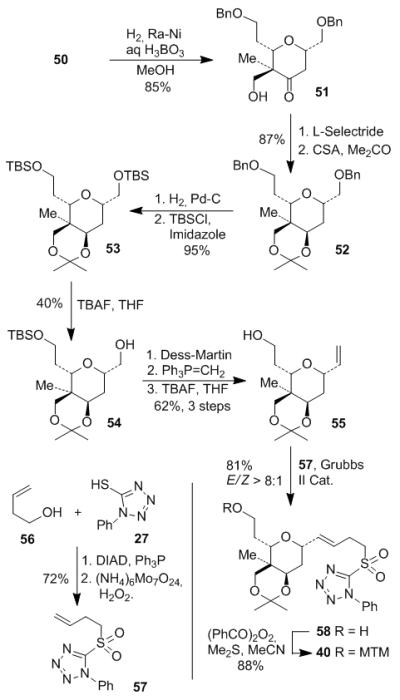
Preparation of sulfone 40 via cross metathesis.
Synthesis of bottom tetrahydropyran
Tetrahydropyran 35 obtained from the hetero-Diels-Alder reaction has now been employed for synthesis of 41 as shown in Scheme 8. Dess-Martin oxidation of alcohol 35 and following Wittig reaction furnished the olefin, of which the primary TBS group was selectively removed with CSA to afford alcohol 59 in 75% yield over 3 steps. Cross metathesis between olefin 59 and bis-TBSO-butene 60 in the presence of Grubbs II catalyst provided olefin 61 in 68% yield (E/Z > 10:1). It was crucial to control the reaction time and catalyst amount. Prolonged reaction time (more than 12 h) or high catalyst loading (more than 10% mol) could cause poor yield and isomerization of the allylic double bond. Alcohol 61 was oxidized to the corresponding aldehyde under Dess-Martin conditions. Horner–Wadsworth–Emmons olefination of the above aldehyde according to Ando’s conditions,[25] with ethyl 2-[di(o-isopropylphenyl)phosphono] propionate 62 provided the trisubstituted Z-olefin 63 in 70% yield for 2 steps. Ester 63 was reduced by Dibal-H to the corresponding alcohol. Following Dess-Martin oxidation gave aldehyde 41 in 91% yield for 2 steps.
Scheme 8.
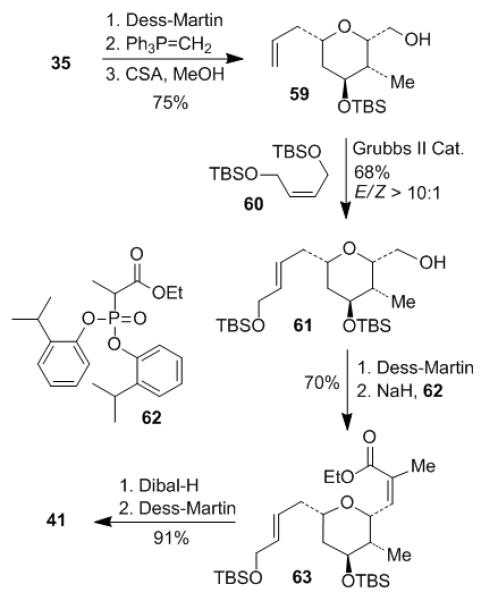
Preparation of aldehyde 41.
Synthesis of phosphonium salt 2
The preparation of phosphonium salt 2 for the lasonolide A side chain is shown in Scheme 9. Phosphonium salt 2 was prepared from esterification of alcohol 64[5] with known acid 65[29]. The resulting ester was converted to bis-TBS-ether 66 by deprotection of the benzylidene group followed by protection of the resulting diol as TBS-ethers. Phosphonium salt 2 was obtained from 66 as described previously.[2]
Scheme 9.
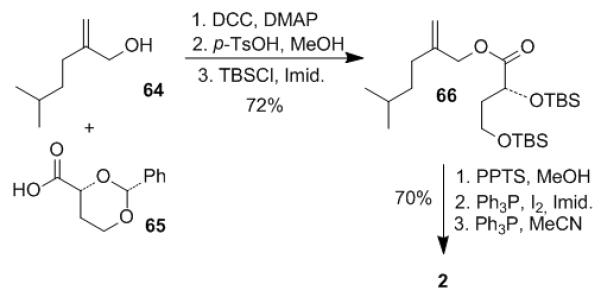
Preparation of phosphonium salt 2.
Synthesis of (−)-lasonolide A: fragments coupling and macrocyclization
With both sulfone 40 and aldehyde 41 in hand, coupling between the two fragments was carried out under Julia-Kocienski conditions[22] using KHMDS as the base, as illustrated in Scheme 10. The resulting tetraene 67 was treated with CSA in MeOH to remove acetonide and primary TBS groups. The two primary hydroxy groups of the resulting triol were then selectively protected with TBSCl to give tris-TBS-ether 68. The free secondary alcohol was reacted with phosphonoacetic acid and DCC and DMAP conditions to provide the corresponding ester. Deprotection of the less hindered allylic primary TBS group by PPTS in MeOH afforded the hydroxy phosphonoacetate 69. Dess-Martin oxidation of alcohol 69 followed by intramolecular HWE olefination[23] furnished macrolactone 39 in 65% yield for 2 steps. Deprotection of MTM ether with HgCl2 in the presence of CaCO3 in aqueous acetonitrile[30] led to the corresponding alcohol, which was oxidized by Dess-Martin periodinane to provide aldehyde 70. Following Wittig olefination with 2-derived phosphorane afforded the TBS-protected lasonolide A with Z-olefin. Final global TBS-deprotection using HF•Py in the presence of excess pyridine furnished (−)-lasonolide A (1, [α]23D −24, c 0.37, CDCl3). The spectroscopic data (1H NMR, 13C NMR, IR, and optical rotation) and HRMS data of synthetic lasonolide A (1) were in agreement with those of the natural product.[1,2]
Scheme 10.
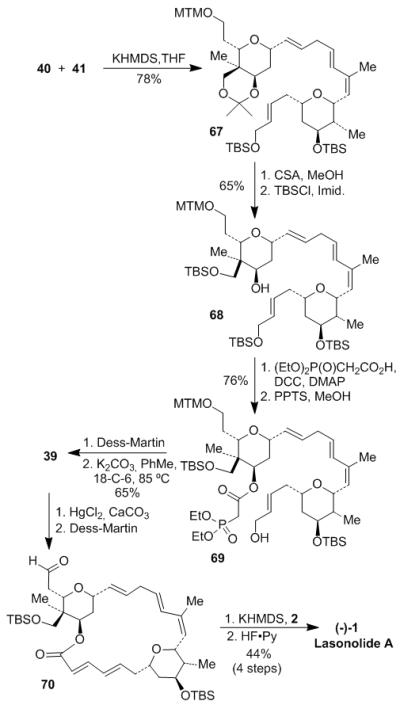
Synthesis of lasonolide A (1).
Conclusion
In summary, we have reported an asymmetric total synthesis of (−)-lasonolide A (1), a potent anticancer agent, with 0.12% overall yield and 32 steps in the longest linear sequence. Our initial approach to the synthesis of highly substituted tetrahydropyran fragments of lasonolide involved an asymmetric alkylation as the key step. Asymmetric alkylation of an α-alkoxy acetimide derivatives bearing an α’-chiral center proceeded with poor stereoselectivity under a variety of reaction conditions. We then devised alternative routes involving a 1,3-dipolar cycloaddition and an asymmetric hetero Diels-Alder reaction for the construction of highly functionalized tetrahydropyran rings of lasonolide A. The top tetrahydropyran ring was synthesized by an intramolecular 1,3-dipolar nitrile oxide cycloaddition to a bicyclic isoxazoline with the construction of quaternary center stereoselectively. The bottom tetrahydropyran ring was assembled by a highly effective Jacobsen catalytic asymmetric hetero Diels-Alder reaction as the key step. The hetero Diels-Alder set three stereocenters in a highly diastereoselective manner. Other key reactions featured in the synthesis include a Lewis acid catalyzed opening of epoxide to form substituted ether stereoselectively, an efficient cross metathesis of functionalized olefins using Grubbs II catalyst, and an intramolecular Horner-Emmons reaction to form the 20-membered macrolide. The synthesis stereoselectively constructred eight of the nine chiral centers of (−)-lasonolide A by asymmetric synthesis. The current synthesis will now pave the way for structure-activity relationship studies and synthesis of lasonolide-derived less complex derivatives as anticancer agents.
Experimental Section
General Methods
All moisture sensitive reactions were carried out under nitrogen or argon atmosphere. Anhydrous solvents were obtained as follows: THF, diethyl ether and benzene, distilled from sodium and benzophenone; dichloromethane, pyridine, triethylamine, and diisopropylethylamine, distilled from CaH2. All other solvents were HPLC grade. Column chromatography was performed with 240-400 mesh silica gel under low pressure of 5-10 psi. TLC was carried out with silica gel 60-F-254 plates visualized under UV light and stained with either phosphomolybdic acid or acidic p-anisaldehyde. 1H NMR spectra were recorded at 300, 400 or 500 MHz with chemical shifts reported in ppm (δ). 13C NMR spectra were recorded at 75, 100 or 125 MHz with chemical shifts reported in ppm (δ). Infrared spectra were recorded as thin films on NaCl plates on a Fourier transform spectrometer. Optical rotations were measured using a sodium (589, D line) lamp polarimeter.
