

Abstract
A medium-throughput murine embryonic stem cell (mESC)-based high-content screening of 17,000 small molecules for cardiogenesis led to the identification of a b-annulated 1,4-dihydropyridine (1,4-DHP) that inhibited Transforming Growth Factor β (TGFβ)/Smad signaling by clearing the type II TGFβ receptor from the cell surface. Since this is an unprecedented mechanism of action, we explored the series' structure activity relationship (SAR) based on TGFβ inhibition, and evaluated SAR aspects for cell-surface clearance of TGFβ receptor II (TGFBR2) and for biological activity in mESCs. We determined a pharmacophore and generated 1,4-DHPs with IC50's for TGFβ inhibition in the nanomolar range (e.g., compound 28, 170 nM). Stereochemical consequences of a chiral center at the 4-position was evaluated, revealing 10- to 15-fold more potent TGFβ inhibition for the (+)- than the (−) enantiomer. This stereopreference was not observed for the low level inhibition against Activin A signaling, and was reversed for effects on calcium handling in HL-1 cells.
Keywords: 1,4-dihydropyridines; enantiomers; TGFβ inhibition; inducers of TGFβ type II receptor degradation; ITD-1; structure-activity-relationship; cardiomyogenesis; murine embryonic stem cells
INTRODUCTION
Developing small molecules as therapeutics for regeneration or repair of damaged tissue in the heart is a long-standing goal of regenerative medicine.1 However, only a small number of chemical mediators of cardiogenesis have been reported, and most are either not very potent or promiscuous in their action.2 We recently reported on a distinct class of condensed 1,4-dihydropyridines (1,4-DHPs) that derived from a medium-throughput phenotypic screen in mouse embryonic stem cells (mESCs).3 The annulated 1,4-DHP “hit” compound 1 was among the most effective candidates for driving mESC-derived mesoderm to cardiac fate.
The 1,4-DHP heterocycle is a frequently found structural feature of various bioactive molecules. Although 1,4-DHPs have received most attention as calcium channel modulators, with nifedipine (2) as the first marketed (1975) calcium antagonist (Figure 1), they are also known to possess vasodilator, antihypertensive, bronchodilator, anti-atherosclerotic, hepatoprotective, anti-tumor, anti-mutagenic, geroprotective, and anti-diabetic properties.7, 8 A few closely related structural analogs of 1 have been reported to have calcium channel modulating activity (e.g., 3, Figure 1).4–6 Along with 1, 1,4-DHPs such as 3 show moderate calcium channel blockade, i.e., typically >100-fold lower potency and 2-5-fold less efficacy (max. effects) than nifedipine-type calcium channel modulators. We previously reported that nifedipine (2) as well as a few 3-type 1,4-DHPs with calcium inhibitory potency did not promote cardiac differentiation from ESCs thus excluding calcium antagonism as the mechanism of action.3 The key mode of action of 1,4-DHP “hit” compound 1 in ESC differentiation is the inhibition of TGFβ signaling. TGFβ is a cytokine that controls many cellular functions that underlie normal and pathological processes, including cell proliferation, differentiation, angiogenesis, and wound healing.9 The TGFβ superfamily of secreted proteins includes three isoforms of TGFβ (i.e., TGFβ-1, TGFβ-2, TGFβ-3) as well as activins, bone morphogenetic proteins (BMPs), myostatin, nodal and other growth and differentiation factors.10 All superfamily members bind a complex of type I and type II transmembrane receptors on the cell surface. For TGFβ, signaling occurs following initial binding to the TGFβ receptor type II (TGFBR2) that recruits TGFβ receptor type I (TGFBR1, activin-like kinase 5, Alk-5) into an active signaling complex that is internalized into the cell concomitant with phosphorylation of the effectors, Smad2 and Smad3. These, in turn complex with Smad4 and in the nucleus recognize and activate transcription from the promoters of TGFβ-responsive genes.10
Figure 1. Chemical structures of 1,4-DHPs.

1 = cardiogenic “hit” from a mESC high-content screen, 2 = calcium channel blocker nifedipine, 3 = b-annulated 1,4-DHPs with weak calcium modulating activity reported in the literature.4–6
The TGFβ pathway has been considered an attractive therapeutic target for a variety of diseases and has been clinically validated for distinct cancer types and fibrosis.11–13 However, TGFβ can either promote or suppress tumor growth and metastasis depending on cell-type, cellular context (micro-environment) and developmental stage of the tumor, and is important for tissue integrity.12 Thus, the generation of an inhibitor with a selective molecular mechanism of action would aid in understanding the potential for therapeutic applications. The 1,4-DHPs reported herein show a distinct mechanism of action because they do not inhibit TGFBR1/2 kinases directly but rather direct TGFβ type II receptors to the proteasome, thereby promoting their selective degradation, hence the name 'inducers of TGFβ type II receptor degradation' (ITDs).3 Given the novel mechanism of action, we report the synthesis and structure-activity relationship (SAR) studies of 50 selected 1,4-DHPs based on the initial “hit” compound 1. The primary SAR studies were conducted based on dose-dependent inhibition of TGFβ signaling. SAR information was then evaluated in a mechanistic assay that quantified the degree of TGFBR2 down-regulation in HEK-293T cells, and in complex mESC-based phenotypic assays that included assays for cardiomyogenesis and mesoderm formation. Herein, we describe key pharmacophoric elements of this subclass of 1,4-DHPs for TGFβ inhibition and provide insight into stereochemical requirements for this activity.
CHEMICAL SYNTHESIS
The 1,4-DHPs were prepared in a library fashion following a procedure described by Ko et al. (2005).14 In a typical preparation, one equivalent of a dimedone derivative 4, one equivalent of the desired aldehyde 5, one equivalent of a β-ketoester 6, one equivalent of ammonium acetate and 0.3 equivalents of iodine were stirred overnight at room temperature in ethanol (Scheme 1A). After an aqueous/organic extraction, compounds were purified by a combination of flash chromatography and PTLC to afford 20–50% yield of the desired compounds 7–30. When the required aldehyde 5 was not commercially available, intermediates 31 were prepared and further modified via Suzuki coupling under microwave conditions to afford compounds 32–43 (Scheme 1B) in overall yields between 35–85%.
Scheme 1.

Synthesis of racemic 1,4-DHPs.
For synthesis of the free carboxylic acid 44, methyl ester 10 was treated with 1M BCl3 in CH2Cl2 to afford 75% of the desired product. N-Methylated 1,4-DHP 45 was prepared by treating 30 with methyl iodide in DMSO.15 A third class of compound synthesized included fully oxidized pyridine analogs of 1,4-DHPs. For example, 46 was synthesized in good yield by treating 30 with periodic acid and sodium nitrite in the presence of wet silica gel.16
Because 1,4-DHPs contain a center of chirality, it was important to prepare the (+)- and (−)-enantiomers and test them separately in functional biological assays. Optically pure (+)- and (−)-enantiomers of selected 1,4-DHPs were prepared based on a modified procedure previously described by Shan et al. (2002) that involved the separation of diastereomeric esters.17 Chiral β-ketoester 51 was prepared in two steps. First, l-threonine was esterified and subsequently treated with 3-nitrobenzoyl chloride to yield benzamide 48. Treatment of 48 with diketene 49 or 2,2,6-trimethyl-4H-1,3-dioxin-4-one (50) afforded the β-ketoester 51 (Scheme 2). Compound 51 was then treated under Hantzsch reaction conditions described in Scheme 1 to afford 52–54. The diastereomeric mixtures 52–54 were obtained in good yield (i.e., 50–70%), but a significant amount of material was lost during chromatographic purification. An isolated yield for a single diastereomer was approximately 10%. Subsequent removal of the chiral ester with DBU (yields: 39–88%) and re-esterification (yields: 76–95%) afforded the desired enantiomers of 1, 23 and 57 in moderate to good yields. For esterification, an alkylation procedure was superior to other methods such as treatment of the acid in alcohol with different catalysts. The 3-(5-indole)-substituted 1,4-DHP enantiomers (i.e., (+)- and (−)-43) were prepared by synthesizing and separating diastereomers of the 3-bromo intermediate 57, and conducting the required Suzuki coupling with 1H-indol-5-ylboronic acid (58) in the last step (Scheme 2).
Scheme 2.

Synthesis of 1,4-DHP enantiomers using a modification of a literature protocol (Shan et al., 2002).17
RESULTS AND DISCUSSION
From over 200 1,4-DHPs that were synthesized and screened against TGFβ in a transient Smad4-binding element (SBE4)-reporter system in HEK-293T cells, about 50 1,4-DHPs were selected for a detailed SAR investigation by examining inhibition of TGFβ-2 signaling. The 50 1,4-DHPs were selected based on their functional activities as TGFβ inhibitors and/or structural features to gain insight into a pharmacophore. The primary assay was TGFβ inhibition and, for key compounds, we evaluated whether the SAR was similar in biologically more complex cardiac differentiation and mesoderm inhibition assays.
To exclude the possibility that TGFβ inhibitory effects of 1,4-DHPs could be mediated through cell toxicity, representative 1,4-DHPs were tested in cell viability assays (i.e., resazurin and ATP quantification assays) and in an acute toxicity assay that indicated cell membrane integrity by quantifying “leakage” of glucose-6-phosphate dehydrogenase (G6PDH) (data not shown). From these experiments, we concluded that in HEK-293T and mESC cells, 1,4-DHPs had no apparent adverse effects on cell viability, as long as the final concentration did not exceed 10 μM. Cellular concentrations of 1,4-DHPs should ideally be between 2.5–7.5 μM for reliable comparison of maximum effects.
Inhibition of TGFβ and Activin A signaling
An SBE4 reporter assay in HEK-293T cells was used to evaluate 1,4-DHPs and controls for inhibition of TGFβ-2 and Activin A signaling in dose-response (i.e., 0.001–5.0 μM). Activin A and TGFβ bind homologous, yet different type I and type II receptors, and signal through common intracellular cascades. All previously reported TGFβ inhibitors bind and inhibit the structurally similar kinase domains and hence indiscriminately block signaling from TGFBR1 (Alk-5) and ActR1 (Alk-4/7).3, 10
In order to gather more detailed SAR information of the synthetic b-annulated 1,4-DHPs, “hit” scaffold 1 was theoretically divided into constituent parts as shown in Figure 2.
Figure 2.
