

Abstract
Innate cell-autonomous antiviral responses are essential first lines of defense against central nervous system infections but may also contribute to neuropathogenesis. We investigated the relationships between innate immunity and neuronal differentiation using an in vitro culture system with human cell lines to analyze cellular responses to the neurotropic alphavirus western equine encephalitis virus. Human neuronal cells displayed a maturation-dependent reduction in virus-induced cytopathology that was independent of autocrine interferon α or β activity. In addition, maturation was associated with enhanced responsiveness to exogenous stimuli, such that differentiated neurons required five- to ten-fold less type I interferon to suppress viral replication or virus-induced cytopathology compared to immature cells, although this enhanced responsiveness extended to only a subset of unique type I interferons. These results demonstrate that maturation-dependent changes in human neuronal cells may be key determinants in the innate immune response to infections with neurotropic alphaviruses.
Keywords: Neurons, Differentiation, Alphavirus, Interferon
Introduction
The innate antiviral immune response is a crucial mechanism to control viral replication prior to the induction of adaptive immunity (Hoebe et al., 2004). Numerous studies have demonstrated that both mice (Aguilar et al., 2005; Grieder and Vogel, 1999; Muller et al., 1994; Ryman et al., 2000; van den Broek et al., 1995) and humans (Casrouge et al., 2006; Dupuis et al., 2003; Zhang et al., 2007) with deficient type I interferon (IFN) responses, a prominent component of the innate immune system (Samuel, 2001), are dramatically more susceptible to serious and often lethal viral infections. Despite its critical role in defense against viral infections, the inappropriate activation of innate immunity in particular cellular microenvironments can also have catastrophic effects on the host. For example, type I IFN-mediated responses can result in cell death as a method to control viral replication or dissemination, but this normally protective response is detrimental when target cells have essential functions but limited renewal capacity, such as mature central nervous system (CNS) neurons (Horner and Gage, 2000). Nevertheless, protection and recovery from CNS infections with neurotropic viruses also depends upon a functional innate immune system (Aguilar et al., 2005; Grieder and Vogel, 1999; Ryman et al., 2000), suggesting that cell type-specific responses may be important determinants of viral pathogenesis (Daffis et al., 2007; Kato et al., 2005; Ma et al., 2006).
To study the interactions between innate immunity and viral pathogenesis within the CNS we use the neurotropic alphavirus western equine encephalitis virus (WEEV), a mosquito-borne enveloped RNA virus that belongs to the Togaviridae family. The neurotropic alphaviruses, which include WEEV and the closely related eastern and Venezuelan equine encephalitis viruses, cause sporadic and epidemic equine and human CNS infections (Deresiewicz et al., 1997; Earnest et al., 1971). The virulent neurotropic alphaviruses are also important members of the growing list of emerging or resurging global public health threats (Gubler, 2002), and are listed as CDC and NIAID category B bioterrorism agents due in part to their neurovirulence, dissemination potential, and lack of effective therapies (Sidwell and Smee, 2003). A detailed understanding of the host-pathogen interactions that occur within the CNS during innate immune responses to neurotropic alphavirus infections may reveal novel targets for the rational design of antiviral drugs, and may also reveal additional genetic determinants that influence susceptibility to viral neuropathogenesis (Casrouge et al., 2006; Dupuis et al., 2003; Zhang et al., 2007).
The molecular mechanisms of alphavirus replication and pathogenesis have been studied extensively with Sindbis virus (SINV), which in humans causes a systemic infection characterized by fever, rash, and arthritis, whereas in mice it infects neurons and produces encephalitis (Griffin, 2001). The alphavirus genome is an 11 to 12-kb single-stranded positive-sense RNA with a 5′ terminal cap and 3′ polyadenylated tail. Genomic RNA is translated into a polyprotein that undergoes regulated autocatalytic processing to form the four nonstructural replicase proteins (nsPs) 1 through 4. During replication via a negative-strand genomic intermediate, alphaviruses produce a 4-kb subgenomic RNA that encodes the structural capsid protein and envelope glycoproteins. SINV infection in most cultured mammalian cells is accompanied by the rapid inhibition of host cell RNA and protein synthesis and eventual cell death (Gorchakov et al., 2005). In contrast, infection of mature rodent CNS neurons can lead to a non-cytopathic persistent infection (Burdeinick-Kerr and Griffin, 2005; Vernon and Griffin, 2005), similar to the response of infected mosquito cells in culture (Griffin, 2001).
Despite the substantial amount of information regarding innate immunity in non-neuronal cells, we have only limited knowledge regarding the initiation, amplification, and effector mechanisms of neuron-specific innate antiviral responses. Gene expression studies have demonstrated the upregulation of multiple IFN-stimulated genes (ISGs) in response to neurotropic virus infections or type I IFN stimulation in vitro and in vivo (Johnston et al., 2001; Labrada et al., 2002; Ousman et al., 2005; Prehaud et al., 2005), and have suggested that neurons may respond differently than non-neuronal cells to type I IFNs (Wang and Campbell, 2005). Furthermore, directed in vivo overexpression of IFNα in the CNS is neurotoxic despite its simultaneous role in protecting against lethal neurotropic viral infections (Akwa et al., 1998). These observations suggest that CNS neurons may employ distinct mechanisms that balance innate antiviral responses to both control virus replication and prevent immune-mediated cellular damage.
In this report we describe the impact of cellular differentiation on the responses of cultured human neuronal cells to both infection with WEEV and stimulation with exogenous type I IFNs. Although neuronal maturity is a well-recognized determinant in the pathogenesis of SINV-induced disease in mice (Labrada et al., 2002; Lewis et al., 1996; Trgovcich et al., 1999), the role of neuronal differentiation in cell-autonomous innate immune responses and type I IFN stimulation remains poorly defined, particularly in the context of infections with virulent neurotropic alphaviruses such as WEEV. We used three different human neuronal cell lines and found a direct correlation between cellular differentiation and partial resistance to WEEV-induced cytopathology, and that maturation-dependent resistance was independent of autocrine type I IFN activity. Furthermore, neuronal differentiation also correlated with heightened responses to type I IFNs, such that five- to ten-fold lower doses were required to induce an antiviral state, although these enhanced responses extended to only a subset of type I IFNs. These results suggest that neuronal maturity is a critical determinant in the cellular response to virulent neurotropic alphavirus infections in humans, and that the process of neuronal differentiation induces changes that alter innate immune response pathway activity.
Results
Human BE(2)-C cells differentiated with retinoic acid (RA) display characteristics of mature neuronal cells and remain susceptible to WEEV infection
We sought to develop a readily available and reproducible in vitro culture model to study cellular differentiation and WEEV pathogenesis in human neurons (Fig. 1). We chose to use the BE(2)-C neuroblastoma cell line, a clone of SK-N-BE cells that has phenotypic characteristics of immature neurons (Peverali et al., 1996), and cultured cells in the presence of 10 μM RA for three weeks to induce differentiation. Immature BE(2)-C cells had a polygonal morphology and grew as a homogeneous monolayer, whereas RA-differentiated cultures contained primarily cells with small perikarya and extensive processes that resembled neuronal axons and dendrites, but also contained a smaller fraction of flattened cells with larger cell bodies and short indistinct processes (Fig. 1A). Similar heterogeneous phenotypes have been described with other RA-differentiated human neuroblastoma cell lines (Encinas et al., 2000; Hill and Robertson, 1997), and potentially represent a transdifferentiation process due to the multipotential nature of neural crest cells from which most neuroblastomas are thought to arise.
Fig. 1.

Differentiated BE(2)-C human neuroblastoma cells are susceptible to WEEV infection. (A) Light microscopy images of BE(2)-C cells cultured in the absence or presence of 10 μM RA for three weeks. (B) Lysates from an equivalent number of control A549 human lung carcinoma cells (lane 1) or BE(2)-C cells cultured in the absence (lane 2) or presence (lane 3) of 10 μM RA were immunoblotted for expression of the neuronal lineage marker NF68, the mature neuronal marker synaptophysin (Synap), or the loading control glyceraldehyde-3-phosphate dehydrogenase (GAPDH). (C) Proliferation of immature (closed circles) and RA-differentiated (open circles) BE(2)-C cells. Cell density was measured by MTT assay and is expressed as the fold-increase from baseline. Initial experiments demonstrated a direct correlation between cell number and MTT signal for both immature and differentiated cells (data not shown). (D) Immunofluorescence microscopy of cells infected with WEEV at an MOI of 10 and processed at 12 hpi. Mock or WEEV-infected immature (upper two rows) or differentiated (lower row) BE(2)-C cells were immunostained with mouse monoclonal antibodies against WEEV virions (middle column) or rabbit polyclonal antibodies against nsP1 (right column). The nuclear stain DAPI is shown in the left column. The arrow indicates an infected differentiated BE(2)-C cell with a small perikaryon and extensive processes characteristic of mature neurons, whereas the arrowhead indicates an infected cell with a larger cell body and shorter processes. (E) Analysis of WEEV nsP and RNA accumulation in infected cells. Lysates from immature (lanes 1-4) and differentiated (lanes 5-8) BE(2)-C cells infected with WEEV at an MOI of 10 were prepared at 6, 12, and 24 hpi and analyzed by immunoblotting with polyclonal rabbit antibodies against WEEV nsPs or monoclonal mouse antibodies against GAPDH. Total RNA from cells described above was analyzed by Northern blotting with a strand-specific 32P-labeled riboprobe that detected both positive-sense WEEV genomic (g) and subgenomic (sg) RNA. The positions of 28S and 16S rRNA are shown on the left, and the ethidium bromide-stained 28S rRNA band in shown as a loading control. Note the detection of the abundant 26S subgenomic WEEV RNA by ethidium bromide staining (arrow). (F) Infectious WEEV production. We infected an equal number of immature (closed circles) and differentiated (open circles) BE(2)-C cells with WEEV at an MOI of 10, harvested supernatants at 6, 12, 24, and 48 hpi, and measured infectious virus titers by plaque assay on Vero cell monolayers. Plaque assay sensitivity was 102 plaque-forming units per ml.
