

Summary
A benefit of multiphoton fluorescence microscopy is the inherent optical sectioning that occurs during excitation at the diffraction-limited spot. The scanned collection of fluorescence emission is incoherent; i.e., no real image needs to be formed on the detector plane. The nearly isotropic emission of fluorescence excited at the focal spot allows for new detection schemes that efficiently funnel all attainable photons to detector(s). We previously showed (Combs et al., 2007) that parabolic mirrors and condensers could be combined to collect the totality of solid angle around the excitation spot for tissue blocks, leading to ~8-fold signal gain. Using a similar approach, we have developed an in vivo Total Emission Detection (epiTED) instrument modified to make non-contact images from outside of living tissue. Simulations suggest that a ~4 fold enhancement may be possible (much larger with lower NA objectives than the 0.95 NA used here) with this approach depending on objective characteristics, imaging depth and the characteristics of the sample being imaged. In our initial prototype, two fold improvements were demonstrated in the mouse brain and skeletal muscle as well as the rat kidney using a variety of fluorophores and no compromise of spatial resolution. These results show this epiTED effectively doubles emission signal in vivo ; thus, it will maintain the image signal to noise ratio (SNR) at two times the scan rate or enable full scan rate at approximately 30% reduced laser power (to minimize photo-damage).
Keywords: Two-photon microscopy, imaging, light collection improvement, epi-detection, brain, kidney, muscle, photon diffusion simulations, scattering
Introduction
Multi-photon fluorescence microscopy (MPFM) has many advantages over fluorescence microscopy techniques that use one photon excitation. These include increased imaging depth (due primarily to the use of near-infrared excitation light), the inherent optical sectioning that occurs by the selective excitation of fluorescence only at the tightly focused spot, and reduced photo-bleaching and phototoxic effects for out of focus planes (Campagnola and Loew, 2003; Denk et al., 1995; Denk et al., 1990; Helmchen and Denk, 2005; Mertz, 2004; Patterson and Piston, 2000; Zipfel et al., 2003). In particular, it is the method of choice for high resolution, deep tissue imaging (for reviews see (Diaspro et al., 2006; Helmchen and Denk, 2005; Masters and So, 2008; Svoboda and Yasuda, 2006; Zinselmeyer et al., 2009). One under-utilized advantage is the fact that the emitted fluorescence originates in a single spot and that all of the emission light can be collected and assigned to a diffraction limited 3D coordinate in the sample during point-scanning imaging without the need for a conjugate pinhole. Although all of the light that exits the sample from the nearly isotropically emitting diffraction limited spot can and should be collected, modern commercial MPFM microscopes are only built to collect emission light coming back through the same objective used for focusing the excitation light. Unfortunately, objectives only capture a small fraction of the available emission light based on their numerical aperture and their design. This is particularly true of the long working distance, low numerical aperture objectives typically used for two-photon excitation fluorescence microscopy (TPFM) imaging. For instance a 20x, 0.95 N.A. objective lens collects less than 15% of the solid angle of fluorescence originating from the tissue (Combs et al., 2007). This is further complicated by the optical properties (absorption, scattering, and scattering anisotropy) of the tissue that can decrease light collection more than predicted by the solid angle alone(Wallace et al. 2001). Therefore, objective based emission collection schemes for MPFM are inefficient and lead to unnecessary use of higher levels of excitation power (which can lead to photo-damage) or slower scan rates to acquire the necessary signal to noise ratio (SNR) for an adequate image.
Several strategies that increase light collection efficiencies from MPFM experiments have been shown to be effective, but each have been shown to have their limitations. These include hybrid objectives and ancillary collection systems that collect light missed by the objective (Combs et al., 2007; Engelbrecht et al., 2009; Vucinic et al., 2006). Theoretically, hybrid objectives have been shown to have high potential for collecting more emission light but require custom design. In terms of external collection systems, fiber optic rings increase light collection efficiency (by over two-fold) but are not as efficient as parabolic mirrors at redirecting the missed emission light and, in current designs, have the additional drawback of touching, and possibly perturbing, the sample. The first parabolic mirror system was shown to enhance light collection by more than eight-fold (Combs et al., 2007). This scheme has the potential to collect the entire solid angle of emission light and therefore was called “Total Emission Detection (TED)”. One drawback to the design was the need to mount the sample between lenses in a confined space due to the orientation of the parabolic mirror, objective lens and a condensing optic on an inverted microscope. While this orientation worked for thin tissue slices and cells, it precluded in vivo imaging in animals or larger ex vivo tissues.
