

Abstract
Mutations in the human ether-a-go-go-related gene (hERG) result in long QT syndrome type 2 (LQT2). The hERG gene encodes a K+ channel that contributes to the repolarization of the cardiac action potential. We have previously shown that hERG mRNA transcripts that contain premature termination codon mutations are rapidly degraded by nonsense-mediated mRNA decay (NMD). In this study, we identified a LQT2 nonsense mutation, Q81X, which escapes degradation by the reinitiation of translation and generates N-terminally truncated channels. RNA analysis of hERG minigenes revealed equivalent levels of wild-type and Q81X mRNA while the mRNA expressed from minigenes containing the LQT2 frameshift mutation, P141fs+2X, was significantly reduced by NMD. Western blot analysis revealed that Q81X minigenes expressed truncated channels. Q81X channels exhibited decreased tail current levels and increased deactivation kinetics compared to wild-type channels. These results are consistent with the disruption of the N-terminus, which is known to regulate hERG deactivation. Site-specific mutagenesis studies showed that translation of the Q81X transcript is reinitiated at Met124 following premature termination. Q81X co-assembled with hERG to form heteromeric channels that exhibited increased deactivation rates compared to wild-type channels. Mutant channels also generated less outward current and transferred less charge at late phases of repolarization during ventricular action potential clamp. These results provide new mechanistic insight into the prolongation of the QT interval in LQT2 patients. Our findings indicate that the reinitiation of translation may be an important pathogenic mechanism in patients with nonsense and frameshift LQT2 mutations near the 5′ end of the hERG gene.
Keywords: cardiac arrhythmias, ion channels, long QT syndrome, patch clamp
1. Introduction
The human ether-a-go-go-related gene (hERG) encodes the pore-forming subunit of a voltage-gated K+ channel, a component of the rapidly-activating delayed rectifier current (IKr) in the heart [1, 2]. hERG channels exhibit unique voltage-dependent properties including rapid inactivation and slow deactivation. Mutations in hERG cause the congenital form of long QT syndrome type 2 (LQT2), an inherited autosomal dominant disorder characterized by prolonged QT interval on the electrocardiogram. Patients with LQT2 are susceptible to severe ventricular arrhythmias that can lead to sudden death [3, 4]. Approximately one-third of identified LQT2 mutations are nonsense, frameshift and splice site mutations that introduce premature termination codons (PTCs) into the coding sequence of hERG [5–9]. We have previously shown that hERG mRNA transcripts that contain PTC mutations are degraded by nonsense-mediated mRNA decay (NMD) [10, 11]. NMD is an evolutionarily conserved RNA surveillance mechanism that recognizes and eliminates PTC-containing transcripts [12, 13]. The degradation of hERG transcripts containing PTC mutations by NMD has been shown to prevent the formation of truncated channel proteins that would otherwise exert a dominant-negative effect on wild-type hERG current [10, 14]. NMD may also increase the pathogenicity of LQT2 by preventing the formation of truncated channels that retain functionality [15, 16].
In mammalian cells, PTCs are distinguished by their physical location relative to the 3′-most exon-exon junction. As opposed to naturally occurring stop codons that usually occur in the terminal exon, nonsense codons located >50 to 55 nt upstream of the 3′-most exon-exon junction target the transcript for degradation by NMD [17]. Several studies, however, have identified PTC mutations near the translation start site that resist NMD when post-termination ribosomes resume scanning and reinitiate translation at downstream methionine codons [18–20].
Several nonsense and frameshift LQT2 mutations have been identified early in the coding sequence of hERG [5–9]. We studied two of these mutations first reported by Splawski et al., Q81X and P141fs+2X [5]. We report that Q81X transcripts escaped NMD by the reinitiation of translation and generated N-terminally truncated channels that exhibited decreased tail current levels and accelerated deactivation rates. Q81X also generated less outward current and transferred less charge during the late phases of action potential repolarization. The reinitiation of translation represents a new mechanism of hERG channel dysfunction in LQT2.
2. Materials and Methods
2.1. hERG minigene and cDNA constructs
The construction of the full-length hERG minigene composed of cDNA from exons 1–10 and genomic DNA from intron 10 to the polyadenylation site has been previously described [16]. The Q81X and P141fs+2X mutations were generated by site-directed mutagenesis using the pAlter in vitro mutagenesis system (Promega, Madison, WI). Wild-type and mutant hERG minigenes and cDNA constructs were subcloned into pcDNA3.1/Hygro (+) expression vectors (Invitrogen, Carlsbad, CA). The Flag and hemagglutinin (HA) epitopes were inserted in frame at the C-terminus of the wild-type and Q81X cDNA constructs. For western blot and immunoprecipitation experiments human embryonic kidney (HEK)293 cells were seeded on 60 mm culture plates and transiently transfected with 1 μg of the hERG minigene or cDNA constructs using the Effectene method (Qiagen, Valencia, CA). For patch clamp studies HEK293 cells were seeded on 35 mm culture plates and transiently transfected with 0.4 μg of the hERG minigene constructs and 0.1 μg of green fluorescent protein cDNA to serve as an indicator. Wild-type and mutant constructs were co-transfected at equimolar concentrations to study the functional properties of heteromeric channels. In immunoprecipitation studies, a HEK293 cell line stably expressing Flag-tagged wild-type hERG was transiently transfected with HA-tagged wild-type or Q81X cDNA constructs. HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum. HL-1 murine cardiomyocytes [21] were transiently transfected with wild-type and Q81X HA-tagged constructs using Lipofectamine 2000 (Invitrogen). HL-1 cells were cultured in Claycomb medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum and 0.1 mM norepinephrine (Sigma-Aldrich).
2.2. RNase protection assay
The level of mRNA was analyzed by the RNase protection assay with a probe spanning 277 nt of exons 12 and 13 as described [16]. The total length of the probe was 409 nt and contained sequences from the pCRII vector at both ends. Yeast RNA was used as a control for the complete digestion of the probes by RNase. The expression level of the hygromycin B resistance gene from the pcDNA3.1/Hygro vector was used as a loading control. The relative intensity of each band was quantified using ImageJ [22].