We characterized the BE(2)-C human neuronal culture system using immunoblotting to examine the expression of neurofilament (NF) 68, a neuronal lineage low molecular weight intermediate filament protein, and synaptophysin, a synaptic vesicle protein and marker of mature differentiated neurons (Fig. 1B). We detected NF68 expression in both immature and RA-differentiated BE(2)-C cells but not in control A549 human lung carcinoma cells, whereas we detected synaptophysin only in RA-differentiated BE(2)-C cells (Fig. 1B, lane 3). Immunofluorescence analysis confirmed the expression of synaptophysin in vesicle-like structures around the cell periphery and the filamentous expression of NF200, a high molecular weight intermediate filament isoform present in mature neurons, in RA-differentiated but not in immature cells (data not shown). Finally, we examined the proliferation of immature and RA-differentiated BE(2)-C cells (Fig. 1C). Differentiation with RA for three weeks significantly decreased cellular proliferation, such that the number of differentiated cells increased less than two-fold over 72 h, whereas immature cells increased almost five-fold over the same time period. Similar results were obtained when we measured 3H-thymidine incorporation over 24 h (data not shown). These results indicated that RA-differentiated BE(2)-C cells had morphological and biochemical characteristics of mature human neuronal cells in culture.
To test the susceptibility of BE(2)-C human neuroblastoma cells to WEEV we generated infectious virions corresponding to strain Cba 87 and examined infected cells by immunofluorescence microscopy (Fig. 1D). After viral infection with a multiplicity of infection (MOI) of 10, greater than 90% of both immature and differentiated BE(2)-C cells expressed WEEV structural proteins (Fig. 1D, middle column) or nsP1 (Fig. 1D, right column) at 12 h post-infection (hpi). In RA-differentiated cultures, both morphologically distinguishable cells were infected (Fig. 1D, bottom row). We also analyzed viral nsP and RNA accumulation in cells up to 24 hpi by western and Northern blotting, respectively (Fig. 1E). WEEV nsP accumulation was evident in both immature and differentiated cells by 6 hpi and corresponded to the appearance of detectable viral genomic and subgenomic RNA (Fig. 1E, lanes 2 and 6). There were no maturation-dependent differences in the maximal accumulation levels of viral nsPs or RNA, although their temporal appearance was slightly delayed in differentiated cells. We obtained similar results with the prototypic Fleming strain of WEEV (data not shown).
We further examined WEEV infection in immature and differentiated BE(2)-C cells by analyzing infectious virion production in a single-step growth assay (Fig. 1F). Virion production after infection with an MOI of 10 plateaued around 12 to 24 hpi in both cell types with maximal titers of approximately 2 × 105 plaque-forming units per ml. Although there was a difference in infectious virion production at 48 hpi between immature and differentiated cells, this observation likely reflected the significant differences in cell viability at this late stage after infection (see below, Fig. 2). Infections with an MOI of 0.1 produced a slight kinetic delay in virion production but no significant change in maximal titers at 24 hpi and did not result in a difference between immature and differentiated BE(2)-C cells (data not shown). We obtained similar results with the Fleming strain of WEEV, although maximal virus titers at 24 hpi were approximately 30-fold higher (data not shown). These results indicated that BE(2)-C cells were highly susceptible to WEEV infections and suggested that neuronal differentiation did not significantly alter WEEV susceptibility and viral replication during the first 24 h after infection.
Fig. 2.
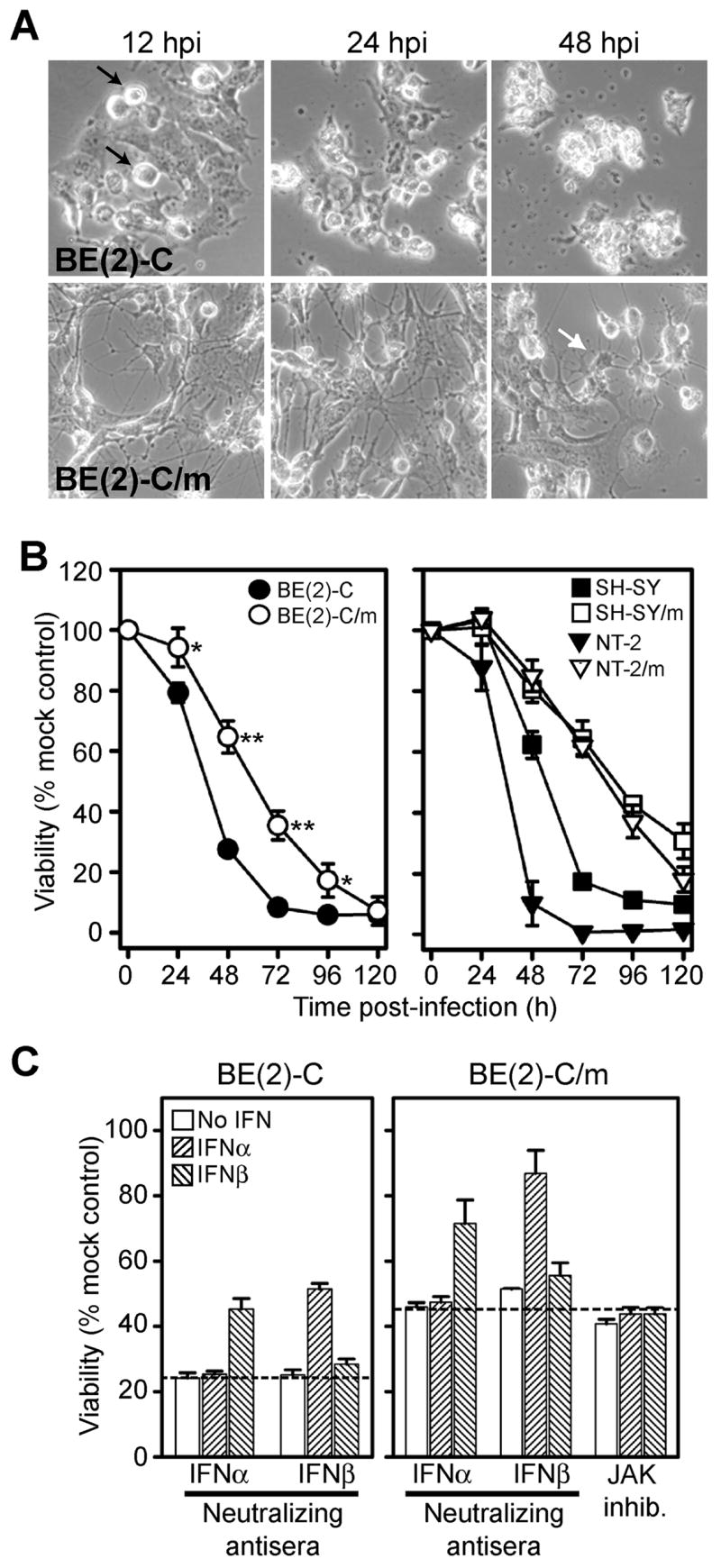
Neuronal differentiation correlates with reduced WEEV-induced cytopathology independent of autocrine IFNα or β activity. (A) Light microscopy images of immature (upper row) and differentiated (lower row) BE(2)-C cells infected with WEEV at an MOI of 0.1. Note the appearance of rounded and pyknotic cells by 12 hpi in immature cells (black arrows), whereas viable cells with morphological features of mature neurons are present even at 48 hpi in differentiated BE(2)-C cells (white arrow). (B) Cell viability in immature (closed symbols) and differentiated (open symbols) BE(2)-C, SH-SY5Y, and NT-2 cells infected with WEEV at an MOI of 0.1 was quantitated by MTT assay. Similar results were obtained with BE(2)-C cells using an MOI of 10 (data not shown). Results were normalized to uninfected control cell viability (*p < 0.005; **p < 0.000005 compared to immature cells). (C) Immature (left graph) and differentiated (right graph) BE(2)-C cells were treated with 20 U per ml IFNα or 10 U per ml IFNβ in the presence of 200 U per ml neutralizing antisera against human IFNα, IFNβ or 5 μM JAK inhibitor, infected with WEEV at an MOI of 0.1, and viability was determined by MTT assay at 48 or 72 hpi for immature and differentiated cells, respectively. JAK inhibitor results are shown only for differentiated BE(2)-C cells, although we obtained similar results with immature cells (data not shown). Results were normalized to uninfected control cell viability, and the hatched lines indicate the percent viability of infected cells treated with control normal rabbit serum.
