

Abstract
Autophagy is an essential, conserved lysosomal degradation pathway that controls the quality of the cytoplasm by eliminating protein aggregates and damaged organelles. It begins when double-membraned autophagosomes engulf portions of the cytoplasm, which is followed by fusion of these vesicles with lysosomes and degradation of the autophagic contents. In addition to its vital homeostatic role, this degradation pathway is involved in various human disorders, including metabolic conditions, neurodegenerative diseases, cancers and infectious diseases. This article provides an overview of the mechanisms and regulation of autophagy, the role of this pathway in disease and strategies for therapeutic modulation.
Autophagy occurs at a basal rate in most cells, eliminating protein aggregates and damaged organelles in order to maintain cytoplasmic homeostasis1. This includes the degradation of dysfunctional mitochondria via mitophagy, a cytoprotective process that limits both the production of reactive oxygen species (ROS) and the release of toxic intramitochondrial proteins.
Autophagy is stimulated during various pathological and physiological states, such as starvation. Starvationinduced autophagy, an evolutionarily conserved response in eukaryotes2, enables the degradation of proteins, carbohydrates and lipids, which allows the cell to adapt its metabolism and meet its energy needs. Indeed, the induction of autophagy in newborn mice has a major role in maintaining energy levels in various tissues after the maternal nutrient supply via the placenta ceases3. Moreover, starvation-induced autophagy has a cytoprotective effect by blocking the induction of apoptosis by mitochondria4.
Autophagy is also essential during development and differentiation1,5. The pre-implantation period after oocyte fertilization requires the autophagic degradation of components of the oocyte cytoplasm6. Moreover, autophagy is crucial for the selective elimination of paternal mitochondria post-fertilization7,8. Autophagy remodels the cytoplasm of erythrocytes, lymphocytes and adipocytes, and thereby contributes to their differentiation1,5.
Autophagy may be dysregulated in several disorders, including metabolic diseases, neurodegenerative disorders, infectious diseases and cancer. In some conditions, autophagy is inhibited and this can occur at different stages of the process to enhance disease, whereas in other cases autophagic activity may be permissive towards pathogenesis. In addition, the induction of autophagy has been shown to increase longevity in a large panel of species (reviewed in REF. 9), thus raising the possibility that ageing and longevity may be therapeutic targets for autophagy induction.
Given these observations, pharmacological approaches to upregulate or inhibit this pathway are currently receiving considerable attention. For example, autophagy upregulation may be of therapeutic benefit in certain neurodegenerative diseases (such as Huntington’s disease), whereas autophagy inhibition is being investigated as a strategy for treating some cancers.
Here, we consider examples of diseases in which autophagy is perturbed, after briefly reviewing the mechanisms and regulation of mammalian autophagy. Potential strategies and agents for therapeutic modulation are also discussed, as well as possible safety concerns and caveats associated with such approaches.
Mechanisms of autophagy
Overview
Autophagy is initiated by the formation of double-membrane-bound vesicles, called autophagosomes, which sequester cytoplasmic material in a nondegradative compartment. Autophagosomes are formed from cup-shaped structures known as phagophores (reviewed in REF. 2) (FIGS 1,2). Once the edges of the phagophores are sealed, the completed autophagosomes have diameters of 300–900 nm and can subsequently receive inputs from the endocytic pathway, which enables acquisition of acidic and degradative capacities. Autophagosomes are formed randomly at multiple locations in the mammalian cytoplasm and need to be trafficked along microtubules towards the microtubule organizing centre to bring them into the proximity of lysosomes, which are clustered at this site. This facilitates the final stage of autophagy, the fusion of autophagosomes or amphisomes with lysosomes, which enables degradation of autophagic cargos and the subsequent recycling of nutrients (to meet metabolic demands) and membranes (to permit ongoing lysosomal functions).
Figure 1. Overview of the regulation of macroautophagy and potential drug targets.

Two major signalling pathways are depicted here: the pathway involving class I phosphoinositide 3-kinase (PI3K), protein kinase B (PKB) and mammalian target of rapamycin complex 1 (mTORC1), and a cyclical mTOR-independent pathway; the basic helix–loop–helix leucine zipper transcription factor EB (TFEB)-mediated pathway is also depicted213. TFEB regulates the expression of the genes involved in the different stages of autophagy between autophagosome formation and cargo degradation (see FIG. 2). In nutrient-rich medium, TFEB is phosphorylated by mTORC1 and is retained in the cytoplasm. In starved cells, TFEB is dephosphorylated and is translocated into the nucleus. TFEB is a potential target for drugs. Retaining TFEB in the cytoplasm would inhibit autophagy, as illustrated in the figure. By contrast, promoting the nuclear translocation of TFEB would stimulate autophagy. Activating the class I PI3K–PKB–mTORC1 pathway by growth factors and amino acids blocks autophagy by inhibiting the initiation of autophagosome formation by the UNC51-like kinase 1 (ULK1) complex. Glucose starvation increases the AMP/ATP ratio, which activates AMP-activated protein kinase (AMPK). This enzyme induces autophagy by inhibiting mTORC1 (via directly targeting elements of the complex) or by phosphorylating and activating tuberous sclerosis 2 (TSC2) in the TSC1–TSC2 complex. Recent data suggest that mTOR interacts with TFEB, as the two proteins are colocalized on lysosomal membranes. Phosphorylation of mTOR inhibits TFEB acivity, whereas ATP-competitive mTOR inhibitors enable TFEB dephosphorylation, thus allowing its nuclear translocation and activation218. AMPK also phosphorylates and thus activates the ULK1 complex. Activators of AMPK (such as metformin) stimulate autophagy, as do inhibitors of mTORC1 (for example, rapamycin or rapalogues) and inhibitors of class I PI3Ks (for example, PI-103). Amino acids activate mTORC1 via RAG GTPases, and also via the vacuolar V-ATPase located in the lysosomal membrane219. Inhibitors of V-ATPases, such as bafilomycin A1, block the maturation of autophagosomes. However, blocking V-ATPase also inhibits the amino-acid-dependent activation of mTORC1, which would have a stimulatory effect on autophagosome formation. Elevation of the intracellular levels of cyclic AMP (cAMP) by adenylyl cyclase downstream of G protein-coupled receptors blocks autophagy by activating the exchange protein directly activated by cAMP (EPAC), the small G protein RAP2B and phospholipase Cε (PLCε). Activation of PLCε results in the production of inositol-1,4,5-trisphosphate (Ins(1,4,5)P3) and, consequently, the release of Ca2+ from the endoplasmic reticulum via the Ins(1,4,5)P3 receptor (Ins(1,4,5)P3R). Influx of Ca2+ into the cytoplasm is also triggered by l-type Ca2+ channel agonists. Drugs acting at the various different steps of the cAMP–EPAC–PLCε– Ins(1,4,5)P3 pathway regulate autophagy. Calpains activated by Ca2+ block autophagy by cleaving and constitutively activating Gsα proteins (also called Gαs), which increases cAMP levels. Thus, inhibitors of calpains would stimulate autophagy in this setting. The binding of B cell lymphoma 2 (BCL-2) to beclin 1 inhibits autophagy, and this effect can be counteracted by BCL-2 homology 3 (BH3) mimetics. DAG, diacylglycerol; IMPase, inositol monophosphatase; InsP, inositol monophosphate; Ins(1,4)P2, inositol-1,4-bisphosphate; PtdIns(4,5)P2, phosphatidylinositol-4,5-bisphosphate; RHEB, RAS homologue enriched in brain (GTP binding protein).
Figure 2. Effects of drugs on the different steps of the autophagic pathway.

The formation of the autophagosome depends on the initiation and/or nucleation of a specific membrane structure known as the phagophore, and on the elongation and closure of the phagophore to form a double-membrane-bound vacuole. The biogenesis of autophagosomes requires the ordered intervention of autophagy-regulated (ATG) proteins that act on different modules. Some of these modules are shown on the figure, including the UNC51-like kinase 1 (ULK1) and phosphoinositide 3-kinase (PI3K) complexes, ATG12 (the ATG12–ATG5 complex) and microtubule-associated protein 1 light-chain 3 (LC3; for example, the LC3–phosphatidylethanolamine conjugates); ATG4B is a protein that is involved in the regulation of LC3 lipidation and delipidation. The ULK1 complex is composed of ULK1 (the mammalian orthologue of yeast Atg1) and RB1-inducible coiled-coil 1 (RB1CC1; also known as FIP200; the mammalian orthologue of yeast Atg13, Atg17 and Atg101). The PI3K complex is composed of beclin 1 (the mammalian orthologue of yeast Atg6 and Atg14), class III PI3K (PIK3C3), PIK3R4 (PI3K regulatory subunit 4) and AMBRA1 (activating molecule in BECN1-regulated autophagy protein 1). Both complexes congregate at the phagophore assembly site to initiate autophagy in response to nutrient starvation. The kinase activity of ULK1 is controlled by the kinase mammalian target of rapamycin (mTOR) in mTOR complex 1 (mTORC1), which is sensitive to rapamycin220. The protein ATG14 appears to have a key role in the targeting of the PI3K complex to the endoplasmic reticulum (ER). How the ULK1 and PI3K complexes are coordinately regulated remains to be elucidated. The production of phosphatidylinositol-3-phosphate (PtdIns3P) by human PIK3C3 recruits the PtdIns3P-binding proteins ATG18 (also known as WIPI1 and WIPI2) and double FYVE domain-containing protein 1 (DFPC1). Cargos can be incorporated into autophagosomes in a non-selective manner (known as bulk autophagy) or in a selective manner (known as selective autophagy). Maturation occurs when the autophagosome fuses with the endolysosomal compartment. The final degradation of cargos occurs in autolysosomes. The activity of the class III PI3K complex can be manipulated by both activators (such as BH3 (BCL-2 homology 3) mimetics) and inhibitors (such as spautin-1; specific and potent autophagy inhibitor 1). PIK3C3, which is a component of the class III PI3K complex, is a potential target for drugs. Some other potential targets are also indicated on the figure. Autophagosome closure can be inhibited by reducing ATG4B activity. Compounds that interfere with LC3-interacting region (LIR) motifs could potentially block the selective recruitment of cargos such as mitochondria, viruses and bacteria. Inhibitors of lysosomal enzymes (such as cystatin B), and lysosomotropic agents that increase the lysosomal pH (such as chloroquine and hydrochloroquine) block the degradative activity of autolysosomes. NDP52, nuclear dot protein 52; p62, ubiquitin-binding protein p62 (also known as sequestosome 1).
Autophagosome formation and regulation
The formation of the autophagosome is a multistep process that includes the biogenesis of the phagophore, followed by its elongation and closure (FIG. 2). More than 15 autophagy-related (ATG) proteins, as well as class III phosphoinositide 3-kinases (PI3Ks; also known as PIK3C3 or VPS34), are required to construct the autophagosome. These ATG proteins appear to be hierarchically recruited at the phagophore assembly site (also known as the pre-autophagosomal structure)10. This process also requires the shuttling of ATG9, the only transmembrane ATG protein, between a peripheral site and the isolation membrane10. Autophagosomes may acquire membrane from multiple sources, including the endoplasmic reticulum (ER)11–13, the mitochondrial outer membrane14 (possibly at mitochondria–ER contact sites) and the plasma membrane15, as well as the Golgi apparatus and post-Golgi compartments16,17. The biogenesis of autophagosomes, including membrane expansion steps, involves SNAREs in yeast and mammalian cells18,19.
The proteins ATG8 and microtubule-associated protein 1 light-chain 3 (LC3; also known as MAP1LC3) appear to have multiple functions in autophagy. In addition to their proposed roles in the expansion and fusion of phagophore edges (reviewed in REF. 1), they can recruit adaptor proteins such as ubiquitin-binding protein p62 (also known as sequestosome 1) and NBR1 to autophagosomes via their LC3-interacting region (LIR) domains, and these adaptors can, in turn, mediate selective autophagy of different cellular structures, protein aggregates and microorganisms20–22 (FIG. 2).
The activity of the autophagic machinery is regulated by upstream signalling (FIG. 1). Many signals, including growth factors, amino acids, glucose and energy status, are integrated by the kinase activity of mammalian target of rapamycin (mTOR)23. The induction of autophagy by the inhibition of mTOR complex 1 (mTORC1), resulting from starvation or rapamycin (also known as sirolimus (Rapamune; Pfizer)) treatment, is conserved from yeast to mammals24,25 and acts upstream of the UNC51-like kinase 1 (ULK1; also known as ATG1) complex. The other mTOR complex, mTORC2, which is not directly inhibited by rapamycin, controls the activity of the transcription factor forkhead box protein O3 (FOXO3). Interestingly, FOXO3 has been shown to stimulate autophagy in muscle cells and this has been correlated with increased transcription of several genes involved in autophagy26. The activity of the class III PI3K complex is also regulated during starvation. The anti-apoptotic protein B cell lymphoma 2 (BCL-2) and other anti-apoptotic members of the BCL-2 family, such as BCL-XL, inhibit autophagy27–29. BCL-2 and BCL-XL bind to beclin 1(BECN1) through a BCL-2 homology 3 (BH3) domain that mediates the docking of BECN1 to the BH3-binding groove of BCL-2 or BCL-XL. In response to starvation, JUN N-terminal kinase 1 (JNK1) phosphorylates BCL-2 to trigger its release from BECN1, which enables autophagy induction30,31. BCL-2 proteins can also be dissociated from BECN1 by alternative mechanisms in response to other stimuli. Some other key autophagy regulators are summarized in TABLE 1, and unconventional roles for autophagic machineries are considered in BOX 1. Below, we consider the roles of autophagy in various key diseases and review emerging strategies for therapeutic intervention.
Table 1.