2.3. Western blot and immunoprecipitation
Western blot analysis and immunoprecipitation were performed as previously described [23, 24]. Flag-tagged and HA-tagged hERG channels from whole cell lysates were subjected to SDS-PAGE, transferred onto nitrocellulose membranes, detected with the anti-HA, anti-Flag or the anti-hERG C-terminus antibody, and visualized with the Plus-ECL (PerkinElmer, Waltham, MA) detection kit. In the immunoprecipitation experiments, hERG channels were immunoprecipitated with anti-Flag antibody and detected by western blot with the anti-HA and the anti-Flag antibody.
2.4. Patch clamp recordings
Membrane currents were recorded in whole cell configuration using an Axopatch-200B patch clamp amplifier. pCLAMP8 software (Molecular Devices, Sunnyvale, CA) was used for data acquisition and analysis of current signals. The action potential clamp was performed using a human ventricular action potential waveform with a sampling rate of 1.04 kHz. Cells were superfused with HEPES-buffered Tyrode solution containing 137 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4). The pipette solution contained 130 mM KCl, 1 mM MgCl2, 5 mM EGTA, 5 mM MgATP, and 10 mM HEPES (pH 7.2). All patch clamp experiments were performed at room temperature except for action potential clamp experiments that were carried out at 35 ± 1°C.
The corrected peak tail current was determined by fitting the deactivation phase with double exponential function and extrapolating to the beginning of the pulse [25]. Current density was obtained by normalizing hERG current to cell capacitance and is expressed as pA/pF. The charge transferred during the action potential clamp was obtained by integrating the area under the current trace. Charge density was obtained by normalizing charge to cell capacitance and is expressed as pC/pF. The voltage-dependence of inactivation was calculated using a three-step protocol as previously described [26–28]. The peak current amplitudes at potentials between −100 and −120 mV were corrected for errors due to deactivation using the following equation as previously described [28]: Icorrected = [(Ipeak − Iend)/(Emem − Erev)] × (40 − Erev), where Ipeak is the peak current during the hyperpolarizing step, Iend is the current level at the end of the hyperpolarizing step, Emem is the test potential, and Erev is the reversal potential. The data were fit using a Boltzmann function. Data analysis was performed using SigmaPlot (San Jose, CA) and are presented as mean ± standard error of the mean (SEM). Statistical comparisons were performed using Student’s t-test and ANOVA as appropriate. The Holm-Sidak method was used in ANOVA to identify significant differences between means. Values of P < 0.05 were considered statistically significant.
3. Results
3.1. hERG transcripts containing the Q81X mutation are resistant to NMD
To test whether PTC mutations occurring early in the coding sequence of hERG were susceptible to NMD we analyzed the mRNA expressed from minigenes containing Q81X or the P141fs+2X LQT2 mutations (Fig. 1). A schematic of the hERG minigene composed of cDNA from exon 1 to exon 10 and genomic DNA from intron 10 to the polyadenylation signal is shown in Fig. 1A. HEK293 cells were transiently transfected with wild-type, Q81X or P141fs+2X minigenes and the presence of mRNA was determined by the RNase protection assay using a probe that hybridized to 277 nt of exons 12 and 13 of the spliced minigene. As shown in Fig. 1B, a similar level of wild-type and Q81X mRNA was detected, while P141fs+2X mRNA levels were significantly reduced compared to wild-type (P < 0.05, ANOVA). The quantitative analysis of the normalized signals revealed that P141fs+2X mRNA was 15 ± 3% of wild-type (n = 4) (Fig. 1C). Because NMD is dependent on protein translation we performed the assay in the presence of the protein synthesis inhibitor cycloheximide. Cycloheximide has been shown to abrogate NMD in PTC-containing transcripts [10, 29]. Treatment with cycloheximide restored the expression of P141fs+2X mRNA to levels comparable to wild-type and Q81X levels but had no effect on the expression of the wild-type and Q81X transcripts. These results suggest that the Q81X mutation, but not the P141fs+2X mutation, is resistant to NMD.
Fig. 1.

Analysis of the Q81X and P141fs+2X mutations using minigene constructs. (A) Structure of the full-length hERG minigene construct. The positions of the wild-type termination codon (TER) and premature termination codons are indicated. (B) Analysis of Q81X and P141fs+2X mRNA. HEK293 cells were transiently transfected with wild-type (WT), Q81X, or P141fs+2X minigenes and the expressed mRNA was analyzed by the RNase protection assay using a probe specific to 277 nt within exons 12 and 13 of hERG. Cells expressing wild-type and mutant minigenes were treated (+) with 100 μg/ml cycloheximide (CHX) for 3 h prior to RNA isolation or left untreated (−).The level of hygromycin resistance gene transcripts (Hygro) served as a loading control. (C) Normalized signals were quantified and plotted as the mean percentage of wild-type ± SEM (n = 4).
3.2. Q81X channels are expressed as truncated proteins
To determine whether Q81X transcripts were translated into hERG channel proteins we performed western blot analysis of the proteins expressed from the mutant minigenes. HEK293 cells were transiently transfected with wild-type, Q81X or P141fs+2X minigenes and the channel proteins were detected using a polyclonal antibody directed against the hERG C-terminus. As shown in Fig. 2A, wild-type channels were expressed as a 135 kDa, core-glycosylated form of the channel that is associated with the endoplasmic reticulum and a 155 kDa, complex-glycosylated channel that is expressed at the cell surface [23]. Q81X channel proteins were expressed at 120 kDa and 140 kDa, suggesting that the core- and complex-glycosylated forms of the mutant channel were truncated compared to wild-type hERG. In contrast to wild-type and Q81X, we did not detect hERG channels in cells transfected with the P141fs+2X minigene. To determine whether Q81X channels could be expressed in heart cells we transiently transfected the HL-1 murine cardiomyocyte cell line with the wild-type and mutant constructs and analyzed protein expression by western blot (Fig. 2B). A similar expression pattern was observed for the wild-type and Q81X channels indicating that cardiac myocytes were also able to generate the mutant channel proteins.
Fig. 2.