Alcohol 13
To a suspension of NaH (60 wt % in mineral oil, 6.12 g, 153 mmol) in THF (200 mL) was added 3-buten-1-ol (10 g, 138 mmol) at 0 °C. After 30 min, benzyl bromide (18.3 mL, 153 mmol) was added. The mixture was allowed to warm to room temperature overnight and quenched with sat. aq. NH4Cl. The aqueous layer was extracted with EtOAc twice. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (pure hexanes-5% EtOAc/hexanes) afforded benzyl ether (21.4 g, 95%) as a clear oil. A solution of above olefin (20.0 g, 124 mmol) in DCM (240 mL) was cooled to −78 °C. O3 was bubbled through the solution until the blue color persisted for 5 min. O2 and N2 were successively bubbled through the solution for 10 min each to purge the remaining ozone. Ph3P (35.6 g, 136 mmol) was added portionwise at −78 °C and the mixture were allowed to warm to room temperature overnight. Evaporation of the solvent and purification of the residue by column chromatography (DCM) afforded 3-benzyloxypropanal 12 (17.0 g, 85%) as a clear oil. 1H NMR (500 MHz, CDCl3) δ 9.78 (s, 1H), 7.26-7.36 (m, 5H), 4.53 (s, 2H), 3.81 (t, J = 6.0, 2H), 2.68 (t, J = 6.0, 2H). To isopropenylmagnesium bromide (220 mL, 110 mmol, 0.5 M solution in THF) was added a solution of 3-benzyloxypropanal 12 (12.0 g, 73 mmol) in 50 mL THF via cannula at 0 °C. After addition, the reaction was stirred 5 min and quenched with sat. aq. NH4Cl followed by 2N HCl at 0 °C. The aqueous layer was extracted twice with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (15% EtOAc/hexanes) provided racemic allylic alcohol 13 (14.1 g, 94%) as a colorless oil. To a solution of allylic alcohol 13 (14.1 g, 68.4 mmol) and (−)-diisopropyl tartrate (6.13 g, 26.2 mmol) in CH2Cl2 (220 mL) with 4Å moleculer sieves (3.6 g) was added Ti(OiPr)4 (5.18 mL, 17.7 mmol) at −20 °C and allowed to stir for 30 min at −20 °C. The reaction then was treated with a solution of tert-butyl hydroperoxide in decane (16.7 mL, 91.7 mmol, 5.5 M) and stirred at −20 °C for 60 h. The reaction was quenched with an aqueous solution of FeSO4 and citric acid at −20 °C and vigorously stirred at 23 °C for 30 min. The above mixture was filtered through celite and the aqueous phase was extracted twice with CH2Cl2. The combined organic phases were concentrated and stirred 1 h with Et2O and 30% NaOH in brine to hydrolyze the DIPT. After phase separation and extraction, the combined organic phases were washed with brine and dried over anhydrous Na2SO4. Purification by column chromatography (15% EtOAc/hexanes) provided alcohol (−)-13 (6.34 g, 45%, 98% ee) as a colorless oil. The ee was determined by 19F NMR of the (−)-MTPA ester of (−)-13. [α]23D −5.54 (c 3.05, CHCl3); IR (neat) 3429, 1099; 1H NMR (500 MHz, CDCl3) δ 7.28-7.34 (m, 5H), 5.01 (s, 1H), 4.85 (s, 1H), 4.51 (s, 2H), 4.24 (dd, J = 7.0, 5.0 Hz, 1H), 3.67 (ddd, J = 9.5, 6.0, 6.0 Hz, 1H), 3.60 (ddd, J = 9.5, 7.0, 5.5 Hz, 1H), 3.33 (s, 1H, OH), 1.85-1.88 (m, 2H), 1.74 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 147.2, 138.1, 128.5, 127.7, 110.7, 74.1, 73.3, 68.4, 34.8, 18.1; HRMS (ESI) [M+Na]+ calcd for C13H18O2Na 229.1205, found 229.1206.
Acid 8
To a suspension potassium hydride (2.68 g, 20.1 mmol, 30 % dispersion in mineral oil) in THF (20 mL) was added a solution of the alcohol (−)-13 (1.38 g, 6.7 mmol) in THF (20 mL) at 0 °C and stirred at 23 °C for 10 min. The reaction was cooled to 0 °C and a solution of bromoacetic acid (1.02 g, 7.27 mmol) in THF (5 mL) was added dropwise. The reaction was warmed to 23 °C overnight and quenched with H2O (50 mL). The mixture was diluted with 2N NaOH and Et2O and separated. The aqueous layer was acidified to pH 2 with concentrated HCl and extracted with CH2Cl2 four times. The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was purified via column chromatography (50% EtOAc/hexanes) to provide acid 8 (1.59 g, 90 %) as a yellow oil. [α]23D −25 (c 7.0, CHCl3); IR (neat) 3031, 1730, 1115; 1H NMR (500 MHz, CDCl3) δ 10.57 (s, 1H, CO2H), 7.26-7.36 (m, 5H), 4.98 (s, 1H), 4.96 (s, 1H), 4.53 (s, 2H), 4.38-4.64 (AB, JAB = 12.0 Hz, ΔνAB = 79.5 Hz, 2H), 3.95 (dd, 1H, J = 8.5, 4.5 Hz, 1H), 3.67 (ddd, 1H, J = 9.5, 7.0, 5.5 Hz, 1H), 3.58 (ddd, J = 9.5, 6.0, 6.0 Hz, 1H), 2.02 (m, 1H), 1.80 (m, 1H), 1.67 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 174.7, 143.0, 137.9, 128.6, 128.4, 128.1, 128.0, 127.8, 114.9, 82.4, 73.1, 67.1, 65.1, 33.6, 16.5; HRMS (ESI) [M+Na]+ calcd for C15H20O4Na 287.1260, found 287.1258.
General procedure for the preparation of acyl-oxazolidinones (14-17)
To a solution of acid 8 (1.30 g, 5.0 mmol) in THF (10 mL) was added iPr2NEt (1.3 mL, 7.5 mmol) and then PivCl (0.8 mL, 6.5 mmol) at −78 °C. The reaction was warmed to 23 °C and stirred for 3 h. In a separate flask, a solution of (4R)-4-isopropyl-oxazolidin-2-one (900 mg, 7.0 mmol) in THF (15 mL) was cooled to −78 °C and treated with nBuLi (4.4 mL, 7.0 mmol, 1.6 M in hexane). The solution of lithiated oxazolidinone was added to the mixed anhydride, pre-cooled at −78 °C, via cannula. The reaction was warmed to 23 °C for 2 h, quenched with sat. aq. NH4Cl, and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography (20% EtOAc/hexanes) to give 1.38 g (74 %) of 16. [α]23D −76 (c 1.8, CHCl3); IR (neat) 1781, 1719, 1120 cm−1; 1 3 H NMR (500 MHz, CDCl3) δ 7.26-7.34 (m, 5H), 4.94 (m, 2H), 4.55 (AB, JAB = 18.0 Hz, ΔνAB = 66.5 Hz, 2H), 4.51 (AB, JAB = 12.0 Hz, ΔνAB = 16.0 Hz, 2H), 4.42 (dt, J = 8.5, 3.5 Hz, 1H), 4.32 (dd, J = 8.5, 8.5 Hz, 1H), 4.25 (dd, J = 8.5, 3.5 Hz, 1H), 3.96 (dd, J = 8.0, 5.5 Hz, 1H), 3.66 (ddd, J = 9.5, 6.5, 6.5 Hz, 1H), 3.57 (ddd, J = 9.5, 6.5, 6.5 Hz, 1H), 2.42 (m, 1H), 2.05 (m, 1H), 1.84 (m, 1H), 1.68 (s, 3H), 0.91 (d, J = 7.0 Hz, 3H), 0.86 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 170.3, 154.0, 143.5, 138.7, 128.3, 127.7, 127.5, 114.8, 81.9, 73.0, 67.7, 67.0, 64.3, 58.2, 33.9, 28.2, 17.9, 16.7, 14.6; HRMS (ESI) [M+Na]+ calcd for C21H29NO5Na 398.1943, found 398.1949.
14
1H NMR (500 MHz, CDCl3) δ 7.18-7.36 (m, 10H), 4.99 (s, 2H), 4.66 and 4.51 (AB, JAB = 18.0 Hz, ΔνAB = 71.0 Hz, 2H), 4.64 (m, 1H), 4.53 (AB, JAB = 12.0 Hz, ΔνAB = 19.0 Hz, 2H), 4.20 (m, 2H), 4.04 (dd, J = 8.0, 5.0 Hz, 1H), 3.72 (m, 1H), 3.61 (m, 1H), 3.28 and 2.80 (ABX, JAB = 13.5 Hz, JAX = 9.5 Hz, JBX = 3.0 Hz, ΔνAB = 241.5 Hz, 2H), 2.10 (m, 1H), 1.89 (m, 1H), 1.73 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.4, 153.4, 143.6, 138.7, 135.1, 129.5, 129.1, 129.0, 128.4, 127.8, 127.5, 127.4, 114.8, 81.8, 73.0, 67.8, 67.3, 67.0, 54.8, 37.7, 34.0, 16.7.