Differentiation of human neuronal cells reduces sensitivity to WEEV-induced cytopathology independent of autocrine IFNα or β activity
We initially investigated whether human neuronal cells showed maturation-dependent cellular responses to WEEV infection by examining virus-induced cytopathic effect (CPE) (Fig. 2). Despite the similar levels of virion production and viral nsP or RNA accumulation between immature and differentiated BE(2)-C cells during the first 24 hpi (Figs. 1E and F), neuronal maturation had a significant impact on the cellular response to virus infection. WEEV-induced CPE was visually evident in immature BE(2)-C cells by 12 hpi and was characterized by the appearance of rounded and pyknotic cells (Fig. 2A, upper left image). Significant CPE had developed in immature cells by 24 hpi, and essentially no viable-appearing cells remained at 48 hpi (Fig. 2A upper right image). In contrast, although CPE was evident in differentiated cells after WEEV infection, the intensity of the response was reduced such that viable cells were still visible at 48 hpi (Fig. 2A, lower right image). When we quantitated cell viability after virus infection by 3-[4,5-dimethylthizol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay, there was a significant decrease in virus-induced CPE in differentiated compared to immature cells up to 96 hpi (Fig. 2B, left graph). Although RA-induced neuronal maturation of BE(2)-C cells was unable to completely abrogate WEEV-mediated CPE, as cell viability at 120 hpi was less than 10% with both cell types, these results indicated that human neuronal cell maturation in vitro was accompanied by an increased resistance to WEEV-induced CPE.
The culture conditions that we used to differentiate BE(2)-2 cells allowed us to directly compare immature and mature neuronal cells grown in similar serum-containing media, with the notable exception of RA. However, RA-differentiated BE(2)-C cultures were not morphologically homogeneous and failed to completely exit the cycle cycle (see Figs. 1A and C), and thus we also used an alternative differentiation protocol that involved a shortened exposure to RA and subsequent treatment with brain-derived neurotropic growth factor (BDNF). This approach has been used with other human neuroblastoma cells to produce BDNF-dependent mature neuronal cells that are post-mitotic but remain viable and functional in RA- and serum-free media (Encinas et al., 2000). We were similarly able to generate morphologically homogenous and non-proliferating cultures with BE(2)-C cells using a sequential RA-BDNF protocol (data not shown). These cultures also showed reduced sensitivity to WEEV-induced CPE with 78% viability at 48 hpi. Although the culture conditions used to produce BDNF-dependent differentiated BE(2)-C cells did not allow us to have a directly comparable immature cell population, these results were consistent with those obtained using RA-only differentiated cells (Fig. 2B, left graph), suggesting that culture heterogeneity or differentiation conditions did not have a significant impact on the maturation-dependent cytopathic responses of BE(2)-C cells to WEEV infection.
Although immortalized cell lines are commonly used as tools to investigate viral pathogenesis, individual transformed cell lines may display aberrant phenotypes. Thus, to further examine the impact of human neuronal maturation on WEEV-induced CPE we analyzed cell viability after virus infection of immature and differentiated cells using two additional independently-derived human cell lines (Fig. 2B, right graph). The SH-SY5Y cell line is a human neuroblastoma that also differentiates into cells with a mature neuronal phenotype in response to RA treatment (Encinas et al., 2000), whereas the NT-2 cell line is a human embryonal carcinoma that differentiates into mature neuronal cells in response to RA, mitotic inhibitors, and selective plating conditions (Pleasure et al., 1992). Differentiated SH-SY5Y and NT-2 cells both displayed phenotypes consistent with mature neuronal cells, including the presence of multiple processes reminiscent of axons and dendrites, expression of mature neuronal markers, and the absence of proliferation over 72 hours (data not shown). When we examined cellular responses to WEEV infection, both differentiated SH-SY5Y and NT-2 cells showed a significant reduction in virus-induced CPE compared to immature cells (Fig. 2B, right graph), similar to results with differentiated BE(2)-C cells (Fig. 2B, left graph), strengthening the conclusion that a direct correlation existed between human neuronal maturity and partial but not complete resistance to virulent neurotropic alphavirus-induced cytopathology.
Autocrine or paracrine type I IFN-mediated amplification of innate immune responses is an important component of non-neuronal cellular responses to virus infection (Samuel, 2001). One hypothesis to explain the maturation-dependent responses of cultured human neuronal cells to WEEV infection is that RA-induced differentiation modulated type I IFN-mediated autocrine activity. Although neurons are generally not considered to be primary producers of type I IFNs during innate immune responses, differentiated human neuronal cells can produce IFNβ in culture (Prehaud et al., 2005), and in vivo data suggest that rodent CNS neurons can also produce type I IFNs during viral encephalitis (Delhaye et al., 2006). We initially examined IFNα and β levels in supernatants of cultured immature and differentiated BE(2)-C cells after WEEV infection by both bioassays and commercially available ELISAs but were unable to detect appreciable levels of either type I IFN (data not shown). We subsequently used antibody neutralization experiments to examine the presence of functional type I IFNs secreted after WEEV infection (Fig. 2C). Immature and differentiated BE(2)-C cells were infected with WEEV at an MOI of 0.1 and simultaneously treated with neutralizing antiserum against human IFNα, IFNβ, or control normal rabbit serum. As positive controls, we treated infected cells with either purified recombinant human IFNα or β in combination with a 10- to 20-fold excess of neutralizing antiserum. We measured viability at 48 and 72 hpi for immature and differentiated cells, respectively, to ensure that baseline viability in the absence of treatment was approximately 30% (see Fig. 2B, left graph). Control samples treated with either exogenous IFNα or β in the absence of the appropriate neutralizing antibody showed a significant reduction in WEEV-induced CPE with both immature (Fig. 2C, left graph) and differentiated (Fig. 2C, right graph) BE(2)-C cells, which was reversed in the presence of the appropriate neutralizing antibody. However, in the absence of exogenous IFNα or β neither antiserum had a significant impact on WEEV-induced CPE irrespective of cellular maturation. In particular, differentiated BE(2)-C cells showed no decrease in viability with IFNα or β neutralization after WEEV infection (Fig. 2C, right graph). Furthermore, treatment of differentiated BE(2)-C cells with a cell-permeable inhibitor of Janus tyrosine kinases (JAKs), which are essential components of the type I IFN-mediated signaling cascade (Schindler et al., 2007), had minimal effect on WEEV-induced CPE in the absence of exogenous type I IFNs (Fig. 2C, right graph). These results suggested that autocrine activity of IFNα or β played a minor if any role in the neuronal maturation-dependent responses of BE(2)-C cells to WEEV infection. However, the results with control IFNα or β also indicated that both immature and differentiated BE(2)-C human neuronal cells could respond to exogenous type I IFNs and activate cellular pathways that reduced WEEV-induced CPE.
Differentiated human neuronal cells display a heightened response to exogenous type I IFN stimulation
We further examined the effects of exogenous type I IFNs on WEEV infection in BE(2)-C cells using IFNα-A/D, a recombinant hybrid universal type I IFN (Fig. 3). Although type I IFNs have antiproliferative activities and can induce apoptosis in some malignant cell lines, treatment of immature or differentiated BE(2)-C cells with 50 U per ml IFNα-A/D for 72 h resulted in 94% and 97% viability compared to untreated cells, respectively, and titration studies indicated that concentrations up to 500 U per ml had minimal impact on cell viability (data not shown). However, IFNα-A/D significantly reduced WEEV-induced CPE at 48 hpi in both immature and differentiated cells (Fig. 3A). This reduction in CPE was evident irrespective of whether cells were pretreated for 24 h prior to infection with an MOI of 0.1 or given a single 50 U per ml dose of IFNα-A/D at the time of infection, although immature cells showed an attenuated response to the pretreatment compared to co-treatment strategy. Furthermore, only the co-treatment strategy effectively suppressed WEEV-induced CPE in immature BE(2)-C cells when we measured viability at 72 hpi (Fig. 3A, left graph), whereas both treatment strategies were equally effective in differentiated cells (Fig. 3A, right graph). We obtained similar results when we used both a higher inoculum (MOI = 10) or the Fleming strain of WEEV (data not shown). We also examined the suppressive activity of IFNα-A/D in WEEV-infected BE(2)-C cells by analyzing infectious virion production (Fig. 3B). Regardless of the initial inoculum, pretreatment with IFNα-A/D suppressed infectious WEEV production at 24 hpi by approximately 100-fold in both immature and differentiated cells. However, the protective effects of IFNα-A/D pretreatment on infectious virus production diminished in immature cells by 48 hpi (Fig. 3B, left graph), whereas virus titers were still suppressed by greater than 10-fold at this time point in differentiated cells (Fig. 3B, right graph). These results indicated that BE(2)-C human neuronal cells responded to exogenous type I IFNs to activate cell-autonomous antiviral responses that reduced virion production and prevented WEEV-induced CPE. Furthermore, they suggested that RA-induced differentiation augmented these responses in part by prolonging their activity after type I IFN stimulation.
Fig. 3.