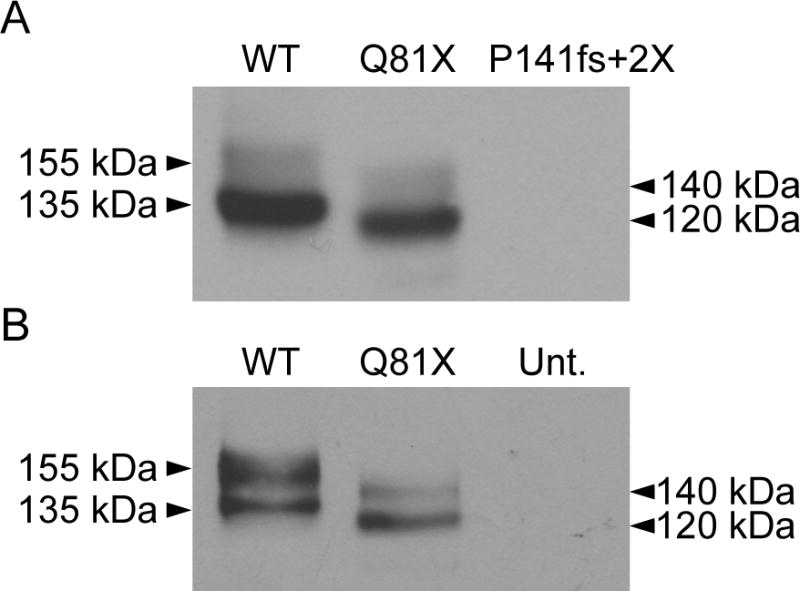
Western blot analysis of wild-type, Q81X, and P141fs+2X channels. (A) HEK293 cells were transiently transfected with wild-type, Q81X, and P141fs+2X hERG minigenes. Proteins from whole cell lysates were subjected to SDS-PAGE and probed with anti-hERG antibody. (B) HL-1 cardiomyocytes were transiently transfected with HA-tagged wild-type and Q81X constructs. Proteins were detected with anti-HA antibody. Mature, fully glycosylated wild-type channels are 155 kDa; immature, core-glycosylated wild-type channels are 135 kDa. Mature, fully-glycosylated Q81X channels are 140 kDa; immature, core-glycosylated Q81X channels are 120 kDa. Untransfected cells, Unt. The results shown are representative of three independent experiments.
3.3. Functional analysis of the truncated Q81X channels
To determine the functional properties of the Q81X channels we performed whole cell patch clamp analysis of HEK293 cells transiently transfected with either the wild-type or mutant minigenes. The voltage-dependent activation of the channels was studied by recording the tail current at −50 mV following a 4 s test potential to voltages ranging between −70 and +60 mV. Representative current traces are shown in Fig. 3A. The current density measured at the end of each activating potential is shown in Fig. 3B. The plots show that wild-type and mutant channels both undergo voltage-dependent activation followed by inactivation at more positive potentials. The averaged maximum outward currents of wild-type (19.9 ± 4.1 pA/pF, n = 11) and Q81X (18.3 ± 3.5 pA/pF, n = 13), measured at +10 mV, were not significantly different (P > 0.05, t-test). The peak tail current amplitudes, normalized to the maximum tail current amplitude, were fit with a Boltzmann function to generate activation curves for wild-type and Q81X (Fig. 3C). The V1/2 and k values of Q81X channels, −12.1 ± 1.4 mV and 7.3 ± 0.5 (n = 13), were not significantly different from those determined for wild-type channels, −13.2 ± 2.3 mV and 8.0 ± 0.5 (n = 11, P > 0.05, t-test). Q81X channels exhibited significantly decreased tail current amplitudes relative to wild-type channels (Fig. 3D). The average tail current densities, following test voltages of +30 mV were 21.7 ± 3.4 pA/pF for wild-type (n = 11) and 11.8 ± 2.3 pA/pF (n = 13, P < 0.05, t-test) for Q81X channels, a 46% decrease. Q81X channels also appeared to deactivate much more rapidly than wild-type channels upon repolarization to −50 mV. Accelerated channel deactivation along with decreased surface density may underlie the decreased tail current density of the mutant channels.
Fig. 3.

Electrophysiological properties of wild-type and Q81X channels. (A) Representative currents recorded from HEK293 cells transiently transfected with wild-type and Q81X minigenes. hERG channels were activated with 4 s test potentials between −70 and +60 mV and tail current was recorded upon repolarization to −50 mV. (B) I–V plot of wild-type and Q81X current density measured at the end of the depolarizing pulse. (C) Activation curves determined using peak tail currents, normalized to maximum tail currents, and fit with a Boltzmann function (solid line). (D) I–V plot of peak tail current densities recorded at −50 mV, following depolarizing test potentials. Data were plotted as mean ± SEM, wild-type (n = 11) and Q81X (n = 13).
To further characterize the deactivation kinetics, channels were activated with a 1 s test potential to +60 mV and the deactivation tail currents were recorded upon repolarization to potentials ranging between −40 and −120 mV. Representative current traces are shown in Fig. 4A. The individual tail current traces were best fit with a double exponential function to determine the fast and slow time constants, τFast and τSlow, of the deactivation rates. The voltage-dependencies of τFast and τSlow are shown in Fig. 4B and Fig. 4C, respectively, and the kinetic properties of the channels at −40 and −120 mV are summarized in Table 1. The deactivation rates of Q81X channels were significantly faster than wild-type channels across all test potentials (wild-type, n = 6, Q81X, n = 7, P < 0.05, ANOVA). At −40 mV the τFast and τSlow of the Q81X channels were decreased by 78% and 84%, and at −120 mV by 74% and 70%, respectively. Comparing the relative amplitudes associated with τFast and τSlow time components revealed that the τFast makes a greater contribution to the wild-type deactivation rate at more negative potentials while it is the dominant component across all test potentials in the Q81X channels (Fig. 4D). At −120 mV the τFast comprised 82% and 87% of the total amplitude of the wild-type and Q81X deactivation rates. At −40 mV the τFast of wild-type hERG comprised significantly less of the total amplitude of the deactivation rate compared with Q81X channels, 29% versus 72%, respectively (P < 0.05, ANOVA). The increase in the deactivation rate is consistent with studies of N-terminally truncated hERG channels [30, 31].
Fig. 4.