15
1H NMR (500 MHz, CDCl3) δ 7.19-7.30 (m, 10H), 5.00 (s, 2H), 4.66 (m, 1H), 4.64 and 4.53 (AB, JAB = 18.0 Hz, ΔνAB = 45.0 Hz, 2H), 4.53 (AB, JAB = 12.0 Hz, ΔνAB = 18.5 Hz, 2H), 4.42 (m, 2H), 4.07 (dd, J = 8.0, 5.5 Hz, 1H), 3.71 (m, 1H), 3.61 (m, 1H), 3.28 and 2.82 (ABX, JAB = 13.5 Hz, JAX = 9.5 Hz, JBX = 3.0 Hz, ΔνAB = 231.5 Hz, 2H), 2.09 (m, 1H), 1.88 (m, 1H), 1.72 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.3, 153.4, 143.5, 138.7, 135.1, 129.5, 129.0, 128.6, 128.4, 128.1, 127.7, 127.5, 114.9, 81.6, 73.0, 67.7, 67.2, 67.0, 54.8, 37.7, 33.9, 16.7.
17
1H NMR (300 MHz, CDCl3) δ 7.25-7.33 (m, 5H), 4.94 (s, 2H), 4.40-4.63 (m, 6H), 3.99 (m, 3H), 3.75 (m, 1H), 3.57 (m, 1H), 2.05 (m, 1H), 1.83 (m, 1H), 1.67 (s, 3H).
General procedure of alkylation for the preparation of α-alkylated acyl-oxazolidinones (18a-21a and 18b-21b)
To a solution of acyl oxazolidinone 16 (1.34 g, 3.57 mmol) in THF was added NaHMDS (4.3 mL, 4.3 mmol, 1 M in THF) at −78 °C. After stirring for 1 h at −78 °C, tert-butyl iodoacetate (1.57 g, 6.5 mmol) was added. After 30 min, the reaction was quenched by the addition of saturated NH4Cl and warmed to 23 °C. The aqueous layer was extracted twice with EtOAc. The combined organic layers were washed with brine and dried over Na2SO4. Concentration in vacuo and purification by column chromatography (15%-25% EtOAc/hexanes) provided 20a (698 mg, 40%) along with its diatereomer 20b (767 mg, 44%).
20a
[α]23D −57 (c 3.7, CHCl3); IR (neat) 1781, 1715, 1154 cm−1; 1 3 H NMR (500 MHz, CDCl3) δ 7.26-7.34 (m, 5H), 5.36 (dd, J = 7.1, 5.2 Hz, 1H), 4.93 (s, 1H), 4.88 (s, 1H), 4.47 (AB, JAB = 12.0 Hz, ΔνAB = 25.9 Hz, 2H), 4.38 (dt, J = 5.8, 4.0 Hz, 1H), 4.16 (d, J = 5.8 Hz, 2H), 3.88 (dd, J = 8.2, 5.3 Hz, 1H), 3.62 (ddd, J = 9.3, 6.8, 6.8 Hz, 1H), 3.55 (ddd, J = 9.4, 6.3, 6.3 Hz, 1H), 2.69 (ABX, JAB = 15.6 Hz, JAX = 7.1 Hz, JBX = 5.2 Hz, ΔνAB = 57.3 Hz, 2H), 2.35 (m, 1H), 1.98 (m, 1H), 1.75 (m, 1H), 1.68 (s, 3H), 1.42 (s, 9H), 0.88 (d, J = 7.0 Hz, 3H), 0.85 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 171.6, 168.8, 153.5, 143.9, 138.8, 128.3, 127.5, 127.4, 114.6, 80.9, 80.5, 72.7, 71.5, 67.1, 63.8, 58.4, 39.2, 33.9, 28.1, 28.0, 17.9, 16.5, 14.7; HRMS (ESI) [M+Na]+ calcd for C27H39NO7Na 512.2625, found 512.2644.
20b
1H NMR (500 MHz, CDCl3) δ 7.26-7.34 (m, 5H), 5.44 (dd, J = 8.5, 5.0 Hz, 1H), 4.96 (s, 1H), 4.78 (s, 1H), 4.42 (AB, JAB = 12.0 Hz, ΔνAB = 18.5 Hz, 2H), 4.33 (dt, J = 8.0, 3.0 Hz, 1H), 4.20 (t, J = 9.0 Hz, 1H), 4.14 (dd, J = 9.0, 3.0 Hz, 1H), 4.04 (dd, J = 8.0, 4.5 Hz, 1H), 3.22-3.50 (m, 2H), 2.55 (ABX, JAB = 16.0 Hz, JAX = 8.0 Hz, JBX = 5.0 Hz, ΔνAB = 40.5 Hz, 2H), 2.28 (m, 1H), 1.98 (m, 1H), 1.73 (m, 1H), 1.61 (s, 3H), 1.39 (s, 9H), 0.86 (d, J = 7.0 Hz, 3H), 0.81 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 172.1, 168.9, 153.8, 144.6, 138.5, 128.3, 127.7, 127.5, 115.3, 83.8, 81.0, 73.2, 72.9, 67.0, 63.5, 58.5, 39.2, 33.6, 28.2, 28.0, 18.0, 16.5, 14.5; HRMS (ESI) [M+Na]+ calcd for C27H39NO7Na 512.2625, found 512.2636.
18a
1H NMR (500 MHz, CDCl3) δ 7.19-7.32 (m, 10H), 5.37 (t, J = 6.5 Hz, 1H), 4.94 (s, 1H), 4.91 (s, 1H), 4.62 (m, 1H), 4.52 (AB, JAB = 12.0 Hz, ΔνAB = 16.5 Hz, 2H), 4.12 (dd, J = 9.5, 3.0 Hz, 1H), 4.07 (t, J = 8.0 Hz, 1H), 3.94 (dd, J = 8.0, 5.5 Hz, 1H), 3.65 (m, 1H), 3.56 (m, 1H), 3.32 (dd, J = 13.0, 3.0 Hz, 1H), 2.66-2.81 (m, 3H), 1.98 (m, 1H), 1.78 (m, 1H), 1.69 (s, 3H), 1.44 (s, 9H).
18b
1H NMR (500 MHz, CDCl3) δ 7.22-7.32 (m, 10H), 5.49 (dd, J = 7.5, 5.5 Hz, 1H), 5.06 (s, 1H), 4.87 (s, 1H), 4.63 (m, 1H), 4.49 (AB, JAB = 12.0 Hz, ΔνAB = 16.5 Hz, 2H), 4.01-4.17 (m, 3H), 3.57 (m, 1H), 3.51 (m, 1H), 3.35 (dd, J = 13.5, 3.5 Hz, 1H), 2.69 (dd, J = 16.0, 5.5 Hz, 1H), 2.58-2.65 (m, 2H), 2.02 (m, 1H), 1.78 (m, 1H), 1.70 (s, 3H), 1.44 (s, 9H).
19a
1H NMR (500 MHz, CDCl3) δ 7.17-7.31 (m, 10H), 5.42 (t, J = 6.5 Hz, 1H), 4.98 (s, 1H), 4.97 (s, 1H), 4.68 (m, 1H), 4.51 (AB, JAB = 11.5 Hz, ΔνAB = 30.5 Hz, 2H), 4.13-4.22 (m, 2H), 4.06 (dd, J = 8.0, 5.5 Hz, 1H), 3.65 (m, 1H), 3.56 (m, 1H), 3.32 (dd, J = 13.0, 3.0 Hz, 1H), 2.62-2.80 (m, 3H), 2.00 (m, 1H), 1.79 (m, 1H), 1.72 (s, 3H), 1.43 (s, 9H).
19b
1H NMR (500 MHz, CDCl3) δ 7.20-7.33 (m, 10H), 5.42 (dd, J = 7.5, 5.0 Hz, 1H), 4.95 (s, 1H), 4.82 (s, 1H), 4.63 (m, 1H), 4.48 (AB, JAB = 12.0 Hz, ΔνAB = 16.5 Hz, 2H), 4.17 (d, J = 5.5 Hz, 2H), 4.07 (dd, J = 8.0, 5.5 Hz, 1H), 3.54 (m, 1H), 3.50 (m, 1H), 3.29 (dd, J = 13.5, 3.0 Hz, 1H), 2.70-2.77 (m, 2H), 2.65 (dd, J = 16.0, 7.5 Hz, 1H), 2.00 (m, 1H), 1.77 (m, 1H), 1.68 (s, 3H), 1.46 (s, 9H).
21a
1H NMR (500 MHz, CDCl3) δ 7.25-7.33 (m, 5H), 5.38 (t, J = 6.5 Hz, 1H), 4.94 (s, 1H), 4.93 (s, 1H), 4.49 (AB, JAB = 12.0 Hz, ΔνAB = 23.0 Hz, 2H), 4.37 (m, 1H), 4.27 (m, 1H), 3.94 (m, 3H), 3.63 (m, 1H), 3.54 (m, 1H), 2.78 and 2.66 (ABX, JAB = 15.5 Hz, JAX = 6.5 Hz, JBX = 6.0 Hz, ΔνAB = 60.0 Hz, 2H), 1.94 (m, 1H), 1.77 (m, 1H), 1.69 (s, 3H), 1.41 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 172.0, 169.1, 143.9, 138.8, 128.3, 127.5, 127.4, 114.5, 81.1, 80.5, 72.7, 71.0, 67.0, 62.4, 42.6, 39.1, 34.0, 28.0, 16.7.
