

Abstract
Over the past decade, biomedical research has witnessed an exponential increase in the throughput of the characterization of biological systems. Here we review the recent progress in large-scale methods to determine protein–protein, genetic and chemical–genetic interaction networks. We discuss some of the limitations and advantages of the different methods and give examples of how these networks are being used to study the evolutionary process. Comparative studies have revealed that different types of protein–protein interactions diverge at different rates with high conservation of co-complex membership but rapid divergence of more promiscuous interactions like those that mediate post-translational modifications. These evolutionary trends have consistent genetic consequences with highly conserved epistatic interactions within complex subunits but faster divergence of epistatic interactions across complexes or pathways. Finally, we discuss how these evolutionary observations are being used to interpret cross-species chemical-genetic studies and how they might shape therapeutic strategies. Together, these interaction networks offer us an unprecedented level of detail into how genotypes are translated to phenotypes, and we envision that they will be increasingly useful in the interpretation of genetic and phenotypic variation occurring within populations as well as the rational design of combinatorial therapeutics.
1 The Promise of Comparative Interaction Networks
In December 1831, the HMS Beagle set sail from Plymouth Sound, England, for a survey expedition in South America. Aboard the HMS Beagle, a young naturalist named Charles Darwin would make observations that would shape his thinking about the origin of new species and revolutionize biology. Centuries later, investments in high-throughput methods are realizing a dream of new voyage. Technological developments, made in the past decade (Fig. 7.1), offer us a chance to experimentally determine the diversity of molecular species and cellular interactions (Fig. 7.2). In analogy to Darwin’s observations of biological diversity, these molecular studies hold the promise of great insight into the evolutionary process. They offer a bridge between the genotype and phenotype and will allow us to study how genetic variability is propagated through molecular structures and interaction networks to have an impact on fitness.
Fig. 7.1. Timeline of Bioinformatics, Genomic and Proteomic developments.

We selected and illustrated here several important landmarks in the development of genomics, proteomics, and bioinformatics, over the past 40 years. Examples of these include: the atlas of protein sequences, published as a book, by the bioinformatics pioneer Margaret Dayhoff; the first protein sequence analysis algorithms like the Needleman–Wunsch algorithm and analysis of gap-penalty costs by Haber and Koshland [87]; the creation of a protein structure repository (P.D.B-Protein Data Bank) that in 1974 contained atomic coordinates for 12 proteins; the first full genome sequences (1982–phage lambda, 1995–E. coli and 1996–budding yeast) along with the creation of the GenBank database that started with 606 sequences; the several technological developments in mass spectrometry (MS) like the first use of MS for peptide sequencing (1966), of electrospray ionization for biomolecules (1984), and novel ion traps like the orbitrap (1999) with increased mass accuracy and resolution, culminating in the first large-scale quantification of protein abundances of an eukaryotic cell (2008)
Fig. 7.2. Timeline for the first large-scale protein–protein, epistatic, and chemical–genetic interaction networks in different species.