Type I IFNs suppress WEEV-induced CPE and virion production in human neuronal cells. (A) WEEV-induced CPE. Immature (left graph) and differentiated (right graph) BE(2)-C cells were either pretreated (pre-Rx) with 50 U per ml IFNα-A/D for 24 h and infected with WEEV at an MOI of 0.1, or treated with a single 50 U per ml IFNα-A/D dose immediately after virion attachment (co-Rx), and cell viability was determined at 48 and 72 hpi by MTT assay. Results were normalized to uninfected control cell viability. (B) Infectious virion production. Immature (left graph) and differentiated (right graph) BE(2)-C cells were pretreated with 50 U per ml IFNα-A/D for 24 h, infected with WEEV at an MOI of 10 or 0.1, and virus titers were determined in culture supernatants at 24 and 48 hpi by plaque assay on Vero cell monolayers. The data from untreated cells represent combined results from both high (10) and low (0.1) MOI infections, as there was no significant effect of viral inoculum at 24 or 48 hpi (data not shown) (*p < 0.05; **p < 0.0005 compared to untreated cells).
To further investigate the impact of neuronal differentiation on type I IFN responsiveness we conducted dose-titration experiments (Fig. 4). Cells were pretreated for 24 h with decreasing concentrations of IFNα-A/D, infected with WEEV at an MOI of 0.1, and viral nsP (Fig. 4A) and RNA (Fig. 4B) levels were analyzed at 24 hpi by quantitative immunoblotting and Northern blotting, respectively. Both immature and differentiated BE(2)-C cells showed dose-dependent responses to IFNα-A/D that resulted in the suppression of WEEV nsP and RNA accumulation. However, RA-induced neuronal differentiation resulted in significantly increased sensitivity to type I IFN stimulation. We calculated the IFNα-A/D doses that produced 50% inhibition (IC50) of WEEV-induced CPE, virion production, and viral nsP and RNA accumulation in both immature and differentiated cells (Table 1). Differentiated BE(2)-C cells had five- to ten-fold lower IC50 values for all parameters compared to immature cells. We also conducted limited dose-titration experiments with BDNF-dependent mature BE(2)-C cells and obtained an IC50 of approximately 5 U per ml for reduction in WEEV-induced CPE, similar to the dose required for RA only-differentiated BE(2)-C cells (Table 1). Furthermore, we conducted dose-titration experiments with immature and differentiated SH-SY5Y and NT-2 cells, and found that both cell lines also displayed maturation-dependent enhancement of type I IFN responses, such that IC50 values for reduction in WEEV-induced CPE were approximately ten- to twenty-fold lower in differentiated compared to immature cells (Table 1). These results indicated that cellular differentiation had a significant impact on type I IFN responsiveness to produce enhanced antiviral activity in cultured human neuronal cells.
Fig. 4.
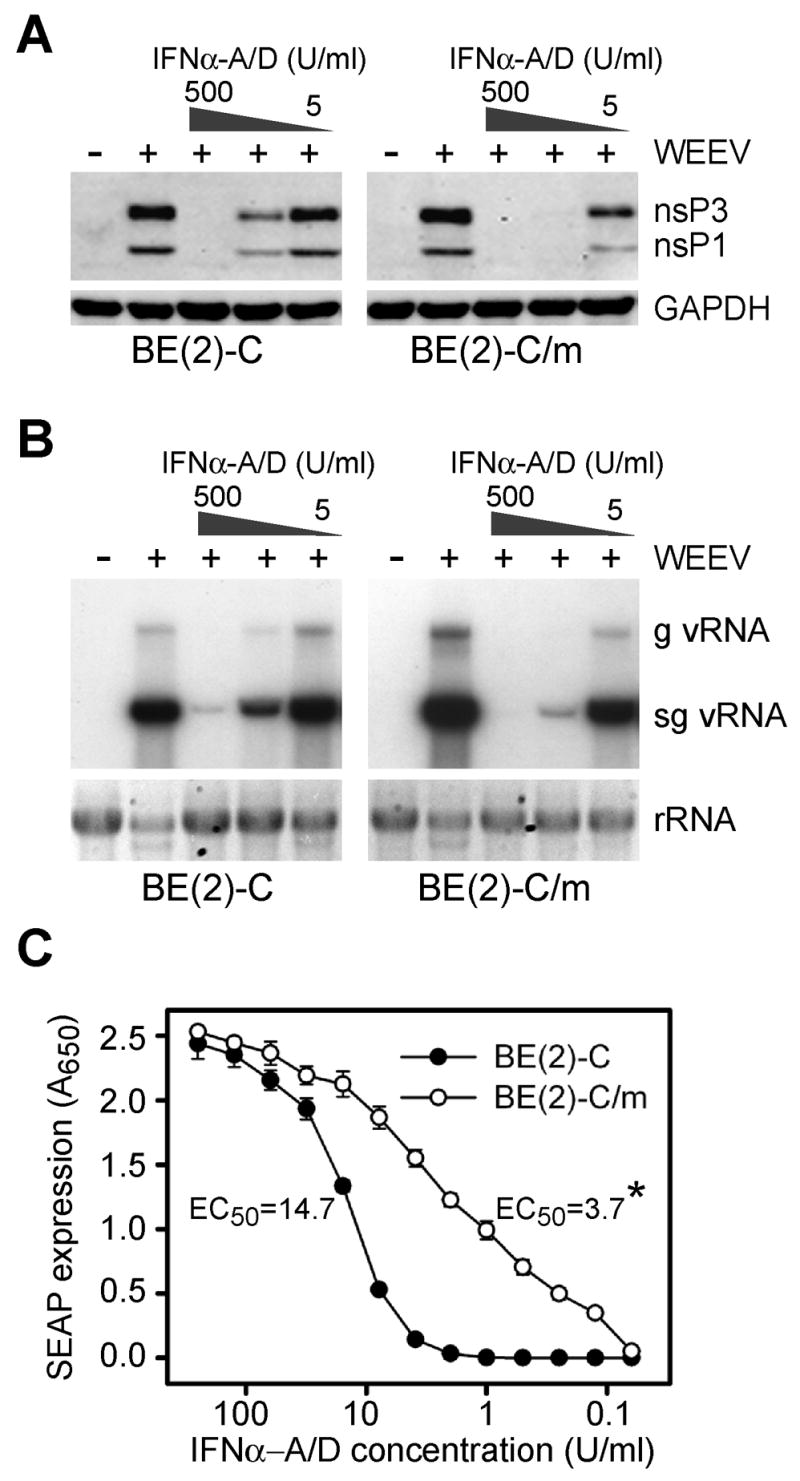
Neuronal differentiation is associated with enhanced responsiveness to type I IFN stimulation. Immature and differentiated BE(2)-C cells were pretreated with decreasing concentrations of IFNα-A/D for 24 h, infected with WEEV at an MOI of 0.1, and samples were analyzed at 24 hpi for WEEV nsP (A) and RNA (B) accumulation by immunoblotting and Northern blotting, respectively, as described in Fig. 1. (C) Dose response of immature (closed circles) or differentiated (open circles) BE(2)-C cells stably transfected with pISRE-SEAP. Reporter gene activity was measured at 24 h after IFNα-A/D stimulation. Calculated concentrations that produced a 50% maximal response in reporter gene activity (EC50) are shown (*p < 0.01 compared to immature BE(2)-C cells).
Table 1.
IFNα-A/D IC50 values with WEEV-infected immature and differentiated human neuronal cells.
| IFNα-A/D IC50 (U/ml)a | |||
|---|---|---|---|
| Cell line | Parameterb | Immature | Differentiated |
| BE(2)-C | Viabilityc | 75 ± 8 | 6 ± 1 |
| Virion production | 59 ± 10 | 7 ± 5 | |
| Viral nsP accumulationd | 28 ± 7 | 3 ± 1 | |
| Viral RNA accumulatione | 56 ± 13 | 12 ± 5 | |
| SH-SY5Y | Viability | 156 ± 7 | 17 ± 3 |
| NT-2 | Viability | >250 | 15 ± 1 |
Results represent the mean ± SEM from three independent experiments except viability values in differentiated NT-2 cells, which were obtained from two independent experiments.
We determined virion production, viral nsP, and viral RNA accumulation values at 24 hpi. The time point for viability determination corresponded to approximately 30 to 40% viability in the absence of IFNα-A/D, which was 48 hpi for immature NT-2 and BE(2)-C cells, 48 and 72 hpi for immature SH-SY5Y cells, 72 hpi for differentiated BE(2)-C cells, and 72 and 96 hpi for differentiated SH-SY5Y and NT-2 cells.
Viability IC50 values for immature and differentiated BE(2)-C cells corresponded to 750 and 60 pg/ml, respectively, based on the IFNα-A/D specific activity of 108 U/mg listed by the manufacturer as determined by antiviral effects against vesicular stomatitis virus in Madin-Darby bovine kidney cells.
Total nsP1 and 3 accumulation assessed by immunoblotting (see Fig. 4A).
Genomic and subgenomic RNA accumulation assessed by Northern blotting (see Fig. 4B).