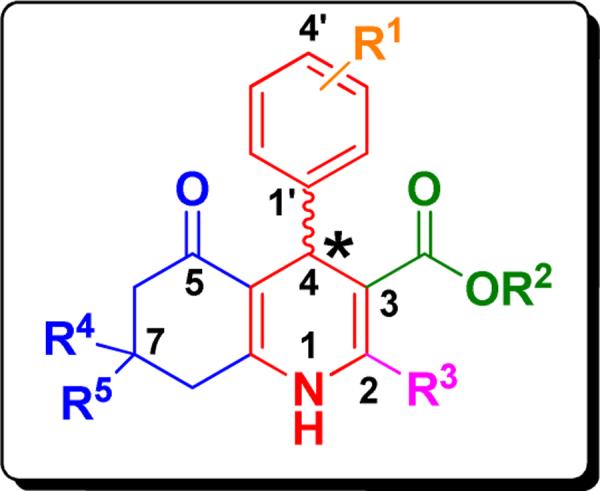
Systematic substructure depiction of b-annulated 1,4-DHPs for regions modified in the SAR studies of TGFβ inhibition.
From the initially evaluated 1,4-DHPs we determined that 4-phenyl substitution of 1,4-DHPs (highlighted in red, Figure 2) was important for TGFβ inhibitory activity, and we thus primarily modified the R1-substituents (orange) of the 4-phenyl moiety, the 3-(green) and 2-(magenta) residues, as well as the condensed ring system highlighted in blue. Finally, the effect of modification of the center of chirality (i.e., 4-position of the 1,4-DHP) was examined to determine the stereoselectivity and SAR features of the key pharmacophore.
Our initial SAR studies focused on the bicyclic core. We found that N-alkylation (45, inactive) largely abrogated TGFβ inhibition potency indicating an H-donor function at position 1 might be important (Figure 3). Aromatization (i.e., full oxidation) to a pyridine derivative (46, inactive) caused complete loss of functional activity underscoring the assumption that an H-donor at the 1-position and a tetrahedral configuration at the 4-position (instead of a planar ring system) were apparently required. Molecular dissection of the annulated ring system led to ring-opened analogs (59, Figure 3) that were devoid of TGFβ inhibitory potency. The evaluation of TGFβ inhibition was extended to include nifedipine (2, Figure 1) that completely lacked TGFβ inhibitory and cardiogenic activities,3 indicating that a) a certain degree of rigidity and b) hydrophobic interactions of a moiety at the annulation site were apparently required. Therefore, the 1,4,5,6,7,8-hexahydroquinoline (i.e., b-annulated 1,4-DHP) scaffold was retained in design and construction of the pharmacophore, and modifications of the R1-R5 residues (Figure 2) were systematically done.
Figure 3.

Illustration of variations in the 1,4-DHP core region.
Table 1a summarizes TGFβ inhibition data of 4'-R1-substituted 1,4-DHPs and clearly showed that bulky aliphatic substituents (9, 32 IC50's = 0.56 and 0.59 μM, respectively) were superior to aromatic ones. 5- or 6-membered heterocycles (11, 12 IC50's = 8.80 and 3.57 μM, respectively) or the bicyclic indole substituent (33 IC50= 4.05 μM) were less potent. The conclusion was that a large 4'-substituent was required for potent TGFβ inhibitory activity and this was supported by the lack of inhibition by 7 or 8 (IC50 >10 μM, < 10% inhibition at 2.5 μM). Presumably, considerable hydrophobic interactions were present in this part of the molecule for potent TGFβ inhibitory activity.
Table 1a.
Effect of the R1 region (4'-substituents) on TGFβ inhibition.a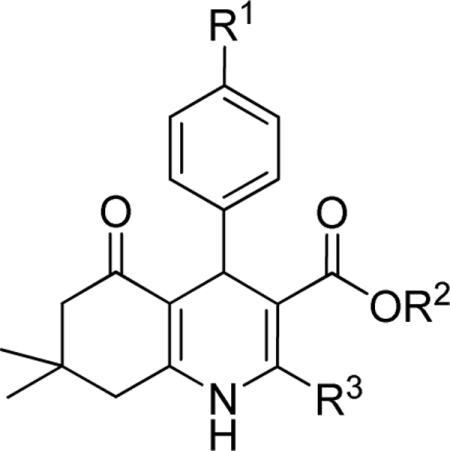
| compd | R1 | R2 | R3 | IC50 (μM) | %inhibition (2.5 μM) |
|---|---|---|---|---|---|
| 7 | F | Me | Me | >10 | 4 ± 2 |
| 8 | H | Me | Me | >10 | 6 ± 4 |
| 9 | tBu | Me | Me | 0.56 ± 0.01 | 79 ± 3 |
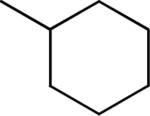
| 32 |
|
Me | Me | 0.59 ± 0.04 | 94 ± 1 |
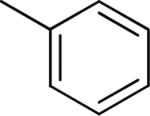
| 10 |
|
Me | Me | 1.49 ± 0.43 | 44 ± 5 |
| 1 |
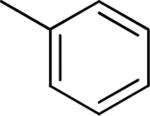
|
Et | Me | 0.85 ± 0.30 | 83 ± 8 |
| 11 b |
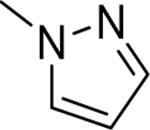
|
Me | Me | 8.80 | 18 |
| 12 |
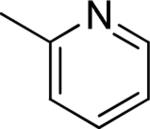
|
Et | Et | 3.57 ± 0.11 | 41 ± 2 |
| 33 |
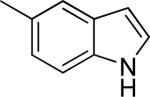
|
Me | Me | 4.05 ± 1.19 | 41 ± 6 |
Data represent means ± SD of n = 2–4 independent experiments.
For this compound a single experiment was done.
A few aromatic substituents of the biphenyl derivative 10 (IC50 = 1.49 μM, Table 1a) were synthesized and tested to examine the effects of steric and electronic interactions (Table 1b). A number of substituents at positions 2”, 3” or 4” maintained or increased the potency for TGFβ inhibition. For example, greater potency was observed with a small substituent at the para-position (i.e., 4”-CH3, 34, IC50 = 0.86 μM, or 4”-CF3, 35, IC50 = 0.73 μM). It is notable that, although the examples are limited, the possibility for torsion/rotation within the biphenyl group (i.e., around the 4'–1” bond) due to the 2'-substituent (e.g., 2”-CH3, 37, IC50 = 0.89 μM) did not apparently affect TGFβ inhibition potency compared to the unsubstituted biphenyl derivative 10.
Table 1b.
Effect of the R1 region (4-biphenyl substituents) on TGFβ inhibition.a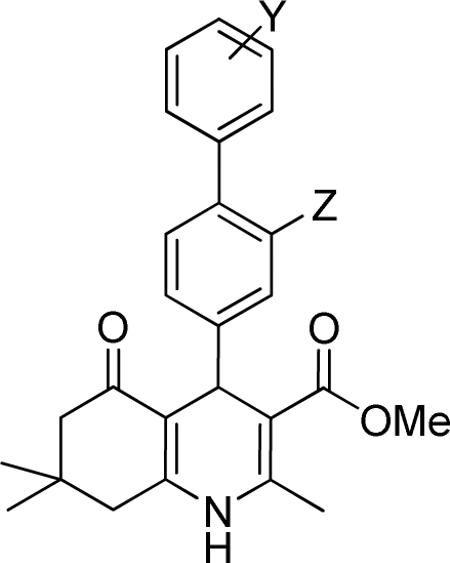
| compd | Y | Z | IC50 (μM) | %inhibition (2.5 μM) |
|---|---|---|---|---|
| 10 | H | H | 1.49 ± 0.43 | 44 ± 5 |
| 13 | H | F | 1.84 ± 0.35 | 59 ± 1 |
| 14 | 4”-CO2Me | H | 1.35 ± 0.16 | 78 ± 1 |
| 34 | 4”-Me | H | 0.86 ± 0.15 | 88 ± 2 |
| 35 | 4”-CF3 | H | 0.73 ± 0.41 | 91 ± 6 |
| 36 | 3”-Cl | H | 1.52 ± 0.18 | 73 ± 1 |
| 37 | 2”-Me | H | 0.89 ± 0.01 | 87 ± 2 |
| 38 | 2”-F | H | 2.06 ± 0.09 | 58 ± 1 |
Data represent means ± SD of n = 2–4 independent experiments.
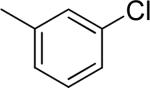
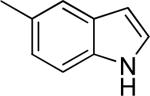
To examine the effect of aryl substituents in greater detail, a small set of 3'-aryl substituted 1,4-DHPs was investigated (Table 1c). Among the compounds examined only the 3'-(5-indolyl) derivative 43 (IC50 = 4.45 μM) showed modest TGFβ inhibition. All other 3'-aryl derivatives examined (39–42) were poorly potent inhibitors (IC50 > 10 μM).
Table 1c.
Effect of the R1 region (3'-phenyl substituents) on TGFβ inhibition.a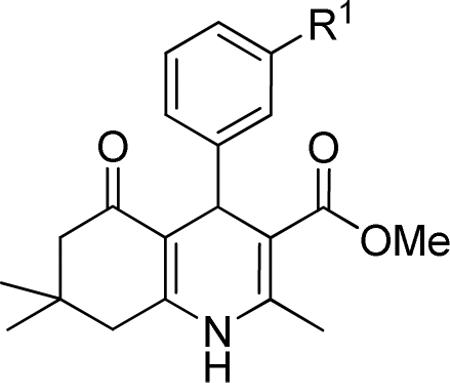
| compd | R1 | IC50 (μM) | %inhibition (2.5 μM) |
|---|---|---|---|
| 39 |
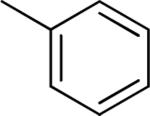
|
>10 | 18 ± 13 |
| 40 |
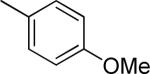
|
>10 | 5 ± 6 |
| 41 |
|
>10 | 27 ± 1 |
| 42 |
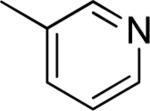
|
>10 | 10 ± 11 |
| 43 |
|
4.45 ± 2.57 | 45 ± 21 |
Data represent means ± SD of n = 2–4 independent experiments.
Another important region for SAR-based optimization of structure 1 was the 3-carboxylic ester moiety. Accordingly, a series of esters were synthesized and tested and revealed that TGFβ inhibition increased with lipophilicity in the order CH3<CH2CH3<CH2CF3<CH2CH(CH3)2 (i.e., 10, 1, 15 and 16 IC50's = 1.49, 0.85, 0.76 and 0.53 μM, respectively) suggesting that a hydrophobic interaction with the cellular target was critical in that region of the molecule. Large R2 groups such as a 2-(4-methoxy)benzyl ester could apparently still be accommodated by the molecular target and afforded potent inhibitors (i.e., 18, IC50 = 0.83 μM). The carboxamide analog 19 (IC50 = 4.06 μM) was also modestly potent but about 4-fold less potent than simple alkyl esters. Consistent with these findings we observed that the free carboxylic acid was not potent (i.e., 44, IC50 > 10 μM).