21b
1H NMR (500 MHz, CDCl3) δ 7.25-7.33 (m, 5H), 5.44 (dd, J = 7.5, 6.0 Hz, 1H), 4.94 (s, 1H), 4.83 (s, 1H), 4.47 (AB, JAB = 12.0 Hz, ΔνAB = 15.5 Hz, 2H), 4.40 (m, 1H), 4.06 (dd, J = 8.5, 5.5 Hz, 1H), 3.97 (m, 2H), 3.54 (m, 1H), 3.47 (m, 1H), 2.72 and 2.61 (ABX, JAB = 16.0 Hz, JAX = 7.5 Hz, JBX = 5.5 Hz, ΔνAB = 56.0 Hz, 2H), 1.97 (m, 1H), 1.76 (m, 1H), 1.64 (s, 3H), 1.43 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 172.3, 168.9, 153.1, 145.0, 138.5, 128.4, 127.7, 127.5, 114.4, 83.1, 81.1, 73.0, 72.5, 66.9, 62.4, 42.5, 39.0, 33.8, 28.1, 16.7.
TBS ether 22
To a solution of acyl oxazolidinone 20a (517 mg, 1.06mmol) in 4 mL THF was added MeOH (70 μL) followed by lithium borohydride (2.11 mL, 2.11 mmol, 1M in THF) at 0 °C. The reaction was stirred at 0 °C for 3 h and then quenched with sat. aq. NH4Cl. The aqueous layer was extracted twice with EtOAc. The organic layers were dried over Na2SO4, concentrated in vacuo, and purified by column chromatography (25% EtOAc/hexanes) to give alcohol (308 mg, 80%) as a clear oil. [α]23D −30 (c 1.9, CHCl3); IR (neat) 3454, 1727, 1155; 1H NMR (500 MHz, CDCl3) δ 7.29-7.35 (m, 5H), 4.94 (s, 1H), 4.89 (s, 1H), 4.52 (AB, JAB = 12.0 Hz, ΔνAB = 18.5 Hz, 2H), 4.08 (dd, J = 9.5, 4.5 Hz, 1H), 3.80 (m, 1H), 3.69-3.75 (m, 2H), 3.45-3.51 (m, 2H), 2.45 (ABX, JAB = 15.0 Hz, JAX = 7.5 Hz, JBX = 6.0 Hz, ΔνAB = 45.7 Hz, 2H), 1.83 (m, 1H), 1.75 (m, 1H), 1.67 (s, 3H), 1.43 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 170.7, 144.7, 137.9, 128.5, 127.9, 127.8, 113.5, 80.6, 79.4, 74.0, 73.0, 66.9, 63.3, 38.7, 33.7, 28.1, 16.8; HRMS (ESI) [M+Na]+ calcd for C21H32O5Na 387.2148, found 387.2137. To a solution of above alcohol (300 mg, 0.82 mmol) in 4 mL DMF was added imidazole (140 mg, 2.06 mmol) followed by tert-butyldimethylsilyl chloride (186 mg, 1.24 mmol) at 0 °C. The reaction was warmed to room temperature and stirred for 2 hours, then quenched with sat. aq. NaHCO3. The aqueous layer was extracted with EtOAc and the combined organic extracts were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (5% EtOAc/hexanes) provided 326 mg (86%) of TBS ether 22. [α]23D − 39 (c 1.0, CHCl3); IR (neat) 1729, 1112, 837; 1 3 H NMR (300 MHz, CDCl3) δ 7.25-7.33 (m, 5H), 4.91 (s, 2H), 4.48 (AB, JAB = 12.0 Hz, ΔνAB = 15.0 Hz, 2H), 4.05 (dd, J = 7.8, 5.1 Hz, 1H), 3.78 (m, 1H), 3.65 (dd, J = 10.8, 4.5 Hz, 1H), 3.44-3.57 (m, 3H), 2.43 (ABX, JAB = 15.5 Hz, JAX = 6.9 Hz, JBX = 5.4 Hz, ΔνAB = 53.5 Hz, 2H), 1.88 (m, 1H), 1.75 (m, 1H), 1.66 (s, 3H), 1.43 (s, 9H), 0.89 (s, 9H), 0.04 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 171.0, 144.6, 138.6, 128.3, 127.5, 127.4, 113.6, 80.1, 79.1, 73.7, 72.9, 67.1, 63.6, 38.9, 34.1, 28.1, 25.9, 18.3, 16.6, −5.4; HRMS (ESI) [M+Na]+ calcd for C27H46O5SiNa 501.3012, found 501.3022.
Alcohol 23
To a suspension of LiAlH4 (45 mg, 1.18 mmol) in Et2O at 0 °C was added a solution of t-butyl ester 22 (300 mg, 0.62 mmol) in Et2O dropwise. After 2 h at 0 °C, aq. potassium sodium tartrate solution was added and the mixture was vigorously stirred for 2 h at room temperature until two clear phases appeared. The aqueous phase was extracted twice with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (20% EtOAc/hexanes) gave 254 mg (99%) of alcohol 21. [α]23D −51 (c 1.7, CHCl3); IR (neat) 3436, 1089, 837; 1H NMR (500 MHz, CDCl3) δ 7.26-7.34 (m, 5H), 4.95 (s, 1H), 4.94 (s, 1H), 4.48 (AB, JAB = 12.0 Hz, ΔνAB = 17.5 Hz, 2H), 4.11 (dd, J = 8.5, 5.5 Hz, 1H), 3.69-3.74 (m, 3H), 3.51-3.56 (m, 3H), 3.47 (dt, J = 9.0, 6.0 Hz, 1H), 1.81-1.87 (m, 2H), 1.72-1.77 (m, 2H), 1.70 (s, 3H), 0.88 (s, 9H), 0.04 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 144.3, 138.5, 128.3, 127.6, 127.5, 114.5, 79.1, 75.7, 73.0, 66.9, 64.1, 60.3, 35.1, 34.1, 25.9, 18.2, 16.5, −5.4; LRMS (ESI) m/z [M+Na]+ 431.4.
Oxime 24
To a solution of alcohol 21 (267 mg, 0.65 mmol) in 4 mL CH2Cl2 was added Dess-Martin periodinane (413 mg, 0.97 mmol) at 0 °C. The reaction was warmed up to room temperature for 6 h and quenched with aq. Na2SO3 and NaHCO3. The aqueous layer was extracted twice with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (20% EtOAc/hexanes) gave 240mg (91%) of aldehyde. 1H NMR (500 MHz, CDCl3) δ 9.73 (s, 1H), 7.33 (m, 5H), 4.93 (s, 2H), 4.48 (AB, JAB = 12.0 Hz, ΔνAB = 16.0 Hz, 2H), 4.06 (dd, J = 7.5, 5.0 Hz, 1H), 3.88 (m, 1H), 3.73 (dd, J = 10.0, 4.0 Hz, 1H), 3.49-3.57 (m, 2H), 3.47 (dt, J = 9.0, 6.0 Hz, 1H), 2.56 (ABMX, JAB = 16.0 Hz, JAX = 7.0 Hz, JBX = 6.0 Hz, JAM = 3.0 Hz, JBM = 2.0 Hz ΔνAB = 47.5 Hz, 2H), 1.87 (m, 1H), 1.74 (m, 1H), 1.64 (s, 3H), 0.88 (s, 9H), 0.04 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 201.7, 144.2, 138.5, 128.4, 127.6, 127.5, 114.4, 79.5, 73.0, 72.4, 66.8, 63.8, 47.0, 34.0, 25.9, 18.2, 16.5, −5.4. The above aldehyde (240 mg, 0.59 mmol) and NaOAc (194 mg, 2.36 mmol) were dissolved in 4 mL EtOH. To the solution was added hydroxylamine hydrochloride (123 mg, 1.77 mmol) at room temperature. The reaction was stirred for 1 h and then partitioned between sat. aq. NaHCO3 and CH2Cl2. The aqueous layer was extracted twice with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo to provide oxime 24 (265 mg, crude, used without further characterization).
Isoxazole 7
The solution of the above oxime 24 in CH2Cl2 (6 mL) was treated with bleach (3 mL, aqueous 4% sodium hypochlorite solution) at room temperature for 4 h. The reaction was quenched with aq. Na2SO3 and NaHCO3. The aqueous layer was extracted twice with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (15% EtOAc/hexanes) gave isoxazoline 7 (230 mg 82% for 2 steps) as clear oil. [α]23D −23 (c 1.9, CHCl3); IR (neat) 3060, 1462, 1100, 837; 1 3 H NMR (500 MHz, CDCl3) δ 7.31 (m, 5H), 4.48 (AB, JAB = 12.0 Hz, ΔνAB = 25.0 Hz, 2H), 4.06-3.86 (AX, JAX = 8.0 Hz, ΔνAX = 102.0 Hz, 2H), 3.68 (ABX, JAB = 11.0 Hz, JAX = 5.0 Hz, JBX = 5.0 Hz, ΔνAB = 37.0 Hz, 2H), 3.52-3.58 (m, 3H), 3.40 (m, 1H), 2.63 (dd, J = 14.5, 3.0 Hz, 1H), 2.27 (dd, J = 14.5, 11.5 Hz, 1H), 1.84 (m, 1H), 1.56 (m, 1H), 1.18 (s, 3H), 0.89 (s, 9H), 0.05 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 161.6, 138.2, 128.4, 127.7, 80.9, 77.7, 76.7, 73.1, 66.3, 65.5, 53.6, 31.7, 25.8, 25.7, 18.3, 15.8, −5.3; HRMS (ESI) [M+Na]+ calcd for C23H37NO4Na 442.2390, found 442.2397.