Major key players in the maturation and degradation of autophagosomes
| Names | Functions | Refs |
|---|---|---|
| Small GTPase RAB7 | RAB7 is required for the fusion of autophagosomes and amphisomes (compartments resulting from the fusion of autophagosomes with endosomes) with lysosomes | 221,222 |
| Small GTPase RAB11 | RAB11 is required for the fusion of autophagosomes with multivesicular bodies | 223 |
| Small GTPase RAB33B | RAB33B is localized in the Golgi; it regulates autophagosome maturation by controlling their fusion with lysosomes | 224 |
| PIK3C3 complex; UVRAG | UVRAG in the PI3K complex interacts with beclin 1 to upregulate the maturation of autophagosomes | 225 |
| Rubicon | Rubicon interacts with UVRAG to downregulate the maturation of autophagosomes | 226, 227 |
| SNAREs: VAMP3, VAMP8 and VTI1B | VAMP3 controls the fusion of autophagosomes with multivesicular bodies; VAMP8 and VTI1B control the fusion of autophagosomes with lysosomes | 228–230 |
| Syntaxin 5 | Syntaxin 5 is required for the late stage of autophagy | 231 |
| ESCRTs | ESCRT proteins mediate the biogenesis of multivesicular bodies and the endocytic sorting of proteins; ESCRT-I, ESCRT-II and ESCRT-III are most likely to be involved in the fusion of autophagosomes with the endolysosomal system | 232–234 |
| DRAM | DRAM is a direct target of p53 and is located in the lysosomal membrane; it may regulate the late stage of autophagy | 235 |
| TFEB | TFEB controls the expression of genes involved in lysosomal biogenesis and in different steps of autophagy; its nuclear transport is controlled by mTORC1 | 213, 218 |
| Microtubules | Microtubules are cytoskeletal elements that are used by autophagosomes to move towards lysosomes | 236–241 |
| V-ATPase | V-ATPase is located in the membrane of acidic compartments; inhibition of V-ATPase by bafilomycin A1 blocks the maturation of autophagosomes; V-ATPase is involved in the amino acid signalling that activates mTORC1 | 219, 242, 243 |
| ATG22 | ATG22 is a permease in yeast that is required for the lysosomal efflux of amino acids | 244 |
| Spinster | Spinster is a putative lysosomal permease that is required for lysosomal reformation | 245 |
ATG22, autophagy-regulated protein 22; DRAM, damage-regulated autophagy modulator; ESCRT, endosomal sorting complex required for transport; mTORC1, mammalian target of rapamycin complex 1; p53, cellular tumour antigen p53; PI3K, phosphoinositide 3-kinase; SNARE, soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein (SNAP) receptor; TFEB, transcription factor EB; UVRAG, UV irradiation resistance-associated gene; VAMP3, vesicle-associated membrane protein 3; V-ATPase, vacuolar ATPase; VTI1B, vesicle transport through interaction with t-SNARE homolog 1B.
Box 1 | Plasticity of autophagosome formation and destination.
The etymology of autophagy — ‘self-eating’ — indicates that the final goal of autophagosomes is to deliver cargo to the lysosome for degradation. Recent studies in yeast have demonstrated that autophagosomes can fuse with the plasma membrane to release their cargo into the extracellular medium214,215. This mechanism sheds some light on the unconventional secretion of proteins. A mechanism of this type in mammalian cells is also involved in exporting the pro-inflammatory cytokine interleukin-1 (REF. 159). This process is dependent on autophagy-related protein 5 (ATG5), the inflammasome, the peripheral Golgi protein Golgi reassembly stacking protein 2 55kDa (GRASP55; also known as GORASP2) and on the small GTPase RAB8A. This autophagy-based secretion mechanism can probably be extended to other proteins and small molecules that modulate the immune response159. Interestingly, the mechanism that leads to the formation of the secretory autophagosomes can be modulated either via the classical biogenesis pathway or via a non-classical pathway (such as that described in yeast) that emerges from a novel compartment for unconventional protein secretion (CUPS)216. Like the conventional site for autophagosome formation, the CUPS contains high levels of the lipid phosphatidylinositol- 3-phosphate (PtdIns3P) as well as ATG8 and ATG9. However, although these two proteins are required for the formation of the phagophore assembly site (also known as the pre-autophagosomal structure), they are not necessary for the formation of the CUPS. It appears that the formation of autophagosomes that are engaged in the degradative pathway is also a matter of plasticity in mammalian cells210. What we already know about the formation of the autophagosome relies on the seminal discovery of Atg genes in yeast10. Most of these genes are conserved and act in an apparent hierarchical manner to form an autophagosome. This classical or canonical pathway can be triggered by amino acids or by serum starvation. However, in other settings — as reviewed in REFs 210,217 — only a subset of ATG proteins are used to form an autophagosome. For example, autophagy may not require some of the components of complex I of phosphoinositide 3-kinase (PI3K), such as beclin 1 and class III PI3K (also known as PIK3C3). However, it is possible that in this setting the lipid PtdIns3P may still have a role. This lipid can be produced by other sources, either from the degradation of phosphatidylinositol polyphosphates such as phosphatidylinositol- 3,4-bisphosphate (PtdIns(3,4)P2), phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) and phosphatidylinositol-3,5-bisphosphate (PtdIns(3,5)P2), or via the activity of class II PI3Ks on phosphatidylinositol phosphates. Deviation from the canonical pathway of autophagosome biosynthesis and the final destination of autophagosomes are emerging concepts that should be taken into consideration in the attempts to develop drugs to target the autophagic pathway.
Autophagy and metabolic disease
Amino acids produced by autophagy in the muscles and liver can be used for hepatic gluconeogenesis32 and can contribute to ATP production by entering the tricarboxylic acid cycle. Degradation of liver lipid droplets by autophagy (called lipophagy) contributes to the generation of free fatty acids that are oxidized in the mitochondria33. Hepatocyte-specific Atg7-knockout mice have elevated levels of hepatic lipids.
In both genetic and dietary mouse models of obesity and insulin resistance, a decrease in hepatic autophagy has been observed34. This has an impact on ER function, including the response to stress. Restoration of ATG7 expression limits obesity-dependent ER stress and rescues insulin resistance and glucose tolerance34, suggesting that pharmacological activation of liver autophagy may be beneficial in patients who suffer from obesity35. By contrast, adipose-specific deletion of autophagy genes decreases white adipose mass and favours the oxidation of free fatty acids by increasing the proportion of brown adipocytes, leading to enhanced insulin sensitivity and a lean body mass36,37. These studies have led to the suggestion that autophagy inhibition may represent a strategy for treating obesity and diabetes. However, the consequences of inhibiting autophagy in mature adipocytes in adult animals (rather than during development) have not been studied.
Moreover, autophagy activation may be beneficial in protecting against metabolic abnormalities in obese animals. For example, exercise induces autophagy in muscle, liver, pancreatic β-cells and adipose tissue, and mice that exhibit increased autophagy in these tissues (in response to exercise) are protected against glucose intolerance, leptin resistance as well as increased serum cholesterol and triglyceride levels induced by a high-fat diet. These benefits are not seen in mice that are unable to induce autophagy after exercise38. Thus, it may not be a coincidence that one of the mainstays of diabetes treatment is the use of AMP-activated protein kinase (AMPK) activators — agents that induce autophagy (as discussed above). Further studies are required to determine whether additional approaches for upregulating autophagy (besides AMPK activators) may also be beneficial in the treatment of metabolic diseases.
Autophagy is involved not only in the regulation of metabolism in peripheral tissues but also in regulating food intake via the brain39,40. These effects are complex and depend on the hypothalamic neurons that are targeted by autophagy knockout models: Atg7 knockout in pro-opiomelanocortin-expressing neurons causes obesity, whereas Atg7 knockout in neurons expressing Agouti-related protein decreases body weight (reviewed in REF. 41). Ultimately, the effects of autophagy on metabolism will need to be deciphered in animal models in which autophagy is modulated within the physiological range, as opposed to analyses in autophagy-null tissues.
Autophagy and neurodegenerative diseases
Most adult-onset neurodegenerative diseases are characterized by the formation of protein inclusions inside neurons. Frequently, these inclusions or aggregates are intracytoplasmic, as is the case in Parkinson’s disease, Alzheimer’s disease (which also has extracellular aggregates) and Huntington’s disease. When these types of conditions are caused by Mendelian mutations, the mutant protein causes disease typically via a toxic gain of function, which is associated with its propensity to aggregate42. Hence, strategies that can limit the toxic load by enhancing the removal of such proteins may have therapeutic potential.
Most neurodegenerative disease-associated proteins that form intracytoplasmic aggregates are autophagy substrates43–45. Inhibition of autophagosome formation or autophagosome–lysosome fusion slows the clearance of both full-length mutant huntingtin (the protein that causes Huntington’s disease) and its more toxic aminoterminal fragments, whereas the wild-type counterparts of these species are still efficiently cleared46. The clearance of these proteins is delayed when autophagy is impaired and, most importantly, their turnover is enhanced when autophagy is upregulated. Indeed, autophagy upregulation decreases the toxic accumulation of different mutant proteins, such as mutant huntingtin (causing Huntington’s disease), mutant ataxin 3 (causing spinocerebellar ataxia type 3) and mutant α-synuclein (causing Parkinson’s disease), and has beneficial effects on disease-associated phenotypes in cell culture as well as Drosophila melanogaster, zebrafish and mouse models43–45,47–50. In addition to facilitating the removal of toxic proteins, autophagy upregulation may reduce neuronal susceptibilities to caspase activation and apoptosis51.
Conversely, conditional knockouts of key autophagy genes in mouse neurons lead to aggregate formation and neurotoxicity52,53. Furthermore, defective autophagy results in the accumulation of mitochondria with an abnormal membrane potential, which are more likely to release pro-apoptotic molecules, as this population of mitochondria is believed to be selectively targeted by mitophagy54. Autophagy may be impaired in neurodegenerative diseases. For instance, autophagy may be impaired at both the levels of autophagosome degradation55 and autophagosome formation56 in Alzheimer’s disease, although these effects may vary according to the relevant genotype of the patients or the stage of the disease.
The accumulation of α-synuclein is a hallmark of Parkinson’s disease, and an excess of this protein is sufficient to cause this condition. Interestingly, excess α-synuclein impairs autophagosome biogenesis in cell cultures and in vivo57, whereas increased autophagy is observed when levels of this protein are reduced58. Mutations in PTEN-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase parkin (also known as PARK2) cause recessive forms of Parkinson’s disease. Recent data suggest that these proteins work together to mediate the clearance of dysfunctional mitochondria via mitophagy59. As disease-associated mutations have impaired mitophagy-related activity, this supports previous assertions that mitochondrial dysfunction may contribute to these forms of Parkinson’s disease. However, the relevance of mitophagy to the common sporadic forms of Parkinson’s disease is still unclear. Furthermore, impaired autophagy is implicated in a rare degenerative form of epilepsy caused by mutations in laforin, called Lafora epilepsy60, as well as in forms of motor neuron disease caused by mutations in dynactin61,62. It is thus likely that neurodegenerative diseases associated with mutations that impair autophagy will have an increased propensity towards aggregate formation and cellular toxicity, and that some of the neuronal stress may be due to defective mitophagy.
Therapeutic strategies and challenges
Autophagy defects can occur at different stages of the pathway in different diseases, and this may influence treatment strategies. Defects in autophagosome formation may be amenable to drugs that enhance autophagosome biogenesis. For example, laforin mutations (causing Lafora epilepsy) impair autophagosome formation by enhancing mTOR activity; mTOR inhibitors (for example, rapamycin) may therefore be beneficial in this context60. Similarly, nitric oxide induction — a frequent occurrence in neurodegenerative diseases — blocks autophagosome formation and this effect can be reversed in Huntington’s disease models by N-l-arginine methyl ester (l-NAME), which inhibits nitric oxide generation63. Indeed, l-NAME induces autophagy, enhances the degradation of mutant huntingtin and alleviates toxicity in in vivo disease models63. However, it may not be beneficial to induce autophagosome formation in certain disease settings: if the mutation or disease prevents the delivery of autophagosomes to lysosomes (which occurs if there are mutations in the dynein apparatus)62; if the mutation results in impaired lysosome activity, which is observed in various lysosomal storage diseases64,65; or in familial Alzheimer’s disease caused by presenilin 1 mutations55. In such cases, increasing autophagosome formation will not necessarily enhance autophagic substrate degradation, and may result in cellular membrane build-up, as the newly formed autophagosome would not be efficiently delivered to lysosomes.
When the initial studies were performed to examine the possibility of upregulating autophagy to enable the clearance of intracytoplasmic aggregation-prone proteins, the only known pharmacological method of inducing autophagy chronically was using rapamycin. However, the side effects of rapamycin (which are unrelated to autophagy) may make it unattractive for use in pre-symptomatic patients who may require long-term therapy. Various screens have identified pathways and compounds that regulate autophagy independently of mTOR. For instance, imidazoline receptor agonists such as clonidine and rilmenidine induce autophagy and have protective effects in cell culture, D. melanogaster and zebrafish models of Huntington’s disease49. Rilmenidine also has protective effects in a mouse model of Huntington’s disease66. Rilmenidine is a safe, centrally acting antihypertensive drug that lowers blood pressure by activating imidazoline receptors in the brain (which are widely distributed) — the same receptors it acts on to induce autophagy.
To date, there have been no reports of deleterious effects associated with specific autophagy upregulation in vivo. Indeed, rapamycin prolongs lifespan in D. melanogaster and in rodents67–69 and, at least in D. melanogaster, these effects are largely autophagy-dependent67. Although it is still not known whether specific upregulation of autophagy is beneficial in mice, deletion of the polyglutamine tract in the wild-type huntingtin protein induces autophagy in mice. This zero glutamine allele is associated with enhanced lifespan in otherwise wildtype mice; it increases lifespan and decreases motor symptoms in a knock-in mouse model of Huntington’s disease50. From a therapeutic perspective, however, constitutive autophagy induction may not be necessary. The therapeutic regimes with rapamycin and rilmenidine in mice have used dosing protocols that are likely to result in pulsatile upregulation of autophagosome formation, with periods of normality between dosing51,66. Thus, intermittent upregulation of autophagy may be effective and associated with fewer side effects in patients.
One major challenge when considering clinical trials for neurodegenerative diseases will be the feasibility of monitoring autophagic flux (not simply autophagosome numbers) in the correct tissue at the correct time. This will be a major challenge in the brain, but it will also be complex in tissues that are likely to be used for obtaining biopsy samples, such as tumours. Simply assaying autophagy in white blood cells or other easily accessible tissues may not be informative as there may be issues associated with tissue access for drugs (for example, the blood–brain barrier or tumour cores) as well as different receptors on different cells; therefore, certain compounds that act in the brain may have no effects in white blood cells.
Autophagy and cancer
Cancer is both the first disease that was linked to a deficiency in autophagy70 and, somewhat paradoxically, the first disease for which clinical trials are being carried out in patients to inhibit autophagy71. This apparent paradox can perhaps be reconciled by the differential effects of autophagy in different stages of tumorigenesis. The prevailing current view is that autophagy functions both as a tumour suppressor pathway that prevents tumour initiation and as a pro-survival pathway that helps tumour cells endure metabolic stress and resist death triggered by chemotherapeutic agents72. The notion of autophagy as a potential target in cancer has been recently discussed in the literature (reviewed in REFS 71–74). Here, we briefly highlight key discoveries and central controversies.
