

Abstract
X-ray crystal structures at 2.9 Å resolution are reported for complexes of catechol diethers 1 and 2 with HIV-1 reverse transcriptase. The results help elucidate the structural origins of the extreme antiviral activity of the compounds. The possibility of halogen bonding between the inhibitors and Pro95 is addressed. Structural analysis reveals key interactions with conserved residues P95 and W229 of importance for design of inhibitors with high potency and favorable resistance profiles.
In a recent report, lead optimization guided by free-energy perturbation (FEP) calculations led to the discovery of extraordinarily potent anti-HIV agents.1 The new compounds, which are non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs), include 1 and 2. Specifically, 1 and 2 yield EC50 values of 55 and 320 pM for inhibition of replication of the wild-type virus in infected human T-cells. For comparison, nevirapine and rilpivirine, well-known approved drugs in the NNRTI class, are 110 nM and 670 pM inhibitors in this assay (Table 1). As detailed in the Supporting Information, we have also confirmed the extreme potency of 1 in cell-free assays for inhibition of reverse transcription; 1, rilpivirine, and nevirapine yield IC50 values 3, 38, and 1060 nM, respectively (Table 1). To help clarify the structural origins of the potency of the catechol diethers, we also report here co-crystal structures of HIV-RT with 1 and 2. It is of added interest to address the possible contribution of halogen bonding to the potency of 1, which arose from the molecular modeling, in addition to other key interactions.1
Table 1.
Activities for Inhibition of HIV-1 Replication (EC50) and Reverse Transcription (IC50).
| ||
|---|---|---|
| compound | EC50 (nM) | IC50 (nM) |
| 1 | 0.055 | 3.0 |
| 2 | 0.320 | ND |
| 3 | 3.2 | 10 |
| 4 | 5.2 | 30 |
| nevirapine | 110 | 1060 |
| rilpivirine | 0.670 | 38 |
Crystals of recombinant RT52A enzyme in complex with 1 and 2 were prepared using similar methods as previously described.2 The best crystals diffracted to amplitudes extending to a resolution of 2.85 Å for 1 and 2.90 Å for 2. Both structures were solved by molecular replacement with Phaser3,4 using the structure of an RT complex with a triazine relative of rilpivirine as a probe model (PDB code: 1S9E).5 Overall, the electron density reveals an “open-cleft” conformation as observed in other RT-NNRTI co-crystal structures (Figure 1).5–10 Root mean square deviations (rmsd) between our structures and several prior ones range from 1.874–3.024 Å (Table S2), suggesting that the general fold is similar and that there are no major conformational changes observed upon binding to the catechol diethers. However, Tyr181 is in the relatively uncommon “down” conformation as anticipated by the modeling1 and observed, for example, in the 2BE2 structure of an iodopyrimidinone NNRTI.7 The 1S9E structure and the 2ZD1 structure for rilpivirine show Try181 in the common “up” conformation.5
Figure 1.

Omit Fo-Fc electron density maps at a contour level of 3.0 σ showing 1 (A) and 2 (B) in the NNBP of HIV-1 RT. Omit Fo-Fc and 2Fo-Fc electron density maps are provided as Supporting Information.
The electron density for the non-nucleoside binding pocket (NNBP) clearly defines the orientation of 1, 2, and the interacting residues (Figures 1, S2). Both 1 and 2 are in the same conformation with only slight variations in flexible regions of the compounds that include the cyanovinyl group and ethoxy linker. The catechol diethers adopt an optimal conformation to make multiple van der Waals interactions with hydrophobic residues of the NNBP (Figure 2). Previously, the NNBP has been described to feature three channels designated as the entrance, groove, and tunnel.11 The cyanovinyl phenyl fragment interacts with P95, L100, V108, Y188, W229, and L234 and is positioned beneath W229, projecting into the tunnel region. The catechol ring is proximal to V106, V108, and Y181, and the Cl or F on C5 protrudes into the entrance channel near K103. The ethoxy linker interacts with L100, V106, F227, and Y318, positioning the terminal uracil ring in the groove near P236. Notably, the C2 carbonyl and adjacent NH of the uracil form hydrogen bonds with the K103 backbone NH and O. K102 adopts a unique rotamer conformation in which the NH3+ atom forms a hydrogen bond with the uracil O4 atom. The observed N-O distances for the hydrogen bonds are ca. 3.50 Å or less (Table S3). The multiple interactions with the uracil are consistent with the diminished activity observed for analogues with alternative heterocycles.1
Figure 2.