Tetrahydropyranone 25
To a solution of isoxazoline 7 (202 mg, 0.48 mmol) in methanol/water (5 mL/1 mL) was added boric acid (120 mg, 1.93 mmol) and a spatula tip (estimated 10-20 mg) of W-2 Raney nickel aqueous suspension. The reaction was placed under hydrogen by repeated (−5 times) evacuation and flushing with H2 gas by means of a balloon attached to a three-way stopcock. The mixture was stirred vigorously for 4 h and then filtered through Celite into a separatory funnel containing sat. aq. NaHCO3 and CH2C12. After separation, the aqueous layer was extracted with CH2Cl2 two more times and the combined organic layers were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (25% EtOAc/hexanes) gave hydroxy ketone 25 (180 mg, 89%) as a clear oil. [α]23D −68 (c 1.4, CHCl3); IR (neat) 3459, 1710, 1113, 837; 1H NMR (500 MHz, CDCl3) δ 7.31 (m, 5H), 4.50 (AB, JAB = 12.0 Hz, ΔνAB = 38.5 Hz, 2H), 3.80-3.86 (m, 2H), 3.62-3.66 (m, 5H), 3.44 (dd, J = 12.0, 6.0 Hz, 1H), 2.68 (m, 1H), 2.57 (b, 1H), 2.26 (dd, J = 14.5, 2.5 Hz, 1H), 1.77-1.83 (m, 2H), 1.04 (s, 3H), 0.89 (s, 9H), 0.05 (d, J = 3.0 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 214.0, 138.4, 128.4, 127.7, 127.6, 77.4, 76.5, 67.2, 65.5, 64.2, 54.1, 41.3, 29.4, 25.8, 18.3, 14.7, −5.2, −5.3; HRMS (ESI) [M+Na]+ calcd for C23H38O5SiNa 445.2387, found 445.2391.
Acetonide 4
To a solution of tetrahydropyranone 25 (34 mg, 0.08 mmol) in 2 mL THF was added L-Selectride (0.24 mL, 0.24 mmol, 1M in THF) at −78 °C. After 1 h, the reaction was quenched with H2O. After warm up to room temperature, 1 N NaOH and 30% H2O2 were added and the mixture stirred for 1h at room temperature. The aqueous layer was extracted with EtOAc twice and the combined organic layers were dried over Na2SO4 and concentrated. Purification by column chromatography (35% EtOAc/hexanes) gave 28 mg (82%) of diol along with its diastereomer (3 mg, 9%). [α]23D −48 (c 3.3, CHCl3); IR (neat) 3391, 1105, 839; 1 3 H NMR (500 MHz, CDCl3) δ 7.32 (m, 5H), 4.52 (AB, JAB = 12.0 Hz, ΔνAB = 16.0 Hz, 2H), 4.09 (dd, J = 9.0, 2.0 Hz, 1H), 3.97 (b, 1H), 3.90 (m, 1H), 3.83 (m, 1H), 3.51-3.66 (m, 7H), 1.82 (m, 1H), 1.68 (m, 1H), 1.59 (m, 1H), 1.52 (m, 1H), 0.89 (s, 9H), 0.73 (s, 3H), 0.04 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 137.9, 128.5, 127.9, 127.8, 75.0, 73.4, 72.9, 71.9, 69.9, 68.7, 66.4, 40.2, 32.3, 30.0, 25.9, 18.4, 15.0, −5.2; HRMS (ESI) [M+Na]+ calcd for C23H40NO5Na 447.2543, found 447.2567. To a solution of above diol (27 mg, 0.05 mmol) in 2 mL acetone was added 2,2-dimethoxypropane (10 μL, 0.1 mmol) and camphorsulfonic acid (0.6 mg, 0.0025 mmol) at room temperature. After stirring for 3 h, the reaction was neutralized with triethylamine (0.1 mL) and concentrated. Purification by column chromatography (10% EtOAc/hexanes) afforded the acetonide 4 (28 mg, 95%). [α]23D − 40 (c 1.4, CHCl3); IR (neat) 1102, 837; 1 3 H NMR (500 MHz, CDCl3) δ 7.33 (m, 5H), 4.51 (AB, JAB = 12.0 Hz, ΔνAB = 33.0 Hz, 2H), 4.19 (dd, J = 10.5, 1.5 Hz, 1H), 3.88 (t, J = 3.0 Hz, 1H), 3.68-3.77 (m, 2H), 3.51-3.64 (m, 5H), 1.80 (m, 1H), 1.74 (m, 1H), 1.64 (m, 1H), 1.46 (m, 1H), 1.44 (s, 3H), 1.42 (s, 3H), 0.89 (s, 9H), 0.74 (s, 3H), 0.04 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 138.8, 128.3, 127.7, 127.4, 98.4, 73.0, 72.8, 72.0, 71.9, 68.4, 66.4, 66.2, 34.6, 29.9, 29.5, 26.0, 18.8, 18.4, 14.8, −5.2; HRMS (ESI) [M+Na]+ calcd for C26H44O5SiNa 487.2856, found 487.2848.
Tetrazole 28
To a solution of 3-butyn-1-ol (385 mg, 5.5 mmol) in THF (15 mL) was added 1-phenyl-1H-tetrazole-5-thiol (1.17 g, 6.6 mmol) , followed by triphenylphosphine (1.73 g, 6.6 mmol) and DIAD (1.27 mL, 6.6 mmol) at 0 °C. The mixture was stirred at room temperature overnight and concentrated in vacuo. The residue was purified by column chromatography (15% EtOAc/Hexanes) to provide the sulfide (1.21g, 97%) as a clear oil. To a solution of above sulfide (219 mg, 0.95 mmol) in EtOH (6 mL) and H2O2 (6 mL, 30%) was added (NH4)6Mo7O24·4H2O (2.35 g, 1.9 mmol) at 0 °C. The reaction was stirred at room temperature overnight and quenched with aq. Na2SO3. The mixtutre was extracted with CH2Cl2. The combined organic phases were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (25% EtOAc/hexanes) afforded sulfone 28 (238 mg, 95%). IR (neat) 3292, 1351, 1146; 1H NMR (500 MHz, CDCl3) δ 7.58-7.64 (m, 5H), 3.91 (t, J = 7.0 Hz, 2H), 2.88 (dt, J = 7.0, 2.7 Hz, 2H), 2.06 (t, J = 2.7 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 153.1, 132.9, 131.6, 129.8, 125.2, 78.1, 71.6, 54.5, 13.4.
Stannane 5
To a solution of alkyne 28 (131 mg, 0.5 mmol) in acetone (2 mL) was added AgOAc (25 mg, 0.15 mmol) followed by NBS (134 mg, 0.75 mmol). The reaction was stirred at room temperature overnight in dark and quenched with H2O. The mixture was extracted with CH2Cl2. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography (25% EtOAc/Hexanes) to provide the bromoalkyne (173 mg, 100%). To a solution of above bromoalkyne (173 mg, 0.5 mmol), Ph3P (14 mg, 0.05 mmol) and Pd(PPh3)4 (29 mg, 0.025 mmol) in THF (4 mL) at −78 °C was added a solution of Bu3SnH (0.31 mL, 1.15 mmol) in THF (2 mL) via syringe pump over 30 min. After stirring at room temperature for 1 h, the mixture was concentrated in vacuo and purified by column chromatography (10% EtOAc/Hexanes) to provide the vinyl stannane 5 (264 mg, 95%) as a E/Z mixture(E/Z 10:1). IR (neat) 1597, 1349, 1152; 1H NMR (500 MHz, CDCl3) δ 7.58-7.62 (m, 5H), 6.14 (d, J = 19.0 Hz, 2H), 5.93 (dt, J = 19.0, 6.0 Hz, 2H), 3.94 (m, 2H), 2.78 (m, 2H), 1.48 (m, 6H), 1.32 (m, 6H), 0.88 (m, 15H); HRMS (EI) [M-Bu]+ calcd for C19H29SN4O2Sn 497.1033, found 497.1032.
Alcohol 29
To a suspension of NaH (60 wt % in mineral oil, 2.14 g, 53.3 mmol) in THF (60 mL) was added 3-butyn-1-ol 26 (1.88 g, 26.8 mmol) at 0 °C. After 30 min, tetrabutylamonium iodide (500 mg, 1.35 mmol) and benzyl bromide (3.5 mL, 29.2 mmol) were added. The mixture was allowed to warm to room temperature overnight and quenched with sat. aq. NH4Cl. The aqueous layer was extracted with EtOAc twice. The combined organic layers were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (pure hexanes-5% EtOAc/hexanes) afforded benzyl ether (4.17g, 97%) as a clear oil. 1H NMR (300 MHz, CDCl3) δ 7.31-7.37 (m, 5H), 4.57 (s, 2H), 3.61 (t, J = 6.9 Hz, 2H), 2.52 (dt, J = 6.9, 2.4 Hz, 2H), 2.01 (t, J = 2.4 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 138.0, 128.4, 127.7, 81.3, 73.0, 69.4, 68.1, 19.9. To a solution of above alkyne (4.17g, 26.1 mmol) in THF (80 mL) was treated nBuLi (1.6M in hexane, 16.3 mL, 26.1 mmol) at −78 °C. After 30 min, propionaldehyde was added. The reaction was stirred at −78 °C for 1 h and quenched with sat. aq. NH4Cl at room temperature. The aqueous phase was extracted with EtOAc twice. The combined organic phases were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (15% EtOAc/hexanes) afforded propargyl alcohol 29 (4.87 g, 86%) as a clear oil. IR (neat) 3400, 1454, 1099; 1H NMR (300 MHz, CDCl3) δ 7.26-7.34 (m, 5H), 4.53 (s, 2H), 4.25 (dt, J = 6.3, 1.8 Hz, 1H), 3.56 (t, J = 6.9 Hz, 2H), 3.08 (br, 1H), 2.51 (dt, J = 6.9, 1.8 Hz, 2H), 1.67 (m, 2H), 0.98 (t, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 137.9, 128.4, 127.7, 82.5, 81.6, 72.8, 68.3, 63.5, 30.9, 20.0, 9.5.