Tumour initiation
Although the role of autophagy in tumour progression is complex, there is a general consensus that autophagy suppresses tumour initiation. Genetic deletion of the autophagy gene BECN1 is associated with enhanced susceptibility to breast, ovarian and prostate cancer in humans70,75, and increased spontaneous malignancies in mice76,77. Mice deficient in ATG4C show increased susceptibility to chemically-induced fibrosarcomas78, and mice with deletion of Atg5 or Atg7 develop benign liver tumours79. Besides this direct evidence that autophagy gene mutations promote tumorigenesis, there is also a strong overlap between oncogenic signalling activation and suppression of autophagy (reviewed in REF. 80). Several tumour suppressor genes, including phosphatase and tensin homolog (PTEN), tuberous sclerosis 1 (TSC1), TSC2 and liver kinase B1 (LKB1), stimulate autophagy either through their inhibitory effects on mTOR and/or their activation of the ULK1 autophagy complex. Conversely, mTOR-activating oncogenic signals — such as oncogenic receptor tyrosine kinases, class I PI3Ks and AKT — inhibit autophagy. Other tumour suppressors (such as deathassociated protein kinase 1 and cyclin-dependent kinase inhibitor 2A (also known as p19ARF)) and oncogenes (such as BCL-2) positively or negatively regulate autophagy, respectively, through their effects on BECN1 (REFS 81,82).
However, the most commonly mutated tumour suppressor gene in human cancer, cellular tumour antigen p53, has both positive effects on autophagy as a nuclear transcription factor and negative effects through its cytoplasmic actions, including an interaction with the human orthologue of yeast Atg17, RB1-inducible coiled-coil 1 (RB1CC1; also known as FIP200)83,84. Mutant forms of p53 that accumulate in the cytoplasm in human cancers suppress autophagy85. The oncogene RAS also has dual functions in autophagy regulation; it can inhibit autophagy through class I PI3K activation86, and it stimulates autophagy perhaps through its effects on RAF1–ERK (extracellular signal-regulated kinase) signalling, RALB signalling or through the upregulation of BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), PMA-induced protein 1 (PMAIP1; also known as NOXA) or BECN1 expression87–89.
Three recent studies have shown that autophagy is essential for RAS-induced malignant cell transformation90– 92. By contrast, another study showed that autophagy gene knockdown enhanced clonogenic survival in cells expressing oncogenic RAS89. It is not yet known whether the requirement for autophagy in the RAS-driven transformation observed in most studies is a unique feature of cells with activated RAS or whether it will also be observed in the context of other activated oncogenes. It is possible that RAS alters metabolism in a unique manner that generates a specific requirement for autophagy in cell proliferation and/or cell survival. One proposed hypothesis is that loss of mitochondrial function mediated by oncogenic RAS-induced mitophagy overcomes the cellular energy deficit resulting from glucose insufficiency87.
Even though autophagy may be required for cellular transformation by RAS (and potentially other oncogenes), there is little doubt that basal autophagy is a bona fide tumour suppressor pathway. Although the precise mechanisms by which autophagy mediates tumour suppression are not completely understood, several pathways are likely to have a contributing role (reviewed in REF. 80). Many of these relate to the ability of autophagy to remove damaged organelles — especially mitochondria — that generate ROS, which in turn promote genotoxic stress as well as pro-inflammatory and pro-tumorigenic signalling. Tumours formed by autophagy-deficient cells (with bi-allelic loss of Atg5 or monoallelic loss of Becn1) display genomic instability and DNA damage, which is in part mediated by ROS93,94. In addition, PARK2 — which encodes an E3 ubiquitin protein ligase that is involved in mitophagy — is a tumour suppressor gene that is frequently mutated in colon cancer, lung cancer and glioblastoma95,96. However, it is not yet known whether its tumour suppressor effects are due to its role in mitophagy or due to mitophagy-independent E3 ligase functions.
Another important mechanism by which basal autophagy may prevent cancer is by controlling the levels of selective autophagy adaptor proteins — such as p62 — that function in pro-tumorigenic signalling: the size of liver tumours in Atg7−/− mice is reduced by simultaneous deletion of p62 (REF. 79); gene targeting of p62 reduces anchorage-independent growth of human hepatocellular carcinoma cells97; and p62−/− mice do not develop RAS-induced lung carcinomas98. The accumulation of p62 in autophagy-deficient cells promotes tumorigenesis through a mechanism that is postulated to involve the role of p62 as a scaffold protein that functions in the activation of the transcription factors nuclear factor-κB and NFE2-related factor 2 (REFS 97,98). In addition, p62 interacts with and activates the mitogenic signalling and autophagy-suppressive molecule mTOR99. Interestingly, SMAD ubiquitylation regulatory factor 1 (SMURF1) — a p62-interacting protein that also has a role in mitophagy100 — is amplified in pancreatic carcinomas101– 103 and its genetic knockdown in human pancreatic carcinoma cells leads to reduced tumour cell invasion and anchorage-independent growth101.
Tumour progression
There is increasing evidence that autophagy may be necessary for tumour progression. The retention of a wild-type Becn1 allele in tumours arising in mice with heterozygous deletion of Becn1 (REF. 76) provided one of the first genetic clues that tumour cells may need to retain autophagy for malignant progression104. This concept has been further underscored by the recent observation that hepatic deletion of Atg7 or Atg5 results in benign but not malignant hepatic tumours79. However, an alternative explanation is that the background mouse strain may not develop the additional mutations needed for malignant transformation. For example, heterozygous Becn1-deficient mice develop pre-neoplastic mammary lesions but not mammary carcinomas, even though Becn1 allelic loss is associated with breast cancer in humans and other malignancies in mice70,76,77. Nonetheless, several other studies also support a role for autophagy in promoting the growth of established tumours in vivo. For example, monoallelic loss of Becn1 almost completely blocked macroscopic renal tumour formation in Tsc2+/− mice105 and delayed tumour development in mice deficient in the ataxia telangiectasia mutated (Atm) gene (Atm−/− mice)106. Bi-allelic deletion of Atg5 or Atg7 impaired tumour growth of RAS-transformed immortalized infant mouse kidney epithelial cells in nude mice90, and Atg5 short hairpin RNA (shRNA) impaired tumour growth of human pancreatic ductal adenocarcinoma cells in a mouse xenograft model107.
The concept underlying these phenotypes is that autophagy provides a survival advantage to tumour cells by enabling them to overcome the metabolic stress that is inherently present in the tumour microenvironment. Consistent with this notion, autophagy is induced by cellular stress — including nutrient, growth factor and oxygen deprivation — and functions to maintain the survival of normal cells, organisms as well as tumour cells in such settings (reviewed in REF. 32).
The pro-oncogenic function of autophagy in established cancer may be context-dependent; not all data are consistent with such a role for autophagy in established tumours. Although autophagy upregulation, in part mediated by the effects of hypoxia-inducible factor 1α (HIF1α) on BNIP3 (REF. 108), may promote tumour cell survival in hypoxic regions within tumours (reviewed in REF. 109), a recent study has shown that autophagy may protect against hypoxia-stimulated tumour growth by decreasing tumour angiogenesis110. In a mouse melanoma xenograft model, heterozygous Becn1-deficient mice display a more aggressive tumour phenotype with increased angiogenesis under hypoxic conditions through a mechanism that is postulated to involve the upregulation of HIF2α (but not HIF1α). Furthermore, several clinicopathological studies have shown a correlation between levels of BECN1 expression and cancer prognosis; low levels of BECN1 expression are associated with worse cancer prognosis in gastric cancer111, colorectal cancer112, pancreatic cancer113, oesophageal cancer114, chondrosarcoma113 and breast cancer111, whereas high levels of BECN1 expression are associated with improved survival in high-grade gliomas115, hepatocellular carcinomas116 and B cell lymphomas117. Although it is not known whether low levels of BECN1 expression directly correlate with low levels of autophagy in these tumours, such studies point to the need for further careful analyses of the relationship between levels of autophagy and tumour progression in different types of tumours. To address this question, there is an urgent need for in vivo models in which autophagy can be selectively regulated at defined stages of tumorigenesis. One caveat in the use of such models is that complete inhibition of autophagy (which results in cell death) may not accurately reflect those phenotypes that are associated with partial inhibition of autophagy.
Autophagy inhibition in cancer therapy
The evolutionarily conserved role of autophagy in promoting cell survival during metabolic stress has stimulated research to determine whether autophagy may promote therapeutic resistance to cytotoxic therapy. The first in vivo data that affirmed this concept involved the demonstration that treatment with the lysosomotropic inhibitor chloroquine (which inhibits autophagosome degradation) enhanced the ability of p53 activation or alkylating agents to induce tumour regression in a mouse model of MYC-induced lymphoma118. However, chloroquine has activities on lysosomal processes that are distinct from autophagy as well as lysosome-independent processes (such as DNA intercalation). There have now been several studies indicating that autophagy inhibition — with 3-methyladenine treatment, genetic knockdown of autophagy genes or chloroquine (or hydroxychloroquine) treatment — sensitizes tumour cells to cell death induced by diverse cytotoxic agents (reviewed in REFS 71,72,74,119). Although many chemotherapeutic agents induce autophagy (at least indirectly by inducing cellular stress), most efforts have focused on using autophagy inhibitors in tumour cell lines with high levels of basal autophagy (such as those with oncogenic RAS mutations) or on using autophagy inhibitors in conjunction with agents that directly stimulate autophagy signalling pathways (such as mTOR inhibitors, dual PI3K and mTOR inhibitors, epidermal growth factor receptor (EGFR) inhibitors and proteasome inhibitors). Accordingly, there are several Phase I/II clinical trials in progress using the lysosome inhibitors chloroquine or hydroxychloroquine in combination with chemotherapy for the treatment of a range of haematological and solid tumours71,74. The poor prognosis of certain tumours (such as pancreatic carcinoma) with oncogenic RAS mutations, coupled with the preclinical data suggesting a role for autophagy in RAS-mediated transformation and tumour growth86,114, has led to the initiation of earlyphase trials of lysosomal inhibitors in patients with RASdriven tumours (reviewed in REF. 119).
In addition, more specific inhibitors of the autophagic machinery (for example, PIK3C3 inhibitors, ATG4B inhibitors and ATG7 inhibitors) are in preclinical development for potential use in cancer clinical trials. Pharmacological inhibition of autophagy adaptor proteins (such as p62) that have a role in pro-tumorigenic signalling, and perhaps of other components of the p62 autophagy adaptor complex, may represent novel strategies for cancer therapy.
Although the rationale for such studies is supported by strong preclinical data, many open questions and controversies remain regarding autophagy as a target in cancer therapy. First, although the available data support the concept that cancer cells exhibiting increased autophagy die in response to lysosomal inhibitors, no study has unequivocally demonstrated that the cytotoxic effects of these agents in cancer cells arise specifically from autophagy inhibition rather than from another effect on lysosomal function or even from lysosome-independent effects. This issue will have crucial relevance in determining whether positive results (if observed) in Phase I/II clinical trials with lysosomotropic agents should be used as a rationale for initiating future clinical trials with agents that directly inhibit the autophagic machinery. Even if lysosomotropic agents exert their antitumour effects by blocking autophagosomal and/or lysosomal fusion, it is possible that this defect in autophagolysosomal fusion is toxic to cells and that similar pro-death effects will not be observed with drugs that block the formation of autophagosomes.
Alternatively, lysosomal inhibitors may potentiate cytotoxicity in cancer cells via autophagy-independent mechanisms. Indeed, some recent studies in glioma and breast cancer cells illustrate a mechanistic dissociation between the actions of lysosomotropic agents and autophagy inhibition in governing sensitivity to cytotoxic therapy. For example, bafilomycin A1 enhanced the cytotoxicity of temozolomide (a DNA alkylating agent) in glioma cells120, whereas shRNA-mediated knockdown of BECN1 and ATG5 protected the same cells against temozolomide-induced death121. Chloroquine sensitized breast cancer cells to death induced by a DNA-damaging agent, a PI3K inhibitor or an mTOR inhibitor, but similar effects were not observed in these cells with Atg12 or Becn1 knockdown or following treatment with a different lysosomal inhibitor, bafilomycin A1 (REF. 111).
Second, there is still controversy as to whether autophagy may represent a mechanism of cell death during chemotherapy. As discussed below, a recent chemical screening study found that ATG7 knockdown was efficient at inhibiting autophagy — but not cell death — induced by 59 compounds that induced both autophagy and cell death122, leading the authors to conclude that “cell death is rarely, if ever, executed by autophagy in human cells”. Yet, there are numerous studies demonstrating that genetic knockdown of autophagy genes blocks tumour cell death induced by oncogenic RAS or by various chemotherapeutic agents (reviewed in REFS 72,123,124); one common theme is that cell death via autophagy is more likely to occur in tumour cells that are deficient in apoptosis or in tumour cells treated with a combination of pan-BCL-2 inhibitors (such as gossypol or GX15-070) and other agents. Further studies are required to delineate more precisely the chemotherapeutic contexts in which autophagy functions as a pro-survival or pro-death mechanism.
Some other potential caveats associated with autophagy inhibition in cancer therapy also warrant consideration. Given the tumour suppressor effects of autophagy (discussed above) and the protective effects of autophagy in other diseases (such as neurodegeneration and infectious diseases, as well as in ageing), there are concerns about whether autophagy inhibition treatment may increase the incidence of secondary tumours or other diseases in patients. Thus, even if short-term benefits on tumour progression are observed, a long duration of patient follow-up may be required before increased secondary malignancies (and other adverse effects with a long latency period), if they occur, are detected. The drugs chloroquine and hydroxychloroquine have been used extensively in the treatment of malaria and systemic lupus erythematosus, and are fairly well tolerated; however, it is not known whether more proximal inhibitors of autophagy will have similar safety profiles. Perhaps the magnitude of autophagy inhibition and/or the duration of therapy used in cancer trials will not be sufficient to observe the predicted adverse effects of systemic autophagy inhibition. Nonetheless, a recent study raises the concern that acute inhibition of autophagy may limit chemotherapy responses by preventing autophagy-dependent anticancer immune responses125. Autophagy-competent (but not autophagydeficient) cells have been shown to release cellular ATP and recruit immune cells into the tumour bed, leading to effective immunogenic cell death and chemotherapeutic responses in mice with intact immune systems.