To initially investigate the underlying mechanisms responsible for the maturation-dependent enhancement of neuronal type I IFN stimulation from receptor engagement to ISG upregulation in BE(2)-C cells, we used a secreted alkaline phosphatase (SEAP) reporter gene linked to a minimal ISG promoter element (Fig. 4C). To avoid the potential confounding effects of cellular differentiation on transfection efficiency, we generated BE(2)-C cells stably transfected with the reporter plasmid and subsequently induced differentiation with RA. Both immature and differentiated BE(2)-C cells with an integrated type I IFN-responsive reporter gene displayed dose-dependent SEAP expression in response to IFNα-A/D stimulation. However, the IFNα-A/D concentration that produced a 50% maximal response differed by four-fold between immature and differentiated cells (Fig. 4C). Furthermore, the shapes of the titration curves differed substantially such that the calculated Hill slopes for immature and differentiated BE(2)-C cells were 1.55 and 0.57, respectively (p < 0.05). Similar results were obtained with immature and differentiated SH-SY5Y cells that stably expressed the IFN-responsive SEAP reporter gene (data not shown). These results indicated that neuronal differentiation enhanced type I IFN signaling pathway activation and ISG promoter stimulation.
Although type I IFNs are frequently treated as a single entity, the human genome contains numerous type I IFN families including 13 α genes encoding 12 distinct subtypes, and single β, ε, κ, ω, and ν genes (Pestka et al., 2004). The α, β, and ω type I IFNs are the best studied and all exert their effects through a common cell surface receptor, although different potencies of distinct type I IFNs with respect to antiviral activity have been described (Foster et al., 1996; Heim et al., 1996; Sperber et al., 1992; Sperber et al., 1993). To further examine the maturation-dependent responses of BE(2)-C cells to type I IFN stimulation and WEEV infection we used a series of purified recombinant type I IFNs (Table 2). Cells were treated with decreasing concentrations of individual type I IFNs for 24 h, infected with WEEV at an MOI of 0.1, and cell viability was measured at 48 and 72 hpi for immature and differentiated cells, respectively. We subsequently calculated weight-based IC50 values to eliminate the confounding effects of the different virus-cell combinations used to determine individual type I IFN specific activities. Several type I IFNs, in particular IFNα8, IFNα10, and IFNβ, showed potent antiviral activity in both immature and differentiated cells infected with WEEV, consistent with previous reports on the preferential antiviral activity of these type I IFNs against picornaviruses (Foster et al., 1996; Heim et al., 1996; Sperber et al., 1993). However, differentiated BE(2)-C cells also showed enhanced responses to several specific type I IFNs, with differences ranging from two- to five-fold for IFNα2a, α8, α14, α16, and IFNω compared to immature cells (Table 2). In addition, not all type I IFNs showed neuronal maturation-dependent differential effects. For example, IC50 values for IFNα5, α6, α10, α17, α21, and IFNω did not differ significantly between immature and differentiated cells. We were unable to determine the significance of potential differences with IFNα1, α4, and α7, as 50% inhibition was not seen with the maximal dose of 1000 pg per ml for immature cells and the IC50 values for differentiated cells was greater than 500 pg per ml. Nevertheless, these results were consistent with the conclusion that neuronal maturation influenced type I IFN responsiveness, but indicated that this differential responsiveness did not apply to all type I IFNs.
Table 2.
Differential effects of type I IFNs on resistance to WEEV-induced cytopathology in immature and differentiated BE(2)-C human neuronal cells.
| IC50 (pg/ml)a | ||
|---|---|---|
| Type I IFNb | Immature | Differentiated |
| IFN-α1 (αD) | >1000 | >1000 |
| IFN-α2a (αA) | 812 ± 114 | 368 ± 77* |
| IFN-α4 (α4b) | >1000 | 623 ± 179 |
| IFN-α5 (αG) | 782 ± 86 | 964 ± 25 |
| IFN-α6 (αK) | 589 ± 119 | 649 ± 213 |
| IFN-α7 (αJ1) | >1000 | 910 ± 81 |
| IFN-α8 (αB2) | 151 ± 5 | 57 ± 28* |
| IFN-α10 (αC) | 230 ± 36 | 116 ± 36 |
| IFN-α14 (αH2) | 830 ± 31 | 344 ± 79* |
| IFN-α16 (αWA) | 732 ± 67 | 239 ± 66* |
| IFN-α17 (αI) | 639 ± 209 | 428 ± 144 |
| IFN-α21 (αAF) | 912 ± 51 | 789 ± 211 |
| IFN-β | 349 ± 85 | 177 ± 24 |
| IFN-ω | >1000 | 210 ± 125 |
Values represent weight-based concentrations that inhibited WEEV-induced cytopathology by 50% at 48 and 72 hpi for immature and differentiated BE(2)-C cells, respectively. Results represent the mean ± SEM from three independent experiments. Specific activities listed by the manufacturer as determined by antiviral effects against vesicular stomatitis virus in Madin-Darby bovine kidney cells (IFNα subtypes), vesicular stomatitis virus in Vero cells (IFNβ), or encephalomyocarditis virus in A549 cells (IFNω), were between 1 to 4 × 108 U/mg for all type I IFNs except IFN-α1 (7.5 × 107 U/mg) and IFN-α21 (6.3 × 108 U/mg). Similar differences between immature and differentiated BE(2)-C cells were obtained when we used specific activity measurements to calculate IC50 values with IFNα subtypes.
Alternative IFNα subtype designations are given in parentheses. All type I IFNs were purified recombinant proteins produced in E. coli except IFN-β, which was produced in mammalian cells.
p < 0.05 compared to immature cells. Indeterminate values above the range of the assay (>1000 pg/ml) were not subjected to statistical analyses.
Discussion
In this study we examined the impact of cellular differentiation on WEEV infection and type I IFN responses in cultured human neuronal cells. We drew four main conclusions. First, human neuronal cells, and in particular immature and differentiated BE(2)-C neuroblastoma cells, were highly permissive to WEEV infection, and RA-induced cellular differentiation did not alter that permissiveness. Second, neuronal differentiation correlated with increased partial resistance to WEEV-induced cytopathology, which was independent of autocrine IFNα or β activity. Third, differentiated neuronal cells displayed a heightened antiviral response to exogenous type I IFNs, which was associated with enhanced ISG promoter activation. And fourth, human neuronal cells showed preferential responsiveness to distinct type I IFNs in the activation of cellular antiviral pathways. These results identify maturation-dependent components of the cell-autonomous innate immune responses utilized by human neuronal cells to combat infections with virulent neurotropic alphaviruses.
The limited availability and unreliability of primary human CNS neuronal cultures prompted us to use immature and differentiated human cell lines as model systems to examine cellular innate immune responses. The ability of RA to induce human neuroblastoma differentiation in culture has been recognized for decades and is frequently used in neurobiology to study cellular maturation and physiology in vitro (Encinas et al., 2000; Hill and Robertson, 1997; Peverali et al., 1996). Retinoids have been used successfully as adjuvant treatment for neuroblastomas in children (Matthay et al., 1999), and studies have shown that they are also important for both normal embryonic (Sockanathan and Jessell, 1998) and adult (Jacobs et al., 2006) neuronal development in vivo, indicating that RA-induced differentiation in culture is a viable approach to study viral pathogenesis in the context of neuronal maturation. Nevertheless, the use of RA to induce cellular differentiation in human neuroblastoma cells as a model culture system is not without potential drawbacks. For example, although the primary approach with BE(2)-C cells described in this report allowed us to directly compare immature and differentiated neuronal cell populations grown in similar conditions with serum-containing culture media, RA-differentiated cultures were not morphologically homogeneous (Figs. 1 and 2). However, we obtained similar results with respect to resistance to WEEV-induced CPE and type I IFN responsiveness with homogenous BDNF-dependent differentiated BE(2)-C cells. Furthermore, the demonstration of neuronal maturation-dependent responses with both SH-SY5Y and NT-2 cells (Fig. 2 and Table 1), two human cell lines unrelated to BE(2)-C cells, supports the hypothesis that enhanced innate immune responses are associated with normal human neuronal development and differentiation. Definitive evidence to support this hypothesis awaits a reliable and defined culture system to examine neuronal differentiation in non-malignant human cells. Recent advances in human embryonic stem cell technology (Wu et al., 2007) may provide an alternative and potentially vital approach to investigate the underlying mechanisms of maturation-dependent and cell-specific innate immune responses to neurotropic viruses.