The purpose of this study was to develop an add-on system for in vivo imaging using a parabolic mirror external light collection system. This system only collects light that comes back in the same hemisphere as the origin of the excitation light and is appropriate for intravital imaging where the animal or tissue is too thick to allow light to be collected in the forward direction. We call this new collection scheme “epi” Total Emission Detection (epiTED). In contrast to the first TED device, the epiTED device consists of an integrating parabola mounted on a microscope invertor (LSM Technologies, Pennsylvania) that surrounds the objective and sends the light to the same detector used for the objective collected light (Fig. 1). This form of TED imaging permits the objective and mirror to approach the sample from above allowing for in vivo imaging. This design has the advantage of not touching the sample and enables the use of highly corrected commercially built objectives for TPFM imaging. Here we show the efficacy of the initial epiTED design for imaging brain, kidney, and skeletal muscle in live animals.
Figure 1. Schematic description of the epiTED Imaging System.
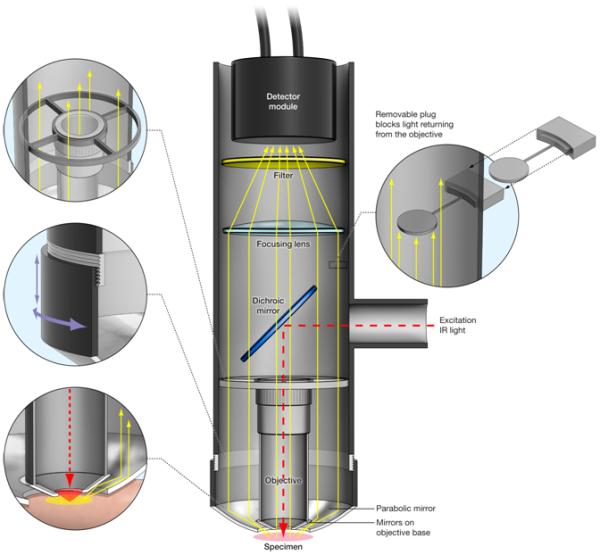
The epiTED system is built around an LSM Technologies objective inverter system. In this scheme the objective inverter is attached to the turret of an inverted microscope (not shown). The pulsed infrared light travels through the inverter to the dicroic mirror positioned in the main tube and through the objective for excitation. Emitted fluorescent light is directed to the detector module through both the objective and the parabola. Aluminum foil tape adhered to the face of the objective allows for somewhat more efficient light collection by the parabola (shown here idealized as small mirrors –see discussion). The left top inset shows the ring holding the objective. The middle left inset shows the focusing mechanism for the parabola. The left bottom inset shows potential paths for the redirected light collected by the parabola. The right inset illustrates the objective blocking element used for testing the amount of emitted light collected solely by the parabola. Details of the elements used in the construction of this device can be found in the Materials and Methods section.
Materials and Methods
epiTED Device Construction
The second generation Total Emission Detection (epiTED) device (Figure 1) was constructed to fit an Olympus IX81 inverted microscope stand and Fluoview 1000 confocal laser scanner adapted for two-photon imaging. Near-infrared pulses from a Newport-SpectraPhysics (Irvine, CA) laser model Maitai Broadband, attenuated by a combination of rotating zero-order half-wave plate and a fixed polarizer, were expanded ~3 times by means of a variable beam expander (56-10-2-8x from Special Optics, Wharton, New Jersey) and launched into an external laser port for the Olympus Fluoview 1000 laser scanning module. All experiments were performed using an Olympus XLUMPlanFl 20x water (0.95 NA) objective. The objective was held in place by a threaded holder as depicted in the top left inset of figure 1. The front surface of the objective (excluding the front lens element) was made reflective by molding a polished aluminum tape around the entire outer surface to help redirect and capture light not collected directly by the parabolic reflector. Most custom parts were manufactured, adapted or supplied by LSM Technologies, Inc. (Stewartstown, PA; www.lsmtech.com). The total epiTED system can be viewed as consisting of three major parts: objective invertor, light collector (parabola) coupled to a detector (wide area PMT), and a sample manipulation stage (not shown in Figure 1). Each of these components was rigidly attached to the floating optical table surface to avoid vibration.