Autophagy activation in cancer therapy
Certain agents that are used as a preventive form of cancer therapy (such as the use of tamoxifen in patients who are at risk of developing familial breast cancer)126 induce autophagy127. Certain epidemiological factors associated with increased cancer incidence reduce autophagy. For example, vitamin D is a potent inducer of autophagy128–130, and patients with low vitamin D levels exhibit an increased risk of developing breast, colon, prostate and other cancers131. Conversely, certain epidemiological factors associated with decreased cancer risk can increase autophagy. For example, exercise induces autophagy38,132 and patients who regularly exercise (more than 150 minutes per week of moderate-intensity exercise) have a decreased risk of breast, prostate, endometrial and colon cancer133–135. Furthermore, the use of metformin (an AMPK activator and autophagy-inducing agent; FIG. 1) in patients with diabetes substantially lowers the risk of cancer relative to that of other antidiabetic drugs136. At present, the association between interventions that reduce cancer incidence and those that induce autophagy is only correlative. Further studies are required to determine whether autophagy upregulation has a mechanistic role in the efficacy of such cancer prevention strategies. If so, the use of more direct activators of autophagy may be a feasible, new and alternative cancer prevention strategy.
Future directions
Ultimately, the question of whether autophagy represents a useful target in cancer prevention or cancer treatment will need to be addressed by conducting clinical trials in patients. Preclinical data have been useful in formulating testable hypotheses; however, there are several conflicting reports regarding the role of autophagy in tumour initiation, tumour progression and cell death decisions during chemotherapy. Some of these may ultimately be reconciled by a better understanding of the diverse set of roles that autophagy has in different oncogenic contexts and in different stages of tumorigenesis. However, there are also limitations to studies in tumour cell lines, mouse xenograft models and targeted mutant mouse models, as well as problems with respect to the specificity of approaches used to modulate (and measure) autophagy, which may account for some of the apparent discrepancies. Therefore, the results of the first round of Phase I/II clinical trials with hydroxychloroquine in cancer will be important in validating or refuting the predictions that have been made based on preclinical data indicating that lysosomal inhibition potentiates tumour regression. If the results of early trials are promising, it will be important to determine the antitumour mechanisms of action of lysosomal inhibitors.
Further preclinical and clinical studies are also warranted to explore the role of autophagy upregulation in cancer prevention, the feasibility of blocking p62 in autophagy-deficient tumours and the possibility of exploiting autophagy as a death pathway in tumour cells. Another important area of future investigation will be to determine whether tumour cell autophagy-dependent survival can be selectively inhibited in tumour cells (while bypassing the potential adverse effects of systemic autophagy inhibition) by targeting tumour cell-specific autophagy activation, such as the platelet-derived growth factor receptor (PDGFR)-induced promotion of HIF1α-dependent hypoxia-selective autophagy137. Moreover, it is likely that emerging research regarding the role of autophagy in cancer stem cells, in epithelial-to-mesencyhmal transition, in DNA damage and cell cycle control, in the regulation of inflammatory signalling and in other aspects of cancer biology will further shift the existing paradigms that dictate our current understanding of autophagy as a target in cancer therapy.
Autophagy and infectious diseases
Autophagic machinery is used in a multipronged defence against microorganisms, including via the selective delivery of microorganisms to degradative lysosomes (a process referred to as xenophagy) and via the delivery of microbial nucleic acids and antigens to endolysosomal compartments for the activation of innate and adaptive immunity138–140. In 1998, enforced neuronal expression of the autophagy gene Becn1 was shown to protect mice against lethal alphavirus encephalitis, providing the first clue that autophagy upregulation may be beneficial in the treatment of infectious diseases141. It is now known that numerous medically important pathogens are degraded in vitro by xenophagy, including: bacteria such as group A Streptococcus pyogenes, Mycobacterium tuberculosis, Shigella flexneri, Salmonella enterica, Listeria monocytogenes and Francisella tularensis; viruses such as herpes simplex virus type 1 (HSV-1) and chikungunya virus; and parasites such as Toxoplasma gondii139,142. There are also in vivo data indicating that autophagy genes have a protective role against numerous pathogens, including L. monocytogenes, M. tuberculosis, S. enterica, T. gondii, HSV-1, Sindbis virus and chikungunya virus139,142–146. Moreover, there are emerging links between host genes that regulate autophagy and host susceptibility to M. tuberculosis infection147,148.
Based on these studies, there is a strong possibility that pharmacological agents that increase autophagy may be effective therapeutic agents for treating certain intracellular bacterial infections, parasitic infections and viral infections. In support of this concept, vitamin D treatment has been shown to inhibit both HIV and M. tuberculosis replication in human macrophages through an autophagy-dependent mechanism149,150. In addition, the antimycobacterial action of standard antituberculous agents is associated with autophagy induction151, raising the possibility that some drugs that are already in clinical use for the treatment of certain infections may be acting, at least in part, via autophagy.
Innate immunity
In addition to enhancing pathogen degradation, the upregulation of autophagy may facilitate optimal regulation of innate immune signalling and enhancement of antigen presentation138–140,152. The interrelationship between autophagy and innate immune signalling is complex; in some contexts, autophagy can enhance type I interferon (IFN) production and innate immune responses by delivering viral nucleic acids to endosomal Toll-like receptors, whereas in other contexts autophagy proteins prevent the innate immune response from being excessive and detrimental, either through direct protein–protein interactions with innate immune signalling molecules or indirectly by controlling cellular levels of ROS production. Despite this complexity, a consensus is emerging that autophagy has a central role in titrating the innate immune response so that it is adaptive rather than maladaptive140. Thus, upregulation of autophagy may be useful in increasing antimicrobial innate immunity while preventing excessive inflammatory responses that can be destructive to the host during infection.
Adaptive immunity
With respect to adaptive immunity, autophagy is involved in the delivery of endogenously synthesized microbial antigens to MHC class II antigen-presenting molecules, leading to the activation of CD4 T lymphocytes140,152. In addition, the autophagy gene ATG5 is required for dendritic cells to process and present extracellular microbial antigens for MHC class II presentation153. It has been reported that autophagy also enhances the presentation of endogenous viral antigens on MHC class I molecules154, and that autophagy in tumour cells is essential for antigen cross-presentation by dendritic cells155. The link between autophagy and CD4 T cell responses suggests that pharmacological induction of autophagy may be beneficial not only in the treatment of acute infection (by enhancing xenophagy, innate immunity and adaptive immunity) but also in enhancing vaccine efficacy. In support of this principle, the targeting of the influenza virus matrix protein 1 (MP1) to autophagosomes via fusion with LC3 enhanced anti-MP1 CD4 T cell responses156, and mice immunized with rapamycin-treated dendritic cells infected with bacille Calmette–Guérin (BCG) or candidate mycobacterial vaccine strains showed enhanced CD4 T cellmediated protection when they were challenged with virulent M. tuberculosis157.
Further studies are warranted to determine whether strategies to augment autophagy-dependent adaptive immune responses could be beneficial in vaccine development. In principle, this may be accomplished in various ways: by enhancing autophagy in cells infected with live attenuated vaccines; by targeting microbial antigens to the autophagosome via fusion with LC3 or an autophagy adaptor (or receptor molecule) that binds to LC3; or by administering antigens that are processed by autophagydependent dendritic cells for MHC class II presentation.
Autophagy-independent functions of autophagy proteins in immunity
There is increasing evidence that autophagy proteins may exhibit diverse functions in innate and adaptive immunity, independently of the autophagy pathway (reviewed in REFS 138,140,158). These functions include: negative regulation of signalling between retinoic acid-inducible gene 1 (RIG1; also known as DDX58) and IFNβ promoter stimulator (IPS); negative regulation of the inflammasome; Toll-like receptor-mediated phagolysosomal maturation, formation and/or release of antimicrobial peptides; secretion of interleukin-1β159; recruitment of immune effectors to intracellular membranes; and mediation of IFNγ-dependent antiviral effects160. Abnormalities in some of these functions, such as aberrant regulation of the inflammasome161 and defects in granule cell exocytosis in Paneth cells162, are thought to be relevant to the pathogenesis of subtypes of Crohn’s disease (a type of inflammatory bowel disease) that are associated with a polymorphism in the autophagy gene ATG16L1.
At present, it is not known whether general stimulation of autophagy will increase the autophagy-independent functions of autophagy proteins in immunity. A more likely long-term strategy may be to devise new agents that mimic the beneficial autophagy-independent functions of autophagy proteins in the control of infectious diseases and autoinflammatory diseases.
Microorganisms co-opt autophagy
In parallel with increasing evidence that autophagy has a role in host defence, there is a growing list of viruses and intracellular bacteria that exploit the host autophagic machinery to enhance their own replication (reviewed in REFS 139,140). These include medically important viruses such as HIV, hepatitis C virus (HCV), hepatitis B virus (HBV), picornaviruses and Dengue virus, as well as intracellular bacteria such as F. tularensis, Coxiella burnetii and Brucella abortus139,140,163–165. Of note, for some viruses such as HIV there is evidence for both an anti- and proviral function of autophagy149,150,166. Until recently, all evidence indicating a promicrobial function of autophagy stemmed from studies in cultured mammalian cell lines. However, recent studies have demonstrated reduced HBV DNA replication in mice with liver-specific knockout of Atg5 (REF. 167), and reduced coxsackie virus replication in mice with pancreatic acinar cell-specific knockout of Atg5 (REF. 168), thus providing a rationale for further studies to explore the targeting of autophagic machinery to treat these infections in patients.
For the list of pathogens that exploit the host autophagic machinery for enhanced replication, the potential dangers of autophagy upregulation or the potential benefits of autophagy inhibition will probably depend on whether the specific pathogen utilizes the complete autophagic pathway or just certain components of the autophagic machinery to enhance its intracellular replication or survival, as well as whether pharmacological targeting manipulates the whole autophagic pathway or only specific components of the autophagic machinery required for pathogen replication. For example, intracellular Brucella abortus survives by promoting the formation of B. abortus-containing vacuoles, which requires the activity of the autophagy-initiating proteins ULK1, BECN1, ATG14L and PIK3C3, but not the activity of the autophagy elongation proteins ATG5, ATG16L1, ATG4B, ATG7 and LC3B163. In this scenario, one might predict that inhibitors of PIK3C3 or signals upstream of ULK1 may exert protective functions, whereas such effects would not be observed with inhibitors of the autophagy protein conjugation systems or inhibitors of autophagosome maturation. By contrast, Dengue virus replication is thought to be enhanced by autophagy-dependent lipid metabolism, which increases cellular β-oxidation and generates ATP164. In this scenario, blocking autophagy at any stage in the pathway might suppress viral replication.
It is prudent to exercise caution in considering the use of autophagy-inducing agents for the treatment of patients with infections that may otherwise be alleviated by autophagy upregulation (such as tuberculosis) if these patients are also co-infected with pathogens that may exploit the autophagy pathway (such as chronic concurrent infections with HCV, HBV and possibly HIV). Another potential concern is the possibility that excess levels of autophagy may exacerbate certain infectious diseases through alternative mechanisms that are independent of pathogen replication. In a recent report, the H5N1 pandemic strain of influenza virus induced autophagic alveolar epithelial cell death, and pharmacological and genetic inhibition of autophagy ameliorated acute lung injury and decreased mortality in H5N1-infected mice without affecting viral replication169.
Promising future therapeutic strategies
A promising future direction in the treatment of infectious diseases is the development of agents that block the activity of specific microbial gene products that antagonize the functions of autophagy (or autophagy proteins) in antimicrobial host defence. Several viral virulence gene products have been shown to block either autophagy initiation or autophagolysosome maturation through their interaction with BECN1 (reviewed in REFS 139,140). These include: the HSV-1 neurovirulence protein ICP34.5; the γ-herpesvirus BCL-2-like proteins encoded by Kaposi’s sarcoma herpesvirus and murine γ-herpesvirus 68 (γHV68); the cytomegalovirus-encoded protein TRS1 (REF. 170); the influenza virus matrix protein M2; and the HIV-1 pathogenic protein Nef. The interaction of some of these viral proteins with BECN1 has been shown to be important in viral pathogenesis in mice; a mutant HSV-1 lacking the BECN1-binding domain of ICP34.5 is substantially attenuated in mouse models of encephalitis171 and of corneal disease172, and a murine γHV68 with a mutant viral BCL-2 that is unable to bind to BECN1 has an impaired ability to maintain chronic infection173. These studies suggest that inhibitors of the interactions between viral proteins that antagonize autophagy and their host autophagy protein targets may be useful for treating diseases such as HSV-1 encephalitis, in which viral antagonism of autophagy is essential for viral virulence.
A related concept emerges from the identification of specific bacterial virulence factors — such as L. monocytogenes actin assembly-inducing protein (ActA) and Shigella flexneri IscB — that enable intracellular bacteria to escape recognition by the autophagic pathway (reviewed in REF. 140). Presumably, inhibition of these bacterial virulence factors would enhance xenophagic degradation of intracellular bacteria and thereby protect the host against diseases caused by such pathogens. The strategy of developing pharmacological inhibitors of microbial antagonists of autophagy is attractive not only because of its potential efficacy in controlling certain viral and intracellular bacterial infections but also because of its potential safety and specificity (in comparison with general strategies to modulate autophagy). By targeting specific microbial virulence factors, such approaches might avoid the potential pitfalls associated with manipulation of systemic autophagy.
Another approach towards harnessing the autophagic pathway for the treatment of infectious diseases is emerging from our rapidly expanding understanding of the machinery involved in the recognition and targeting of viruses and intracellular bacteria to the autophagosome. Several adaptor molecules have been identified that selectively target microorganisms for autophagy by binding to ubiquitin-associated bacteria and/or viral nucleocapsids, as well as to the autophagosome-associated protein LC3, through a LIR domain; these include p62, NBR1, nuclear dot protein 52 (NDP52) and optineurin140,174. In addition, SMURF1 binds to alphavirus nucleocapsid and targets it to autophagosomes through a mechanism that may involve its C2 phospholipid-binding domain100. Moreover, the function of certain microbial adaptor proteins can be regulated by phosphorylation; for example, TANK-binding kinase 1 phosphorylates optineurin, thus enhancing its LC3-binding affinity and the autophagic clearance of cytosolic S. enterica174. This suggests that strategies to augment the activity of optineurin or other autophagy adaptors may be effective in enhancing the xenophagic degradation of intracellular pathogens. Future research to further delineate the mechanisms of regulating the function of autophagy adaptors should lay the groundwork for the preclinical development of agents that selectively enhance microbial autophagy for the treatment of certain infectious diseases. One potential caveat of this approach is that many of the autophagy adaptors are not specific for pathogens and they also function in the selective autophagy of damaged mitochondria and other host cell components; thus, such strategies may have additional unwanted effects on host cell function.