The results with WEEV infection of immature and differentiated human neuronal cells in culture shared some similarities with rodent neuronal culture systems that were developed to examine SINV neuropathogenesis (Burdeinick-Kerr and Griffin, 2005; Vernon and Griffin, 2005), but there were also significant differences. Differentiated rat CSM14.1 cells (Zhong et al., 1993) are less susceptible to SINV-induced cytopathology (Vernon and Griffin, 2005), consistent with our results using differentiated human neuronal cells (Fig. 2). However, differentiated rodent neurons demonstrate little CPE up to six days after SINV infection (Vernon and Griffin, 2005), whereas differentiated human neuronal cells, although more resistant than immature cells, still developed significant CPE in response to WEEV infection after four to five days (Fig. 2). Furthermore, differentiated CSM14.1 cells are resistant to SINV infection and incompletely differentiated mouse NSC34 cells (Cashman et al., 1992) show partial resistance to SINV infection (Burdeinick-Kerr and Griffin, 2005; Vernon and Griffin, 2005), whereas we found no significant differences between immature and differentiated BE(2)-C cells in the susceptibility to WEEV infection and replication (Fig. 1). These discrepant observations may be due to host differences in the intrinsic neuronal antiviral responses between cultured rodent and human neurons or secondary to differential alphavirus virulence, as the 633 strain of SINV used in the rodent culture system is avirulent in adult mice (Tucker et al., 1993), whereas the Cba 87 strain of WEEV used in this report is highly virulent in adult animals (Schoepp et al., 2002). The rodent neuronal culture system has also been used to demonstrate the important role of IFNγ in noncytolytic cytokine-mediated clearance of SINV (Burdeinick-Kerr and Griffin, 2005). Although we did not examine the effects of IFNγ with the human neuronal cell culture systems described in this report, prolonged treatment with IFNα-A/D did not completely eliminate WEEV-induced CPE in differentiated BE(2)-C cells (data not shown), suggesting that type I IFNs are insufficient to completely control virus replication and that type II IFNs or other innate or adaptive immune system components are required for clearance of WEEV from infected human neuronal cells.
The underlying molecular mechanisms that impart a decreased sensitivity of differentiated human neuronal cells to WEEV-induced CPE are unknown. For rodent neurons, cellular differentiation is accompanied by an increase in the resistance to SINV-induced suppression of cellular translation (Vernon and Griffin, 2005), a prominent component of the cellular response that ultimately results in cell death in some mammalian cell lines (Gorchakov et al., 2005). In contrast to rodent cells, we saw no differences in WEEV-induced suppression of cellular translation between immature and differentiated BE(2)-C cells up to 12 hpi (data not shown). Results with BE(2)-C cells also suggested that autocrine type I IFN activity was not involved in the intrinsic neuronal maturation-dependent responses to WEEV infection, as we were unable to detect the secretion of functional IFNα or β after viral infection (Fig. 2). The absence of type I IFN production by BE(2)-C cells after WEEV infection was in contrast to published results demonstrating the production of IFNβ by differentiated NT-2 cells after rabies virus infection (Prehaud et al., 2005). However, in addition to the difference in cell lines, the level of IFNβ produced by rabies virus-infected NT-2 cells was below the detection sensitivity of our assays. Furthermore, differentiated NT-2 cells did not produce IFNβ after herpes simplex virus infection (Prehaud et al., 2005), suggesting that neuronal type I IFN production may be virus specific.
We hypothesize that changes in the activation and potentiation of type I IFN-independent innate immune pathways play an important role in the maturation-dependent responses that ultimately result in enhanced survival after neurotropic alphavirus infection of differentiated neuronal cells. Furthermore, we hypothesize that these maturation-dependent responses are not restricted to alphaviruses, as we have also seen reduced CPE in differentiated BE(2)-C cells infected with either vesicular stomatitis virus (Rhabdoviridae) or La Crosse virus (Bunyaviridae) (J. Farmer and D. Miller, unpublished data). In support of the innate immune pathway hypothesis, preliminary experiments have demonstrated that differentiated BE(2)-C cells show enhanced innate immune system activation in response to stimulation with the double-stranded RNA mimetic polyinosinic-polycytidylic acid (D. Peltier and D. Miller, unpublished data). On the basis of our differentiation protocol, one potential innate immune system component that may be involved in this altered responsiveness is the cytosolic pattern recognition receptor retinoic acid-induced gene I (RIG-I) (Yoneyama et al., 2004), which was so named due to its upregulation in promyelocytic leukemia cells during RA-induced differentiation (Liu et al., 2000). However, we did not identify a significant effect of prolonged RA stimulation on basal RIG-I expression levels in BE(2)-C cells (data not shown), suggesting that RA may have differential stimulatory effects on leukemia and neuroblastoma cells. Experiments are currently in progress using both targeted and global genetic profiling studies to identify candidate innate immune system components whose expression is altered during human neuronal cell differentiation.
In addition to the maturation-dependent but type I IFN-independent responses associated with increased survival after WEEV infection, our studies uncovered a heightened responsiveness of differentiated human neuronal cells to exogenous type I IFNs (Figs. 3 and 4, Tables 1 and 2). This observation suggests that in the mature CNS a maximal antiviral response is obtained in neuronal cells with exposure to minimal amounts of type I IFNs, and perhaps other cytokines, thereby reducing the potential for immune-mediated damage due to excessive inflammation (Akwa et al., 1998). Furthermore, the observation of the differential responsiveness of human neuronal cells to distinct type I IFNs (Table 2) suggests that targeted type I IFN production may also play an important yet unrecognized role in the overall innate immune response to neurotropic virus infections within the CNS. We are currently examining the underlying mechanisms responsible for the increased responsiveness of differentiated human neuronal cells to type I IFN stimulation. The significant differences in the Hill slopes between immature and differentiated BE(2)-C cells expressing a type I IFN-responsive reporter gene (Fig. 4C) suggest that alterations in protein-protein interactions, such as receptor subunit modulation, may be involved. Additional detailed studies will further define the impact of neuronal differentiation on individual steps, from receptor engagement to specific ISG upregulation, in the type I IFN-mediated amplification of innate immune responses in the CNS.
Materials and Methods
Cell culture and neuronal differentiation
We obtained the human BE(2)-C and SH-SY5Y neuroblastomas, human NT-2 embryonal carcinoma, and African green monkey kidney Vero cells from the American Type Culture Collection (Manassas, VA). We cultured BE(2)-C and Vero cells at 37°C in a humidified atmosphere with 5% CO2 in high glucose (4.5 g per L) formulation Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 4 mM L-glutamine, 10 units penicillin per ml, 10 μg streptomycin per ml, and 5% bovine grown serum (HyClone, Logan, UT) (complete DMEM). We cultured SH-SY5Y cells in RPMI-1640 media supplemented with 10% bovine growth serum and the antibiotics listed above, and we maintained undifferentiated NT-2 cells in DMEM with 10% fetal bovine serum.
We routinely differentiated BE(2)-C cells according to established procedures (Peverali et al., 1996). Briefly, we added 10 μM all-trans RA to complete DMEM and changed culture media three times weekly for three weeks until cell proliferation slowed and cells developed a morphology consistent with mature neuronal cells (see Fig. 1). We routinely passed BE(2)-C cells two to three times by trypsinization during the differentiation period and plated cells into their final tissue culture vessels at least 48 h prior to initiating infection or treatment experiments. To facilitate a direct comparison between cell types, we used the measured proliferative capacity of immature and differentiated BE(2)-C cells (see Fig. 1C) to obtain equal cell densities of 105 cells per ml at either the time of infection or treatment with type I IFNs. This cell density resulted in a surface area confluence of approximate 50–80% for both cell types. For growth factor-dependent differentiation of BE(2)-C cells, we cultured cells in the presence of 10 μM RA in complete DMEM for five days followed by serum-free DMEM with 50 ng per ml BDNF without RA for an additional seven days. We differentiated SH-SY5Y cells with 10 μM RA for one week as previously described (Encinas et al., 2000), and differentiated NT-2 cells using the six week RA-mitotic inhibitor sequential regimen as previously described (Pleasure et al., 1992).
Viruses
Michael Parker at the U.S. Army Medical Research Institute of Infectious Diseases generously provided the full-length WEEV cDNA clone pWE2000 (Schoepp et al., 2002), which was derived from the WEEV strain Cba 87, an epizootic strain from Argentina that is both neurovirulent and neuroinvasive in mice (Bianchi et al., 1993) and cynomolgus macaques (Reed et al., 2005). To generate infectious virions we transfected Vero cells with T7 RNA polymerase-synthesized transcripts generated in vitro from Not I-linearized pWE2000, and 48 h after transfection harvested tissue culture supernatants and further expanded virus stocks by two passages in Vero cells at a low MOI to minimize the formation of defective interfering particles. The final virus stock titer was 5 × 106 plaque-forming units per ml as quantitated by plaque assay on Vero cell monolayers. We also obtained the prototypic North American Fleming strain of WEEV from the CDC (Arbovirus Diseases Branch, Fort Collins, CO) and generated virus stocks in Vero cells that had titers approximately ten-fold higher than pWE2000-derived virus. We used pWE2000-derived infectious virus corresponding to strain Cba 87 for all experiments unless otherwise noted.
Antibodies
To generate polyclonal rabbit antisera against individual WEEV nsPs we expressed full-length nsP1 and nsP3 and the carboxy terminal fragment of nsP2 in E. coli, separated partially purified inclusion bodies by SDS-PAGE, and immunized rabbits with pulverized gel slices (Harlan Bioproducts for Sciences, Inc., Madison, WI). We tested rabbit antisera for reactivity and specificity by immunoblotting with lysates from mock and WEEV-infected Vero cells (data not shown). We purified nsP-specific rabbit immunoglobulin from antisera by Staphylococcus aureus protein A affinity chromatography and used these purified preparations for immunoblotting and immunofluorescence analyses.