The objective invertor consisted of a system of optics to transfer the excitation light from the turret of the microscope to the sample and to redirect the emission light up to a wide area PMT detector. The optical parts consisted of a 2” diameter right-angle periscope with a pair of relay lenses and an adjustable elliptical mirror (MC1 from Edmund Optics, Barrington, NJ) and had its exit side entering a section of a vertical 3” SM3 aluminum tube (Thorlabs, Newton, NJ) modified to have 1” diameter entrance hole on its side. A short-pass IR-reflecting dichroic elliptical mirror (custom p/n DSPXR675 from Chroma Technology Corp., Brattleboro, VT) was custom mounted at 45 degrees to reflect excitation light downward into the back aperture of the objective. This mirror mount had adjustment controls to center the excitation beam on the objective. Below the dichroic mirror, the threaded objective holder was precisely centered and aligned inside the SM3 tube by means of four metal pins to minimize obstruction of upward-directed multi-photon generated emission light. Above the mirror a slot in SM3 tube was cut to allow insertion of ~1” dia. light stop that effectively blocked all fluorescence and scattered light collected by the objective alone (top right insert in figure 1). This light stop was used to quantitatively determine the amount of light collected by the parabola alone by blocking the light collected by the objective.
The upward directed light from the parabolic reflector and objective back aperture was refocused by means of a large plano-convex lens (part number LA1002-A, diameter = 75mm, f = 150mm, Thorlabs, Newton, NJ) on a wide-area PMT (see below) supported by an aluminum collar electrically isolated from PMT surface. An insert between the PMT and the lens allowed for mounting of a 2” dia. optical filters to further discriminate fluorescence from scattered excitation light. Typically only one 710 nm short-pass filter (e.g. 700HSP from Omega Optical, Brattleboro, VT) was necessary to produce high-contrast images of scattering samples and tissue when excitation laser was tuned above 800 nm. The wide area PMT from Hamamatsu (H2431/R2083) was biased typically at −1.6 kV using a stable high voltage power supply (PS350/5000V-25W from Stanford Research Systems, Inc.). Its analog signal output was terminated with 500 kΩ resistor at an available input of an A/D channel of Fluoview 1000 extension board.
The parabolic reflector was coated inside with Al-MgF2 and its vertex was cut to the focal point (p/n P10-11, outer diameter = 78 mm, inner diameter at focus = 41 mm, f = 10.2 mm, Opti-forms, Temecula, CA). It was fitted inside the SM3 extension tube, so that it could be raised or lowered about 1” from the extreme positions to focus the collected light. The best focus position for the parabola was found experimentally by observing the brightest signal from the sample.
The sample stage which accommodated home-built animal support systems resided on a three dimensional translation flexure stage (NanoMax-TS MAX303 from Thorlabs, Newton, NJ) equipped with two manual differential micrometers for coarse XY positioning and one stepper motor (DRV001 from Thorlabs, Newton, NJ) for fine Z (vertical) optical slicing. This system having bidirectional repeatability of ~0.5 μm was computer-controlled through a BSC101 driver which was set up to execute a preset amount of travel (typically 1.5 μm) every time a TTL level trigger was received from the microscope scanner at the end of a frame acquisition. For fast frame rates an additional pause at the end of a frame was included to allow time for the Z-motor movement. The whole assembly was in turn mounted on a sturdy post with a coarse vertical translation mechanism to allow space for sample mounting and animal preparation.