Deactivation kinetics of wild-type and Q81X channels. (A) Representative currents recorded from HEK293 cells transiently transfected with wild-type and Q81X minigenes. hERG channels were activated with a 1 s test potential to +60 mV and the deactivation tail currents were recorded upon repolarization to potentials ranging between −40 and −120 mV. The tail current was fit with a double exponential function and the fast and slow components of the deactivation time constants of wild-type and Q81X channels are shown in (B) and (C). The time constants for the fast and slow components of the deactivation rate were significantly faster for Q81X channels at all test potentials (P < 0.05, ANOVA). (D) Relative contribution of the fast component of the deactivation time constants at −40 and −120 mV for wild-type and Q81X channels. All values were plotted as mean ± SEM, wild-type (n = 6) and Q81X (n = 7). *, P < 0.05 compared to Q81X, ANOVA.
Table 1.
Voltage-dependence of deactivation of wild-type hERG and Q81X
| Deactivation, at −40 mV | Wild-Type | Q81X | Wild-Type + Q81X |
|---|---|---|---|
| τFast (ms) | 245.5 ± 23.0 (6) | 54.2 ± 1.1a (7) | 141.4 ± 17.0a (6) |
| τSlow (ms) | 1761.1 ± 100.5 | 290.3 ± 25.3a | 1233.9 ± 43.6a |
| A Fast/(A Fast + A Slow) | 0.29 ± 0.02 | 0.72 ± 0.03a | 0.51 ± 0.06a |
| Deactivation, at −120 mV | |||
| τFast (ms) | 38.7 ± 10.9 | 10.0 ± 1.1a | 31.0 ± 3.0 |
| τSlow (ms) | 222.7 ± 55.4 | 67.5 ± 7.8a | 140.9 ± 10.0 |
| A Fast/(A Fast + A Slow) | 0.82 ± 0.07 | 0.87 ± 0.03 | 0.81 ± 0.06 |
| Inactivation | |||
| V1/2 (mV) | −57.6 ± 1.7 (7) | −61.7 ± 3.0 (5) | −57.3 ± 4.4 (5) |
| k (mV) | −20.5 ± 0.3 | −28.0 ± 0.9a | −22.8 ± 1.4 |
| τRecovery at −60 mV (ms) | 6.4 ± 0.5 (12) | 2.8 ± 0.2a (11) | 4.6 ± 0.5a (8) |
Data were listed as mean ± SEM. The numbers of cells are indicated in brackets.
Indicates a significant difference compared to wild-type hERG (P < 0.05, ANOVA).
We also characterized the voltage-dependence of inactivation using a three-step protocol as previously described [26]. Briefly, channels were activated by a 0.5 s pulse to +40 mV, a second step to potentials between −120 and +40 mV allowed the channels to recover from inactivation, and the relative number of channels competent to activate was determined by a third step to +40 mV. To minimize channel deactivation at negative test potentials we set the time of the hyperpolarizing pulse to 5 ms for Q81X and 20 ms for wild-type channels [27, 32]. Representative current traces are shown in Fig. 5A and the kinetic properties are summarized in Table 1. The peak tail currents were plotted as a function of voltage and fit with a Boltzmann function (Fig. 5B). The voltage at which 50% of the wild-type channels had recovered from inactivation was −57.6 ± 1.7 mV (n = 7), which was not significantly different from the mutant channels −61.7 ± 3.0 mV (n = 5, P > 0.05, t-test). We determined the time constants of the recovery from inactivation from the deactivation voltage clamp protocol shown in Fig. 4. The time constants were determined by fitting the initial 10 ms of the rising phase of the deactivation voltage clamp protocol with a single exponential function. Due to the rapid rates of deactivation, the recovery from inactivation could only be accurately determined at test potentials positive to −60 mV [27]. We found that the mutant channels recovered from inactivation significantly faster than the wild-type channels across all potentials. At −60 mV the time constant for recovery from inactivation was 6.4 ± 0.5 ms (n = 12) for wild-type and 2.8 ± 0.2 ms for the Q81X mutant (n = 11, P < 0.05, ANOVA). A similar result was observed in the recovery from inactivation of N-terminally truncated hERGb channels compared with the full-length hERG channel [27].
Fig. 5.

Voltage-dependent inactivation of wild-type and Q81X channels. (A) Representative currents from HEK293 cells transiently transfected with wild-type and Q81X minigenes. Current was recorded using a three-pulse protocol: channels were activated with a 0.5 s pulse to +40 mV followed by either a 20 ms or 5 ms hyperpolarizing pulse to potentials between −120 and +40 mV, the inactivation currents were recorded following a final pulse to +40 mV. (B) The corrected peak currents (described in the method), normalized to maximum currents, were plotted versus voltage and fit with a Boltzmann function. Values were plotted as mean ± SEM, wild-type (n = 7) and Q81X (n = 5).
3.4. The Q81X mutation escapes NMD by the reinitiation of translation
The observation that Q81X channels exhibit increased rates of deactivation, together with the decreased molecular weight of the mutant channel led to the hypothesis that translation is reinitiated following premature termination. An analysis of the hERG coding sequence revealed three in-frame methionine codons downstream of the Q81X mutation, M124, M133, and M137, that could potentially serve as sites of reinitiation (Fig. 6A). To determine whether translation was reinitiated at these sites following termination at Q81X, we mutated the downstream methionine codons to valine codons in a Q81X cDNA construct and analyzed hERG expression by western blot (Fig. 6B). When all three methionine residues downstream of Q81X were mutated to valine the expression of the truncated channels was completely abolished. This result indicates that downstream methionine codons at residue 124, 133, or 137 are required for reinitiation and supports the hypothesis that translation reinitiation allows the PTC-containing transcripts to escape NMD. Truncated hERG channels were expressed when M133 and M137 were mutated to valine, indicating that M124 alone is able to reinitiate translation. The expression of truncated channels from constructs containing Q81X and M124V mutations suggests that M133 or M137 may contribute to translation reinitiation. These results support the conclusion that the increased deactivation rates of the Q81X channels are the result of minimally deleting the first 123 residues of the hERG N-terminus. This is the first report of a LQT2 mutation that generates N-terminally truncated hERG channels.