Next, the effect of the size of the 1,4-DHP-core side chain at position 2 was examined. As shown in Table 3, increasing the bulk of the R3-position did not apparently affect the potency of TGFβ inhibition. For example, the 3-propyl substituted 1,4-DHP (i.e., 20, IC50 = 0.83 μM) was equipotent as the 3-methyl-substituted analog (i.e.,1, IC50 = 0.85 μM).
Table 3.
Effect of the R3 region on TGFβ inhibition.a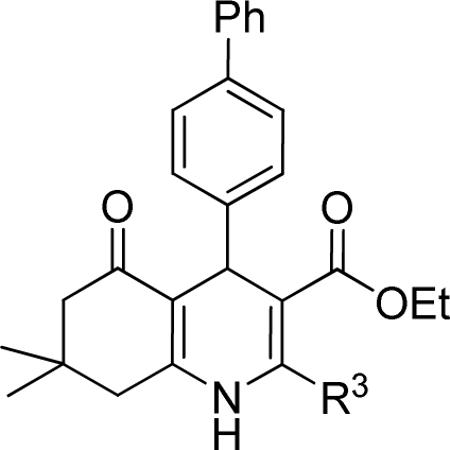
| compd | R3 | IC50 (μM) | %inhibition (2.5 μM) |
|---|---|---|---|
| 1 | Me | 0.85 ± 0.30 | 83 ± 8 |
| 20 | nPr | 0.83 ± 0.09 | 88 ± 1 |
Data represent means ± SD of n = 2–4 independent experiments.
As described above, the annulated region of the 1,4-DHP appeared to be important for TFGβ inhibition. Accordingly, the R4,5 region of 1 was modified. As shown in Table 4, mono- and di-substitution as well as size and branching of alkyl residues at the 7-position influenced the potency of TGFβ inhibition. Potency increased for mono-substituted (R5) compounds in the order of increasing size CH3<CH2CH3<CH2CH2CH3 (i.e., 21<22<23, IC50's = 2.73, 0.79 and 0.53 μM, respectively). A further increase in size of the R5 group by introducing phenyl-substituents resulted in analogs that were less potent (i.e., 26 and 27 IC50's = 1.92 and 3.97 μM, respectively).
Table 4.
Effect of the R4,5 region on TGFβ inhibition.a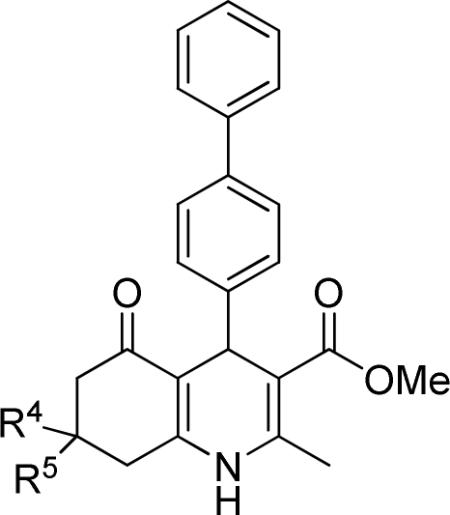
| compd | R4 | R5 | IC50 (μM) | %inhibition (2.5 μM) |
|---|---|---|---|---|
| 10 | Me | Me | 1.49 ± 0.43 | 44 ± 5 |
| 21 | H | Me | 2.73 ± 0.75 | 48 ± 6 |
| 22 | H | Et | 0.79 ± 0.15 | 92 ± 1 |
| 23 | H | nPr | 0.53 ± 0.20 | 96 ± 2 |
| 24 | H | iPr | 0.94 ± 0.44 | 83 ± 8 |
| 25 | H | iBu | 0.59 ± 0.21 | 94 ± 2 |
| 26 | H | Ph | 1.92 ± 0.20 | 61 ± 1 |
| 27 | H | 4-(N(Me)2)Ph | 3.97 ± 0.94 | 40 ± 6 |
Data represent means ± SD of n = 2–4 independent experiments.
Mono- and di-methyl substitution (i.e., 10, 21 IC50's = 1.49 and 2.73 μM) did not alter the potency of TGFβ inhibition. This was even more apparent when the maximum inhibitory effects (44% and 48% inhibition, for 10 and 21, respectively) were examined.
To elaborate the most potent TGFβ inhibitors, optimization of TGFβ inhibition potency in the 1,4-DHP series required combining optimal substituents from various first generation compounds. A “mix-and-match”-approach to optimizing compounds was taken to examine the concept of improving potency by additive substituent effects. Thus, a 1,4,5,6,7,8-hexahydroquinoline was designed that comprised a bulky aliphatic substituent in the 4'-(4-phenyl) position and the single 7-(n-propyl) residue in combination with an ethyl or iso-butylcarboxylic ester. Indeed, TGFβ inhibition potency was further increased with this substitution pattern, because both ethyl (i.e., 28) and butyl (i.e., 29) carboxylic ester derivatives had IC50 values of 170 nM. Optimized second generation structures 28 and 29 were almost as potent as the reported TGFBR1 kinase inhibitor SB-431542 that had an IC50 of 66 nM in the TGFβ assay (Figure 4) or GW788388 (IC50 = 93 nM in a cell-based TGFβ assay)18 or LY364947 (IC50 = 40 nM in a cell-based TGFβ assay)3, 19.
Figure 4. Effect of stereochemistry of 1,4-DHPs 1, 23 and 43 on TGFβ and Activin A inhibition.

A) Dose-response profiles of (+)- and (−) enantiomers for TGFβ and Activin A inhibition; B) Table of TGFβ and Activin A inhibition parameters, data represent means ± SD of n = 2–5 independent experiments. SB = SB-431542, a literature reported TGFβ inhibitor (Alk 4/5/7 inhibitor).21
An important determinant of the potency of 1,4-DHPs on TGFβ signaling inhibition was stereopreference of the molecules based on a center of chirality at the 4-position. Along these lines, it is well-recognized that chirality was crucial for the pharmacological properties of calcium channel modulating 1,4-DHPs. For example, the (−)-(S)-enantiomer of BAY K8644 is approximately 10-fold more potent as a calcium channel agonist compared to the (+)-(R)-antipode that acts as an antagonist.20
To understand the structural basis for the stereopreference of the 1,4-DHP enantiomers on TGFβ inhibition, several enantiomeric pairs of 1,4-DHPs were synthesized and tested. To examine a possible range of stereoselective function, three DHPs were selected that covered a broad range of TGFβ inhibition potency and cardiogenic activity. Accordingly, 1,4-DHPs 1, 23 and 43 were chosen as probe molecules and both enantiomers of each were prepared in >96%ee as described above. Compounds 1 (TGFβ IC50 = 0.85 μM) and 23 (TGFβ IC50 = 0.53 μM) were among the most potent compounds prepared, whereas 43 (TGFβ IC50 = 4.45 μM) represented a moderately potent 1,4-DHP derivative.
Figure 4 summarizes the effect of the compounds on TGFβ inhibition relative to Activin A inhibition. The functionally active (+)-enantiomers were 10- to 15-fold more potent than their (−)-antipodes, with IC50's of 0.46 μM for (+)-1 and 0.40 μM for (+)-23 compared to 6.90 μM and 4.06 μM for their (−)-enantiomers (Figure 4B). Stereopreference for and potency of Activin A inhibition, however, was much less than inhibition of TGFβ suggesting a different, presumably non-selective mode of action. Interestingly, in contrast to 1 and 23, neither enantiomer of 43 showed any selectivity for TGFβ or Activin A inhibition. These findings indicated that the substitution pattern of the 4-phenyl group was not only critical with regards to potency of TGFβ inhibition (Tables 1a–c) but also for pathway selectivity (i.e., selective inhibition of TGFβ over Activin A). In the case of 43, the 3'-(indol-5-yl) substituent decreased TGFβ inhibitory potency as well as selectivity over Activin A and diminished the selective biological activity of the (+)-and(−)-enantiomers. For 1 and 23, (+)-stereochemistry at the 4-position was apparently critical for greater potency in TGFβ inhibition. For compounds 21–27 (Table 4), a second center of chirality at the 7-position was present. However, based on data for 7-mono and 7,7-disubstituted 1,4-DHPs it appeared unlikely that this position would be as crucial for stereoselective binding as that of the 4-position. Thus, we did not investigate a role of the stereochemistry of the 7-position on TGFβ or Activin A inhibition.
In summary, the key pharmacophoric structural elements for TGFβ inhibition included a 1,4,5,6,7,8-hexahydroquinoline core, a 4-phenyl substituent with bulky aliphatic residues at the 4'-position and a lipophilic carboxylic ester. Evaluation of the center of chirality at the 4-position revealed that TGFβ inhibition was associated with the (+)-enantiomer. Moreover, with regards to pathway selectivity for TGFβ versus Activin A, the substitution pattern of the 4-phenyl group was found to be critical. Our previous findings for instance showed that a 4-(4”-trifluoromethyl)biphenyl substituted DHP (compound 35) exhibited significantly greater pathway selectivity than a 4-(4”-methyl)biphenyl substituted analog (compound 34).3 Here, we observed that 3'-substitution decreased TGFβ inhibitory potency as well as selectivity over Activin A and diminished the selective biological activity of the (+) and (−)-enantiomers.