Live Animal Imaging
In vivo imaging of microglia through a thinned skull
Microglia were imaged in heterozygous C57BL/6 Cx3cr1GFP/+ mice (Jackson Laboratories) through a surgically thinned skull as described (Davalos et al., 2005; Kim et al., 2009). Briefly, mice were anesthetized intraperitoneally with ketamine (100 mg/kg body weight), xylazine (13 mg/kg body weight), and acepromazine malate (2 mg/kg body weight) in PBS. For transcranial imaging, a circle, approximately 1-2 mm in diameter, was drilled into the parietal bone using a small rotary drill under a dissecting microscope. The area was further thinned manually with a microsurgical blade to a thickness of ~20 μm. Mouse temperature was maintained with a 37°C warming blanket. Whole ex vivo brain, from the same mouse type, was also imaged following death and extraction from the skull for comparison to the thinned skull preparation. In both types of experiments, GFP was excited with 860nm excitation light and the emission was collected as outlined above. Fast bi-directional scanning (1 μs pixel dwell times) was used to reduce motion artifacts. Z-stack data was acquired with a 1.5μm inter-slice interval.
Rat kidney blood vessels labeled with di-8-ANEPPS
Rats (male Wistars, 250-300 g, Charles River) were anesthetized with 5% isoflurane, shaved with small animal clippers, and placed in the supine position on a heated platform. Remaining fur was removed with a topical depilatory agent, followed by cleaning of the skin with 70% ethanol. Isoflurane anesthesia was maintained at 1.5% for the remainder of the experiment. With an incision in the neck, slightly left of the midline, and blunt dissection of underlying connective tissue, the jugular vein was visualized. A 20G catheter was secured inside the vessel, permitting administration of di-8-ANEPPS (Invitrogen), a fluorescent probe sensitive to cellular membrane potential. Working concentration of ANEPPS, assuming 70 mL/kg blood volume, was approximately 50 μM. Following injection, the cannula and jugular vein were ligated to prevent blood loss. With a U-shaped incision, the abdominal cavity was opened and internal viscera were displaced laterally to expose the right kidney. The kidney was unsheathed by removal of its superficial fascia and mobilized by dissection of the perirenal fat pad. The anesthetized, free-breathing animal then was transferred to the microscope stage and positioned with the kidney directly beneath the objective. The kidney was placed into a cup-like restraining device, custom-made to minimize breathing-induced motion during imaging without altering normal blood flow. The dye di-8-ANEPPS was excited with 860nm excitation light and the emission was collected as outlined above. Z-stack data was acquired with a 1.5mm inter-slice interval. In total, with preparation and imaging, total experimental duration was 60-90 minutes.
Mouse Skeletal Muscle with DsRed labeled Actin
A comparable setup was employed for imaging male mice expressing DsRed under the actin promoter (Jackson Labs, stock #005441). Briefly, mice were anesthetized with isoflurane and placed in the supine position on a heated pad. Fur on the right hind limb was cleared using a topical depilatory agent, followed by removal of skin and fascia from underlying musculature. After transfer to the microscope stage, the animal was positioned and its hind limb rotated to orient the vastus lateralis towards the objective. A small metal clamp around the metatarsals and compression of the lower right abdominal quadrant reduced motion artifacts during imaging. DsRed was excited with 860nm excitation light and the emission was collected as outlined above. Fast bi-directional scanning (1us pixel dwell times) as used to reduce motion artifacts. Z-stack data was acquired with a 1.5mm inter-slice interval.