Fig. 6.

Western blot analysis of translation reinitiation. (A) Schematic illustrating putative reinitiation sites downstream of the Q81X mutation. The partial cDNA sequence of hERG exon 3 reveals three downstream methionine codons (underlined) which are numbered according to the hERG cDNA sequence. (B) Western blot analysis of HEK293 cells transiently transfected with wild-type and the Q81X, Q81X+M124V, Q81X+M133V+M137V, and Q81X+M124V+M133V+M137V cDNA constructs. Proteins were detected with anti-hERG antibody. The results shown are representative of three independent experiments.
3.5. Q81X alters the inactivation and deactivation kinetics of heteromeric channels
Because the rates of channel deactivation and recovery from inactivation for the Q81X channels was significantly faster than those of wild-type channels, we determined whether the N-terminally truncated channels altered the functional properties of wild-type channels in HEK293 cells co-transfected with equimolar amounts of wild-type and Q81X minigenes. We used the deactivation voltage clamp protocol described in Fig. 4 to obtain the deactivation time constants as well as the time constants for the rate of recovery from inactivation at test potentials positive to −60 mV. Representative deactivation current traces are shown in Fig. 7A. Analysis of the deactivation kinetics indicated that both the slow and fast components of the deactivation time constant were intermediate between wild-type and Q81X channels (Fig. 7B, Table 1) and were significantly faster than wild-type rates at more positive test potentials. The τFast of heteromeric channels was significantly decreased compared with that of wild-type at test potentials positive to −100 mV and the τSlow was significantly decreased at test potentials positive to −70 mV (P < 0.05, ANOVA). The time constants of the recovery from inactivation were also intermediate between wild-type and mutant channels across all test potentials. At −60 mV the time constant for the recovery from inactivation was 4.6 ± 0.5, which was significantly faster than wild-type hERG (P < 0.05, ANOVA). These results strongly suggest that wild-type and Q81X channels co-assemble to generate heteromeric channels with altered kinetic properties.
Fig. 7.

Dominant-negative effects of Q81X. (A) Representative current trace from HEK293 cells transiently co-transfected with equimolar amounts of wild-type and Q81X minigenes. hERG current was recorded using the protocols in the legend of Fig. 4. (B) Voltage-dependence of the slow (circles) and fast (diamonds) components of the deactivation time constants for the heteromeric wild-type + Q81X channels are indicated as the black symbols (n = 6). The fast and slow components of deactivation of wild-type channels from Fig. 4 are shown as white symbols for comparison. The time constants for the fast component of the deactivation rate were significantly faster for Q81X channels at test potentials positive to −100 mV and for the slow components of the deactivation rate at test potentials positive to −70 mV (P < 0.05, ANOVA).
To demonstrate the physical association between wild-type hERG and Q81X we performed immunoprecipitation studies using differentially tagged wild-type and mutant hERG constructs. HEK293 cells stably expressing Flag-tagged wild-type channels were transiently transfected with HA-tagged wild-type or Q81X cDNA constructs. The expression of both wild-type and mutant channels was confirmed by western blot analysis using anti-HA and anti-Flag antibodies. Co-assembly of wild-type and mutant hERG channels was determined by immunoprecipitation with anti-Flag antibody followed by western blot analysis with anti-HA antibody (Fig. 8). The membrane was also probed with anti-Flag antibody demonstrating the efficiency of immunoprecipitation of the Flag-tagged channels. The co-assembly between wild-type and Q81X was observed in the immature and the mature forms of the hERG protein indicating that heteromeric channels are able to undergo complex glycosylation and traffic to the cell-surface. These results indicate that the Q81X mutation alters the gating properties of heteromeric hERG channels formed by co-assembly of wild-type and Q81X channels.
Fig. 8.
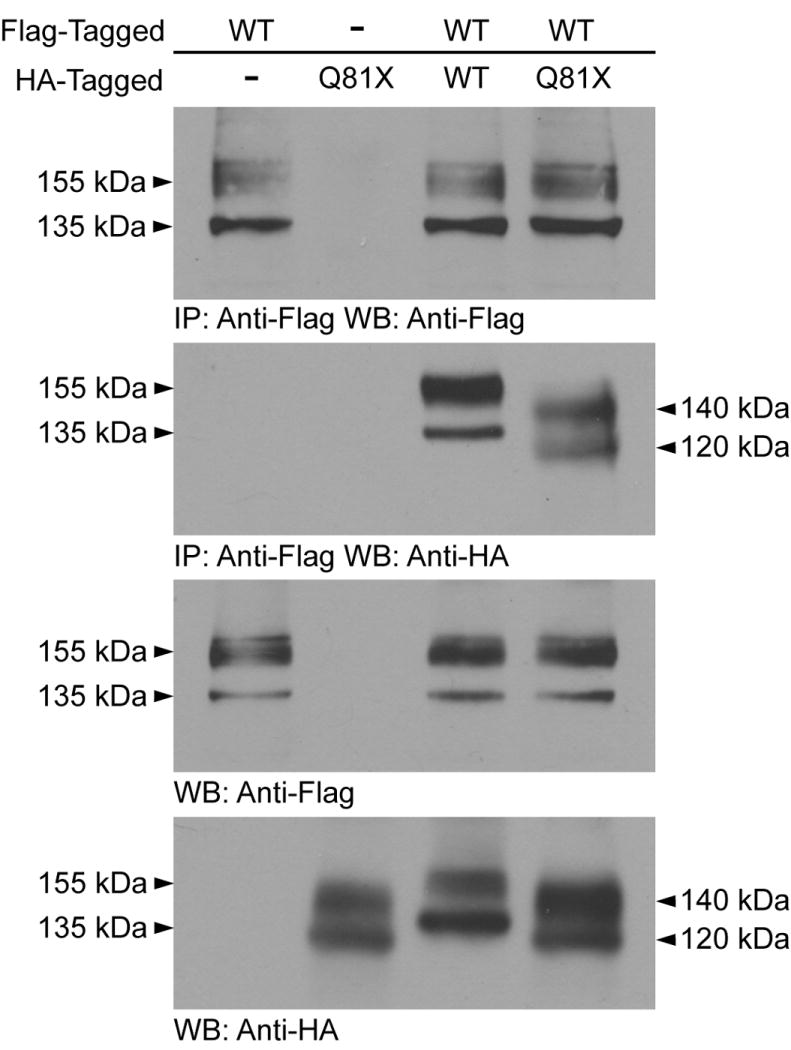
Co-assembly of wild-type and Q81X channels. HA-tagged Q81X cDNA constructs were transiently transfected into HEK293 cells stably expressing Flag-tagged wild-type hERG channels. Cell lysates were subjected to immunoprecipitation using anti-Flag, followed by western blot with anti-HA and anti-Flag antibody. HEK293 cells stably expressing Flag-tagged wild-type or transiently expressing HA-tagged Q81X were used as controls. The lower two panels are inputs and are probed with anti-Flag and anti-HA antibody. The results shown are representative of three independent experiments.