Biological activity in secondary functional assays correlates with TGFβ inhibition but not with calcium antagonism
We next asked if the SAR profiles of the 1,4-DHPs for TGFβ inhibition correlated with their stem cell activity. To address this question, we focused primarily on the enantiomers of 1, 23 and 43, as well as selected 1,4-DHPs that covered a wide range of TGFβ inhibition potency. In the mESC-based phenotypic assays we made use of the biphasic role that we discovered for this class of DHPs, i.e., 1) inhibition of mesoderm formation and, consequently, inhibition of cardiogenesis when 1,4-DHPs were administered to cells early during differentiation (days 1–3) and 2) promotion of cardiogenesis when given during later phases of differentiation (days 3–5).3
Effects of 1,4-DHP enantiomers on cardiogenesis in mESCs
1,4-DHPs were administered to mESCs on days 3–5 of differentiation and alpha-myosin heavy chain-green fluorescent protein α-MHC-GFP) levels – as an index of cardiogenesis – were quantified by imaging on day 10. Figure 5A shows a representative dose-response curve for cardiogenesis for (+)- and (−)-enantiomers of 1. Based on the increase in fluorescence intensity at day 10, the amount of α-MHC-GFP was significantly increased for (+)-1 compared to (−)-1. Stimulation of cardiomyocyte differentiation by (+)-1 reached a maximum at 5 μM and then decreased with increasing concentration of (+)-1. This type of dose-response curve has also been observed for other cardiogenic compounds (i.e., inhibitors of the canonical Wnt pathway) in human ESCs.22,23 We speculate this may arise due to a number of contributions including very strong TGFβ inhibition that interferes with cell differentiation or proliferation. It is unlikely that the sigmoidal effect on cell growth was due to cell toxicity because overall cell viability was normal, as assessed by viability assays in mESCs and HEK-293T cells.3 Accordingly, determination of meaningful SAR information (i.e., IC50 or EC50 values) deduced from 10-day phenotypic read-outs were complex to interpret but normalized data at lower concentrations afforded reliable and reproducible data. It was apparent that the optimum concentration for 1,4-DHPs to promote cardiogenesis in mESCs was in the range from 2.5–7.5 μM for (+)-1.
Figure 5. Characterization of 1,4-DHP enantiomers in secondary functional assays.

A) Cardiogenesis in Myh6-GFP mESC cells: lower panel illustrates representative whole-well images of mESCs on day 10 of differentiation after treatment with (+)- or (−)-enantiomers of 1 compared to media and DMSO controls, upper panel shows the corresponding plot after image analysis (mean ± SD, normalized to DMSO = 1-fold); B) Cardiogenesis in Myh6-GFP mESC cells: (+)-or (−)-enantiomers of 23 and 43 compared to (rac)-1 at 2.5 μM (mean ± SD, normalized to DMSO = 1-fold); C) TGFBR2 degradation (down regulation) in HEK-293T cells: % HA-tagged TGFBR2 positive cells after treatment with (+)-or (−)-enantiomers of 1, 23 or 43 at 3 μM doses (mean ± SD, normalized to DMSO = 100%); D) Mesoderm inhibition with J1 Brachyury/T-GFP mESC cells: % T-GFP positive cells after treatment with (+)- or (−)-enantiomers of 1, 23 or 43 at 1 and 3 μM (mean ± SD, normalized to DMSO = 100%); E) 3D correlation plot: x axis, TGFβ/Smad inhibition values (log IC50's), y-axis: mesoderm inhibition with J1 Brachyury/T-GFP mESC cells at 3 μM 1,4-DHP doses (% Brachyury/T-GFP positive cells, normalized to DMSO = 100%), z-axis: TGFBR2 degradation values at 3 μM 1,4-DHP doses (% HA-TGFBR2 positive cells, normalized to DMSO = 100%); F) Calcium transient peak values in HL-1 cells: comparison of Ca2+ transient peak values (Fluo4 levels, average pixel intensity) in the presence of (+) and (−)-enantiomers of 1 and 23 (mean ± SEM).
Similar to the enantiomers of 1, selective stimulation of cardiomyogenesis was observed for the (+)-enantiomer of 23 compared to the (−)-enantiomer of 23 (Figure 5B). Moreover, data in Figure 5B suggested a difference in efficacy of cardiogenesis for the 1,4-DHPs 1, 23 and 43 that correlated with their potency for TGFβ inhibition.
Effects of 1,4-DHP enantiomers on TGFBR2 degradation
To examine if the SAR profile for TGFβ inhibition correlated with that for receptor clearance, we studied the clearance of TGFBR2 from the membrane using an extracellular hemagglutinin (HA)-tagged TGFBR2 in HEK-293T cells. Compared to DMSO-treated control cells, through live immunostaining of the extracellular HA-tag, flow cytometry analysis allowed quantification of cell surface TGFBR2 in 1,4-DHP-treated HEK-293T cells. In the presence of 2.5 μM of 1 or 23, the (+)-enantiomers selectively induced a greater clearance of TGFBR2 from the cell surface compared to their (−)-antipodes (Figure 5C). In good agreement with the data of Figure 4, the enantiomers of 43 showed no detectable stereopreference for TGFBR2 clearance (Figure 5C). In summary, the TGFBR2 clearance data (Table 5C) was in excellent agreement with TGFβ inhibition data (Figure 4) for 1, 23 and 43 with regard to potency and stereopreference of (+)-versus (−)-enantiomers.
Effects of 1,4-DHP enantiomers on mesoderm formation in mESCs
To further validate the ability of 1,4-DHPs to functionally inhibit TGFβ, the effect of 1, 23 or 43 and their enantiomers on mesoderm formation was examined using a J1 mESC line with a stably integrated Brachyury/T promoter-eGFP reporter construct (Brachyury/T is a marker for mesoderm).24 Treatment of mESCs on day 1–2 showed a dose-dependent inhibition of mesoderm formation based on a decrease in Brachyury/T-GFP-positive cells on day 4 (Figure 5D). Inhibition of mesoderm formation was selective regarding stereochemistry for 1 and 23 and followed the selectivity of TGFβ inhibition (i.e., (+)-enantiomer > (−)-enantiomer). In contrast to the lack of selective TGFβ inhibition in the SBE4/Smad reporter assay (Figure 4), compound 43 showed a slight stereopreference of the (+)-enantiomer for mesoderm inhibition (Figure 5D). We conclude that, at least for 43, mesoderm inhibition at day 1 occurs by a distinct and presumably more complex mechanism than that for TGFβ inhibition in both the reporter gene assay (Figure 4) and the induction of proteasomal TGFBR2 degradation (Figure 5C).
Correlation of TGFβ/Smad4 signaling, TGFBR2 degradation and mesoderm inhibition
A 3D-correlation plot depicting TGFβ inhibition (log IC50's, x-axis), inhibition of Brachyury/T-positive cells (%, y-axis) and TGFBR2 clearance from the cell surface by DHP compounds (%HA-TGFBR2 positive cells, z-axis) was generated for 13 selected 1,4-DHPs that covered a broad range of biological activity (i.e., from inactive to highly potent) as well as pairs of (+)- and (−)-enantiomers (i.e., enantiomers of 1, 23, 43) (Figure 5E). Here, the stereopreference of enantiomers of 1 and 23 was apparent as the less active (−)-enantiomers (grey dots) can be found in the upper right corner, whereas the highly potent (+)-enantiomers (red dots) are clustered in the lower left corner. In contrast, the 43 enantiomers are present in the center of the graph underscoring their lack of stereopreference and modest potency in all assays examined. The robust overall correlation from these biologically and technically distinct functional assays validated the SAR profile of 1,4-DHPs as TGFβ inhibitors, considering that the common thread of these studies was inhibition of TGFβ signaling.
Effects of 1,4-DHPs on calcium transients
We also evaluated the (+)- and (−)-enantiomers of 1 and 23 for modulation of calcium transients in HL-1 cells (immortalized murine atrial cardiomyocytes) because it has been reported that 1,4-DHP enantiomers can have opposing effects on calcium channels (i.e., agonistic or antagonistic activity20). Figure 5F shows the calcium transient peak values in the presence of 10 μM 1 or 23 enantiomers or nifedipine (NFP, 2). The results showed that the (+)-enantiomers (i.e., (+)-1, 44% inhibition, (+)-23, 58% inhibition) inhibited calcium transients less effectively than nifedipine (i.e., 66% inhibition), whereas the (−)-enantiomers were stronger inhibitors (i.e., (−)-1, 87% inhibition, (−)-23, 85% inhibition). The stereopreference observed for TGFβ inhibition (in 3 different assays, Figure 5E) and cardiomyogenesis (Figure 5A) is thus inversely proportional to the inhibition of cellular calcium transients. Inhibition of calcium transients was about 3–5-fold greater for the (−)-enantiomers compared to the (+)-enantiomers of 1 and 23. Because the (−)-enantiomers examined were not cardiomyogenic, this finding supports the suggestion that TGFβ inhibition and not calcium inhibition is largely responsible for the mechanism of action for 1,4-DHPs in mESC cardiogenesis. The opposite stereopreference results for calcium inhibition vice versa reinforce the view that calcium inhibition is not a mechanism of action for 1,4-DHP cardiomyogenesis.
CONCLUSIONS
1,4-DHP 1 was a potent “hit” in a medium throughput mESC-based screen of small molecules for the ability to drive cardiomyocyte formation from mESCs. An SAR model was constructed for a b-annulated subclass of 1,4-DHPs based primarily on the ability of 1,4-DHPs to inhibit TGFβ/Smad signaling through induction of TGFβ receptor type II degradation (hence the name ITDs). Key structural features of the 1,4-DHP pharmacophore included a 1,4,5,6,7,8-hexahydroquinoline core, a 4-phenyl substituent with bulky aliphatic residues at the 4'-position and a lipophilic carboxylic ester. The substitution pattern of the 4-phenyl group was found critical not only for potent TGFβ inhibition, but also for selectivity in favor of TGFβ signaling versus the closely-related Activin A pathway. In contrast to alkyl- and aryl-substitution at the 4'-position, 3'-substitution decreased TGFβ inhibitory potency as well as selectivity over Activin A and diminished the selective biological activity of the (+)- and (−)-enantiomers. Evaluation of the center of chirality at the 4-position for certain selected 1,4-DHPs revealed that potent cardiogenesis activity was most commonly associated with the (+)-enantiomer. Interestingly, this stereopreference of 1,4-DHP enantiomers was reversed for inhibition of calcium transients in HL-1 cells, with the (−)-enantiomers showing a greater inhibition compared to the (+)-antipodes. In addition, a correlation of data from secondary functional assays in mESCs (i.e., mesoderm inhibition assay) as well as for TGFβ receptor II degradation with IC50 values (TGFβ/Smad inhibition) confirmed and validated the SAR model.
There have been a number of small molecule TGFβ inhibitors reported in the literature to target TGFBR1 kinase (Alk-5) selectively. However, these all show similar potency against signaling by Activin A or other growth factors that act through the closely related Alk4 receptor (and Alk7 in most cases), thus, the 1,4-DHPs are the first selective TGFβ inhibitors to our knowledge. Also, the selective targeting of TGFBR2 for clearance from the cell surface is a novel mechanism of action. Although TGFBR2 trafficking to the proteasome occurs during receptor cycling, it does so with TGFBR1, and we could also not find any reports of small molecule or biological reagents that dissociate proteasomal trafficking of TGFBR2 and TGFBR1. Thus, in summary, the 1,4-DHPs described herein provide conceptually novel chemical probes that may be useful to explore the role of TGFBR2-mediated signaling in basic biology and for the molecular basis of disease. The 1,4-DHP molecules not only represent useful reagents for cardiac regenerative medicine, but also provide excellent probes for studying TGFβ signaling in various biological systems.