Considerations for clinical trials
Given the central role of the autophagic machinery in controlling infection and immunity (either through the classical autophagy pathway or through autophagy-independent functions of autophagy proteins), it will be important for all clinical trials using autophagy inhibitors to carefully monitor the incidence, prevalence and severity of infectious diseases, autoimmune diseases and inflammatory diseases. Conversely, patients enrolled in clinical trials with autophagy inducers, such as patients who are at risk of developing neurodegenerative diseases, may experience protection against mycobacterial infections and other infectious diseases in which autophagy has a central role in host defence.
Pharmacological manipulation of autophagy
For medical purposes, it may be valuable to identify drugs that induce or inhibit autophagy (some examples from above are summarized in TABLE 2, their sites of action illustrated in FIGS 1,2 and possible uses listed in TABLE 3). Notably, many of the compounds being considered are still under investigation or are tool compounds and may not therefore be suitable for clinical use, although they do illustrate potentially druggable points in the autophagic pathway. Inducers may have particular value in certain neurodegenerative diseases, some infectious diseases and in cytoprotection. As discussed above, it has been suggested that autophagy inhibition may be valuable in cancers. However, this is largely based on studies using the lysosomotropic drug chloroquine or its derivatives, which affect all acidic compartments in cells and have many autophagy-independent effects72. Some of the agents discussed are in clinical trials (for example, lysosomotropic drugs for certain cancers and rilmenidine for Huntington’s disease); however, a deeper understanding of the roles of autophagy and autophagy modulators is still required for many of the conditions in which there may be therapeutic possibilities. Furthermore, as many of these drugs have autophagy-independent effects, one may need to be cautious before inferring that all of their effects are autophagy-dependent (although this may not necessarily preclude the consideration of these drugs for therapeutic purposes).
Table 2.
Selected drugs and compounds that modulate autophagy
| Compounds | Company | Structure | Mechanism of action | Refs |
|---|---|---|---|---|
| Autophagy inducers | ||||
| Rapamycin | Sigma-Aldrich | 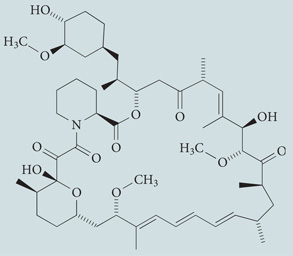 |
Induces autophagy by inhibiting mTORC1 | 44,49, 246 |
| PP242 | Sigma-Aldrich | 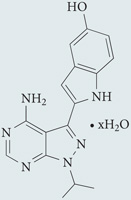 |
Induces autophagy by inhibiting mTORC1 | 247 |
| Torin 1 | Tocris Bioscience | 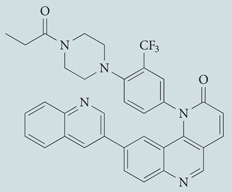 |
Directly inhibits both mTORC1 and mTORC2 | 176 |
| Metformin | Sigma-Aldrich | 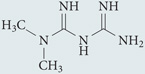 |
Upregulates AMPK, which promotes autophagy by inducing ULK1 phosphorylation | 248,249 |
| BH3 mimetics (ABT-737) | Selleckchem | 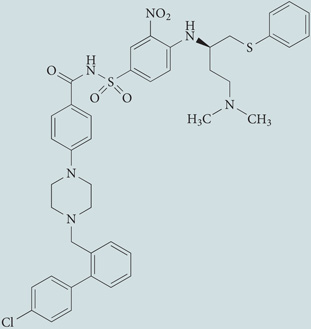 |
Disrupts the inhibitory interaction between the BH3 domain of beclin 1 and BCL-2, stimulating the beclin 1-dependent allosteric activation of the pro-autophagic lipid kinase PIK3C3 | 250 |
| Xestospongin B | Sigma-Aldrich | 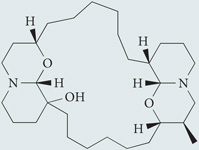 |
Disrupts the molecular complex formed by the Ins(1,4,5)P3 receptor and beclin 1 | 251 |
| l-NAME | Cayman Chemical | 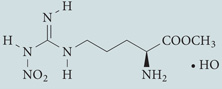 |
Decreases nitric oxide formation to induce autophagy | 63 |
| Rilmenidine* | Tocris Bioscience | 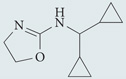 |
Lowers cAMP levels | 49,66 |
| Clonidine | Sigma-Aldrich | 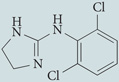 |
Lowers cAMP levels to induce autophagy | 49 |
| PI-103 hydrochloride | Tocris Bioscience | 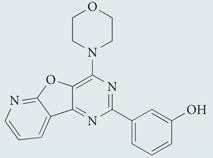 |
Highly selective class I PI3K inhibitor and ATP-competitive mTOR inhibitor | 179 |
| Lithium, L-690330 | Enzo Life Sciences | 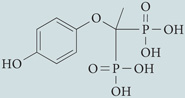 |
Lower inositol and Ins(1,4,5)P3 levels | 182 |
| Carbamazepine | Sigma-Aldrich | 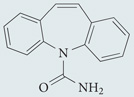 |
Lowers inositol and Ins(1,4,5)P3 levels | 49,182 |
| Resveratrol | Sigma-Aldrich | 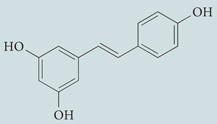 |
Activates sirtuin 1 (histone deacetylase) and inhibits 70 kDa ribosomal protein S6 kinase | 252–254 |
| Verapamil | Sigma-Aldrich | 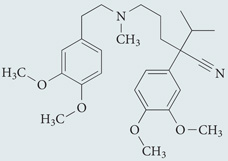 |
Lowers intracytosolic Ca2+ levels | 49 |
| EGFR antagonists, erlotinib hydrochloride | Roche | 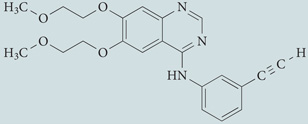 |
Inhibits the PI3K–AKT–mTOR signalling pathway | 255,256 |
| Sodium valproate | Sigma-Aldrich | 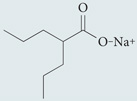 |
Lowers inositol and Ins(1,4,5)P3 levels | 49,182 |
| Spermidine | Sigma-Aldrich | Postulated to affect expression of ATG genes | 186 | |
| Vinblastine | Sigma-Aldrich | 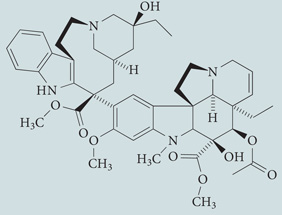 |
Inhibits microtubule formation | 257 |
| Nocodazole | Sigma-Aldrich | 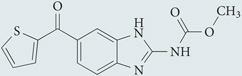 |
Inhibits microtubule formation and blocks autophagosome–lysosome fusion | 258,259 |
| Bafilomycin A1 | Merck Millipore | 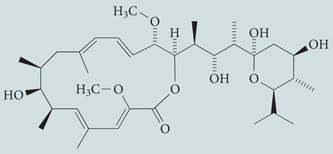 |
Specific inhibitor of V-ATPase; inhibits autophagosome–lysosome fusion | 44,243 |
| Chloroquine‡ | Sigma-Aldrich | 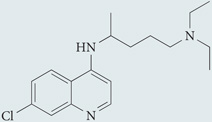 |
Inhibits autophagosome–lysosome fusion | 4,118 |
| Hydroxychloroquine‡ | Sigma-Aldrich | 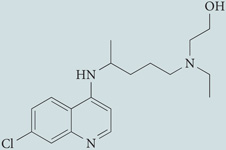 |
Inhibits autophagosome–lysosome fusion | 60,118 |
| Spautin-1 | Cellagen Technology | 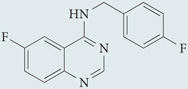 |
Lowers beclin 1 levels by promoting its ubiquitylation | 200 |
AMPK, AMP-activated protein kinase; ATG, autophagy-related; BCL-2, B cell lymphoma 2; BH3, BCL-2 homology 3; cAMP, cyclic AMP; EGFR, epidermal growth factor receptor; Ins(1,4,5)P3, inositol-1,4,5-trisphosphate; l-NAME, N-l-arginine methyl ester; mTOR, mammalian target of rapamycin; mTORC1, mTOR complex 1; PI3K, phosphoinositide 3-kinase; PIK3C3, class III PI3K (also known as VPS34); spautin-1, specific and potent autophagy inhibitor 1; ULK1, UNC51-like kinase 1; V-ATPase, vacuolar ATPase.
Phase I safety trial underway.
These drugs are undergoing Phase I/II clinical trials as autophagy inhibitors in patients with different types of cancers. See the ClinicalTrials.gov website for a list of US National Institutes of Health (NIH)-sponsored clinical trials.
Table 3.
Autophagy-modulating strategies and possible therapeutic targets
| Indications or targets | Therapeutic strategy | Refs |
|---|---|---|
|
Enhance autophagy |
9, 38, 43–45, 47–49, 138–148 |
|
Modify adaptor proteins to increase substrate affinities: for example, phosphorylation of optineurin to enhance S. enterica clearance | 174 |
|
Inhibit bacterial or viral virulence factors that perturb autophagy | 140 |
|
Inhibit autophagy |
71, 72, 74, 90, 104, 105, 107, 165 |
|
Inhibit specific autophagy proteins | 163 |
|
Downregulate p62 | 97, 98 |
ActA, Listeria monocytogenes actin assembly-inducing protein; HSV-1, herpes simplex virus type 1; ICP34.5, HSV-1 neurovirulence protein ICP34.5; p62, ubiquitin-binding protein p62.
mTOR complex 1
Drugs (or signals) that modulate autophagy can be considered in two categories, depending on whether or not they act via mTOR (FIG. 1). mTORC1 is inhibited by rapamycin and its analogues (CCI-779, RAD001 and AP23573), which induce autophagy in yeast, mammalian cell lines, primary cultures and in vivo175. Studies have revealed that rapamycin does not inhibit mTORC1 completely, leading to the concept that certain functions of mTORC1 are rapamycin-resistant (such as cap-dependent translation and the phosphorylation of eukaryotic translation initiation factor 4E-binding protein 1 (EIF4EBP1; also known as 4EBP1). These studies have identified two selective ATP-competitive smallmolecule mTOR inhibitors, PP242 and Torin 1, that directly inhibit both mTORC1 and mTORC2 (REF. 176). The maximal autophagy induction mediated by ATPcompetitive inhibitors appears to be higher than that seen with rapamycin, although rapamycin can induce autophagy at lower concentrations than ATP-competitive inhibitors in certain cell types177.
The activity of mTOR is regulated by the class I PI3K– AKT pathway. PI-103 is a molecule within the new class of dual mTOR and class I PI3K inhibitors. This combinatorial inhibition appears to be very effective as it addresses the negative feedback mechanism between mTORC1 and the PI3K–AKT pathway: rapamycin inhibits mTORC1 but this releases the negative feedback mechanism such that PI3K–AKT signalling is enhanced175,178. As expected, PI-103 has been shown to be a strong inducer of autophagy179. Although PI-103 itself does not appear to have suitable structural properties for clinical development, it could stimulate the development of this new class of dual inhibitors175. It is important to note that mTOR affects many processes that are distinct from autophagy, and AKT inhibition may result in increased susceptibility to cell death. Although this may be attractive in the context of treating cancers, AKT inhibition may prove to be a liability if it is used for treating neurodegenerative diseases.
Targeting AMPK activity
As discussed above, AMPK activates autophagy via at least two mechanisms: mTOR inhibition and direct ULK1 activation180 (FIG. 1). Metformin, a widely used antidiabetic agent, activates AMPK and induces autophagy181. As discussed above, it is possible that some of the effects of this drug in diabetes may be mediated via autophagy, and the use of metformin may therefore be worth considering for conditions in which autophagy upregulation is beneficial.
Phosphatidylinositol signalling pathway
Intracellular inositol and inositol-1,4,5-trisphosphate (Ins(1,4,5)P3) levels negatively regulate autophagy via an mTOR-independent mechanism (FIG. 1). Autophagy can be induced via this pathway using various mood-stabilizing drugs such as lithium, carbamazepine and valproic acid182. Subsequent studies have suggested that at least part of the autophagyinducing effects of these drugs are mediated via a reduction in mitochondrial uptake of Ca2+ released by the Ins(1,4,5)P3 receptor, which causes a mild defect in mitochondrial respiration and AMPK activation183. However, one should bear in mind that these drugs have additional targets; for example, valproic acid is a histone deactylase inhibitor. Interestingly, carbamazepine has been shown to have beneficial effects in a mouse model of α1 antitrypsin deficiency, and these effects may be at least partly mediated via autophagy induction184.
cAMP–EPAC–PLCε–Ins(1,4,5)P3 and Ca2+–calpain–GSα pathways
Recent screens of US Food and Drug Administration (FDA)-approved drugs and pharmacological probes have identified numerous mTOR-independent autophagy modulators acting on targets such as the imidazoline receptor (for example, clonidine and rilmenidine), l-type Ca2+ channels (for example, verapamil) and calpains (for example, calpain inhibitors)49. These compounds appear to be linked in a potential cyclic fashion via the pathway involving cyclic AMP, exchange protein directly activated by cAMP (EPAC), phospholipase Cε (PLCε) and Ins(1,4,5)P3 as well as the pathway involving Ca2+, calpain and the G protein Gsα (REF. 49). As many of the drugs acting on this pathway are used in the clinic (for example, clonidine and rilmenidine have been used to treat hypertension, and verapamil is used to treat hypertension, cluster headaches, cardiac arrhythmias and angina) and have favourable safety profiles, they may represent a tractable set of compounds for therapeutic applications in diseases that may benefit from autophagy induction.
Other autophagy-modulating drugs
In addition to the compounds described above, numerous other compounds have been described that induce autophagy, including resveratrol185, spermidine186, EGFR antagonists187, BH3 mimetics (which decrease the inhibitory interactions of BCL-2 and BCL-XL with BECN1)29,188 and l-NAME63 (which blocks nitric oxide formation). The precise mechanisms by which some of these compounds (for example, l-NAME) induce autophagy are still unclear, and many of these drugs and their targets affect processes that are distinct from autophagy.