Ketone 30
To a suspension of LiAlH4 (1.69 g, 44.6 mmol) in THF (60 mL) was added a solution of alcohol 29 (4.87 g, 22.3 mmol) in THF (20 mL) via cannula at 0 °C. The reaction was stirred at room temperature for 24 h and quenched with H2O (2 mL) at 0°C. 2 N HCl was added and the mixture was stirred for 2 h at room temperature until two clear phases appeared. The aqueous phase was extracted with EtOAc twice. The combined organic phases were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (20% EtOAc/hexanes) afforded allylic alcohol (4.56 g, 93%) as a clear oil. IR (neat) 3400, 1454, 1098; 1H NMR (500 MHz, CDCl3) δ 7.26-7.34 (m, 5H), 5.62 (dt, J = 15.5, 6.5 Hz, 1H), 5.51 (dd, J = 15.5, 6.5 Hz, 1H), 4.49 (s, 2H), 3.91 (dt, J = 6.5, 6.5 Hz, 1H), 3.50 (t, J = 6.5 Hz, 2H), 2.84 (br, 1H), 2.35 (dt, J = 6.5, 6.5 Hz, 2H), 1.43-1.60 (m, 2H), 0.89 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 138.4, 135.1, 128.4, 127.8, 127.7, 127.6, 74.1, 72.8, 69.8, 32.7, 30.0, 9.9. To a solution of above alcohol (4.56 g, 20.7 mmol) in CH2Cl2 (60 mL) was added PCC (8.95 g, 41.4 mmol) at 0 °C. The reaction was stirred at room temperature for 3 h. The mixture was filtered though Celite eluting with Et2O and the filtrate was concentrated in vacuo. Purification by column chromatography (15% EtOAc/hexanes) afforded enone 30 (3.47 g, 77%) as a clear oil. IR (neat) 1673, 1632, 1101; 1H NMR (500 MHz, CDCl3) δ 7.26-7.31 (m, 5H), 6.84 (dt, J = 16.0, 7.0 Hz, 1H), 6.15 (d, J = 16.0 Hz, 1H), 4.51 (s, 2H), 3.58 (t, J = 6.5 Hz, 2H), 2.56 (q, J = 7.5, 2H), 2.51(dt, J = 6.5, 6.5 Hz, 2H), 1.09 (t, J = 7.5, 3H); 13C NMR (125 MHz, CDCl3) δ 200.9, 143.4, 138.1, 131.5, 128.4, 127.7, 73.1, 68.3, 33.2, 32.9, 8.1; HRMS (ESI) [M+Na]+ calcd for C14H18O2Na 241.1205, found 241.1206.
Silyl enolate 10
To a solution of enone 30 (3.25 g, 14.9 mmol) in Et2O (40 mL) was added Et3N (4.18 mL, 29.8 mmol) and triethylsilyl triflate (4.04 mL, 17.9 mmol) at −78 °C. The reaction was stirred at 0 °C for 3 h. The mixture was poured into sat. aq. NaHCO3 and the aqueous layer was extracted with hexanes twice. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (5% EtOAc/hexanes) afforded silyl enolate 10 (4.40 g, 90%) as a clear oil. IR (neat) 1698, 1628, 1117; 1H NMR (500 MHz, CDCl3) δ 7.29-7.36 (m, 5H), 5.95 (d, J = 15.5 Hz, 1H), 5.80 (dt, J = 15.0, 7.5 Hz, 1H), 4.76 (q, J = 7.0, 1H), 4.53 (s, 2H), 3.54 (t, J = 6.5 Hz, 2H), 2.43 (dt, J = 7.0, 7.0 Hz, 2H), 1.67 (d, J = 7.0 Hz, 3H), 1.02 (t, J = 8.0, 9H), 0.73 (q, J = 8.0, 6H); 13C NMR (125 MHz, CDCl3) δ 149.5, 138.5, 130.6, 128.4, 127.7, 127.6, 124.3, 107.9, 73.0, 70.2, 32.8, 11.4, 6.9, 5.5.
Pyranone (32)
To a mixture of diene 10 (2.26 g, 6.6 mmol), 2-(tert-butyldimethylsilyloxy)acetaldehyde 11 (1.73 g, 9.9 mmol) and powdered 4Å molecular sieves (1.2 g) was added catalyst (1S, 2R)-31 (300 mg, 0.66 mmol) under argon. The reaction mixture was stirred for 40 h at room temperature and then filtered through a short pad of silica gel, eluting with 25% EtOAc/Hexanes. The filtrate was concentrated and the residue was dissolved in THF (15 mL). At 0 °C, acetic acid (0.75 mL, 13 mmol) and TBAF (1M in THF, 9.8 mL, 9.9 mmol) were added and the mixture was allowed to stir for 30 min. The mixture was diluted with EtOAc and washed with NaHCO3 and brine. The organic phase was dried over Na2SO4, filtered and concentrated in vacuo. Purification by column chromatography (10% EtOAc/hexanes) provided the pyranone 32 (1.84 g, 71%) as a yellow oil. [α]23D 20 (c 2.0, CHCl3); IR (neat) 1717, 1119, 839; 1H NMR (500 MHz, CDCl3) δ 7.28-7.36 (m, 5H), 4.49 (AB, JAB = 12.0 Hz, ΔνAB = 14.0 Hz, 2H), 3.81 (m, 1H), 3.71 (m, 2H), 3.59 (m, 3H), 2.55 (m, 1H), 2.45 (dd, J = 14.5, 12.5 Hz, 1H), 2.26 (d, J = 14.5 Hz, 1H), 1.92 (m, 1H), 1.84 (m, 1H), 1.10 (d, J = 7.0 Hz, 3H), 0.88 (s, 9H), 0.05 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 211.3, 138.3, 128.4, 127.7, 78.7, 74.3, 73.1, 66.1, 62.0, 46.3, 44.6, 36.5, 25.8, 18.2, 10.6, −5.3, −5.5; HRMS (ESI) [M+Na]+ calcd for C22H36OSiNa 415.2281, found 415.2299.
Reduction of ketone 32
To a solution of ketone 32 (1.84 g, 4.69 mmol) in CH2Cl2 (20 mL) was added DIBAL-H (5.6 mL, 5.58 mmol, 1 M in CH2Cl2) at −78 °C. After 1 h, the reaction was quenched with sat. aq. potassium sodium tartrate and stirred for 2 h at room temperature. The aqueous layer was extracted with CH2Cl2 twice and the combined organic layers were dried over Na2SO4 and concentrated. Purification by column chromatography (20%, 25% EtOAc/hexanes) gave alcohol 33 (592 mg, 32%) along with cis-alcohol 34 (1.18 g, 64%).
33
[α]23D −12 (c 1.1, CHCl3); IR (neat) 3434, 1094, 837; 1 3 H NMR (500 MHz, CDCl3) δ 7.28-7.36 (m, 5H), 4.63 (s, 2H), 3.95 (dt, J = 6.5, 2.5 Hz, 1H), 3.89 (m, 1H), 3.83 (m, 1H), 3.64 and 3.48 (ABX, JAB = 10.0, JAX = 6.5, JBX = 6.5 Hz, ΔνAB = 80.0, 2H), 3.59 (t, J = 6.5 Hz, 2H), 2.36 (br, 1H), 1.79 (m, 1H), 1.72 (m, 2H), 1.60 (m, 1H), 1.47 (m, 1H), 0.91 (s, 9H), 0.86 (d, J = 5.5 Hz, 3H) 0.07 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 138.5, 128.4, 127.7, 127.7, 74.4, 73.0, 70.5, 69.5, 67.0, 63.6, 36.3, 35.8, 34.5, 25.9, 18.3, 10.8, −5.1, −5.4; HRMS (ESI) [M+H]+ calcd for C22H39OSi 375.2618, found 375.2635.
34
1H NMR (500 MHz, CDCl3) δ 7.30-7.38 (m, 5H), 4.53 (s, 2H), 3.92 (d, J = 11.7 Hz, 1H), 3.69 (m, 1H), 3.62 (m, 2H), 3.57 (m, 2H), 3.46 (m, 1H), 2.07 (m, 1H), 1.87 (m, 1H), 1.80 (m, 1H), 1.75 (m, 1H), 1.66 (m, 1H), 1.42 (m, 1H), 0.93 (s, 9H), 0.87 (d, J = 6.9 Hz, 3H) 0.09 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 138.5, 128.3, 127.6, 127.5, 78.8, 73.2, 73.0, 70.9, 66.7, 63.1, 36.0, 35.5, 35.3, 25.8, 18.2, 4.6, −5.3, −5.5; HRMS (ESI) [M+H]+ calcd for C22H39OSi 375.2618, found 375.2610.