3.6. Q81X decreases resurgent outward current during ventricular action potential clamp
LQT2 missense mutations which cause accelerated deactivation rates have been shown to decrease the outward current through hERG channels during the repolarization phase resulting in action potential prolongation [32]. To characterize the effect of the Q81X mutation during the ventricular action potential, we performed action potential clamp using a waveform recorded from a human ventricular myocyte. Fig. 9A shows the action potential waveform and the current traces recorded from representative HEK-293 cells transfected with wild-type, Q81X, or both hERG constructs. Plotting the averaged current density versus time (Fig. 9B) revealed no significant difference in the current levels during the plateau of the action potential. However, during the late phases of the action potential, the mutant channels exhibited a significant decrease in outward current levels compared to wild-type channels. We also plotted the averaged current density as a function of the action potential voltage (Fig. 9C). Although the peak current densities of Q81X and heteromeric channels were not significantly decreased relative to wild-type hERG both the channels exhibited a significant positive shift in the peak potentials (Fig. 9D, P < 0.05, ANOVA). The positive shift in the peak potential of the mutant channels presumably results from the rapid recovery from inactivation observed in these channels. To determine the charge transferred during the action potential clamp, we integrated the area under the current trace. The total charges transferred during the entire action potential were not significantly different among mutant and wild-type channels. However, the charges transferred during the late phase of the action potential, corresponding to voltages more negative than −40 mV, were significantly decreased in Q81X and heteromeric channels compared to wild-type channels (Fig. 9E, P < 0.05, ANOVA). These results are consistent with previous studies of hERG channels with LQT2 missense mutation in the N-terminal Per, Arnt and Sim (PAS) domain [32].
Fig. 9.

hERG current during ventricular action potential clamp. (A) The ventricular action potential clamp waveform (upper trace) used to generate the representative current traces of wild-type hERG, Q81X and wild-type + Q81X channels (lower traces). (B) Plot of the averaged current density versus time during the ventricular action potential clamp. (C) Plot of the averaged current density as a function of action potential voltage. (D) Peak current density plotted as a function of peak potential. (E) Plot of the charge density transferred during the late phases of repolarization of the ventricular action potential clamp at potentials negative to −40 mV. Data were plotted as mean ± SEM, wild-type (n = 6), Q81X (n = 4), wild-type + Q81X (n = 5), *, P < 0.05 compared to wild-type hERG, ANOVA.
4. Discussion
In the present study, we describe a mechanism of long QT syndrome in which hERG transcripts containing the Q81X nonsense mutation escape NMD by the reinitiation of translation resulting in the generation of N-terminally truncated channels. The full-length hERG minigenes used in this study permitted a comprehensive analysis of this novel mechanism at the mRNA, protein and functional levels. The resistance to NMD and the subsequent translation of Q81X transcripts were shown by the persistence of PTC-containing mRNA in the RNase protection assay, the detection of truncated channel proteins by western blot, and the current recordings from the Q81X channels. The mutant channels exhibited decreased tail current density, accelerated deactivation kinetics, reduced resurgent outward current and co-assembled with wild-type hERG to form heteromeric channels with altered gating properties. The reinitiation of translation represents a new mechanism of hERG channel dysfunction in LQT2.
The NMD surveillance mechanism has evolved to eliminate PTCs, thereby, preventing the expression of potentially dangerous truncated proteins. NMD is dependent on pre-mRNA splicing and the exon-junction protein complex (EJC) that is deposited 20–24 nt upstream of each exon-exon junction. The EJC is normally displaced during the pioneer round of translation, but premature termination leaves the complex intact which triggers NMD [33]. It has been proposed that PTCs occurring <50 nt upstream of the last exon-exon junction or within the terminal exon are immune to degradation by NMD [17]. Certain PTC mutations occurring near the translational start site have also been shown to escape NMD [34]. Translation may be reinitiated following premature termination when initiation factors are still associated with the ribosome, allowing the ribosome to resume scanning for alternate start codons. Generation of N-terminally truncated proteins by translation reinitiation has been reported in a growing number of genes associated with inherited diseases including the BRCA1, ATP7A and NEMO genes [35–37]. Q81X represents the first LQT2 mutation known to generate N-terminally truncated hERG channels.
Translation reinitiation was confirmed by the inhibition of protein synthesis following the mutation of three methionine codons downstream of Q81X to valine. Our experimental results were consistent with in silico analysis using the NetStart 1.0 server [38] which provides a prediction of the likelihood of translation based on an artificial neural network. Scores generated by NetStart 1.0 range between 0.0 and 1.0 and a score greater than 0.5 represents a probable start site. M124 was scored at 0.85 indicating a strong likelihood of translation initiation (for comparison the hERG M1 was scored at 0.64). The downstream M133 and M137 codons received lower scores, 0.42 and 0.47, respectively. Following M137, the next in-frame start codon is M218, and although its predicted score is 0.75, it is unable to reinitiate translation of transcripts containing the Q81X or the P141fs+2X mutations. Three lines of evidence lead to the conclusion that M124 is a preferred site of translation reinitiation in the Q81X mutant. First, M124 is the first in-frame methionine codon following the Q81X mutation, second, the in silico analysis predicts a very high translation initiation score, and third, the results of our mutagenesis study indicating that M124 alone can be used to synthesize truncated channels.