Image Processing
All image analysis was performed by custom written software in the IDL (RSI Inc., Boulder, CO) or the MATLAB (Mathworks, Natick, Massachusetts) programming languages. In all cases gain was calculated from the integrated signal from each imaging plane for the imaging system as a whole (parabola and objective) and with parabola alone (objective emission collection blocked) data sets. Here, gain is defined as the ratio of the total light collected by the total system to the light collected by the objective alone (total system minus the parabola collected light). For all z-stack data the integrated signal from each plane was first threshold corrected (average taken from a region of background pixels plus 2 standard deviations above the mean background) and correlated in space to match common regions of interest between images acquired with the whole system and with the objective back aperture space blocked as described above. Skeletal muscle time-lapse data required more processing to remove motion artifacts and noise due to the relatively high PMT gains that were necessary to produce the images. For the skeletal muscle images shown in Figure 2, both time-series (whole system and objective blocked data sets) were averaged temporally to improve signal to noise. To remove periodic electronic noise coming from the PMT and its amplifier, both series were examined in three-dimensional frequency domain to identify noise signal, and these frequencies were suppressed. To correct for non-linear galvo-mirror motion coming from bi-directional scanning, images were deinterlaced using a polynomial fit that minimized mean square error between sequential lines. To correct for motion between frames, images that were out of plane, as measured by two dimensional cross correlation, were dropped, and remaining images were processed using a deformable registration algorithm described previously (Hermosillo et al., 2002; Schroeder et al., 2010). Registered image data was cropped to eliminate edge artifacts of registration process, and time series was fit using principal component analysis (ChefdHotel et al., 2002; Schroeder et al., 2010). All components that accounted for more than 1% of data variance were used in reconstruction of composite image. Calculated images corresponding to the intensity of light from the objective only were reconstructed using the difference in intensity between the image from (parabola and objective combined) and the intensity of the (parabola-only) image.
Figure2. Comparison of the parabola and objective contributions to the total signal detected in three different tissues imaged in vivo.

(Top Row), Maximum projection TPFM of GFP labeled microglia from a mouse brain (100 individual slices imaged at 1.5 μm intervals). The Combined (total epiTED system) and Parabola images were acquired separately as detailed in the Materials and Methods section. The objective image was calculated from the percent difference between the Combined and Parabola difference. The scale bar represents 30μm. (Middle Row), Single plane image of di-8- ANNEPS labeled blood vessels in an intact kidney imaged in vivo. The objective image was calculated as described above. The scale bar represents 60μm. (Bottom Row) Single plane in vivo image captured in the skeletal muscle of a mouse expressing DsRed under the actin promoter. The scale bar represents 30μm.The false color scalebar to the right of the image shows increasing intensity values from the bottom up (blue through white). Other details of image processing can be found in the Materials and Methods section.
Numerical Simulation of Light Propagation and Expected Gain
We used the same underlying Monte Carlo algorithms as in our previous paper (Combs et al., 2007) to model the behavior of emitted fluorescent photons and determine the gain of the instrument. The measurements were calculated based on the optical properties of brain tissue at the emission wavelength given in (Van Staveren et al., 1991). The main difference was that photons were only collected in the epi-plane, and the volume was extended so as to better represent a semi-infinite medium. This included implementing an absorbing boundary condition at the non-epi boundaries rather than a partial reflection based on the mediums Refractive Index.
Results
Brain, Kidney and Skeletal Muscle Imaging In Vivo
The results in figures 2, 3, and 4 show that the prototype epiTED device significantly increased the amount of emission light collected in all tissue types that were tested. Signal gains varied according to the tissue type and the imaging depth in the sample. Images from the mouse brain (maximum projection of a z-stack taken from an extracted brain) taken with the parabola and the whole system can be seen in the top panels of Figures 2 and 3. In this example the gain in light collection (defined as the amount of light collected by the parabola and objective divided by that in the objective alone) was 1.9 fold (52% of the total light was collected from the objective and 48% from the parabola). The gain as a function of imaging depth curve is shown in Figure 4A for both the extracted brain and for imaging through a thinned skull of a living mouse. In the extracted brain the gain ranged from a factor of 2.9 to 1.6 fold. In the thinned skull the gain was much less ranging from 1.8 to 1.2. It should be noted that the largest gains observed in the extracted brain occurred near the top of the sample. In the thinned skull images were first taken at a depth that was approximately 20μm (imaged through the approximately 20μm of skull bone). It is likely that the morphology of the thinned skull preparation (reduced solid angle for collection) prevented a significant portion of the light from reaching the parabola.
Figure 3. All pixel histograms of intensity values from the example images presented in Figure 2.

These all pixel histograms were generated from the representative images shown in figure 2 in the exact layout as appear in figure 2. A log scale was used for the Y-axis.
Figure 4. Gain versus imaging depth for various tissues imaged in vivo.