Alcohol 35
To a solution of alcohol 33 (592 mg, 1.50 mmol) in CH2Cl2 (8 mL) was added 2,6-lutidine (0.35 mL, 3.0 mmol) followed by tert-butyl dimethylsilyl triflate (0.41 mL, 1.8 mmol). The reaction was stirred at room temperature for 2 hours, and quenched with sat. aq. NaHCO3. The aqueous layer was extracted with CH2Cl2 and the combined organic extracts were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (5% EtOAc/hexanes) provided TBS ether (755 mg, 99%) as a clear oil. [α]23D −1.3 (c 1.3, CHCl3); IR (neat) 1099, 837; 1 3 H NMR (500 MHz, CDCl3) δ 7.26-7.34 (m, 5H), 4.50 (AB, JAB = 12.0 Hz, ΔνAB = 14.5 Hz, 2H), 3.96 (dt, J = 7.0, 2.0 Hz, 1H), 3.92 (m, 1H), 3.82 (m, 1H), 3.61 and 3.46 (ABX, JAB = 10.0, JAX = 7.5, JBX = 6.5 Hz, ΔνAB = 75.0, 2H), 3.60 (t, J = 6.5 Hz, 2H), 1.79 (m, 1H), 1.70 (m, 2H), 1.58 (m, 1H), 1.38 (m, 1H), 0.91 (s, 9H), 0.89 (s, 9H), 0.85 (d, J = 7.5 Hz, 3H) 0.05 (s, 6H), 0.04 (d, J = 4.0 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 138.7, 128.3, 127.6, 127.4, 74.2, 73.0, 71.0, 69.7, 67.2, 63.4, 36.4, 36.0, 35.2, 25.8, 18.2, 18.1, 10.7, −4.8, −5.1, −5.4; LRMS (ESI) m/z [M+Na]+ 531.9. To a solution of above benzyl ether (755 mg, 1.48 mmol) in MeOH (15 mL) was added Pd/C (78 mg, 0.074 mmol, 10 wt. %) and stirred under H2 balloon at room temperature for 20 h. The reaction suspension was filtered through Celite and concentrated. Purification by column chromatography (15% EtOAc/hexanes) provided alcohol 35 (600 mg, 97%) as a clear oil. [α]23D +14 (c 1.5, CHCl3); IR (neat) 3436, 1097, 836; 1H NMR (500 MHz, CDCl3) δ 4.00 (m, 2H), 3.80 (m, 3H), 3.60 and 3.45 (ABX, JAB = 10.5, JAX = 7.0, JBX = 6.5 Hz, ΔνAB = 78.0 Hz, 2H), 1.73 (m, 1H), 1.66 (m, 2H), 1.59 (m, 1H), 1.32 (m, 1H), 0.89 (s, 9 H), 0.88 (s, 9 H), 0.85 (d, J = 7.0 Hz, 3H), 0.05 (s, 6H), 0.04 (d, J = 4.0 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 74.6, 74.1, 70.7, 63.5, 62.3, 37.4, 36.1, 35.0, 25.8, 25.8, 18.2, 18.0, 10.8, −4.9, −5.3, −5.5; LRMS (ESI) m/z [M+H]+ 419.5.
Ester 36
To a solution of DMSO (122 μL, 1.73 mmol) in CH2Cl2 (3 mL) was added oxalyl chloride (75 μL, 0.86 mmol) at −78 °C. After 20 min, the alcohol 7 (61 mg, 0.14 mmol) was added. The reaction mixture was stirred at −78 °C for 1 h and quenched with Et3N (0.4 mL, 2.88 mmol). After stirring at −78 °C for 30 min, the mixture was warmed to room temperature and stirred for another 1 h. The mixture was poured into 0.2 N HCl. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine and dried over Na2SO4. Filtration and concentration in vacuo gave the crude aldehyde which was used directly for next step without further purification. A solution of above crude aldehyde and (carbethoxymethylene)triphenylphosphorane (200 mg, 0.57 mmol) in CH2Cl2 (5 mL) was heated to reflux for 20 h. The resulting mixture was cooled to room temperature, filtered through a short silica gel pad eluting with 25% EtOAc/Hexanes. The filtrate was concentrated and the residue was purified by column chromatography (5% EtOAc/Hexanes) to provide ester 36 (57 mg, 84% for 2 steps) as a clear oil. [α]23D +7.6(c 1.1, CHCl3); IR (neat) 1723, 1072, 838; 1H NMR (500 MHz, CDCl3) δ 6.96 (dt, J = 15.5, 7.0 Hz, 1H), 5.85 (d, J = 15.5 Hz, 1H), 4.16 (q, J = 7.0 Hz, 2H), 3.96 (dt, J = 7.0, 2.5 Hz, 1H), 3.87 (m, 1H), 3.80 (m, 1H), 3.62 and 3.45 (ABX, JAB = 10.0, JAX = 7.0, JBX = 6.5 Hz, ΔνAB = 84.5, 2H), 2.36 (m, 1H), 2.27 (m, 1H), 1.67 (m, 1H), 1.55 (m, 1H), 1.33 (m, 1H), 1.26 (t, J = 7.0 Hz, 3H), 0.88 (s, 9 H), 0.86 (s, 9 H), 0.82 (d, J = 7.0 Hz, 3H), 0.03 (d, J = 1.0 Hz, 6H), 0.02 (d, J = 3.0 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 166.5, 145.6, 123.0, 74.4, 71.0, 70.9, 63.4, 60.1, 38.7, 35.9, 34.7, 25.8, 25.8, 18.2, 18.0, 14.3, 10.6, −4.9, −5.2, −5.4; HRMS (ESI) [M+Na]+ calcd for C25H50O5Si2Na 509.3095, found 509.3112.
Aldehyde 37
To a solution of 36 (135 mg, 0.28 mmol) in MeOH (3 mL) was added camphorsulfonic acid (12.4 mg, 0.054 mmol) at room temperature. After stirring for 30 min, the reaction was poured into sat. aq. NaHCO3 and extracted with CH2Cl2. The organic layers were dried over Na2SO4 and concentrated. Purification by column chromatography (20% EtOAc/hexanes) afforded the alcohol (100 mg, 97%) as a clear oil. [α]23D +20 (c 1.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.95 (dt, J = 15.5, 7.0 Hz, 1H), 5.85 (d, J = 15.5 Hz, 1H), 4.16 (q, J = 7.0 Hz, 2H), 4.01 (dt, J = 9.0, 2.5 Hz, 1H), 3.90 (m, 1H), 3.76 (m, 1H), 3.63 and 3.41 (ABX, JAB = 11.5, JAX = 9.0, JBX = 3.5 Hz, ΔνAB = 111.7, 2H), 2.38 (m, 1H), 2.32 (m, 1H), 2.07 (br, 1H), 1.55 (m, 2H), 1.35 (m, 1H), 1.26 (t, J = 7.0 Hz, 3H), 0.87 (s, 9 H), 0.82 (d, J = 7.0 Hz, 3H), 0.01 (d, J = 1.5 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 166.4, 145.2, 123.3, 75.1, 70.9, 70.8, 64.4, 60.2, 38.7, 36.8, 34.5, 25.8, 18.0, 14.3, 11.4, −4.9. To a solution of DMSO (0.2 mL, 2.7 mmol) in CH2Cl2 (4 mL) was added oxalyl chloride (0.12 mL, 1.35 mmol) at −78 °C. After 20 min, the above alcohol (100 mg, 0.27 mmol) was added. The reaction mixture was stirred at −78 °C for 1 h and quenched with Et3N (0.56 mL, 4.05 mmol). After stirred at −78 °C for 30 min, the mixture was warmed to room temperature and stirred for another 1 h. The mixture was poured into 1 N HCl. The aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine and dried over Na2SO4. Filtration and concentration in vacuo gave the crude aldehyde 37 (100mg, 100%) which was used directly for next step without further purification. 1H NMR (500 MHz, CDCl3) δ 9.63 (s, 1H), 6.99 (dt, J = 15.5, 7.0 Hz, 1H), 5.89 (d, J = 16.0 Hz, 1H), 4.35 (d, J = 3.0 Hz, 1H), 4.17 (q, J = 7.0 Hz, 2H), 3.95 (m, 1H), 3.84 (m, 1H), 2.48 (m, 1H), 2.38 (m, 1H), 2.00 (m, 1H), 1.64 (m, 1H), 1.39 (m, 1H), 1.27 (t, J = 7.0 Hz, 3H), 0.87 (m, 12 H), 0.04 (d, J = 3.0 Hz, 6H).
Preparation of vinyl iodides (38 and 6)
To a suspension of ethyl triphenylphosphonium iodide (167 mg, 0.4 mmol) in THF (3 mL) was added nBuLi (0.25 mL, 0.4 mmol, 1.6 M in hexane) at 23 °C. After 5 min, above homogenous solution was transferred into a solution of iodine (101 mg, 0.4 mmol) in THF (4 mL) at −78 °C. The resulting suspension was vigorously stirred for 5 min and warmed to −20 °C. NaHMDS (0.4 mL, 0.4 mmol, 1 M in THF) was added at −20 °C to produce a red solution, followed by stirring for 5 min and addition of a solution of aldehyde 37 (73 mg, 0.197 mmol). After 10 min at −20 °C, the reaction was quenched with sat. aq. NH4Cl. The aqueous phase was extracted with EtOAc twice. The combined organic phases were dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (5% EtOAc/hexanes) afforded E-vinyl iodide 38 (28 mg, 31%) and Z-vinyl iodide 6 (11 mg, 12%).