Conversely, chloroquine is a lysosomotropic agent that impairs autophagosome degradation as well as other lysosomal degradation pathways. Furthermore, lysosomotropic agents have effects on diverse cellular compartments that require an acidic pH, and this may cause complex perturbations in cells in addition to autophagy arrest; however, the possible enhanced toxicity mediated by such drugs (that have both autophagy-blocking and other effects) may be beneficial in certain cancers.
Drugs with unforeseen autophagy-blocking effects
In addition to studies that have revealed autophagy-modulating drugs of potential therapeutic benefit in various diseases, similar studies have identified drugs that have unforeseen and possibly toxic autophagy-inhibiting effects. In many neurodegenerative diseases, such as Huntington’s disease, oxidative stress is increased, and antioxidants have therefore been proposed as a rational therapeutic strategy. Indeed, there are at least two major trials of antioxidants underway for Huntington’s disease. However, a diverse range of antioxidant drugs inhibit autophagy in vivo but exacerbate mutant protein aggregation and toxicity in animal models189. Thus, the potential benefits of ROS scavengers in neurodegenerative diseases may be compromised by their autophagy-blocking properties.
An alternative strategy that has been considered for neurodegenerative diseases associated with intracellular aggregate formation is to boost the chaperone activity of cells to decrease protein aggregation. This can be achieved using heat shock protein 90 (HSP90) inhibitors, such as geldanamycin, which induce HSP chaperones via the heat shock response190. Recent data suggest that HSP90 inhibition may impair autophagy191, which may partly explain the minimal efficacy of this strategy in a mouse model of Huntington’s disease192.
Another drug that has been identified as an autophagy inhibitor is the macrolide antibiotic azithromycin, which has been widely used for its anti-inflammatory properties in patients with cystic fibrosis193 and, similarly to bafilomycin A1, can block autophagosome maturation69,194. Recent epidemiological evidence indicates that the use of azithromycin in patients with cystic fibrosis may be accompanied by an increasing incidence of drug-resistant non-tuberculous mycobacterial infection69,194,195. Moreover, azithromycin inhibited intracellular killing of drug-resistant mycobacteria in macrophages and enhanced pulmonary disease in drugresistant mycobacteria-infected mice through a mechanism that is postulated to involve azithromycin-mediated blockade of lysosomal acidification, autophagic maturation and phagosomal degradation69. Although further studies are warranted to confirm a causal relationship among chronic azithromycin use, blockade of lysosomal acidification and patient susceptibility to drug-resistant mycobacterial infections, these data highlight the need for cautious monitoring of the prevalence of infectious diseases in patient populations that receive chronic treatment with lysosomotropic agents (such as patients in oncology trials with hydroxychloroquine).
Autophagy drug screens
In recent years there has been a rapid increase in reports in the literature on drugs that affect autophagy. Although this has revealed many drugs of potential clinical utility, it is important to review studies carefully before any conclusions are made. The tools we use to measure autophagy are imperfect and frequently require multiple orthogonal assays to allow robust conclusions to be made about their effects. For instance, many screens have relied on LC3 vesicle count as a primary read-out, as this correlates with autophagosome numbers. This count can increase when autophagy is induced if autophagosome formation exceeds degradation196. Conversely, LC3 vesicle numbers also increase if autophagosome degradation is impaired. Another commonly used read-out for autophagy studies involves measurements of the levels of p62, an endogenous autophagy substrate. Analyses of p62 levels may be confounded by agents that regulate its transcription197. This caveat may be addressed by using stable inducible cell lines overexpressing p62 and by measuring p62 clearance after switching off transgene expression198.
Comparisons between different reported screens have yielded both concordant and discordant findings (reviewed in REF. 199). In some cases, apparently discrepant results can be explained by LC3-based assays that have not accounted for the effects of flux and have misinterpreted LC3 vesicle counts or increases in LC3-II as being simply due to increased autophagosome formation. Other reasons for discrepant results include the fact that drugs can have different effects on autophagy if different concentrations are used; for instance, a drug that induces autophagy at a low concentration (at which it has relatively good target specificity) could block autophagy at a high concentration (as a result of additional targets being engaged).
Recent compound screens have revealed important insights into the biology of autophagy. For instance, ‘spautin-1’ (specific and potent autophagy inhibitor 1)200 has been identified and shown to inhibit autophagy by blocking two proteins, ubiquitin-specific peptidase 10 (USP10) and USP13, that target the BECN1-containing PIK3C3 complex, thereby enhancing the degradation of PIK3C3 complexes200. BECN1 also regulates the stability of the proteins USP10 and USP13, which might explain the novel observation200 that BECN1 regulates p53 levels, as USP10 mediates p53 deubiquitylation. These data suggest that BECN1 haploinsufficiency may contribute to tumorigenesis by reducing the levels of the tumour suppressor gene p53.
For many compounds that have been reported to modulate autophagy, the targets are still unclear. It is therefore desirable to confirm whether the proposed targets of autophagy-modifying drugs act as one would predict when they are modulated genetically (for example, by knockdown or overexpression); it is also desirable to ascertain whether the drugs modulate target activity at physiological concentrations, and whether the drugs act primarily via a particular target (for example, by showing that the drugs do not modulate autophagy when the target is knocked down or knocked out).
Future directions
The development of more specific autophagy modulators is vital, both for therapeutic applications and for their use as chemical probes to allow the acute modulation of this process for cell biology and physiological studies. Such reagents will be crucial for inferring the direct effects of autophagy on biological and disease processes by limiting the influences of autophagy-independent effects. Various strategies have been proposed. The first involves inhibitors of the class III PI3K (PIK3C3), the crystal structure of which exhibits a smaller ATP-binding pocket than that of class I PI3K isoforms201, indicating that structure-based design may be used to develop compounds that have considerably higher selectivity for class III PI3Ks than for class I PI3Ks. The two ubiquitin-like conjugation systems that are crucial for the structural formation of autophagosomes may also be amenable to the development of specific inhibitors. ATG7 (an E1-activating enzyme), ATG3 and ATG10 (E2-conjugating enzymes), and the ATG12–ATG5 conjugate (an E3-like ligase) may be suitable targets for structure-based drug design202,203. Such approaches may also be used to exploit related strategies that have been developed for targeting ubiquitin and ubiquitin-like E1 enzymes using semi-synthetic protein inhibitors. ATG4B, the cysteine protease that cleaves ATG8 at its carboxy-terminal to allow phosphatidylethanolamine conjugation, may also be a suitable candidate for structure-based drug design. Although the active site cleft of ATG4B is masked in the free form of the protein, thus eliminating this region as a target for inhibitors, possible targets for the design of specific inhibitors have been identified and include the inhibitory loop of ATG4B or the substrate ubiquitin core binding site204,205.
However, even the design of specific inhibitors of ATG4 proteins may not guarantee the absence of autophagy-independent off-target effects, as recent data suggest that autophagic machinery may have roles in seemingly distinct processes that do not appear to require conventional autophagosomes, such as osteoclast function206, phagocytosis207, entosis208 and mitochondrial cell death209. Conversely, it also seems as though inhibition of certain autophagy core proteins, including PIK3C3, BECN1 and ULK1, does not necessarily ablate autophagy completely210.
In principle, one may be able to influence the clearance of autophagy substrates by acting not only on the autophagy pathway itself but also on processes influencing substrate recruitment100 as well as on lysosomal activity211. Substrate recruitment may be influenced by altered activities of autophagy adaptor molecules that help to recruit substrates to autophagosomes212. This may allow enhanced clearance of a repertoire of selected substrates. Although this may be achievable, in principle, by altering the levels of such adaptors, it is possible that post-translational modifications of these adaptors may be a more effective means of influencing substrate recognition, and this approach may also be more amenable to targeting with drugs. An elegant example of this phenomenon (discussed above) is the observation that phosphorylation of optineurin, which can act as an autophagy receptor, promoted the selective autophagy of ubiquitin-coated cytosolic S. enterica. In a similar mode, the PINK1–PARK2 machinery is thought to regulate selective autophagic degradation of mitochondria with disrupted membrane potentials. A recent genome-wide screen identified many genes involved in the selective clearance of the Sindbis virus capsid protein; some of these genes also regulated mitochondrial degradation after depolarization100. This suggests that there may be many different types of adaptors and regulatory mechanisms that are amenable to perturbation, and also that this type of mechanism may ultimately affect a range of selective autophagic substrates.
One other step that may enhance the degradation of autophagic substrates is at the level of the lysosome. This may be achieved by modulating the activity of transcription factor EB (TFEB), a master transcriptional modulator that influences both lysosomal biogenesis and autophagy213. This may be a tractable target, as TFEB activity is modulated by phosphorylation. Another way to enhance autophagic substrate degradation could be via the depletion of the endogenous lysosomal cathepsin inhibitor cystatin B. Indeed, this strategy appears to enhance autophagic substrate clearance and reduce amyloid-β pathology in a mouse model of Alzheimer’s disease211.
Although this article has focused on modulating autophagy using drugs, it should be noted that there may be other ways of inducing autophagy, some of which could also be beneficial to health. These include dietary restriction, exercise and possibly gene therapy routes (potentially transducing tissues with vectors that enhance or block autophagy in some instances). As we know that various hormones (such as insulin) affect autophagy, we also need to consider non-cell-autonomous modes of autophagy regulation, particularly in the whole-body context.
Acknowledgements
D.C.R. is supported by the Wellcome Trust (Principal Fellowship), a Wellcome Trust/Medical Research Council Strategic Award on Neurodegenerative Diseases, the UK Medical Research Council and the National Institute for Health Research Biomedical Research Unit in Dementia at Addenbrooke’s Hospital, Cambridge, UK. B.L. is supported by US National Institutes of Health (NIH) grants RO1 CA109618 and U54AI057156. P.C. is supported by INSERM, Université Paris-Sud 11, Agence Nationale pour la Recherche and Institut National du Cancer. The authors are grateful to F. Menzies and W. Hochfeld for technical assistance.
Glossary
- Autophagosomes
Double-membrane vesicles that engulf cytoplasmic contents for delivery to the lysosome.
- Phagophore
A cup-shaped, double-membrane autophagic precursor structure.
- Microtubule organizing centre
A structure near the nucleus of eukaryotic cells, from which microtubules emerge.
- Class III phosphoinositide 3-kinases
(PI3Ks; also known as PIK3C3 or VPS34). Enzymes that produce phosphatidylinositol-3-phosphate (PtdIns3P).
- SNAREs
Soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein (SNAP) receptors; these molecules enable vesicle fusion events.
- Selective autophagy
Autophagic processes involving the preferential recruitment of specific substrates, in contrast to random bulk cytoplasmic sequestration.
- Gluconeogenesis
A metabolic pathway that generates glucose from non-carbohydrate carbon sources, thus preventing blood sugar levels from becoming too low.
- Pancreatic β-cells
Insulin-producing cells in the pancreas.
- Dynactin
A component of the dynein motor complex.
- Tumour suppressor genes
Genes that protect cells from one of the steps along the pathway to cancer.
- Autophagy adaptor proteins
Proteins that mediate selective autophagy: for example, ubiquitin-binding protein p62 (also known as sequestosome 1).
- Short hairpin RNA
Small RNAs that form hairpins that can induce sequence-specific silencing in mammalian cells through RNA interference.
- Lysosomotropic agents
Compounds that accumulate preferentially in lysosomes; many weak bases are lysosomotropic agents.
- Encephalitis
Inflammation in the brain.
- Interferon
A type of protein made and released by host cells in response to bacteria, viruses and tumour cells.
- MHC class II
Major histocompatibility complex (MHC) class II; molecules that are found on antigen-presenting cells and lymphocytes. The antigens presented by MHC class II molecules are derived from extracellular proteins.
- MHC class I
Major histocompatibility complex (MHC) class I; molecules that are found on the surfaces of all nucleated cells and present peptides derived from cytosolic proteins.
- Paneth cells
Cells in the small intestine that assist in antibacterial defence.
- Autophagosome maturation
The processes occurring after the completion of autophagosome closure that enable the delivery of the autophagosome to — and its degradation in — the lysosome.
Footnotes
Competing interests statement
The authors declare competing financial interests: see Web version for details.
FURTHER INFORMATION
David C. Rubinsztein’s homepage: http://www.cimr.cam.ac.uk/investigators/rubinsztein/index.html
Patrice Codogno’s homepage: http://www.ipbs.fr/?Dr-Patrice-Codogno-Autophagie
Beth Levine’s homepage: http://www4.utsouthwestern.edu/idlabs/Levine/Levine_intro.htm
ClinicalTrials.gov website: http://clinicaltrials.gov
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Contributor Information
David C. Rubinsztein, Email: dcr1000@cam.ac.uk.
Patrice Codogno, Email: patrice.codogno@u-psud.fr.
Beth Levine, Email: beth.levine@utsouthwestern.edu.
References
- 1.Ravikumar B, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 2.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kuma A, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. This study illustrates the importance of autophagy in newborn mammals as a process that protects against starvation in the period before breast feeding is established.
- 4.Boya P, et al. Inhibition of macroautophagy triggers apoptosis . Mol. Cell. Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nature Cell Biol. 2010;12:823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsukamoto S, et al. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321:117–120. doi: 10.1126/science.1154822. [DOI] [PubMed] [Google Scholar]
- 7.Al Rawi S, et al. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- 8.Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–1144. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- 9.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 10.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 11.Axe EL, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi-Nishino M, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nature Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 13. Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–1185. doi: 10.4161/auto.5.8.10274. References 11, 12 and 13 provide strong support for a role of the ER in autophagosome biogenesis.
- 14. Hailey DW, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. This study suggests that mitochondria contribute membrane to autophagosomes during starvation.
- 15. Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nature Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. This study provides evidence that the plasma membrane is involved in the formation of autophagosome precursor structures.
- 16.Mari M, et al. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010;190:1005–1022. doi: 10.1083/jcb.200912089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohashi Y, Munro S. Membrane delivery to the yeast autophagosome from the Golgi–endosomal system. Mol. Biol. Cell. 2010;21:3998–4008. doi: 10.1091/mbc.E10-05-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nair U, et al. SNARE proteins are required for macroautophagy. Cell. 2011;146:290–302. doi: 10.1016/j.cell.2011.06.022. This study shows how SNARE proteins are involved in autophagosome biogenesis in yeast.
- 19. Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011;146:303–317. doi: 10.1016/j.cell.2011.06.023. This paper describes how SNAREs regulate mammalian autophagy by aiding in the expansion of the membranes of autophagosome precursors.