EXPERIMENTAL SECTION
General
All reagents and solvents were of the purest grade available from commercial sources and used as received. Synthetic materials were purified using a flash column chromatography system (CombiFlash Rf 200, Teledyne ISCO, Lincoln) and PTLC (Preparative Thin Layer Chromatography) with UV indicator. NMR spectra were recorded at 300 MHz (1H) on a Varian Mercury 300 (at HBRI, San Diego), Bruker ARX 300 (at Christian-Albrechts University Kiel) or at 500 MHz (1H) and 125 MHz (13C) on a Bruker AMX 500 II (NuMega Resonance Lab, San Diego). Chemical shifts were reported as ppm (δ) relative to the solvent (CDCl3 at 7.26 ppm, CD3OD at 3.31 ppm, DMSO-d6 at 2.50 ppm) or TMS as internal standard. Optical rotations were recorded on a Jasco P-1010 polarimeter (not thermostated). Low resolution mass spectra were obtained using Hitachi M-8000 mass spectrometer with an ESI source. Purity of final compounds was determined with a Hitachi 8000 LC-MS using reverse phase chromatography (C18 column, 50 × 4.6 mm, 5 μm, Thomson Instrument Co., Oceanside). Compounds were eluted using a gradient elution of 95/5 to 5/95 (A/B) over 5 min at a flow rate of 1.5 mL/min, where solvent A was water with 0.05% TFA and solvent B was acetonitrile with 0.05% TFA. For purity analysis, peak area percent for the TIC (Total Ion Count) at 254 nm and retention time (tR in minutes) were provided. Purity of synthetic final products was ≥ 95%.
Multiwell plates were from Greiner Bio-One (Monroe, USA); Recombinant human TGFβ-2 from Merck-Millipore (Billerica, USA) and Activin A from R&D Systems (Minneapolis, USA) were purchased; Gelatin, fibronectin, SB-431542, Claycomb medium, streptomycin, L-glutamine, norepinephrine, Hoechst 33342 were purchased from Sigma-Aldrich (St- Louis, USA); FBS, HEPES buffer and probenecid were from Life Technologies.
Chemistry
General procedure for the preparation of racemic 1,4-DHPs (1, 7–31)
Compounds were prepared in a library fashion following a procedure described by Ko et al. (2005).14 In a typical reaction, 28 mg (0.2 mmol) of dimedone 4, 0.2 mmol of the desired aldehyde 5, 0.2 mmol of β-ketoester 6, 15 mg (0.2 mmol) of ammonium acetate and 15 mg (0.06 mmol) of iodine were stirred overnight at room temperature in 0.5 mL of ethanol. Solvent was evaporated and the residue was dissolved in 1 mL of ethyl acetate. The organic layer was washed with 0.5 mL of a saturated solution of NaS2O3, dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified by PTLC with hexanes/EtOAc to afford 20–50% yield of the final compound.
Methyl 4-(Biphenyl-4-yl)-2-methyl-5-oxo-7-(n-propyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (23)
The title compound was obtained according to the general procedure, but replacing dimedone by 5-(n-propyl)cyclohexane-1,3-dione and using 4-biphenylcarboxaldehyde as the aldehyde. Because of the presence of two centers of chirality (4- and 7-positions), a mixture of diastereomers was obtained. 1H NMR (500 MHz, CDCl3): δ 0.85–0.89 (m, 3H), 1.18–1.34 (m, 4H), 1.98–2.48 (m, 5H), 2.40 + 2.41 (2 × s, 3H), 3.63 (s, 3H), 5.11 + 5.15 (2 × s, 1H), 6.04 + 6.11 (2 × s, 1H), 7.20 (t, J = 8.4 Hz, 1H), 7.33–7.44 (m, 6H), 7.52 (br d, J = 7.9 Hz, 2H); LRMS calcd for C27H29NO3 415.22 [M]+, found 416.35 [M + H]+. HPLC purity 95.8%, tR = 7.03 min.
Ethyl 4-(4-tert-Butylphenyl)-2-methyl-5-oxo-7-(n-propyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (28)
The title compound was obtained according to the general procedure, but replacing dimedone by 5-(n-propyl)cyclohexane-1,3-dione and methyl acetoacetate by ethyl acetoacetate, and using 4-tert-butylbenzaldehyde as the aldehyde. Because of the presence of two centers of chirality (4- and 7-positions) a mixture of diastereomers was obtained. 1H NMR (500 MHz, CDCl3): δ 0.85–0.91 (m, 3H), 1.19 (t, J = 7.3 Hz, 2H), 1.28 (s, 9H), 1.28–1.36 (m, 4H), 1.98–2.09 (m, 2H), 2.17–2.23 (m, 2H), 2.27–2.44 (m, 1H), 2.36 (s, 3H), 4.07 (q, J = 7.3 Hz, 2H), 5.02 + 5.06 (2 × s, 1H), 5.80 + 5.85 (2 × s, 1H), 7.18 (br s, NH), 7.19 (br s, 4H); LRMS calcd for C26H35NO3 409.26 [M]+, found 410.48 [M + H]+. HPLC purity 95.1%, tR = 9.11 min.
Isobutyl 4-(4-tert-butylphenyl)-2-methyl-5-oxo-7-(n-propyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (29)
The title compound was obtained according to the general procedure, but replacing dimedone by 5-(n-propyl)cyclohexane-1,3-dione and methyl acetoacetate by isobutyl acetoacetate, and using 4-tert-butylbenzaldehyde as the aldehyde. Because of the presence of two centers of chirality (4- and 7-positions), a mixture of diastereomers was obtained. 1H NMR (500 MHz, CDCl3): δ 0.76–0.80 (m, 6H), 0.85–0.91 (m, 3H), 1.25 (s, 9H), 1.28–1.36 (m, 4H), 1.81–1.87 (m, 1H), 1.96–2.07 (m, 2H), 2.16–2.21 (m, 1H), 2.28–2.44 (m, 1H), 2.37 (s, 3H), 3.71–3.76 (m, 1H), 3.81–3.85 (m, 1H), 5.02 + 5.06 (2 × s, 1H), 5.70 + 5.75 (2 × s, 1H), 7.19 (br s, NH), 7.20 (br s, 4H); LRMS calcd for C28H39NO3 437.3 [M]+, found 438.75 [M + H]+. HPLC purity 95.1%, tR = 9.11 min.
General procedure for Suzuki coupling of 1,4-DHPs (32–43)
In a typical reaction, to a 2–5 mL microwave vial, 50 mg of bromo intermediate 31, 25 mg of desired boronic acid, 17 mg of palladium tetrakis, and 0.15 mL of a 2 M Na2CO3 in water in dioxane/water (2/1.5 mL) was heated in a microwave for 10 minutes at 150 °C. Solvents were evaporated, the crude mixture dissolved in ethyl acetate and washed with water, dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by PTLC with hexanes/EtOAc to afford 35–85% yield of the final compound.
Methyl 4-(4-Cyclohexylphenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (32)
The title compound was obtained according to the general procedure, using cyclohexylboronic acid as the boronic acid. 1H NMR (500 MHz CDCl3): δ 0.95 (s, 3H), 1.07 (s, 3H), 1.18–1.21 (m, 2H), 1.34 (t, J = 10.3 Hz, 4H), 1.71–1.85 (m, 4H), 2.16–2.39 (m, 4H), 2.35 (s, 3H), 3.62 (s, 3H), 3.75 (t, J = 6.4 Hz, 1H), 5.03 (s, 1H), 5.95 (s, 1H), 7.01 (d, J = 6.9 Hz, 2H), 7.17 (d, J = 6.9 Hz, 2H); LRMS calcd for C26H33NO3 407.25 [M]+, found 408.07 [M + H]+. HPLC purity 100 %, tR = 8.06 min.
Methyl 4-(3-(1H-Indol-5-yl)phenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (43)
The title compound was obtained according to the general procedure using 1H-indol-5-ylboronic acid as the boronic acid. 1H NMR (500 MHz, CDCl3): δ 0.97, 1.08 (2 × s, 3H), 2.15–2.32 (m, 4H), 2.38 (s, 3H), 3.63 (s, 3H), 5.16 (s, 1H), 6.05 (br s, 1H), 6.58 (br t, 1H), 7.20 – 7.29 (m, 6H), 7.56 (mc, 1H), 7.78 (mc, 1H), 8.30 (br s, 1H); LRMS calcd for C28H28N2O3 440.21 [M]+, found 463.27 [M + Na]+. HPLC purity 97.2 %, tR = 6.40 min.
1,4-DHP ester hydrolysis. 4-(Biphenyl-4-yl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylic acid (44)
10 mL of BCl3 (1M DCM solution) was added to a cooled solution of 1 g (2.49 mmol) of 10 in 20 mL dry DCM. The mixture was stirred at room temperature overnight and diluted in ice water/EtOAc. The organic layer was concentrated and the product purified by column chromatography with DCM/MeOH (95/5). Yield: 700 mg (75%) of a white solid. 1H NMR (500 MHz, DMSO-d6): δ 0.86 (s, 3H), 1.02 (s, 3H), 2.02–2.40 (m, 4H), 3.33 (s, 3H), 4.88 (s, 1H), 7.20–7.59 (m, 9H), 8.89 (s, NH), 11.70 (br s, 1H, COOH); LRMS calcd for C25H25NO3 387.18 [M]+, found 343.42 [M-COOH]+. HPLC purity 99.1 %, tR = 4.61 min.
1,4-DHP N-Methylation. Methyl 4-(Biphenyl-4-yl)-2-ethyl-1,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (45)
This synthesis was done according to a modified protocol of Cozzi et al. (1993).15 50 mg (0.12 mmol) of 30 was dissolved in 0.2 mL DMSO. 8 μL (1.1 equiv) of methyl iodide and 7 mg (1 equiv) of KOH were added and the mixture was stirred at room temperature for 72 hours in a sealed reaction vessel. The reaction mixture was diluted with 3 mL of EtOAc, washed once with water (0.5 mL), dried with Na2SO4 and purified by PTLC (Hexane/EtOAc 1:1, Rf = 0.7) to give a pale yellow solid. 1H NMR (300 MHz, CDCl3): δ 1.03 (s, 3H), 1.08 (s, 3H), 1.25 (t, J = 9.0 Hz, 3H), 2.28 (d, J = 1.5 Hz, 2H), 2.54 (d, J = 1.5 Hz, 2H), 3.26 (s, 3H), 3.67 (s, 3H), 5.28 (s, 1H), 7.24–7.44 (m, 7H), 7.54 (d, J = 9.0 Hz, 2H); LRMS calcd for C28H31NO3 429.23 + [M]+, found 430.75 [M + H]+. HPLC purity 99.7 %, tR = 8.40 min.