Stereo view of the crystal structure for 1 complexed with HIV-RT. Multiple contacts with residues in the NNBP are apparent; the dashed lines highlight the hydrogen bonds with K102 and K103. The corresponding illustration for 2 is nearly identical (Figure S3).
It may also be noted that in spite of the differences in the core structures of 1/2 and rilpivirine, the positioning of the cyanovinylphenyl groups is the same yielding beneficial contacts with W229. The effect is evident in comparing the activity of 1 in the T-cell assay (55 pM) versus that of its analogue with chlorine replacing the cyanovinyl group (20 nM).1 The high potency of 1 and 2 undoubtedly also includes contributions from the hydrogen bonds with K102 and K103.
Most significantly, the halocyanovinylphenyl (HCVP) group forms numerous van der Waals contacts in the tunnel region with P95 and W229, both conserved residues near or part of the primer grip.12, 13 Based on in vitro mutational and clinical studies, P95 and W229 are immutable residues in the NNBP essential for preserving reverse transcriptase activity.13–15 Thus, the HCVP group may be an advantageous moiety compensating for potential lost interactions with NNBP mutations conferring resistance.
Another intriguing notion raised by the modeling1 was the possible contribution of halogen bonding between the carbonyl oxygen of P95 and the chlorine atom in the terminal phenyl ring of 1 (Figure 3). Halogen bonding can occur between Lewis bases and chlorine, bromine, and iodine atoms.16 It features a favorable electrostatic interaction between the Lewis base, e.g., a carbonyl oxygen, and the backside of the halogen, which has a region depleted in electron density, a “σ hole”.16 For simple gas-phase complexes, halogen bonding becomes more favorable in progressing from Cl to Br to I. There is essentially perfect agreement between the crystal structure and the predicted structure for the complex of 1 (Figure 3 in ref. 1) including the conformations of the cyanovinyl and uracilylethoxy side chains and the hydrogen bonding with K103. However, an issue with the modeling is that classical force fields, which represent a carbonyl oxygen and an aryl chlorine as single particles with partial negative charges, cannot reproduce the attractive electrostatics of halogen bonds. This was subsequently remedied for the OPLS force fields by addition of a partial positive charge to represent the σ hole.17 FEP results then predicted relative free energies of binding (ΔGb) of 0.0, −3.7, and −3.5 kcal/mol for 3, 1, and 4 with HIV-RT and of −2.6 kcal/mol for the iodo analogue.17 Thus, the binding and activity are computed to peak with the chloro analogue 1. Addition of the partial positive charge on Cl was found to be favorable by 1 – 1.5 kcal/mol; however, a range of distances (3.6 – 4.5 Å) for the Cl-P95 halogen bond was sampled in the Monte Carlo FEP simulations for 1. This range is greater than the optimal separation of ca. 3.1 Å for PhCl, PhBr, or PhI interacting with an amide carbonyl oxygen.17–18
Figure 3.