- 20.Noda NN, et al. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells. 2008;13:1211–1218. doi: 10.1111/j.1365-2443.2008.01238.x. [DOI] [PubMed] [Google Scholar]
- 21.Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 2011;80:125–156. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- 22.Kraft C, Reggiori F, Peter M. Selective types of autophagy in yeast. Biochim. Biophys. Acta. 2009;1793:1404–1412. doi: 10.1016/j.bbamcr.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Kim DH, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 24.Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem. 1995;270:2320–2326. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- 25.Noda T, Ohsumi Y. Tor a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 26.Mammucari C, et al. FoxO3 controls autophagy in skeletal muscle. in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 27. Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. This study describes BCL-2 proteins as key negative regulators of autophagy that act via interactions with BECN1.
- 28.Erlich S, et al. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy. 2007;3:561–568. doi: 10.4161/auto.4713. [DOI] [PubMed] [Google Scholar]
- 29.Maiuri MC, et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wei Y, Pattingre S, Bassik M, Sinha S, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. This paper describes how JNK1-mediated phosphorylation of BCL-2 during starvation results in dissociation of the interaction between BCL-2 and BECN1, and mediates autophagy induction.
- 31.Pattingre S, et al. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 2009;284:2719–2728. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Codogno P, Meijer AJ. Autophagy: a potential link between obesity and insulin resistance. Cell Metab. 2010;11:449–451. doi: 10.1016/j.cmet.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 36.Singh R, et al. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Invest. 2009;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, et al. Adipose-specific deletion of autophagyrelated gene 7 ( atg7 ) in mice reveals a role in adipogenesis. Proc. Natl Acad. Sci. USA. 2009;106:19860–19865. doi: 10.1073/pnas.0906048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. He C, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. This paper describes exercise as a novel physiological inducer of autophagy and demonstrates a role for autophagy in the exercise-induced beneficial effects on metabolism.
- 39.Kaushik S, et al. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011;14:173–183. doi: 10.1016/j.cmet.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IκB kinase β (IKKβ)/NF-κB pathway. J. Biol. Chem. 2011;286:32324–32332. doi: 10.1074/jbc.M111.254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rubinsztein DC. Autophagy — alias self-eating — appetite and ageing. EMBO Rep. 2012;13:173–174. doi: 10.1038/embor.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubinsztein DC. The roles of intracellular proteindegradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 43.Berger Z, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 44. Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. This is the first description of autophagy as a process that clears intracytoplasmic aggregation-prone proteins, and provides data in cell culture suggesting that autophagy induction may be a therapeutic strategy for certain neurodegenerative diseases.
- 45.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. α-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 46.Shibata M, et al. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 2006;281:14474–14485. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- 47.Menzies FM, et al. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain. 2010;133:93–104. doi: 10.1093/brain/awp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spencer B, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Williams A, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nature Chem. Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. This paper describes a large-scale drug screen that identified a series of mTOR-independent autophagy inducers that have protective effects in cell, D. melanogaster and zebrafish models of Huntington’s disease.
- 50.Zheng S, et al. Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet. 2010;6:e1000838. doi: 10.1371/journal.pgen.1000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ravikumar B, Berger Z, Vacher C, O’Kane CJ, Rubinsztein DC. Rapamycin pre-treatment protects against apoptosis. Hum. Mol. Genet. 2006;15:1209–1216. doi: 10.1093/hmg/ddl036. [DOI] [PubMed] [Google Scholar]
- 52.Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 53. Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. References 52 and 53 describe how conditional loss of autophagy in the mammalian nervous system leads to cell death and aggregation of endogenous proteins.
- 54.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nature Rev. Mol. Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee JH, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimerrelated PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pickford F, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winslow AR, Rubinsztein DC. The Parkinson disease protein α-synuclein inhibits autophagy. Autophagy. 2010;7:429–431. doi: 10.4161/auto.7.4.14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corrochano S, et al. α-synuclein levels affect autophagosome numbers in vivo and modulate Huntington disease pathology. Autophagy. 2012;8:431–432. doi: 10.4161/auto.19259. [DOI] [PubMed] [Google Scholar]
- 59.Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012;125:795–799. doi: 10.1242/jcs.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aguado C, et al. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum. Mol. Genet. 2010;19:2867–2876. doi: 10.1093/hmg/ddq190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Puls I, et al. Mutant dynactin in motor neuron disease. Nature Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 62.Ravikumar B, et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nature Genet. 2005;37:771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- 63.Sarkar S, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raben N, Shea L, Hill V, Plotz P. Monitoring autophagy in lysosomal storage disorders. Meth. Enzymol. 2009;453:417–449. doi: 10.1016/S0076-6879(08)04021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Settembre C, et al. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 66.Rose C, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2010;19:2144–2153. doi: 10.1093/hmg/ddq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bjedov I, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster . Cell Metab. 2010;11:35–46. doi: 10.1016/j.cmet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Renna M, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J. Clin. Invest. 2011;121:3554–3563. doi: 10.1172/JCI46095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. This paper describes BECN1 as a mammalian autophagy gene that is associated with tumour suppression.
- 71.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol. Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nature Rev. Clin. Oncol. 2011;8:528–539. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- 73.Mah LY, Ryan KM. Autophagy and cancer. Cold Spring Harb. Perspect. Biol. 2012;4:a008821. doi: 10.1101/cshperspect.a008821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Amaravadi RK, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011;17:654–666. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aita VM, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 76.Qu X, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl Acad. Sci. USA. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. References 76 and 77 demonstrate that BECN1 is a haploinsufficient tumour supressor gene.
- 78.Marino G, et al. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/ autophagin-3. J. Biol. Chem. 2007;282:18573–18583. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 79.Takamura A, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morselli E, et al. Oncosuppressive functions of autophagy. Antioxid. Redox Signal. 2011;14:2251–2269. doi: 10.1089/ars.2010.3478. [DOI] [PubMed] [Google Scholar]
- 81.He C, Levine B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010;22:140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pimkina J, Humbey O, Zilfou JT, Jarnik M, Murphy ME. ARF induces autophagy by virtue of interaction with Bcl-xl. J. Biol. Chem. 2009;284:2803–2810. doi: 10.1074/jbc.M804705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tasdemir E, et al. A dual role of p53 in the control of autophagy. Autophagy. 2008;4:810–814. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
- 84.Morselli E, et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle. 2011;10:2763–2769. doi: 10.4161/cc.10.16.16868. [DOI] [PubMed] [Google Scholar]
- 85.Morselli E, et al. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle. 2008;7:3056–3061. doi: 10.4161/cc.7.19.6751. [DOI] [PubMed] [Google Scholar]
- 86.Furuta S, Hidaka E, Ogata A, Yokota S, Kamata T. Ras is involved in the negative control of autophagy through the class I PI3-kinase. Oncogene. 2004;23:3898–3904. doi: 10.1038/sj.onc.1207539. [DOI] [PubMed] [Google Scholar]
- 87.Kim JH, et al. Involvement of mitophagy in oncogenic K-Ras-induced transformation: overcoming a cellular energy deficit from glucose deficiency. Autophagy. 2011;7:1187–1198. doi: 10.4161/auto.7.10.16643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu SY, et al. Ras-related tumorigenesis is suppressed by BNIP3-mediated autophagy through inhibition of cell proliferation. Neoplasia. 2011;13:1171–1182. doi: 10.1593/neo.11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 90.Guo JY, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim MJ, et al. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 2011;286:12924–12932. doi: 10.1074/jbc.M110.138958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lock R, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell. 2011;22:165–178. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mathew R, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mathew R, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. This study describes how autophagy may limit tumour formation by preventing the accumulation of p62.
- 95.Poulogiannis G, et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl Acad. Sci. USA. 2010;107:15145–15150. doi: 10.1073/pnas.1009941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Veeriah S, et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nature Genet. 2010;42:77–82. doi: 10.1038/ng.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Inami Y, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011;193:275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Duran A, et al. The signaling adaptor p62 is an important NF-κB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 99.Duran A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Orvedahl A, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. doi: 10.1038/nature10546. This study describes a genome-wide screen to identify novel mediators of the selective autophagy of substrates, including the Sindbis virus capsid protein and damaged mitochondria.
- 101.Kwei KA, et al. SMURF1 amplification promotes invasiveness in pancreatic cancer. PLoS ONE. 2011;6:e23924. doi: 10.1371/journal.pone.0023924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Birnbaum DJ, et al. Genome profiling of pancreatic adenocarcinoma. Genes Chromosomes Cancer. 2011;50:456–465. doi: 10.1002/gcc.20870. [DOI] [PubMed] [Google Scholar]
- 103.Loukopoulos P, et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci. 2007;98:392–400. doi: 10.1111/j.1349-7006.2007.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Edinger AL, Thompson CB. Defective autophagy leads to cancer. Cancer Cell. 2003;4:422–424. doi: 10.1016/s1535-6108(03)00306-4. [DOI] [PubMed] [Google Scholar]
- 105.Parkhitko A, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc. Natl Acad. Sci. USA. 2011;108:12455–12460. doi: 10.1073/pnas.1104361108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Valentin-Vega YA, et al. Mitochondrial dysfunction in ataxia-telangiectasia. Blood. 2012;119:1490–1500. doi: 10.1182/blood-2011-08-373639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yang S, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bellot G, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains . Mol. Cell. Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nature Rev. Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee SJ, Kim HP, Jin Y, Choi AM, Ryter SW. Beclin 1 deficiency is associated with increased hypoxiainduced angiogenesis. Autophagy. 2011;7:829–839. doi: 10.4161/auto.7.8.15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maycotte P, et al. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–212. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li BX, et al. The expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancers. Autophagy. 2009;5:303–306. doi: 10.4161/auto.5.3.7491. [DOI] [PubMed] [Google Scholar]
- 113.Kim HS, et al. Clinicopathologic correlation of beclin-1 expression in pancreatic ductal adenocarcinoma. Pathol. Res. Pract. 2011;207:247–252. doi: 10.1016/j.prp.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 114.Chen Y, Lu Y, Lu C, Zhang L. Beclin-1 expression is a predictor of clinical outcome in patients with esophageal squamous cell carcinoma and correlated to hypoxia-inducible factor (HIF)-1α expression. Pathol. Oncol. Res. 2009;15:487–493. doi: 10.1007/s12253-008-9143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pirtoli L, et al. The prognostic role of Beclin 1 protein expression in high-grade gliomas. Autophagy. 2009;5:930–936. doi: 10.4161/auto.5.7.9227. [DOI] [PubMed] [Google Scholar]
- 116.Ding ZB, et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008;68:9167–9175. doi: 10.1158/0008-5472.CAN-08-1573. [DOI] [PubMed] [Google Scholar]
- 117.Huang JJ, et al. Beclin 1 expression predicts favorable clinical outcome in patients with diffuse large B-cell lymphoma treated with R-CHOP. Hum. Pathol. 2011;42:1459–1466. doi: 10.1016/j.humpath.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 118.Amaravadi RK, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mancias JD, Kimmelman AC. Targeting autophagy addiction in cancer. Oncotarget. 2011;2:1302–1306. doi: 10.18632/oncotarget.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kanzawa T, et al. Role of autophagy in temozolomideinduced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 121.Voss V, et al. The pan-Bcl-2 inhibitor (–)-gossypol triggers autophagic cell death in malignant glioma. Mol. Cancer Res. 2010;8:1002–1016. doi: 10.1158/1541-7786.MCR-09-0562. [DOI] [PubMed] [Google Scholar]
- 122.Shen S, et al. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene. 2011;30:4544–4556. doi: 10.1038/onc.2011.168. [DOI] [PubMed] [Google Scholar]
- 123.Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kaza N, Kohli L, Roth KA. Autophagy in brain tumors: a new target for therapeutic intervention. Brain Pathol. 2012;22:89–98. doi: 10.1111/j.1750-3639.2011.00544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Michaud M, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. This study describes how autophagy enables the immunogenic release of ATP from dying cells, which enhances the efficacy of antineoplastic chemotherapies.
- 126.Cuzick J, et al. Preventive therapy for breast cancer: a consensus statement. Lancet Oncol. 2011;12:496–503. doi: 10.1016/S1470-2045(11)70030-4. [DOI] [PubMed] [Google Scholar]
- 127.Bursch W, et al. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: the role of autophagy. Carcinogenesis. 1996;17:1595–1607. doi: 10.1093/carcin/17.8.1595. [DOI] [PubMed] [Google Scholar]
- 128.Yuk JM, et al. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe. 2009;6:231–243. doi: 10.1016/j.chom.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 129.Wang J, Lian H, Zhao Y, Kauss MA, Spindel S. Vitamin D3 induces autophagy of human myeloid leukemia cells. J. Biol. Chem. 2008;283:25596–25605. doi: 10.1074/jbc.M801716200. [DOI] [PubMed] [Google Scholar]
- 130.Wang RC, Levine B. Calcipotriol induces autophagy in HeLa cells and keratinocytes. J. Invest. Dermatol. 2011;131:990–993. doi: 10.1038/jid.2010.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fleet JC, DeSmet M, Johnson R, Li Y. Vitamin D and cancer: a review of molecular mechanisms. Biochem. J. 2012;441:61–76. doi: 10.1042/BJ20110744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Grumati P, et al. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI deficient muscles. Autophagy. 2011;7:1415–1423. doi: 10.4161/auto.7.12.17877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cummings SR, et al. Prevention of breast cancer in postmenopausal women: approaches to estimating and reducing risk. J. Natl Cancer Inst. 2009;101:384–398. doi: 10.1093/jnci/djp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Friedenreich CM, Orenstein MR. Physical activity and cancer prevention: etiologic evidence and biological mechanisms. J. Nutr. 2002;132:3456S–3464S. doi: 10.1093/jn/132.11.3456S. [DOI] [PubMed] [Google Scholar]
- 135.Harriss DJ, et al. Lifestyle factors and colorectal cancer risk (2): a systematic review and meta-analysis of associations with leisure-time physical activity. Colorectal Dis. 2009;11:689–701. doi: 10.1111/j.1463-1318.2009.01767.x. [DOI] [PubMed] [Google Scholar]
- 136.Decensi A, et al. Metformin and cancer risk in diabetic patients: a systematic review and metaanalysis. Cancer Prev. Res. 2010;3:1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 137.Wilkinson S, O’Prey J, Fricker M, Ryan KM. Hypoxia-selective macroautophagy and cell survival signaled by autocrine PDGFR activity. Genes Dev. 2009;23:1283–1288. doi: 10.1101/gad.521709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Virgin HW, Levine B. Autophagy genes in immunity. Nature Immunol. 2009;10:461–470. doi: 10.1038/ni.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liang XH, et al. Protection against fatal Sindbis virus encephalitis by Beclin, a novel Bcl-2-interacting protein. J. Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Joubert P, et al. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 2012;209:1029–1047. doi: 10.1084/jem.20110996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sumpter R, Jr, Levine B. Autophagy and innate immunity: triggering, targeting and tuning. Semin. Cell Dev. Biol. 2010;21:699–711. doi: 10.1016/j.semcdb.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Orvedahl AO, et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mazanillo P, Watson RO, Cox JS. Detection of mycobacterial DNA activates ubiquitin-mediated autophagy and is essential for control of M. tuberculosis infection. Cell. (in the press) [Google Scholar]
- 146. Gutierrez MG, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. This paper describes for the first time that mycobacteria are autophagy substrates.