1,4-DHP Oxidation. Methyl 4-(Biphenyl-4-yl)-2-ethyl-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydroquinoline-3-carboxylate (46)
The title compound was prepared using a literature protocol for the DHP oxidation.16 A mixture of 41 mg (0.1 mmol) of 30, 45 mg (2 equiv) HIO4, 14 mg (2 mmol) of Na2NO2 and 50 mg of wet silica gel was stirred in DCM at room temperature overnight. After filtration of the silica gel, the residue was purified by PTLC (hexanes/EtOAc, 7/3). 1H NMR (CDCl3): δ 1.14 (s, 6H), 1.34 (t, J = 9.0 Hz, 3H), 2.50 (s, 2H), 2.84 (q, J = 9.0 Hz, 2H), 3.13 (s, 2H), 3.49 (s, 3H), 7.20 (d, J = 8.4 Hz, 1H), 7.50–7.35 (m, 4H), 7.66–7.60 (m, 4H); LRMS calcd for C27H27NO3 413.20 [M]+, found 414.35 [M + H]+. HPLC purity 99.9 %, tR = 8.70 min.
Synthesis of 1,4-DHP single enantiomers of 1, 23 and 43
A synthetic procedure by Shan et al. (2002) was used in a modified form.17 Below, we provide experimental details for the preparation of (+)-1 and (−)-1, whereas data for 23 and 43 enantiomers can be found in the Supporting Information.
(2S,3R)-Methyl 2-(3-nitrobenzamido)-3-(3-oxobutanoyloxy)butanoate (51)
Method a) 10 g (35 mmol) of the L-threonine-derived starting material 4817 was dissolved in 20 mL of dry THF and 3.8 mL of diketene (49, 50 mmol) was added while stirring. The mixture was stirred at room temperature and 214 mg of 4-DMAP (1.75 mmol, 5 mol%) was added carefully (exothermic reaction). Stirring was continued overnight after which the organic solvent was evaporated to dryness, taken up with DCM and washed with 1% aqueous HCl and brine. The organic phase was dried (Na2SO4), concentrated in a vacuum and purified by flash chromatography (SiO2, hexane/EtOAC) to give 10 g of a colorless oil (78%, crystallized upon standing for few days). Method b) 3 g (10.7 mmol) of the L-threonine starting material 48 was dissolved in 7 mL of xylenes and 1.52 g of 2,2,6-trimethyl-4H-1,3-dioxin-4-one (50, 10.7 mmol) was added. The mixture was heated to 150 °C for 30 min while stirring vigorously. After evaporation, the crude mixture was purified by flash chromatography (SiO2, hexane/EtOAc) to yield 2.2 g of a colorless oil (86%). TLC: Rf = 0.36 (hexane/EtOAc, 1:1). 1H NMR (500 MHz, CDCl3): δ 1.39 (d, J = 6.4 Hz, 3H), 2.29 (s, 2H), 3.53 (d, J = 8.4 Hz, 2H), 3.80 (s, 3H), 4.00 (dd, J = 8.9 and 2.5 Hz, 1H), 5.57 (qd, J = 6.9 and 2.5 Hz, 1H), 7.48 (d, J = 8.9 Hz, NH), 7.68 (t, J = 7.7 Hz, 1H), 8.01 (br s, OH), 8.30 (br d, J = 7.9 Hz, 1H), 8.39 (br d, J = 8.4 Hz, 1H), 8.80 (t, J = 2.0 Hz, 1H); LRMS calcd for C16H18N2O8 366.11 [M]+, found 265.95 [M – C4H5O3(acetoacetate) + H]+. HPLC purity 95.3 %, tR = 6.47 min.
General protocol for synthesis and resolution of diastereomers 52–54
A mixture of 2.56 g (7 mmol) of (2S,3R)-Methyl 2-(3-nitrobenzamido)-3-(3-oxobutanoyloxy)butanoate (51), 1.28 g of 4-biphenylcarboxaldehyde (5) (7 mmol), 981 mg of dimedone (4) (7 mmol), 540 mg of dry NH4OAc (7 mmol) and 533 mg of iodine (2.1 mmol) were stirred overnight in 4 mL of EtOH at room temperature. 150 mL of EtOAc was added to the brown slurry and washed with aqueous saturated Na2S2O3 (2×) and brine (1×). The organic layer was dried (Na2SO4) and concentrated in a vacuum to afford 5.8 g of a yellow solid foam. The crude mixture was purified by flash chromatography (SiO2) using a short column with a hexane/EtOAc gradient (20–60%) followed by a second gravity flow chromatography on a larger column (250 g SiO2) with toluene/EtOAc (2:1) as the eluent to separate the diastereomers.
(2R,3S)-4-Methoxy-3-(3-nitrobenzamido)-4-oxobutan-2-yl 4-(Biphenyl-4-yl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate Diastereomers (52a,b)
Diastereomer 52a
Yield: 550 mg (12%). TLC: Rf = 0.32 (tol/EtOAc, 1:1); α_D^23= +67.8 (c = 0.4, CHCl3); %de = ≥99% (1H NMR according to 4-CH, 2-CH3 and COOCH3); 1H NMR (500 MHz, CDCl3): δ 0.89 (s, 3H), 1.07 (s, 3H), 1.38 (d, J = 6.4 Hz, 3H), 2.18–2.34 (m, 4H), 2.49 (s, 3H), 3.59 (s, 3H), 4.74 (dd, J = 4.1 and 8.6 Hz, 1H), 5.10 (s, 1H), 5.54–5.59 (dq, 4.1 and 6.5 Hz, 1H), 5.94 (br s, NH), 6.51 (br d, J = 8.6 Hz, 1H), 7.14–7.36 (m, 10H), 7.79 (dt, J = 1.1 and 7.7 Hz, 1H), 8.14 (ddd, J = 1.0, 2.3 and 8.4 Hz, 1H), 8.40 (br t, J = 1.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 18.1, 20.1, 27.4, 29.6, 33.0, 35.9, 41.3, 51.0, 52.9, 57.5, 69.2, 104.2, 112.9, 122.8, 125.5, 126.3, 126.9, 127.1, 127.3, 128.0, 128.8, 129.8, 133.0, 135.4, 128.9, 140.7, 145.7, 146.4, 147.7, 148.3, 165.7, 166.8, 170.1, 195.9; LRMS calcd for C37H37N3O8 651.26 [M]+, found 652.08 [M + H]+ and 371.28 [M – C12H13N2O6 (4-methoxy-3-(3-nitrobenzamido)-4-oxobutan-2-oyl) + H]+. HPLC purity 99.8 %, tR = 8.23 min.
Diastereomer 52b
Yield: 650 mg (14%). TLC: Rf = 0.28 (tol/EtOAc, 1:1); α_D^23= +24.4 (c = 0.4 in CHCl3); %de = ≥99% (1H NMR according to 4-CH, 2-CH3 and COOCH3); 1H NMR (500 MHz, CDCl3): δ 0.90 (s, 3H), 1.08 (s, 3H), 1.15 (d, J = 6.4 Hz, 3H), 2.16–2.34 (m, 4H), 2.45 (s, 3H), 3.81 (s, 3H), 4.91 (dd, J = 8.7 and 3.5 Hz, 1H), 5.00 (s, 1H), 5.39–5.44 (dq, J = 3.5 and 6.4 Hz, 1H), 5.90 (s, NH), 6.99 (d, J = 8.7 Hz, 1H), 7.14–7.42 (m, 9H), 7.50 (t, J = 8.0 Hz, 1H), 8.05 (dt, J = 1.1 and 7.7 Hz, 1H), 8.25 (ddd, J = 1.0, 2.3 and 8.2 Hz, 1H), 8.65 (br t, J = 1.9 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 17.3, 19.9, 27.4, 29.5, 33.0, 36.5, 41.5, 50.9, 53.2, 57.2, 70.5, 104.7, 112.8, 123.0, 125.6, 126.5, 127.0, 127.2, 128.3, 128.9, 130.2, 132.9, 135.6, 139.0, 141.0, 145.7, 146.1, 147.5, 148.5, 165.7, 166.6, 170.4, 195.7; LRMS calcd for C37H37N3O8 651.26 [M]+, found 652.19 [M + H]+ and 652.35 [M + H]+ and 371.28 [M – C12H13N2O6 (4-methoxy-3-(3-nitrobenzamido)-4-oxobutan-2-oyl) + H]+. HPLC purity 97.5%, tR = 8.04 min.
General procedure for the synthesis of carboxylic acids 44, 55, 56
0.406 mmol of each diastereomer was dissolved and stirred in 11 mL of MeOH. 186 μL of DBU (1.24 mmol) was added dropwise and then the solution turned yellow. Stirring was continued for another 3 h at room temperature until all starting material was consumed as determined by TLC. After evaporation of the solvent, the residue was taken up in 40 mL of water, acidified to pH 2–3 with 1 N HCl (fine precipitate formed), and the title compound was extracted with EtOAc (3×). The organic phase was dried (Na2SO4) and concentrated to dryness.
4-(Biphenyl-4-yl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylic acid Enantiomers (44a,b)
The title enantiomers were obtained according to the general procedure, starting from 52 with 87–88% yields. The crude mixture was purified by flash chromatography (SiO2, DCM/MeOH: 0–5%) to afford 137 mg for 44a (87%) and 139 mg for 44b (88%) as off-white solids. TLC: Rf = 0.14 (DCM/MeOH, 95:5). LC/MS and 1H NMR were identical for both enantiomers and the same as the racemic compound (see above for 44).
General procedure for the synthesis of carboxylic ester enantiomers 1, 23, 57
137 mg (0.353 mmol) of the respective carboxylic acid enantiomer (44), 56.8 mg of K2CO3 and 45 mg of ethyl bromide (0.41 mmol) was stirred in 10 mL of dry DMF overnight at room temperature. For work-up, 50 mL of water was added and the mixture extracted with EtOAc (3×). The organic layer was washed with water (1× 20 mL) and brine (1× 20 mL), dried (Na2SO4) and concentrated to dryness. The crude mixture (>95% pure by TLC) was further purified by flash chromatography (SiO2, hexanes/EtOAc: 0–60%).