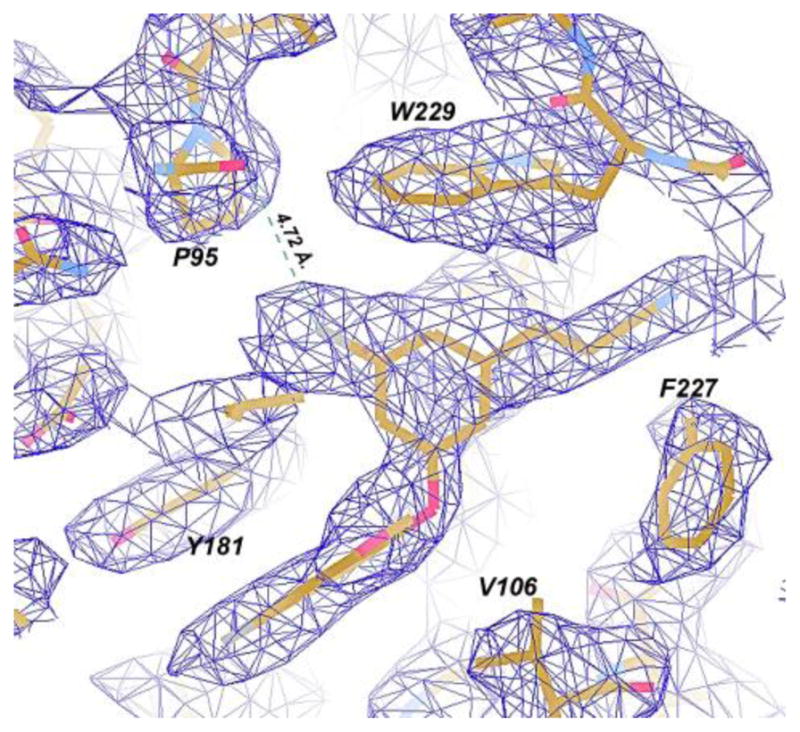
Omit Fo-Fc electron density (contour level at 3.0 σ) for the RT52A:1 complex. Dashed lines represent the interaction distance between the P95 carbonyl and Cl-group, which is 4.72Å.
We have now further addressed experimentally the halogen bonding issue. First, the bromo analogue 4 was synthesized and its activities were evaluated (Table 1). In general, the EC50 and IC50 results correlate well, e.g., 1 is more active than rilpivirine by a factor of 12 in both cases. For the catechol diether series (3, 1, and 4), the activities are indeed greatest for the chloro analogue; however, the fluoro analogue 3 is more active than the bromide 4 in contrast to the expectation from the computed ΔGb values.16 The issue could be further addressed with a direct binding assay. However, a notable point is that if halogen bonding is operative, the I > Br > Cl >F order is not guaranteed since the heavier halogens do require more space, which can lead to greater entropic penalties on complexation. Secondly, the crystallography has provided a value for the Cl – P95 O separation of 4.72 Å (Figure 3), with a coordinate uncertainty of ± 0.41 Å, which is on the high edge of the range sampled in the FEP simulations. The C-Cl…O angle of 132° is also significantly bent from the ideal linear geometry.16–18 While a strong halogen bond is not observed, there may be some electrostatic benefit from the Cl – P95 O interaction for 1.
In summary, crystal structures for the extraordinarily potent anti-HIV agents 1 and 2 show striking complementarity between the conformation of the inhibitors and the NNBP with substantial extensions into the three channels that characterize the binding site. The activity of the catechol diethers has been further assessed by determination of the inhibition of the activity of HIV-RT, which was found to parallel the cell-based results and to confirm the extreme potency of 1. The contribution to the activity of 1 from halogen bonding has received attention from the structural analyses and addition of data for the bromo analogue 4. The crystallography shows that the halogen bonding is not optimal for the present compounds, which raises the intriguing possibility of designing even more potent agents with favorable resistance profiles. From this analysis, a design strategy emerges to improve interactions with P95 and maintain interactions with W229, both residues important for the assembly and activity of HIV-RT and are thus not prone to mutation.13–15
Supplementary Material
Acknowledgments
Gratitude is expressed to the National Institutes of Health (AI44616, GM32136, GM49551) for support and to Prof. Eddy Arnold for discussions.
Footnotes
No competing financial interests have been declared.
The two structures have been deposited in the RCSB Protein Data Bank with codes 4H4M and 4H4O.