- 147.Intemann CD, et al. Autophagy gene variant IRGM-261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog. 2009;5:e1000577. doi: 10.1371/journal.ppat.1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kumar D, et al. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis . Cell. 2010;140:731–743. doi: 10.1016/j.cell.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 149.Campbell GR, Spector SA. Hormonally active vitamin D3 (1α, 25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J. Biol. Chem. 2011;286:18890–18902. doi: 10.1074/jbc.M110.206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Campbell GR, Spector SA. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012;8:e1002689. doi: 10.1371/journal.ppat.1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim JJ, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. 2012;11:457–468. doi: 10.1016/j.chom.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 152.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lee HK, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32:227–239. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.English L, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nature Immunol. 2009;10:480–487. doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li Y, et al. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008;68:6889–6895. doi: 10.1158/0008-5472.CAN-08-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Jagannath C, et al. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nature Med. 2009;15:267–276. doi: 10.1038/nm.1928. This study describes how autophagy increases the efficacy of antituberculosis vaccination.
- 158.Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr. Opin. Immunol. 2012;24:21–31. doi: 10.1016/j.coi.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dupont N, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30:4701–4711. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hwang S, et al. Nondegradative role of Atg5–Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon γ. Cell Host Microbe. 2012;11:397–409. doi: 10.1016/j.chom.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 162. Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. This study describes how compromised function of ATG16L1 and disrupted autophagy in the intestine may result in Crohn’s disease-like pathology, providing support for a role of this pathway and/or ATG16L1 in this disease.
- 163.Starr T, et al. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe. 2012;11:33–45. doi: 10.1016/j.chom.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010;8:422–432. doi: 10.1016/j.chom.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li J, et al. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 2011;85:6319–6333. doi: 10.1128/JVI.02627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kyei GB, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tian Y, Sir D, Kuo CF, Ann DK, Ou JH. Autophagy required for hepatitis B virus replication in transgenic mice. J. Virol. 2011;85:13453–13456. doi: 10.1128/JVI.06064-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Alirezaei M, Flynn CT, Wood MR, Whitton JL. Pancreatic acinar cell-specific autophagy disruption reduces coxsackievirus replication and pathogenesis in vivo . Cell Host Microbe. 2012;11:298–305. doi: 10.1016/j.chom.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sun Y, et al. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci. Signal. 2012;5:ra16. doi: 10.1126/scisignal.2001931. [DOI] [PubMed] [Google Scholar]
- 170.Chaumorcel M, et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J. Virol. 2012;86:2571–2584. doi: 10.1128/JVI.05746-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Orvedahl A, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 172.Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J. Virol. 2009;83:12164–12171. doi: 10.1128/JVI.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.E X, et al. Viral Bcl-2-mediated evasion of autophagy aids chronic infection of γherpesvirus 68. PLoS Pathog. 2009;5:e1000609. doi: 10.1371/journal.ppat.1000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Wild P, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. This paper describes how phosphorylation of an autophagy receptor protein may increase its substrate affinity.
- 175.Liu Q, Thoreen C, Wang J, Sabatini D, Gray NS. mTOR mediated anti-cancer drug discovery. Drug Discov. Today Ther. Strateg. 2009;6:47–55. doi: 10.1016/j.ddstr.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycinresistant functions of mTORC1. J. Biol. Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nyfeler B, et al. Relieving autophagy and 4EBP1 from rapamycin resistance. Mol. Cell. Biol. 2011;31:2867–2876. doi: 10.1128/MCB.05430-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Raynaud FI, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–5850. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- 179.Degtyarev M, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol. 2008;183:101–116. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks . Mol. Cell. Biol. 2012;32:2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Buzzai M, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 182. Sarkar S, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005;170:1101–1111. doi: 10.1083/jcb.200504035. This study was the first to demonstrate a mammalian autophagy pathway that was independent of mTOR.
- 183.Cardenas C, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hidvegi T, et al. An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 185.Morselli E, et al. Caloric restriction and resveratrol promote longevity through the sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Eisenberg T, et al. Induction of autophagy by spermidine promotes longevity. Nature Cell Biol. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 187.Li X, Fan Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1α and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010;70:5942–5952. doi: 10.1158/0008-5472.CAN-10-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Maiuri MC, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-XL. Autophagy. 2007;3:374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 189.Underwood BR, Rubinsztein DC. Spinocerebellar ataxias caused by polyglutamine expansions: a review of therapeutic strategies. Cerebellum. 2008;7:215–221. doi: 10.1007/s12311-008-0026-z. [DOI] [PubMed] [Google Scholar]
- 190.Sittler A, et al. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum. Mol. Genet. 2001;10:1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 191.Joo JH, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol. Cell. 2011;43:572–585. doi: 10.1016/j.molcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Labbadia J, et al. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J. Clin. Invest. 2011;121:3306–3319. doi: 10.1172/JCI57413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zarogoulidis P, et al. Macrolides: from in vitro antiinflammatory and immunomodulatory properties to clinical practice in respiratory diseases. Eur. J. Clin. Pharmacol. 2011;68:479–503. doi: 10.1007/s00228-011-1161-x. [DOI] [PubMed] [Google Scholar]
- 194.Esther CR, et al. Chronic Mycobacterium abscessus infection and lung function decline in cystic fibrosis. J. Cyst. Fibros. 2010;9:117–123. doi: 10.1016/j.jcf.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Roux AL, et al. Multicenter study of prevalence of nontuberculous mycobacteria in patients with cystic fibrosis in France. J. Clin. Microbiol. 2009;47:4124–4128. doi: 10.1128/JCM.01257-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rubinsztein DC, et al. In search of an “autophagomometer”. Autophagy. 2009;5:585–589. doi: 10.4161/auto.5.5.8823. [DOI] [PubMed] [Google Scholar]
- 197.Jain A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Larsen KB, et al. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy. 2010;6:784–793. doi: 10.4161/auto.6.6.12510. [DOI] [PubMed] [Google Scholar]
- 199.Renna M, Jimenez-Sanchez M, Sarkar S, Rubinsztein DC. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J. Biol. Chem. 2010;285:11061–11067. doi: 10.1074/jbc.R109.072181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Liu J, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–234. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Miller S, et al. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science. 2010;327:1638–1642. doi: 10.1126/science.1184429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Noda NN, et al. Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol. Cell. 2011;44:462–475. doi: 10.1016/j.molcel.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 203.Taherbhoy AM, et al. Atg8 transfer from Atg7 to Atg3: a distinctive E1-E2 architecture and mechanism in the autophagy pathway. Mol. Cell. 2011;44:451–461. doi: 10.1016/j.molcel.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kumanomidou T, et al. The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J. Mol. Biol. 2006;355:612–618. doi: 10.1016/j.jmb.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 205.Sugawara K, et al. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. J. Biol. Chem. 2005;280:40058–40065. doi: 10.1074/jbc.M509158200. [DOI] [PubMed] [Google Scholar]
- 206. DeSelm CJ, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev. Cell. 2011;21:966–974. doi: 10.1016/j.devcel.2011.08.016. This paper describes how components of the autophagic machinery are required for aspects of secretion that may be autophagy-independent.
- 207.Sanjuan MA, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 208.Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nature Cell Biol. 2011;13:1335–1343. doi: 10.1038/ncb2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Radoshevich L, et al. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell. 2010;142:590–600. doi: 10.1016/j.cell.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nature Rev. Mol. Cell Biol. 2011;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 211.Yang DS, et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy. 2011;7:788–789. doi: 10.4161/auto.7.7.15596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Dupont N, Temime-Smaali N, Lafont F. How ubiquitination and autophagy participate in the regulation of the cell response to bacterial infection. Biol. Cell. 2010;102:621–634. doi: 10.1042/BC20100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Settembre C, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. This study describes how the transcription factor TFEB coordinately regulates both lysosomal biogenesis and autophagy.
- 214.Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010;188:527–536. doi: 10.1083/jcb.200911154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J. Cell Biol. 2010;188:537–546. doi: 10.1083/jcb.200911149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bruns C, McCaffery JM, Curwin AJ, Duran JM, Malhotra V. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J. Cell Biol. 2011;195:979–992. doi: 10.1083/jcb.201106098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Proikas-Cezanne T, Codogno P. Beclin 1 or not Beclin 1. Autophagy. 2011;7:671–672. doi: 10.4161/auto.7.7.14877. [DOI] [PubMed] [Google Scholar]
- 218.Settembre C, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Zoncu R, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. This paper advances the understanding of amino acid sensing by showing that amino acids are detected within the lysosome by the vacuolar V-ATPase.
- 220.Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 222.Jager S, et al. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 223.Fader CM, Sanchez D, Furlan M, Colombo MI. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in K562 cells. Traffic. 2008;9:230–250. doi: 10.1111/j.1600-0854.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- 224.Itoh T, et al. Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol. Biol. Cell. 2008;19:2916–2925. doi: 10.1091/mbc.E07-12-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Liang C, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nature Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 226.Matsunaga K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nature Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 227.Zhong Y, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nature Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Atlashkin V, et al. Deletion of the SNARE vti1b in mice results in the loss of a single SNARE partner, syntaxin 8. Mol. Cell. Biol. 2003;23:5198–5207. doi: 10.1128/MCB.23.15.5198-5207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Fader CM, Sanchez DG, Mestre MB, Colombo MI. TI-VAMP/VAMP7 and VAMP3/ cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim. Biophys. Acta. 2009;1793:1901–1916. doi: 10.1016/j.bbamcr.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 230.Furuta N, Fujita N, Noda T, Yoshimori T, Amano A. Combinational soluble N -ethylmaleimidesensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Mol. Biol. Cell. 2010;21:1001–1010. doi: 10.1091/mbc.E09-08-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Renna M, et al. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J. Cell Sci. 2011;124:469–482. doi: 10.1242/jcs.076489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Filimonenko M, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 2007;179:485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lee JA, Beigneux A, Ahmad ST, Young SG, Gao FB. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007;17:1561–1567. doi: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 234.Rusten TE, et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr. Biol. 2007;17:1817–1825. doi: 10.1016/j.cub.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 235.Crighton D, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 236.Aplin A, et al. Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J. Cell Physiol. 1992;152:458–466. doi: 10.1002/jcp.1041520304. [DOI] [PubMed] [Google Scholar]
- 237.Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 2006;281:36303–36316. doi: 10.1074/jbc.M607031200. [DOI] [PubMed] [Google Scholar]
- 238.Kochl R, Hu XW, Chan EY, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–145. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- 239.Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9:574–587. doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008;33:109–122. doi: 10.1247/csf.08005. [DOI] [PubMed] [Google Scholar]
- 241.Geeraert C, et al. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 2010;285:24184–24194. doi: 10.1074/jbc.M109.091553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Mousavi SA, et al. Effects of inhibitors of the vacuolar proton pump on hepatic heterophagy and autophagy. Biochim. Biophys. Acta. 2001;1510:243–257. doi: 10.1016/s0005-2736(00)00354-0. [DOI] [PubMed] [Google Scholar]
- 243.Yamamoto A, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 244.Yang Z, Huang J, Geng J, Nair U, Klionsky DJ. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol. Biol. Cell. 2006;17:5094–5104. doi: 10.1091/mbc.E06-06-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Rong Y, et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl Acad. Sci. USA. 2011;108:7826–7831. doi: 10.1073/pnas.1013800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nature Genet. 2004;36:585–595. doi: 10.1038/ng1362. This paper describes how mTOR inhibition induces autophagy and ameliorates the toxicity of the huntingtin mutation in D. melangaster and mouse models of Huntington’s disease.
- 247.Feldman ME, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. This study describes ULK1 as a direct target of both mTOR and AMPK.
- 249.Meley D, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006;281:34870–34879. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 250.Malik SA, et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene. 2011;30:3918–3929. doi: 10.1038/onc.2011.104. [DOI] [PubMed] [Google Scholar]
- 251.Vicencio JM, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009;16:1006–1017. doi: 10.1038/cdd.2009.34. [DOI] [PubMed] [Google Scholar]
- 252.Jeong JK, et al. Autophagy induced by resveratrol prevents human prion protein-mediated neurotoxicity. Neurosci. Res. 2012;73:99–105. doi: 10.1016/j.neures.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 253.Opipari AW, Jr, et al. Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res. 2004;64:696–703. doi: 10.1158/0008-5472.can-03-2404. [DOI] [PubMed] [Google Scholar]
- 254.Armour SM, et al. Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging. 2009;1:515–528. doi: 10.18632/aging.100056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Gorzalczany Y, et al. Combining an EGFR directed tyrosine kinase inhibitor with autophagy-inducing drugs: a beneficial strategy to combat non-small cell lung cancer. Cancer Lett. 2011;310:207–215. doi: 10.1016/j.canlet.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 256.Han W, et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS ONE. 2011;6:e18691. doi: 10.1371/journal.pone.0018691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Punnonen EL, Reunanen H. Effects of vinblastine, leucine, and histidine, and 3-methyladenine on autophagy in Ehrlich ascites cells. Exp. Mol. Pathol. 1990;52:87–97. doi: 10.1016/0014-4800(90)90061-h. [DOI] [PubMed] [Google Scholar]
- 258.Webb JL, Ravikumar B, Rubinsztein DC. Microtubule disruption inhibits autophagosomelysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. Int. J. Biochem. Cell Biol. 2004;36:2541–2550. doi: 10.1016/j.biocel.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 259.Reunanen H, Marttinen M, Hirsimaki P. Effects of griseofulvin and nocodazole on the accumulation of autophagic vacuoles in Ehrlich ascites tumor cells. Exp. Mol. Pathol. 1988;48:97–102. doi: 10.1016/0014-4800(88)90048-2. [DOI] [PubMed] [Google Scholar]