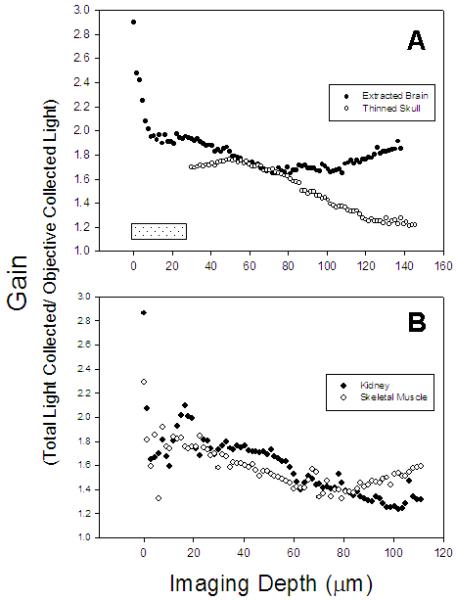
(A) Measured gain versus depth for GFP-tagged microglia imaged in extracted brain (closed circles) and through a thinned skull (open circles). The hatched rectangle shows the approximate thickness of the skull that was imaged through in the thinned skull preparation. (B) Measured gain versus depth for di-8-labelled blood vessels in rat kidney (closed circles) and actin-DsRed (open circles) in mouse skeletal muscle.
The middle and the last row of Figures 2 and 3 show images and the raw data from tissues that are more absorptive of visible light than brain. The representative kidney images are from a slice approximately 50μm into an intact kidney. In this slice the gain was 1.9 fold (53% of the total light was collected from the objective and 47% from the parabola). The representative skeletal muscle image was taken at a depth of approximately 30μm. In this slice the gain was 1.7 fold (60% of the total light was collected from the objective and 40% from the parabola). Figure 4 shows the gain versus imaging depth for both kidney and skeletal muscle imaged in vivo. The gain in the kidney ranged from 2.9 to 1.2 fold, whereas in the skeletal muscle the gain ranged from 2.3 to 1.3 fold.
Numerical Simulations
Numerical simulations suggest that an increase in light collection of over 6.5 and 4 fold is possible with 0.8 NA and 0.95 NA water objectives, respectively, and a parabola radius of at least 2 mm (Figure 5A and 5B) in brain tissue. The benefit of using the epiTED decreases with two-photon source depth but at a minimum the simulations predict a gain of at least 4 fold even for the 0.95 NA objective at the deepest depth simulated. Figure 5A shows that parabola aperture sizes below 2mm radius are not optimal for capturing all of the light missed by the objective.
Figure 5. Results of numerical simulations of epiTED light collection efficiency in brain tissue.
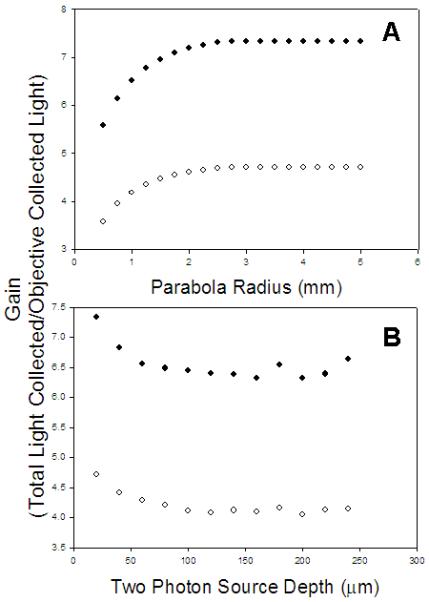
(A) Gain by parabola radius for a 0.95 NA water immersion objective lens (open circles) and a 0.8 NA water immersion lens (closed circles) at an imaging depth of 20 microns from the surface of the tissue (B) Gain by imaging depth at a fixed parabola radius of 5mm for a 0.95 NA water immersion objective lens (open circles) and a 0.8 NA water immersion lens (closed circles).
Discussion
Our findings demonstrate that epiTED can be used to increase the efficiency of fluorescence emission collection in multi-photon experiments performed in vivo. This was shown in three different tissue types in living animals using a relatively high NA (0.95) water immersion lens with a long working distance (2 mm), demonstrating that the TED imaging concept can be applied not only to thin slices and cellular imaging but also to intravital imaging. The main advantage of epiTED is that it can be attached directly to the objective and can enhance collection efficiency by a factor of two or more.