Full details on the crystallography, synthesis and characterization of 4, and enzymatic assays are provided. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Bollini M, Domaoal RA, Thakur VV, Gallardo-Macias R, Spasov KA, Anderson KS, Jorgensen WL. J Med Chem. 2011;54:8582. doi: 10.1021/jm201134m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauman JD, Das K, Ho WC, Baweja M, Himmel DM, Clark AD, Jr, Oren DA, Boyer PL, Hughes SH, Shatkin AJ, Arnold E. Nucleic Acids Res. 2008;36:5083. doi: 10.1093/nar/gkn464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCoy AJ. Acta Crystallogr D Biol Crystallogr. 2007;63:32. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das K, Clark AD, Jr, Lewi PJ, Heeres J, De Jonge MR, Koymans LM, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B, Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R, De Bethune MP, Boyer PL, Clark P, Hughes SH, Janssen PA, Arnold E. J Med Chem. 2004;47:2550. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 6.Das K, Bauman JD, Clark AD, Jr, Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. Proc Natl Acad Sci USA. 2008;105:1466. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Himmel DM, Das K, Clark AD, Jr, Hughes SH, Benjahad A, Oumouch S, Guillemont J, Coupa S, Poncelet A, Csoka I, Meyer C, Andries K, Nguyen CH, Grierson DS, Arnold E. J Med Chem. 2005;48:7582. doi: 10.1021/jm0500323. [DOI] [PubMed] [Google Scholar]
- 8.Kertesz DJ, Brotherton-Pleiss C, Yang M, Wang Z, Lin X, Qiu Z, Hirschfeld DR, Gleason S, Mirzadegan T, Dunten PW, Harris SF, Villasenor AG, Hang JQ, Heilek GM, Klumpp K. Bioorg Med Chem Lett. 20:4215. doi: 10.1016/j.bmcl.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 9.Ren J, Esnouf RM, Hopkins AL, Warren J, Balzarini J, Stuart DI, Stammers DK. Biochemistry. 1998;37:14394. doi: 10.1021/bi981309m. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Smerdon SJ, Jager J, Kohlstaedt LA, Rice PA, Friedman JM, Steitz TA. Proc Natl Acad Sci USA. 1994;91:7242. doi: 10.1073/pnas.91.15.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ekkati AR, Bollini M, Domaoal RA, Spasov KA, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2012;22:1565. doi: 10.1016/j.bmcl.2011.12.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia Q, Radzio J, Anderson KS, Sluis-Cremer N. Protein Sci. 2007;16:1728. doi: 10.1110/ps.072829007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Auwerx J, Van Nieuwenhove J, Rodriquez-Barrios F, de Castro S, Velázquez S, Ceccherini-Silberstein F, De Clerq E, Camarasa MJ, Perno CF, Gago F, Balzarini J. FEBS Lett. 2005;579:2294. doi: 10.1016/j.febslet.2005.02.077. [DOI] [PubMed] [Google Scholar]
- 14.Pelemans H, Esnouf R, De Clercq E, Balzarini J. Mol Pharmacol. 2000;57:954. [PubMed] [Google Scholar]
- 15.Ceccherini-Silberstein F, Gago F, Santoro M, Gori C, Svicher V, Rodriguez-Barrios F, d’Arrigo R, Ciccozzi M, Bertoli A, d’Arminio Monforte A, Balzarini J, Antinori A, Perno CF. J Virol. 2005;79:10718. doi: 10.1128/JVI.79.16.10718-10729.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For reviews, see: Politzer P, Murray JS, Clark T. Phys Chem Chem Phys. 2010;12:7748. doi: 10.1039/c004189k.Metrangolo P, Murray JS, Pilati T, Politzer P, Resnati G, Terraneo G. Cryst Growth Des. 2011;11:4238.
- 17.Jorgensen WL, Schyman P. J Chem Theory Comput. 2012;8:3895. doi: 10.1021/ct300180w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardegger LA, Kuhn B, Spinnler B, Anselm L, Ecabert R, Stihle M, Gsell B, Thoma R, Diez J, Benz J, Plancher JM, Hartmann G, Banner DW, Haap W, Diederich F. Angew Chem Int Ed. 2011;50:314. doi: 10.1002/anie.201006781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.