(E)-38
1H NMR (500 MHz, CDCl3) δ 6.96 (dt, J = 16.0, 7.0 Hz, 1H), 6.19 (dd, J = 7.5, 1.5 Hz, 1H), 5.86 (d, J = 16.0 Hz, 1H), 4.64 (dd, J = 8.0, 2.0 Hz, 1H), 4.18 (q, J = 7.0 Hz, 2H), 3.91 (m, 1H), 3.82 (m, 1H), 2.42 (s, 3H), 2.37 (m, 1H), 2.31 (m, 1H), 1.57 (m, 2H), 1.32 (m, 1H), 1.28 (t, J = 7.0 Hz, 3H), 0.92 (d, J = 7.5 Hz, 3H), 0.89 (s, 9H), 0.03 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 166.5, 145.2, 140.8, 123.3, 96.1, 72.9, 71.3, 70.6, 60.2, 39.6, 38.7, 33.8, 28.6, 25.8, 18.0, 14.3, 11.3, −4.8, −4.9; HRMS (ESI) [M+Na]+ calcd for C21H37O4ISiNa 531.1404, found 531.1416.
(Z)-6
1H NMR (500 MHz, CDCl3) δ 6.98 (dt, J = 16.0, 7.0 Hz, 1H), 5.87 (d, J = 16.0 Hz, 1H), 5.60 (dd, J = 7.0, 1.5 Hz, 1H), 4.55 (dd, J = 7.0, 2.5 Hz, 1H), 4.18 (q, J = 7.0 Hz, 2H), 3.96 (m, 1H), 3.82 (m, 1H), 2.51 (s, 3H), 2.38 (m, 1H), 2.32 (m, 1H), 1.78 (m, 1H), 1.58 (m, 1H), 1.32 (m, 1H), 1.28 (t, J = 7.0 Hz, 3H), 0.92 (s, 9H), 0.91 (d, J = 7.5 Hz, 3H), 0.06 (d, J = 16.5 Hz, 6H); HRMS (ESI) [M+Na]+ calcd for C21H37O4ISiNa 531.1404, found 531.1406.
Acknowledgements
Financial support this work is provided in part by the National Institutes of Health and Purdue University. We thank Mr. Chun-xiao Xu for assistance with NMR experiments.
Dedicated to Professor E. J. Corey with admiration and appreciation on the occasion of his 80th birthday
References
- [1].Horton PA, Koehn FE, Longley RE, McConnell OJ. J. Am. Chem. Soc. 1994;116:6015. [Google Scholar]
- [2]a).Lee E, Song HY, Kang JW, Kim D-S, Jung C-K, Joo JM. J. Am. Chem. Soc. 2002;124:384. doi: 10.1021/ja017265d. [DOI] [PubMed] [Google Scholar]; b) Song HY, Joo JM, Kang JW, Kim DS, Jung CK, Kwak HS, Park JH, Lee E, Hong CY, Jeong S, Jeon K, Park JH. J. Org. Chem. 2003;21:8080. doi: 10.1021/jo034930n. [DOI] [PubMed] [Google Scholar]
- [3]b).Kang SH, Kang SY, Choi H, Kim CM, Jun H, Youn J. Synthesis. 2004;7:1102. [Google Scholar]; b) Kang SH, Kang SY, Kim CM, Choi H, Jun H, Lee BM, Park CM, Jeong JW. Angew. Chem., Int. Ed. 2003;42:4779. doi: 10.1002/anie.200352016. [DOI] [PubMed] [Google Scholar]
- [4].Yoshimura T, Yakushiji F, Kondo S, Wu X, Shindo M, Shishido K. Org. Lett. 2006;8:475. doi: 10.1021/ol0527678. [DOI] [PubMed] [Google Scholar]
- [5].Ghosh AK, Gong G. Org. Lett. 2007;9:1437. doi: 10.1021/ol0701013. [DOI] [PubMed] [Google Scholar]
- [6]a).Hart DJ, Patterson S, Unch JP. Synlett. 2003;9:1334. [Google Scholar]; b) Nowakowski M, Hoffmann HMR. Tetrahedron Lett. 1997;38:1001. [Google Scholar]; c) Beck H, Hoffmann HMR. Eur. J. Org. Chem. 1999:2991. [Google Scholar]; d) Gurjar MK, Kumar P, Rao B. Venkateswara. Tetrahedron Lett. 1996;37:8617. [Google Scholar]; e) Gurjar MK, Chakrabarti A, Rao B. Venkateswara, Kumar P. Tetrahedron Lett. 1997;38:6885. [Google Scholar]; f) Dalgard JE, Rychnovsky SD. Org. Lett. 2005;7:1589. doi: 10.1021/ol050270s. [DOI] [PubMed] [Google Scholar]; g) Sawant KB, Ding F, Jennings MP. Tetrahedron Lett. 2006;47:939. [Google Scholar]; h) Sawant KB, Ding F, Jennings MP. Tetrahedron Lett. 2007;48:5177. [Google Scholar]
- [7].Gao Y,Y, Klunder JM, Hanson RM, Masamune H, Ko SY, Sharpless KB. J. Am. Chem. Soc. 1987;109:5765. [Google Scholar]
- [8].Evans DA, Ennis MD, Mathre DJ. J. Am. Chem. Soc. 1982;104:1737. [Google Scholar]
- [9].For precedent examples, see: Crimmins MT, Emmitte KA, Katz JD. Org. Lett. 2000;2:2165. doi: 10.1021/ol006091m. Crimmins MT, Emmitte KA. J. Am. Chem. Soc. 2001;123:1533. doi: 10.1021/ja005892h. Lee E, Choi SJ, Kim H, Han H, Kim Y, Min S, Son S, Lim S, Jang W. Angew. Chem., Int. Ed. 2002;41:176. doi: 10.1002/1521-3773(20020104)41:1<176::aid-anie176>3.0.co;2-#.
- [10].Dess DB, Martin JC. J. Am. Chem. Soc. 1991;113:7277. [Google Scholar]
- [11].Yokoshima S, Ueda T, Kobayashi S, Sato A, Kuboyama T, Tokuyama H, Fukuyama T. J. Am. Chem. Soc. 2002;124:2137. doi: 10.1021/ja0177049. [DOI] [PubMed] [Google Scholar]
- [12].Curran DP. J. Am. Chem. Soc. 1983;105:5826. [Google Scholar]
- [13].Stille JK. Angew. Chem., Int. Ed. Engl. 1986;25:508. [Google Scholar]
- [14].Zhang HX, Guibe F, Balavoine G. J. Org. Chem. 1990;55:1857. [Google Scholar]
- [15].Mitsunobu O. Synthesis. 1981:1. [Google Scholar]
- [16].Schultz HS, Freyermuth HB, Buc SR. J. Org. Chem. 1963;28:1140. [Google Scholar]
- [17].Liu K, Yan S, Wu Y, Yao Z. J. Org. Chem. 2002;67:6758. doi: 10.1021/jo026056o. [DOI] [PubMed] [Google Scholar]
- [18]a).Dossetter AG, Jamison TF, Jacobsen EN. Angew. Chem., Int. Ed. 1999;38:2398. doi: 10.1002/(sici)1521-3773(19990816)38:16<2398::aid-anie2398>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; b) Gademann K, Chavez DE, Jacobsen EN. Angew. Chem., Int. Ed. 2002;41:3059. doi: 10.1002/1521-3773(20020816)41:16<3059::AID-ANIE3059>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- [19].Chen J, Wang T, Zhao K. Tetrahedron Lett. 1994;35:2827. [Google Scholar]
- [20]a).Meng D, Bertinato P, Balog A, Su D, Kamenecka T, Sorensen E, Danishefsky SJ. J. Am. Chem. Soc. 1997;119:10073. [Google Scholar]; b) Smith AB, III, Condon SM, McCauley JA. Acc. Chem. Res. 1998;31:35. [Google Scholar]; c) Marshall JA, Johns BA. J. Org. Chem. 1998;63:7885. doi: 10.1021/jo971900+. [DOI] [PubMed] [Google Scholar]; d) Sawada D, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2000;122:10521. [Google Scholar]
- [21].Crews P, Rodriquez J, Jaspars M. Organic Structure Analysis. Oxford University Press; Oxford, UK: 1998. [Google Scholar]
- [22]a).Baudin JB, Hareau G, Julia SA, Ruel O. Tetrahedron Lett. 1991;32:1175. [Google Scholar]; b) Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998;1:26. [Google Scholar]
- [23].Maryanoff BE, Reitz AB. Chem. Rev. 1989;89:863. [Google Scholar]
- [24].Chatterjee AK, Choi T, Sanders DP, Grubbs RH. J. Am. Chem. Soc. 2003;125:11360. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- [25].Ando K. J. Org. Chem. 1998;63:8411. [Google Scholar]
- [26].Hoffmann HMR, Brandes A. Tetrahedron. 1995;51:155. [Google Scholar]
- [27].Scholl M, Ding S, Lee C, Grubbs RH. Org. Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- [28].Medina JC, Salomon M, Kyler KS. Tetrahedron Lett. 1988;29:3773. [Google Scholar]
- [29].Ma X, Saksena R, Chernyak A, Kovac P. Org. Biomol. Chem. 2003;1:775. doi: 10.1039/b211660j. [DOI] [PubMed] [Google Scholar]
- [30].Corey EJ, Bock MJ. Tetrahedron Lett. 1975;16:3269. [Google Scholar]