Ethyl 4-(Biphenyl-4-yl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate Enantiomers (1a,b)
Enantiomer 1a
Yield: 140 mg (95%) of a pale-yellow solid. TLC: Rf = 0.33 (hexanes/EtOAc, 1:1); α_D^23= −70.3 (c = 0.4 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 0.96 (s, 3H), 1.09 (s, 3H), 1.23 (t, J = 7.2 Hz, 3H), 2.15–2.39 (m, 4H), 2.38 (s, 3H), 4.08 (q, J = 7.2 Hz, 2H), 5.10 (s, 1H), 6.49 (br s, NH), 7.25–7.54 (m, 9H); 13C NMR (125 MHz, CDCl3): δ 14.3 (OCH2CH3), 19.5 (2-CH3), 27.3 (7-CH3), 29.4 (7-CH3), 32.3 (7-C), 36.5 (4-CH), 41.3 (8-CH2), 50.7 (6-CH2), 59.9 (OCH2), 106.2 (3-C), 112.3 (4a-C), 126.8 (ArCH), 127.0 (ArCH), 128.4 (ArCH), 128.6 (ArCH), 138.8 (ArC), 141.3 (ArC), 143.2 (2-C), 146.1 (1'-C), 147.7 (8a-C), 167.4 (COOMe), 195.4 (CO); LRMS calcd for C27H29NO3 415.22 [M]+, found 416.68 [M + H]+. HPLC purity 99.0%, tR = 8.19 min.
Enantiomer 1b
Yield: 137 mg (93%) as a pale-yellow solid. TLC: Rf = 0.33 (hexanes/EtOAc, 1:1); α_D^23= +68.05 (c = 0.4 in CHCl3); NMR data was identical to 1a; LRMS calcd for C27H30NO3 415.22 [M]+, found 416.62 [M + H]+. HPLC purity 100%, tR = 8.28 min.
In both cases, the optical purity of (+)-1a and (−)-1b was determined to be ≥96%ee by 1H NMR spectroscopy. In this experiment 1.5 mg (+)Eu(hfc)3 dissolved in 50 μL (CDCl3) was added to 1.5 mg of the respective enantiomer in 550 μL (CDCl3). NMR resonances of the 2-CH3 and 7-CH3 groups as well as for the 4-CH group shifted differently upon addition of the chiral reagent for each enantiomer. The shifted resonances of the 7-methyl groups, however, were best suited to estimate the %ee (see also Supporting Information).
Biology
SMAD-4 binding element (SBE-4)-based transient luciferase reporter gene assay (TGFβ/Activin A assay)
An SBE-4-firefly luciferase plasmid25 was co-transfected with a TK-driven Renilla luciferase plasmid (for data normalization) into HEK-293T (culture media: DMEM, 2% FBS). Cells were transfected in bulk with SBE-4 and Rluc plasmids. After incubation for 12–14 h cells were counted, plated on 96-well plates at 25,000 cells/well (media: DMEM, 1% FBS), and incubated for 2 hours before addition of test compound or DMSO or TGFβ-2 or Activin A (10 ng/mL) addition. Cells were treated with different amounts of 1,4-DHP test compounds (0.001–5 μM) and the data was compared to cells treated with DMSO as a control, non-treated cells or SB-431542-treated cells. Each condition was done in triplicate. Plates were incubated for 20–22 hours, media was aspirated and Promega cell lysis buffer was added. Firefly and Renilla luciferase activities were determined using the Dual Luciferase Assay Kit (Promega, Madison, USA) according to the manufacturer instructions. The assay conditions were optimized to give a dynamic range of typically >100-fold pathway induction and Z' values between 0.75 and 0.95. Data was derived from a minimum of two independent experiments unless otherwise stated. GraphPad Prism 5 software was used for data evaluation: log(inhibitor)/normalized response for TGFβ assays, log(inhibitor)/response for Activin A assays.
TGFBR2 degradation assay
The assay was done as described previously.3 Briefly, in a 6-well format a plasmid expressing extracellularly hemagglutinin (HA)-tagged TGFBR226 was transfected in HEK-293T cells. Cells were allowed to recover from transfection for >6 h. 1,4-DHPs were added at a final concentration of 3 μM and cells were incubated for another 20 h before analysis by flow cytometry. Cell populations were dissociated to single cell suspensions and analyzed on a LSR Fortessa for HA-positive cells, using an anti-HA antibody (Covance, San Diego) and an APC-labeled secondary antibody (Life Technologies). Data analysis was performed using FlowJo (Tree Star, Ashland).
Mouse embryonic stem cell culture and differentiation assays
The assays were done as described previously.3,27 Here, brief descriptions of the assays are provided. mESC cardiogenesis assay: For the HCS assay with the mESC lines CGR8 carrying a Myh6-GFP reporter,27,28 cells were seeded in LIF-free DMEM (containing 10% FBS) with high glucose in black 384-well μClear plates to allow differentiation. On day 3 of differentiation different amounts of 1,4-DHPs (and controls) were added and media was changed every other day, thereby attaining a compound exposure window from days 3–5. On days 10 or 11 of differentiation cells were analyzed by image analysis on an INCell 1000 system (GE Healthcare, Little Chalfont) and quantified with Cyteseer software (Vala Sciences, San Diego). mESC mesoderm inhibition assay: For the mesoderm inhibition assay J1 Brachyury/T-GFP reporter cells were used as suspension cultures and were conducted in serum-containing media. Embryoid bodies (EBs) were allowed to form in differentiation media and were incubated in the presence of 1,4-DHPs (and controls) at day 1 of differentiation, just before the initiation of mesoderm. Analysis was done by flow cytometry on day 4. Cell populations were dissociated to single cell suspensions and analyzed on a FACSCanto for GFP-positive cells. Data analysis was performed using FlowJo.
Calcium transient assay
HL-1 cells were used in these studies, and represent an immortalized murine atrial cell line that can be serially passaged while retaining a differentiated cardiac phenotype and displaying spontaneous contractions.29 For experiments, cells were seeded on 96-well glass bottom plates coated with 0.02% gelatin and 5 μg/mL fibronectin at a density of 25,000 cells/well, and were maintained in Claycomb Medium supplemented with 10% FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, 0.1 mM norepinephrine. For Ca2+ analysis, cells were loaded with 200 ng/mL Hoechst 33342 and Fluo-4 for 30 minutes at 37 °C followed by 30 minutes at room temperature. Fluo-4 NW Calcium Assay Kit (LifeTechnologies/Molecular Probes) was prepared according to the manufacturers' instructions in 1× Hanks Balanced Salt Solution containing 20 mM HEPES buffer and 2.5 mM probenecid. After washing twice with Tyrode's solution (140 mM NaCl, 6 mM KCl, 1 mM MgCl2, 5 mM HEPES, 2 mM CaCl2, 10 mM glucose, pH 7.4), cells were incubated with indicated compounds diluted in Tyrode's solution for 10 minutes at room temperature before beginning Ca2+ recordings. Ca2+ transient recordings were done using a Kinetic Image Cytometer IC 100 (Vala Sciences, San Diego).30 Video streams of 10 seconds of the Fluo-4 green channel were collected for each well at 33 frames per second. Autofocus by the IC 100 was done prior to video acquisition using the nuclear channel. All image capture was conducted with a 20× 0.50 numerical aperture (NA) objective. Cytometric calcium kinetic parameters were determined by image analysis using CyteSeer software.
Supplementary Material
Table 2.
Effect of the R2 region (carboxylic esters and amides) on TGFβ inhibition.a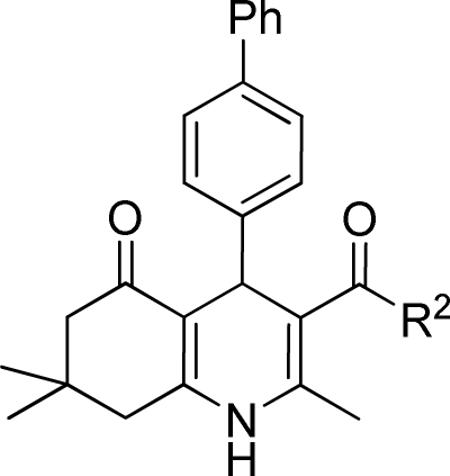
| compd | R2 | IC50 (μM) | %inhibition (2.5 μM) |
|---|---|---|---|
| 44 | OH | >10 | 21 ± 11 |
| 10 | OMe | 1.49 ± 0.43 | 44 ± 5 |
| 1 | OEt | 0.85 ± 0.30 | 83 ± 8 |
| 15 | OCH2CF3 | 0.76 ± 0.22 | 85 ± 4 |
| 16 | OiBu | 0.53 ± 0.09 | 87 ± 1 |
| 17 | OiPentyl | 1.46 ± 0.43 | 71 ± 4 |
| 18 |
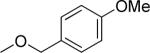
|
0.83 ± 0.40 | 72 ± 8 |
| 19 |
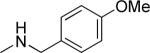
|
4.06 ± 1.03 | 40 ± 1 |
Data represent means ± SD of n = 2–4 independent experiments.
Table 5.
Effect of SAR-optimized substitution pattern on TGFβ inhibition.a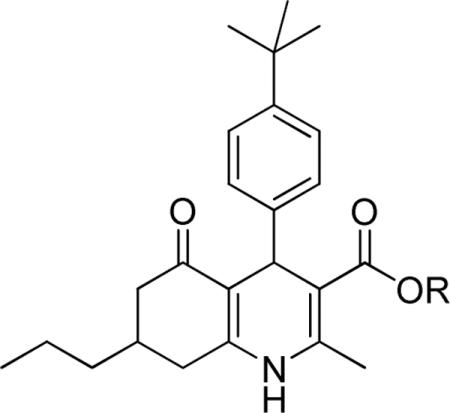
| compd | R | IC50 (μM) | %inhibition (2.5 μM) |
|---|---|---|---|
| 28 | Et | 0.17 ± 0.05 | 98 ± 1 |
| 29 | iBu | 0.17 ± 0.02 | 94 ± 1 |
Data represent means ± SD of n = 2–4 independent experiments.
ACKNOWLEDGMENT
We thank Ms. Alyssa Morgosh at HBRI for her technical assistance with preparation of compounds, Drs. Zebin Xia and Marcia I. Dawson (SBMRI) for helpful discussions and Dr. Kim Janda (The Scripps Research Institute) for providing access to a polarimeter.