As expected, the highest gains were observed in brain imaging (Fig 4A). We assume that this is due to less light absorption than in the other tissues and because brain is a high light scattering tissue that allows many forward-directed photons to scatter back into a detectable zone for the optics in the epiTED system. The thinned skull preparation provided much less increase in gain than did the extracted brain. This is likely due to the bone absorption of emitted light outside of a narrow cone delimited by the boundaries of the thinned region in the skull. Although exposed brain imaging showed the best results, other more absorptive tissues like kidney and skeletal muscle showed a twofold improvement in light collection as well (Figure 4B).
The observed gains in light collection could lead to decreased laser excitation powers (by a factor of the square root of the gain for the TPFM excitation mode) and/or an increase in temporal resolution (lower pixel dwell times necessary for integration of the signal). The gains observed in this work ranged from 2.9 to 1.2 fold increase in light collection (average ~ 2 fold). This would enable a decrease in laser power by 42% or could allow for ~ 2 fold increase in imaging speed (in photon counting mode) depending on the characteristics of the fluorophores. Both of these factors are crucial for many in vivo imaging applications. Lower laser powers would decrease photo-toxicity and background signal. Faster imaging can decrease the chance of motion artifacts during imaging and/or allow for monitoring faster physiological events in vivo by limiting the need for signal averaging. When no averaging and minimum dwell times are used, for example in resonance scanners (Leybaert et al., 2005), the benefit is limited to the improvement in signal to noise alone.
The epiTED design has advantages over the hybrid objective and fiber optic external detection schemes in their published forms. First, the potential gain is inversely related to the NA of the objectives (Combs et al., 2007). Here we have shown that emission collection gains of two or more are possible even when using a relatively high NA (0.95) objective. This is similar to the gains found in the aforementioned studies (Engelbrecht et al., 2009; Vucinic et al., 2006) where 0.8 NA objectives were used. This may be of further importance when considering co-localization studies or multi-photon STED (Stimulated Emission Depletion Microscopy) where two color excitation is necessary and the use of highly corrected, wider acceptance angle lenses is important. Although similar gains are predicted for hybrid objective systems (Vucinic et al., 2006), the epiTED device is much simpler in design and could be retrofitted to commercially available objectives. Further, in theory the epiTED device should be superior to the ancillary fiber optic ring collection due to the light losses inherent in the solid angle interrupted by the fiber optic ring system (non-collecting regions due to fiber NA and cladding-limited packing). This can be seen from a comparison of the numerical simulations (Figures 5A and 5B). Engelbrecht et al. 2009 predict a maximum gain of < 4 fold for a 0.8 NA water immersion lens, which is less than the gain calculated here for a higher NA water immersion lens. Our simulations suggest a gain of greater than 7 fold is possible using a comparable NA objective (Figure 5A). As already mentioned, another benefit of the epiTED system is that it does not contact the tissue or sample being imaged. This gives added room for tissue access and manipulation and does not cause potential physiological perturbation due to the pressure of contact by the fiber optic ring.
The gains observed in the prototype epiTED system are considerably less than predicted by the numerical simulations (Figures 5A and 5B). This is most likely due to light loss at the face of the objective or in the conjugate cylindrical body of the system leading to the detector. It is likely that the greatest loss mechanism could be related to either absorption of photons in the lens outside the collectible NA (the front element of the objective) or reflection of some of the emitted light back into the tissue from the objective face. The current epiTED design has the objective face and chamfer body simply wrapped in reflective aluminum foil. Instead, a small oblique conical mirror on the face of the objective would be more efficient in redirecting such light sideways to the parabola, esp. for longer working distance objectives.
In summary, we demonstrate that the newly constructed epiTED device significantly increases TPFM fluorescence light collection, which has the potential to markedly improve in vivo imaging. The advantages of this device are that it does not touch the tissue, has a similar or higher light gain potential when compared to other systems, and is easy to implement using the standard optics of an inverted TPFM microscope. It is likely that future developments will include a compact version of this device for TPFM microscopes in both the upright and inverted configurations.