The N-terminus of hERG contains several regions that contribute to the maintenance of slow channel deactivation. These regions include an initial unordered segment comprised of residues 2–9, an amphiphatic α-helix formed by residues13–23 and a helix-loop-helix motif Per, Arnt and Sim (PAS) domain that encompasses residues 26–135 [39–41]. The importance of these regions was determined by functional studies showing that N-terminally truncated hERG channels exhibited increased rates of deactivation relative to the full-length channel [30, 31, 39, 42]. Early studies suggested that deactivation was regulated by interactions between the N-terminus and the S4-S5 linker [39, 43, 44]. Recent studies have provided strong evidence that deactivation is regulated by interactions between N-terminus and the cytosolic C-terminus of the channel. FRET studies revealed that a PAS domain fragment interacted with the cytoplasmic C-terminus of hERG and was sufficient to restore the regulation of deactivation of LQT2 mutant channels [45]. Muskett et al. recently reported an acidic patch on the cyclic nucleotide-binding homology domain as a putative interface for the binding of the N-terminus [40]. Several LQT2 missense mutations within the hERG N-terminus exhibit similar accelerated deactivation properties to N-terminally truncated channels [28, 32]. The deletion of these N-terminal domains in Q81X channels explains the observed increase in deactivation rates.
The mutant channel generated less outward current during the later phase of repolarization in ventricular action potential clamp studies. Two factors may contribute to the loss of repolarizing current: increased rates of deactivation and decreased numbers of channels expressed at the cell surface. Decreased surface density of hERG may arise from reduced efficiencies of translation reinitiation or by trafficking deficiencies of the N-terminally truncated channels. Future studies will be necessary to determine the relative contribution of these factors to the loss of repolarizing current and to elucidate the precise mechanism of cardiac action potential prolongation. The Q81X channels also exhibited a significant positive shift in the peak potential during the action potential clamp indicating that the mutant channels peak earlier during the action potential. This shift may arise from the increased rates of recovery from inactivation in the mutant channels. The earlier peak of the mutant channel did not result in a significant change in the total charge transferred during the action potential. Rather, we observed a significant decrease in the charge transferred during the late phases of action potential repolarization caused by a decrease in the outward current. The present results suggest that generation of smaller outward current and transfer of less charge during the repolarization phase of the cardiac action potential contribute to the prolongation of the QT interval in affected individuals.
Previous studies have identified several pathogenic mechanisms of LQT2 mutations. These mechanisms include defective trafficking and assembly, abnormal gating or kinetics, the disruption of channel permeability, and degradation of PTC-containing transcripts by NMD. The reinitiation of translation by LQT2 nonsense mutations represents a novel mechanism to generate dysfunctional hERG channels. Identification of reinitiation of translation by nonsense mutations may have important implications for the clinical phenotype in LQT2. For example, NMD is often associated with a mild clinical phenotype arising from haploinsufficiency and it is likely that the translation reinitiation mechanism that allows LQT2 mutations to escape this surveillance pathway and generate N-terminally truncated hERG channels may give rise to more severe pathogenic phenotypes.
In summary, our finding that the LQT2 nonsense mutation Q81X escapes NMD by the reinitiation of translation has increased our understanding of mechanisms that cause long QT syndrome. Translation reinitiation may represent a new pathogenic role of early LQT2 nonsense and frameshift mutations in the hERG gene. Furthermore, the characterization of a naturally occurring mutation that generates N-terminally truncated hERG channels provides biological significance for studies of the structure and function of the hERG N-terminus.
Research Highlights.
The LQT2 nonsense mutation Q81X is resistant to nonsense-mediated mRNA decay.
N-terminally truncated Q81X channels are generated by translation reinitiation.
Q81X channels exhibit increased rates of deactivation.
Q81X channels generate less outward current during action potential repolarization.
Translation reinitiation is a novel mechanism of long QT syndrome.
Acknowledgments
We thank Dr. Gui-Rong Li in the University of Hong Kong for providing the human ventricular action potential waveform. This study was supported in part by NIH grant HL-68854 (ZZ). MRS is supported by NIH training grant T32HL094294. ZZ is an Established Investigator of the American Heart Association.
Footnotes
Disclosures
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 2.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–5. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz PJ, Periti M, Malliani A. The long Q-T syndrome. Am Heart J. 1975;89:378–90. doi: 10.1016/0002-8703(75)90089-7. [DOI] [PubMed] [Google Scholar]
- 4.Sanguinetti MC. HERG1 channelopathies. Pflugers Arch. 2010;460:265–76. doi: 10.1007/s00424-009-0758-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–85. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 6.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–17. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 7.Napolitano C, Priori SG, Schwartz PJ, Bloise R, Ronchetti E, Nastoli J, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294:2975–80. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 8.Nagaoka I, Shimizu W, Itoh H, Yamamoto S, Sakaguchi T, Oka Y, et al. Mutation site dependent variability of cardiac events in Japanese LQT2 form of congenital long-QT syndrome. Circ J. 2008;72:694–9. doi: 10.1253/circj.72.694. [DOI] [PubMed] [Google Scholar]