We obtained mouse monoclonal antibodies against WEEV from ATCC (catalog No. VR-1251AF), mouse monoclonal antibodies against NF68 and synaptophysin from Sigma (St. Louis, MO), mouse monoclonal antibodies against GAPDH from Santa Cruz Biotechnology (Santa Cruz, CA), and neutralizing rabbit antisera against human IFNα and β from R&D Systems (Minneapolis, MN). We obtained all secondary antibodies for immunoblotting and immunofluorescence staining from Jackson Immunoresearch (West Grove, PA).
Type I IFNs, JAK inhibitor, and IFN-responsive reporter plasmid
We used recombinant human IFNα-A/D (Sigma), which is a hybrid universal type I IFN that can substitute for all IFNα subtypes, IFNβ, and IFNω (Sigma product literature) for all experiments unless otherwise indicated. For select experiments we used purified individual recombinant human type I IFNs, including 12 IFNα isoforms, IFNβ, and IFNω (R&D Systems). We obtained the cell-permeable JAK inhibitor Pyridone 6 from Calbiochem (San Diego, CA) and the type I IFN-responsive plasmid pISRE-SEAP and Quanti-Blue substrate from InvivoGen (San Diego, CA).
Plaque assay
We used Vero cells and 12 well tissue culture plates for all plaque assays. We incubated subconfluent Vero cell monolayers with culture supernatants serially diluted in sterile phosphate buffered saline (100 mM sodium chloride, 50 mM sodium phosphate, pH 7.4) with 0.2% bovine serum albumin for 60 min at 37°C, removed virus and overlaid cells with complete DMEM containing 0.6% SeaPlaque agarose (Cambrex, Rockland, ME) preheated to 50°C. We allowed the agarose to solidify at room temperature for 30 min, supplemented cultures with a 50% volume of complete DMEM, and continued incubation at 37°C for 36 to 48 h. After plaques were visible we added formaldehyde to 9.25% for 30 min to fix cells and inactivate infectious virus, gently removed the media and agarose overlay, and stained monolayers with 0.1% crystal violet in 20% methanol. We air dried plates and counted plaques in duplicate samples to calculate infectious virus titers.
Immunofluorescence analysis
We cultured cells on either polyethyleneimine-coated or Lab-Tek II CC2 eight-well chamber slides (Nalge Nunc, Rochester, NY), washed cells twice with Tris-buffered saline (TBS) (100 mM sodium chloride, 50 mM Tris, pH 7.2) and fixed cells with 2% paraformaldehyde for 30 min at room temperature. We blocked slides with TBS containing 0.1% Tween 20 and 1% goat serum for 1 h, incubated with primary antibodies diluted in blocking buffer overnight, washed extensively in TBS with 0.1% Tween 20, and incubated with fluorescein- or Texas Red-labeled secondary goat antibodies for 1 h. We stained nuclei with 0.5 μg per ml 4,6-diamidino-2-phenyindole (DAPI) and mounted slides as previously described (Miller et al., 2001). We obtained digital light and fluorescent images using a Zeiss Axiophot-2 microscope and CCD camera and prepared final images with Adobe Photoshop software.
Immunoblot and Northern blot analyses
We prepared protein samples and performed immunoblot analyses as previously described (Kampmueller and Miller, 2005) with the following modifications. We removed media from adherent cell cultures, washed cells twice with TBS, and lysed cells directly in tissue culture vessels with reducing SDS-PAGE sample buffer (62.5 mM Tris [pH 6.8], 2% SDS, 5% glycerol, 14.4 mM 2-mercaptoethanol, 0.02% bromophenol blue). We used peroxidase-conjugated secondary antibodies and developed immunoblots with an enhanced chemiluminescence reagent solution containing 100 mM Tris (pH 8.5), 1.25 mM luminol, 0.2 mM p-coumaric acid, and 0.001% hydrogen peroxide.
We isolated total RNA with TRIzol reagent (Invitrogen, Carlsbad, CA) and stored RNA samples at −80°C until analysis. We denatured samples in glyoxal buffer (Cambrex) prior to separation in 1% agarose gels in 90 mM Tris-borate/2 mM EDTA buffer and subsequent transfer to ZetaProbe nylon membranes (Bio-Rad, Hercules, CA). We prepared strand-specific 32P-labeled riboprobes using an in vitro transcription system (Promega, Madison, WI) with 32P-UTP according to the manufacturer’s instructions. We constructed the template plasmid by inserting a PCR-generated fragment corresponding to viral nucleotides 7551 to 7850 from pWE2000 into pGEM-4Z (Promega). Primer sequences are available upon request. The sequence used for probe generation encompassed the 5′ region of the WEEV capsid protein coding sequence, and thus antisense riboprobes efficiently detected both genomic and subgenomic positive-sense viral RNA. We hybridized membranes with 32P-labeled riboprobes in a solution containing 50 mM phosphate (pH 6.8), 0.75 M sodium chloride, 75 mM sodium citrate, 1% SDS, 50% formamide, 5X Denhardt’s solution, and 0.1 mg of salmon sperm DNA per ml at 62°C overnight, washed extensively with a solution containing 0.1% SDS, 15 mM sodium chloride, and 1.5 M sodium citrate, and detected radioactive signals by autoradiography with Blue Light AutoRad film (ISC BioExpress, Kaysville, UT).
We obtained digital chemiluminescent and film images using an Alpha Innotech Fluorchem 8900, quantitated band intensities with AlphaEaseFC software, and prepared final images with Adobe Photoshop software. When image cropping was necessary, all panels were taken from the same exposure of the blot and all contrast adjustments to the initial image were done prior to cropping.
Viability assays
We used MTT assays to determine cell viability after virus infection. We incubated mock and infected cell cultures with 0.5 mg MTT per ml for 1 h at 37°C, added an equal volume of stop solution containing isopropanol with 10% Triton X-100 and 0.1 N hydrochloric acid to lyse cells, inactivate virus, and solubilize formazan crystal, and measured the absorbance at 595 nm. We calculated cell viability as the percent A595 compared to untreated or uninfected controls.
Statistical analyses
We used a two-tailed Student’s t-test assuming unequal variances for all statistical comparisons, and considered a p-value < 0.05 as statistically significant. We used log10-transformed data for statistical comparisons of virus titers, and a four parameter logistic sigmoidal regression analysis to calculate Hill slope values. We present results that are representative of at least three independent experiments, where quantitative data represent the mean ± SEM from those experiments.
Acknowledgments
We thank Donna Gschwend, Leslie Goo, Bethany Sprader, and Allison Simms for assistance, David Sherman and all laboratory members for their helpful comments on the research and manuscript, and Michael Parker for generously providing reagents. The NIH/NIAID Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (RCE) Program sponsored this work. The authors acknowledge membership within and support from the Region V ‘Great Lakes’ RCE (NIH award 1-U54-AI-057153). The MSTP training grant T32-GM007863 supported DCP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilar PV, Paessler S, Carrara AS, Baron S, Poast J, Wang E, Moncayo AC, Anishchenko M, Watts D, Tesh RB, Weaver SC. Variation in interferon sensitivity and induction among strains of eastern equine encephalitis virus. J Virol. 2005;79(17):11300–11310. doi: 10.1128/JVI.79.17.11300-11310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H, Whitton JL, Bloom FE, Campbell IL. Transgenic expression of IFN-α in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol. 1998;161(9):5016–5026. [PubMed] [Google Scholar]
- Bianchi TI, Aviles G, Monath TP, Sabattini MS. Western equine encephalomyelitis: virulence markers and their epidemiologic significance. Am J Trop Med Hyg. 1993;49(3):322–328. doi: 10.4269/ajtmh.1993.49.322. [DOI] [PubMed] [Google Scholar]
- Burdeinick-Kerr R, Griffin DE. γ interferon-dependent, noncytolytic clearance of Sindbis virus infection from neurons in vitro. J Virol. 2005;79(9):5374–5385. doi: 10.1128/JVI.79.9.5374-5385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194(3):209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Senechal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Heron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314(5797):308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- Daffis S, Samuel MA, Keller BC, Gale M, Diamond MS. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and independent mechanisms. PLoS Pathog. 2007;3(7):e106. doi: 10.1371/journal.ppat.0030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, Michiels T. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci U S A. 2006;103(20):7835–7840. doi: 10.1073/pnas.0602460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deresiewicz RL, Thaler SJ, Hsu L, Zamani AA. Clinical and neuroradiographic manifestations of eastern equine encephalitis. N Engl J Med. 1997;336(26):1867–1874. doi: 10.1056/NEJM199706263362604. [DOI] [PubMed] [Google Scholar]
- Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, Al Ghonaium A, Tufenkeji H, Frayha H, Al-Gazlan S, Al-Rayes H, Schreiber RD, Gresser I, Casanova JL. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33(3):388–391. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- Earnest MP, Goolishian HA, Calverley JR, Hayes RO, Hill HR. Neurologic, intellectual, and psychologic sequelae following western encephalitis. A follow-up study of 35 cases. Neurology. 1971;21(9):969–974. doi: 10.1212/wnl.21.9.969. [DOI] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Cena V, Gallego C, Comella JX. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem. 2000;75(3):991–1003. doi: 10.1046/j.1471-4159.2000.0750991.x. [DOI] [PubMed] [Google Scholar]