ACKNOWLEDGEMENTS
We thank Brad Bobbitt of LSM Technologies and Michael Naumann of the Laboratory of Cardiac Energetics, NHLBI, NIH for assisting in the construction and modifications of the epiTED device. We also wish to thank Alan Hoofring and Ethan Tyler for help in preparing figure 1 in this work, and Bernd Zinselmeyer for insightful discussions.
References
- Campagnola PJ, Loew LM. Second-harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues and organisms. Nat Biotechnol. 2003;21:1356–60. doi: 10.1038/nbt894. [DOI] [PubMed] [Google Scholar]
- ChefdHotel C, et al. Flows of diffeomorphisms for multimodal image registration. Proc. IEEE Int. Symp. Biomed. Imag. 2002:753–756. [Google Scholar]
- Combs CA, et al. Optimization of multiphoton excitation microscopy by total emission detection using a parabolic light reflector. J Microsc. 2007;228:330–7. doi: 10.1111/j.1365-2818.2007.01851.x. [DOI] [PubMed] [Google Scholar]
- Davalos D, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–8. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Denk W, et al. Two-photon molecular excitation in laser-scanning microscopy. In: Pawley JB, editor. Handbook of biological confocal microscopy. Plenum Press; New York: 1995. pp. 445–458. [Google Scholar]
- Denk W, et al. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–6. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Diaspro A, et al. Multi-photon excitation microscopy. Biomed Eng Online. 2006;5:36. doi: 10.1186/1475-925X-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelbrecht CJ, et al. Enhanced fluorescence signal in nonlinear microscopy through supplementary fiber-optic light collection. Opt Express. 2009;17:6421–35. doi: 10.1364/oe.17.006421. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2:932–40. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- Hermosillo G, et al. Variational Methods for Multimodal Image Matching. Int. J. Comp. Vis. 2002;50:329–343. [Google Scholar]
- Kim JV, et al. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature. 2009;457:191–5. doi: 10.1038/nature07591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leybaert L, et al. A simple and practical method to acquire geometrically correct images with resonant scanning-based line scanning in a custom-built video-rate laser scanning microscope. J Microsc. 2005;219:133–40. doi: 10.1111/j.1365-2818.2005.01502.x. [DOI] [PubMed] [Google Scholar]
- Masters BR, So PTC. Handbook of Biomedical Nonlinear Optical Microscopy. Oxford University Press; Oxford: 2008. [Google Scholar]
- Mertz J. Nonlinear microscopy: new techniques and applications. Curr Opin Neurobiol. 2004;14:610–6. doi: 10.1016/j.conb.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Piston DW. Photobleaching in two-photon excitation microscopy. Biophys J. 2000;78:2159–62. doi: 10.1016/S0006-3495(00)76762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JL, et al. Subcellular Motion Compensation for Minimally Invasive Microscopy, in vivo: Evidence for Oxygen Gradients in Resting Muscle. Circ. Res. 2010 doi: 10.1161/CIRCRESAHA.109.211946. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 2006;50:823–39. doi: 10.1016/j.neuron.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Van Staveren HJ, et al. Light scattering in intralipid 10% in the wavelength range of 400-1100nm. Appl. Opt. 1991;l30:4507–4514. doi: 10.1364/AO.30.004507. [DOI] [PubMed] [Google Scholar]
- Vucinic D, et al. Hybrid reflecting objectives for functional multiphoton microscopy in turbid media. Opt Lett. 2006;31:2447–9. doi: 10.1364/ol.31.002447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace VP, et al. Two-photon microscopy in highly scattering tissue. In: Periasamy A, editor. Methods in cellular imaging. Oxford University press; Oxford: 2001. pp. 180–199. [Google Scholar]
- Zinselmeyer BH, et al. Chapter 16. Two-photon microscopy and multidimensional analysis of cell dynamics. Methods Enzymol. 2009;461:349–78. doi: 10.1016/S0076-6879(09)05416-0. [DOI] [PubMed] [Google Scholar]
- Zipfel WR, et al. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol. 2003;21:1369–77. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]