Funding Sources We acknowledge the NIH (R01HL059502 and R33HL088266 to MM) and the California Institute for Regenerative Medicine (CIRM, RC1-000132 to MM), the Leducq Transatlantic Alliance (SHAPEHEART to MM) and the T Foundation (to JRC) for their generous financial support. We gratefully acknowledge postdoctoral fellowships from the CIRM (TG-0004) and American Heart Association (to EW) and German Research Foundation (DFG, SCHA1663/1-1 and SCHA1663/2–1 to DS).
ABBREVIATIONS USED
- 1,4-DHP
1,4-dihydropyridine
- TGFβ
Transforming Growth Factor β
- TGFBR2
TGFβ receptor type II
- ITD
inducer of TGFβ type II receptor degradation
- SAR
structure-activity-relationship
- SBE4
Smad-4 binding element
- HEK-293
human embryonic kidney cells 293
- mESCs
murine embryonic stem cells
Footnotes
Author Contributions The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Analytical data for 1, 7–22, 24–30 and 33–42; synthetic procedures and analytical data for the (+)- and (−)-enantiomers of 23, 43 and synthetic intermediates 48–57; 1D and 2D NMR spectra of 1 (Figures S1–3), 1H NMR spectra of 52a,b diastereomers (Figure S4); Representative 1H NMR spectra of (+)Eu(hfc)3 shift reagent experiments for (rac)-1, (+)- and (−) 1 (Figures S5–7); Representative calcium transient curves (HL-1 cells) for (+)- and (−)-enantiomers of 1 and 23; This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Willems E, Lanier M, Forte E, Lo F, Cashman J, Mercola M. A chemical biology approach to myocardial regeneration. J. Cardiovasc. Transl. Res. 2011;4(3):340–350. doi: 10.1007/s12265-011-9270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willems E, Bushway PJ, Mercola M. Natural and synthetic regulators of embryonic stem cell cardiogenesis. Pediatr. Cardiol. 2009;30(5):635–642. doi: 10.1007/s00246-009-9409-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willems E, Teixeira JC, Schade D, Cai W, Reeves P, Bushway PJ, Lanier M, Walsh C, Kirchhausen T, Izpizua Belmonte JC, Cashman J, Mercola M. Small molecule-mediated TGFβ Type II receptor degradation promotes cardiomyogenesis in embryonic stem cells. Cell Stem Cell. 2012;11(2):242–252. doi: 10.1016/j.stem.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rose U. Hexahydroquinolinones with calcium-modulatory effects--synthesis and pharmacologic action. Arch. Pharm. (Weinheim) 1990;323(5):281–286. doi: 10.1002/ardp.19903230506. [DOI] [PubMed] [Google Scholar]
- 5.Aydin F, Safak C, Simsek R, Erol K, Ulgen M, Linden A. Studies on condensed 1,4-dihydropyridine derivatives and their calcium modulatory activities. Pharmazie. 2006;61(8):655–659. [PubMed] [Google Scholar]
- 6.Takahashi D, Oyunzul L, Onoue S, Ito Y, Uchida S, Simsek R, Gunduz MG, Safak C, Yamada S. Structure-activity relationships of receptor binding of 1,4-dihydropyridine derivatives. Biol. Pharm. Bull. 2008;31(3):473–479. doi: 10.1248/bpb.31.473. [DOI] [PubMed] [Google Scholar]
- 7.Safak C, Simsek R. Fused 1,4-dihydropyridines as potential calcium modulatory compounds. Mini Rev. Med. Chem. 2006;6(7):747–755. doi: 10.2174/138955706777698606. [DOI] [PubMed] [Google Scholar]
- 8.Edraki N, Mehdipour AR, Khoshneviszadeh M, Miri R. Dihydropyridines: evaluation of their current and future pharmacological applications. Drug Discov. Today. 2009;14(21–22):1058–1066. doi: 10.1016/j.drudis.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Derynck R, Miyazono K. TGFβ and the TGFβ Family. In: Derynck R, Miyazono K, editors. The TGFβ Family. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2008. pp. 29–43. [Google Scholar]
- 10.Wharton K, Derynck R. TGFbeta family signaling: novel insights in development and disease. Development. 2009;136(22):3691–3697. doi: 10.1242/dev.040584. [DOI] [PubMed] [Google Scholar]
- 11.Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 2004;3(12):1011–1022. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
- 12.Nagaraj NS, Datta PK. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin. Investig. Drugs. 2010;19(1):77–91. doi: 10.1517/13543780903382609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-beta signaling in fibrosis. Growth Factors. 2011;29(5):196–202. doi: 10.3109/08977194.2011.595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ko S, Sastry MNV, Lin C, Yao C-F. Molecular iodine-catalyzed one-pot synthesis of 4-substituted-1,4-dihydropyridine derivatives via Hantzsch reaction. Tetrahedron Lett. 2005;46:5771–5774. [Google Scholar]
- 15.Cozzi P, Carganico G, Fusar D, Grossoni M, Menichincheri M, Pinciroli V, Tonani R, Vaghi F, Salvati P. Imidazol-1-yl and pyridin-3-yl derivatives of 4-phenyl-1,4-dihydropyridines combining Ca2+ antagonism and thromboxane A2 synthase inhibition. J. Med. Chem. 1993;36(20):2964–2972. doi: 10.1021/jm00072a017. [DOI] [PubMed] [Google Scholar]
- 16.Zolfigol MA, Bagherzadehb M, Niknamc K, Shirinid F, Mohammadpoor-Baltorke I, Choghamarania AG, Baghbanzadeha M. Oxidation of 1,4-Dihydropyridines under Mild and Heterogeneous Conditions Using Solid Acids. J. Iran. Chem. Soc. 2006;3(1):73–80. [Google Scholar]
- 17.Shan R, Howlett SE, Knaus EE. Syntheses, Calcium Channel Agonist-Antagonist Modulation Activities, Nitric Oxide Release, and Voltage-Clamp Studies of 2-Nitrooxyethyl 1,4-Dihydro-2,6-dimethyl-3-nitro-4-(2-trifluoromethylphenyl)pyridine-5-carboxylate Enantiomers. J. Med. Chem. 2002;45(4):955–961. doi: 10.1021/jm010394k. [DOI] [PubMed] [Google Scholar]
- 18.Gellibert F, de Gouville AC, Woolven J, Mathews N, Nguyen VL, Bertho-Ruault C, Patikis A, Grygielko ET, Laping NJ, Huet S. Discovery of 4-{4-[3-(pyridin-2-yl)-1H-pyrazol-4-yl]pyridin-2-yl}-N-(tetrahydro-2H-pyran-4-yl)benzamide (GW788388): a potent, selective, and orally active transforming growth factor-beta type I receptor inhibitor. J. Med. Chem. 2006;49(7):2210–2221. doi: 10.1021/jm0509905. [DOI] [PubMed] [Google Scholar]
- 19.Gellibert F, Woolven J, Fouchet MH, Mathews N, Goodland H, Lovegrove V, Laroze A, Nguyen VL, Sautet S, Wang R, Janson C, Smith W, Krysa G, Boullay V, De Gouville AC, Huet S, Hartley D. Identification of 1,5-naphthyridine derivatives as a novel series of potent and selective TGF-beta type I receptor inhibitors. J. Med. Chem. 2004;47(18):4494–4506. doi: 10.1021/jm0400247. [DOI] [PubMed] [Google Scholar]
- 20.Franckowiak G, Bechem M, Schramm M, Thomas G. The optical isomers of the 1,4-dihydropyridine BAY K 8644 show opposite effects on Ca channels. Eur. J. Pharmacol. 1985;114(2):223–226. doi: 10.1016/0014-2999(85)90631-4. [DOI] [PubMed] [Google Scholar]
- 21.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002;62(1):65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 22.Lanier M, Schade D, Willems E, Tsuda M, Spiering S, Kalisiak J, Mercola M, Cashman JR. Wnt inhibition correlates with human embryonic stem cell cardiomyogenesis: a structure-activity relationship study based on inhibitors for the Wnt response. J. Med. Chem. 2012;55(2):697–708. doi: 10.1021/jm2010223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willems E, Spiering S, Davidovics H, Lanier M, Xia Z, Dawson M, Cashman J, Mercola M. Small-molecule inhibitors of the Wnt pathway potently promote cardiomyocytes from human embryonic stem cell-derived mesoderm. Circ. Res. 2011;109(4):360–364. doi: 10.1161/CIRCRESAHA.111.249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kita-Matsuo H, Barcova M, Prigozhina N, Salomonis N, Wei K, Jacot JG, Nelson B, Spiering S, Haverslag R, Kim C, Talantova M, Bajpai R, Calzolari D, Terskikh A, McCulloch AD, Price JH, Conklin BR, Chen HS, Mercola M. Lentiviral vectors and protocols for creation of stable hESC lines for fluorescent tracking and drug resistance selection of cardiomyocytes. PLoS One. 2009;4(4):e5046. doi: 10.1371/journal.pone.0005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jonk LJ, Itoh S, Heldin CH, ten Dijke P, Kruijer W. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J. Biol. Chem. 1998;273(33):21145–21152. doi: 10.1074/jbc.273.33.21145. [DOI] [PubMed] [Google Scholar]
- 26.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat. Cell Biol. 2003;5(5):410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 27.Bushway PJ, Mercola M. High-throughput screening for modulators of stem cell differentiation. Methods Enzymol. 2006;414:300–316. doi: 10.1016/S0076-6879(06)14017-3. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi T, Lord B, Schulze PC, Fryer RM, Sarang SS, Gullans SR, Lee RT. Ascorbic acid enhances differentiation of embryonic stem cells into cardiac myocytes. Circulation. 2003;107(14):1912–1916. doi: 10.1161/01.CIR.0000064899.53876.A3. [DOI] [PubMed] [Google Scholar]
- 29.Claycomb WC, Lanson NA, Jr., Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U.S.A. 1998;95(6):2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerignoli F, Charlot D, Whittaker R, Ingermanson R, Gehalot P, Savtchenko A, Gallacher DJ, Towart R, Price JH, McDonough PM, Mercola M. High throughput measurement of Ca(2+) dynamics for drug risk assessment in human stem cell-derived cardiomyocytes by kinetic image cytometry. J. Pharmacol. Toxicol. Methods. 2012 doi: 10.1016/j.vascn.2012.08.167. in press. ( http://dx.doi.org/10.1016/j.vascn.2012.08.167) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.