- Foster GR, Rodrigues O, Ghouze F, Schulte-Frohlinde E, Testa D, Liao MJ, Stark GR, Leadbeater L, Thomas HC. Different relative activities of human cell-derived interferon-α subtypes: IFN-α8 has very high antiviral potency. J Interferon Cytokine Res. 1996;16(12):1027–1033. doi: 10.1089/jir.1996.16.1027. [DOI] [PubMed] [Google Scholar]
- Gorchakov R, Frolova E, Frolov I. Inhibition of transcription and translation in Sindbis virus-infected cells. J Virol. 2005;79(15):9397–9409. doi: 10.1128/JVI.79.15.9397-9409.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieder FB, Vogel SN. Role of interferon and interferon regulatory factors in early protection against Venezuelan equine encephalitis virus infection. Virology. 1999;257(1):106–118. doi: 10.1006/viro.1999.9662. [DOI] [PubMed] [Google Scholar]
- Griffin DE. Alphaviruses. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SS, editors. Fields Virology. 4. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 917–962. [Google Scholar]
- Gubler DJ. The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res. 2002;33(4):330–342. doi: 10.1016/s0188-4409(02)00378-8. [DOI] [PubMed] [Google Scholar]
- Heim A, Stille-Seigener M, Pring-Akerblom P, Grumbach I, Brehm C, Kreuzer H, Figulla HR. Recombinant interferons β and γ have a higher antiviral activity than interferon-α in coxsackievirus B3-infected carrier state cultures of human myocardial fibroblasts. J Interferon Cytokine Res. 1996;16(4):283–287. doi: 10.1089/jir.1996.16.283. [DOI] [PubMed] [Google Scholar]
- Hill DP, Robertson KA. Characterization of the cholinergic neuronal differentiation of the human neuroblastoma cell line LA-N-5 after treatment with retinoic acid. Brain Res Dev Brain Res. 1997;102(1):53–67. doi: 10.1016/s0165-3806(97)00076-x. [DOI] [PubMed] [Google Scholar]
- Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol. 2004;5(10):971–974. doi: 10.1038/ni1004-971. [DOI] [PubMed] [Google Scholar]
- Horner PJ, Gage FH. Regenerating the damaged central nervous system. Nature. 2000;407(6807):963–970. doi: 10.1038/35039559. [DOI] [PubMed] [Google Scholar]
- Jacobs S, Lie DC, DeCicco KL, Shi Y, DeLuca LM, Gage FH, Evans RM. Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc Natl Acad Sci U S A. 2006;103(10):3902–3907. doi: 10.1073/pnas.0511294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston C, Jiang W, Chu T, Levine B. Identification of genes involved in the host response to neurovirulent alphavirus infection. J Virol. 2001;75(21):10431–10445. doi: 10.1128/JVI.75.21.10431-10445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmueller KM, Miller DJ. The cellular chaperone heat shock protein 90 facilitates Flock House virus RNA replication in Drosophila cells. J Virol. 2005;79(11):6827–6837. doi: 10.1128/JVI.79.11.6827-6837.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23(1):19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Labrada L, Liang XH, Zheng W, Johnston C, Levine B. Age-dependent resistance to lethal alphavirus encephalitis in mice: analysis of gene expression in the central nervous system and identification of a novel interferon-inducible protective gene, mouse ISG12. J Virol. 2002;76(22):11688–11703. doi: 10.1128/JVI.76.22.11688-11703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, Wesselingh SL, Griffin DE, Hardwick JM. Alphavirus-induced apoptosis in mouse brains correlates with neurovirulence. J Virol. 1996;70(3):1828–1835. doi: 10.1128/jvi.70.3.1828-1835.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TX, Zhang JW, Tao J, Zhang RB, Zhang QH, Zhao CJ, Tong JH, Lanotte M, Waxman S, Chen SJ, Mao M, Hu GX, Zhu L, Chen Z. Gene expression networks underlying retinoic acid-induced differentiation of acute promyelocytic leukemia cells. Blood. 2000;96(4):1496–1504. [PubMed] [Google Scholar]
- Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lu J, Kosaras B, Sidman RL, Volpe JJ, Vartanian T. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol. 2006;175(2):209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, Gerbing RB, Reynolds CP. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med. 1999;341(16):1165–1173. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- Miller DJ, Schwartz MD, Ahlquist P. Flock House virus RNA replicates on outer mitochondrial membranes in Drosophila cells. J Virol. 2001;75(23):11664–11676. doi: 10.1128/JVI.75.23.11664-11676.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Ousman SS, Wang J, Campbell IL. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J Virol. 2005;79(12):7514–7527. doi: 10.1128/JVI.79.12.7514-7527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- Peverali FA, Orioli D, Tonon L, Ciana P, Bunone G, Negri M, Della-Valle G. Retinoic acid-induced growth arrest and differentiation of neuroblastoma cells are counteracted by N-myc and enhanced by max overexpressions. Oncogene. 1996;12(2):457–462. [PubMed] [Google Scholar]
- Pleasure SJ, Page C, Lee VM. Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J Neurosci. 1992;12(5):1802–1815. doi: 10.1523/JNEUROSCI.12-05-01802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehaud C, Megret F, Lafage M, Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of β interferon. J Virol. 2005;79(20):12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed DS, Larsen T, Sullivan LJ, Lind CM, Lackemeyer MG, Pratt WD, Parker MD. Aerosol exposure to western equine encephalitis virus causes fever and encephalitis in cynomolgus macaques. J Infect Dis. 2005;192(7):1173–1182. doi: 10.1086/444397. [DOI] [PubMed] [Google Scholar]
- Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. α/β interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol. 2000;74(7):3366–3378. doi: 10.1128/jvi.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14(4):778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282(28):20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- Schoepp RJ, Smith JF, Parker MD. Recombinant chimeric western and eastern equine encephalitis viruses as potential vaccine candidates. Virology. 2002;302(2):299–309. doi: 10.1006/viro.2002.1677. [DOI] [PubMed] [Google Scholar]
- Sidwell RW, Smee DF. Viruses of the Bunya- and Togaviridae families: potential as bioterrorism agents and means of control. Antiviral Res. 2003;57(1–2):101–111. doi: 10.1016/s0166-3542(02)00203-6. [DOI] [PubMed] [Google Scholar]
- Sockanathan S, Jessell TM. Motor neuron-derived retinoid signaling specifies the subtype identity of spinal motor neurons. Cell. 1998;94(4):503–514. doi: 10.1016/s0092-8674(00)81591-3. [DOI] [PubMed] [Google Scholar]
- Sperber SJ, Gocke DJ, Haberzettl C, Kuk R, Schwartz B, Pestka S. Anti-HIV-1 activity of recombinant and hybrid species of interferon-α. J Interferon Res. 1992;12(5):363–368. doi: 10.1089/jir.1992.12.363. [DOI] [PubMed] [Google Scholar]
- Sperber SJ, Hunger SB, Schwartz B, Pestka S. Anti-rhinoviral activity of recombinant and hybrid species of interferon α. Antiviral Res. 1993;22(2–3):121–129. doi: 10.1016/0166-3542(93)90090-6. [DOI] [PubMed] [Google Scholar]
- Trgovcich J, Aronson JF, Eldridge JC, Johnston RE. TNFα, interferon, and stress response induction as a function of age-related susceptibility to fatal Sindbis virus infection of mice. Virology. 1999;263(2):339–348. doi: 10.1006/viro.1999.9913. [DOI] [PubMed] [Google Scholar]
- Tucker PC, Strauss EG, Kuhn RJ, Strauss JH, Griffin DE. Viral determinants of age-dependent virulence of Sindbis virus for mice. J Virol. 1993;67(8):4605–4610. doi: 10.1128/jvi.67.8.4605-4610.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Broek MF, Muller U, Huang S, Zinkernagel RM, Aguet M. Immune defence in mice lacking type I and/or type II interferon receptors. Immunol Rev. 1995;148:5–18. doi: 10.1111/j.1600-065x.1995.tb00090.x. [DOI] [PubMed] [Google Scholar]
- Vernon PS, Griffin DE. Characterization of an in vitro model of alphavirus infection of immature and mature neurons. J Virol. 2005;79(6):3438–3447. doi: 10.1128/JVI.79.6.3438-3447.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Campbell IL. Innate STAT1-dependent genomic response of neurons to the antiviral cytokine α interferon. J Virol. 2005;79(13):8295–8302. doi: 10.1128/JVI.79.13.8295-8302.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Xu J, Pang ZP, Ge W, Kim KJ, Blanchi B, Chen C, Sudhoff TC, Sun YE. Integrative genomic and functional analyses reveal neuronal subtype differentiation bias in human embryonic stem cell lines. Proc Natl Acad Sci U S A. 2007;104(34):13821–13826. doi: 10.1073/pnas.0706199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5(7):730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- Zhang S, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku C, Casrouge A, Zhang X, Barreiro L, Leonard J, Hamilton C, Lebon P, Heron B, Vallee L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova J. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317(5844):1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- Zhong LT, Sarafian T, Kane DJ, Charles AC, Mah SP, Edwards RH, Bredesen DE. bcl-2 inhibits death of central neural cells induced by multiple agents. Proc Natl Acad Sci U S A. 1993;90(10):4533–4537. doi: 10.1073/pnas.90.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]