- 9.Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gong Q, Zhang L, Vincent GM, Horne BD, Zhou Z. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation. 2007;116:17–24. doi: 10.1161/CIRCULATIONAHA.107.708818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zarraga IG, Zhang L, Stump MR, Gong Q, Vincent GM, Zhou Z. Nonsense-mediated mRNA decay caused by a frameshift mutation in a large kindred of type 2 long QT syndrome. Heart Rhythm. 2011;8:1200–6. doi: 10.1016/j.hrthm.2011.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- 13.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 14.Gong Q, Keeney DR, Robinson JC, Zhou Z. Defective assembly and trafficking of mutant HERG channels with C-terminal truncations in long QT syndrome. J Mol Cell Cardiol. 2004;37:1225–33. doi: 10.1016/j.yjmcc.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, et al. Recurrent intrauterine fetal loss due to near absence of HERG: clinical and functional characterization of a homozygous nonsense HERG Q1070X mutation. Heart Rhythm. 2008;5:553–61. doi: 10.1016/j.hrthm.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong Q, Stump MR, Zhou Z. Inhibition of nonsense-mediated mRNA decay by antisense morpholino oligonucleotides restores functional expression of hERG nonsense and frameshift mutations in long-QT syndrome. J Mol Cell Cardiol. 2011;50:223–9. doi: 10.1016/j.yjmcc.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998;23:198–9. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- 18.Kozak M. Constraints on reinitiation of translation in mammals. Nucleic Acids Res. 2001;29:5226–32. doi: 10.1093/nar/29.24.5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rinne T, Clements SE, Lamme E, Duijf PH, Bolat E, Meijer R, et al. A novel translation re-initiation mechanism for the p63 gene revealed by amino-terminal truncating mutations in Rapp-Hodgkin/Hay-Wells-like syndromes. Hum Mol Genet. 2008;17:1968–77. doi: 10.1093/hmg/ddn094. [DOI] [PubMed] [Google Scholar]
- 20.Neu-Yilik G, Amthor B, Gehring NH, Bahri S, Paidassi H, Hentze MW, et al. Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. RNA. 2011;17:843–54. doi: 10.1261/rna.2401811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95:2979–84. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abramoff M, Magelhaes P, Ram S. Image Processing with ImageJ. Biophotonics International. 2004;11:36–44. [Google Scholar]
- 23.Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem. 1998;273:21061–6. doi: 10.1074/jbc.273.33.21061. [DOI] [PubMed] [Google Scholar]
- 24.Stump MR, Gong Q, Zhou Z. Multiple splicing defects caused by hERG splice site mutation 2592+1G>A associated with long QT syndrome. Am J Physiol Heart Circ Physiol. 2011;300:H312–8. doi: 10.1152/ajpheart.00818.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sale H, Wang J, O’Hara TJ, Tester DJ, Phartiyal P, He JQ, et al. Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b-specific mutation associated with Long-QT syndrome. Circ Res. 2008;103:e81–95. doi: 10.1161/CIRCRESAHA.108.185249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–6. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- 27.Larsen AP, Olesen SP, Grunnet M, Jespersen T. Characterization of hERG1a and hERG1b potassium channels-a possible role for hERG1b in the I (Kr) current. Pflugers Arch. 2008;456:1137–48. doi: 10.1007/s00424-008-0476-7. [DOI] [PubMed] [Google Scholar]
- 28.Gianulis EC, Trudeau MC. Rescue of aberrant gating by a genetically encoded PAS (Per-Arnt-Sim) domain in several long QT syndrome mutant human ether-a-go-go-related gene potassium channels. J Biol Chem. 2011;286:22160–9. doi: 10.1074/jbc.M110.205948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inoue K, Khajavi M, Ohyama T, Hirabayashi S, Wilson J, Reggin JD, et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004;36:361–9. doi: 10.1038/ng1322. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Trudeau MC, Zappia AM, Robertson GA. Regulation of deactivation by an amino terminal domain in human ether-a-go-go-related gene potassium channels. J Gen Physiol. 1998;112:637–47. doi: 10.1085/jgp.112.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schonherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol. 1996;493 (Pt 3):635–42. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J, Zou A, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J Biol Chem. 1999;274:10113–8. doi: 10.1074/jbc.274.15.10113. [DOI] [PubMed] [Google Scholar]
- 33.Ishigaki Y, Li X, Serin G, Maquat LE. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell. 2001;106:607–17. doi: 10.1016/s0092-8674(01)00475-5. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Maquat LE. Evidence that translation reinitiation abrogates nonsense-mediated mRNA decay in mammalian cells. EMBO J. 1997;16:826–33. doi: 10.1093/emboj/16.4.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buisson M, Anczukow O, Zetoune AB, Ware MD, Mazoyer S. The 185delAG mutation (c.68_69delAG) in the BRCA1 gene triggers translation reinitiation at a downstream AUG codon. Hum Mutat. 2006;27:1024–9. doi: 10.1002/humu.20384. [DOI] [PubMed] [Google Scholar]
- 36.Puel A, Reichenbach J, Bustamante J, Ku CL, Feinberg J, Doffinger R, et al. The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am J Hum Genet. 2006;78:691–701. doi: 10.1086/501532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paulsen M, Lund C, Akram Z, Winther JR, Horn N, Moller LB. Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites. Am J Hum Genet. 2006;79:214–29. doi: 10.1086/505407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pedersen AG, Nielsen H. Neural network prediction of translation initiation sites in eukaryotes: perspectives for EST and genome analysis. Proc Int Conf Intell Syst Mol Biol. 1997;5:226–33. [PubMed] [Google Scholar]
- 39.Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M, Mackinnon R. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell. 1998;95:649–55. doi: 10.1016/s0092-8674(00)81635-9. [DOI] [PubMed] [Google Scholar]
- 40.Muskett FW, Thouta S, Thomson SJ, Bowen A, Stansfeld PJ, Mitcheson JS. Mechanistic Insight into Human ether-a-go-go-related Gene (hERG) K+ Channel Deactivation Gating from the Solution Structure of the EAG Domain. J Biol Chem. 2011;286:6184–91. doi: 10.1074/jbc.M110.199364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng CA, Hunter MJ, Perry MD, Mobli M, Ke Y, Kuchel PW, et al. The N-terminal tail of hERG contains an amphipathic alpha-helix that regulates channel deactivation. PLoS One. 2011;6:e16191. doi: 10.1371/journal.pone.0016191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. J Gen Physiol. 1996;107:611–9. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanguinetti MC, Xu QP. Mutations of the S4–S5 linker alter activation properties of HERG potassium channels expressed in Xenopus oocytes. J Physiol. 1999;514 (Pt 3):667–75. doi: 10.1111/j.1469-7793.1999.667ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Myers CD, Robertson GA. Dynamic control of deactivation gating by a soluble amino-terminal domain in HERG K(+) channels. J Gen Physiol. 2000;115:749–58. doi: 10.1085/jgp.115.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gustina AS, Trudeau MC. A recombinant N-terminal domain fully restores deactivation gating in N-truncated and long QT syndrome mutant hERG potassium channels. Proc Natl Acad Sci U S A. 2009;106:13082–7. doi: 10.1073/pnas.0900180106. [DOI] [PMC free article] [PubMed] [Google Scholar]