We selected from the literature the first articles describing large-scale protein–protein, epistatic, and chemical–genetic interaction networks for several model organisms (E. coli, S. cerevisiae, S. pombe, the fly D. melanogaster, the worm C. elegans, and human). We illustrate here the timeline in which these studies were conducted with additional information provided in Table 7.1. Y2H, PPIs derived from yeast-two-hybrid; AP-MS, PPIs derived from affinity tag-purification followed by mass spectrometry
In the past decade, the availability of genome sequences for several model organisms allowed us to look for patterns via comparative studies (reviewed by Koonin [1]). What has become known as comparative genomics has brought us tremendous insight into the evolutionary process. Among many other things, we have learned about the rate of gene birth/death [2], the timing and impact of whole genome duplication events [3, 4], the forces shaping genome complexity [5], and the origins of multicellularity and functional diversity [6, 7]. Comparing different genomes allows us also to better identify the coded functional elements and what constrains their evolution [8, 9]. More recently, large-scale global efforts were initiated to determine the genetic variability of human individuals with the hope of identifying the genetic underpinnings of human disease [10].
In the same way that comparative genomics has resulted in an impressive leap forward in our understanding of genome evolution, we argue that combining and comparing different cellular interaction data are crucial for our understanding of the evolutionary process. We review here different high-throughput methods to determine interaction networks, their limitations, and how they are being used in comparative studies. We will focus primarily on comparative analysis of protein–protein, epistatic, and chemical–genetic interaction networks and we will not report on transcriptional regulatory networks as these have been extensively covered elsewhere [11, 12]. In the context of evolutionary systems biology, these networks give us a first glimpse of how different molecular entities cooperate as a system to perform specific cellular functions and how these interactions diverge over time. Importantly, the characterization of the same cellular function (i.e. system) across different species let us study to what extent natural selection constraints phenotypes (ex. response to stress, accurate cell division, etc) and the particular molecular implementations of these functions in different species. We believe that these evolutionary studies have also practical applications in the development of therapeutic strategies since we gain an understanding not just of what are the essential elements (e.g., proteins) but the essential combinations of elements that are specific to pathogen (or diseased state) but not host.
2 Comparative Protein–Protein Interaction Networks
Proteins exert their function most often as part of larger molecular assemblies or complexes. Each protein may be part of multiple complexes, and these complexes themselves may interact. Consequently protein function cannot be understood in isolation, and analyzing the network of possible protein–protein interactions (PPIs) is a required step in the study of any biological system.
Over the past few years, different methods have been applied to map PPIs in large scale. Most approaches developed to date fall generally in two camps: the yeast-two-hybrid [13] or protein complementation methods [14] and the affinity tag-purification/mass spectrometry (AP-MS) approach [15]. These different experimental methods are best suited to capture different types of interactions. While the AP-MS approaches are best suited to report co-complex associations, the yeast-two-hybrid and protein complementation assays identify pair-wise interactions and can be more sensitive in characterizing direct and low-affinity interactions. Furthermore, unlike genome DNA sequencing, where there is a clear and static end-goal, mapping all of the PPIs in a species is an ill-defined problem. Interactions are not binary all-or-none events; they can change due to temporal or environmental factors [16] and their binding affinities span many orders of magnitude. For these reasons, defining accuracy and coverage (or completeness) of an interactome is challenging, and a careful statistical combination of methods is likely to be required to fully determine the repertoire of PPIs in a species [17].
Despite some of these limitations, strategies to interrogate the protein interaction networks have been applied to several model organisms (Fig. 7.2 and Table 7.1). In parallel to these studies curation efforts have collected, in a standardized format, information on PPIs obtained from these high-throughput studies and from small-scale studies reported in the literature [18] and made these available in public databases. As an example, at the present time, the BioGRID database (http://thebiogrid.org/) reports over 200,000 PPIs.
Table 7.1.
List of the first large-scale protein–protein, epistatic, and chemical–genetic interaction networks in different species. We collected from the literature the first large-scale networks reported for different model organisms. For each study we report the method used, the species studied, and the total number of interactions identified or quantified
| Interaction type | Method | Species | Interactions total (higher confidence) | Year | Reference |
|---|---|---|---|---|---|
| Protein–protein | Yeast-two-hybrid | S. cerevisiae | 183 | 2000 | [88] |
| S. cerevisiae | 957 | 2000 | [89] | ||
| D. melanogaster | 20,405 (4780) | 2004 | [90] | ||
| C. elegans | ~ 4,000 | 2004 | [91] | ||
| H. sapiens | 3,186 (911) | 2005 | [92] | ||
| H. sapiens | ~ 2,800 | 2005 | [93] | ||
| AP-MS | S. cerevisiae | 3,610 | 2002 | [94] | |
| S. cerevisiae | 7,123 | 2002 | [95] | ||
| S. cerevisiae | 589 protein assemblies | 2002 | [96] | ||
| H. sapiens | 6,463 (2.251) | 2007 | [97] | ||
| E. coli | 5,254 | 2005 | [98] | ||
| D. melanogaster | 556 protein assemblies | 2011 | [99] | ||
| HIV-H. sapiens | 497 | 2011 | [100] | ||
| Epistatic | SGA | S. cerevisiae | ~ 4,000 | 2001 | [46] |
| E-MAP | S. cerevisiae | ~ 180,000 quantifications | 2005 | [48] | |
| RNAi | C. elegans | ~ 65,000 quantifications | 2006 | [59] | |
| E-MAP | S. pombe | ~ 118,000 quantifications | 2008 | [57] | |
| SGA | S. pombe | ~ 49,000 quantifications | 2008 | [58] | |
| RNAi | D. melanogaster | ~ 70,000 quantifications | 2011 | [60] | |
| Chemical-genetic | Pooled growth | S. cerevisiae | 6 conditions X 5,916 genes | 2002 | [44] |
| Colony size | S. pombe | 21 conditions X 438 genes | 2010 | [76] | |
| Colony size | E. coli | 324 conditions X 3,979 mutants | 2010 | [69] |
The availability of these PPI networks has revealed some surprising findings regarding the evolution of these interactions. Seminal studies of protein homology, across species, led to the hypothesis that gene-expression changes are the main driving force for the generation of phenotypic diversity [19]. Since species with distinct observable phenotypes (e.g., human and chimp) have orthologous proteins that are almost identical, King and Wilson (among others) suggested that the phenotypic changes should be due to changes in the noncoding regions [19]. However, PPIs have been observed to diverge rapidly after gene-duplication, suggesting that modest changes in coding regions can result in substantial changes in interaction networks [20–22]. Andreas Wagner and others have shown that after gene duplication paralogous proteins diverge rapidly in their interactions, in proportion to the time since duplication [20–22]. However, despite the rapid divergence observed for all protein interactions, studies of protein complexes (a subset of all PPIs) have shown a remarkably different evolutionary pattern. In fact, co-complex membership is highly conserved across distantly related species. Van Dam and colleagues [23] have noted that 90% of human co-complex interactions are conserved in S. cerevisiae when both proteins have identifiable orthologs in yeast. Additional studies have convincingly shown that protein complexes tend to evolve mostly by duplication and divergence of their subunits. For example, Pereira-Leal and colleagues have observed that over 30% protein complexes in budding yeast contain duplicated gene pairs [24–26]. That is, complexes do not tend to grow by gaining interaction partners from previously existing proteins (e.g., through rewiring). Consistent with this observation, cores of homologous complexes that are more likely to have been present in an ancestral state are enriched in paralogous interacting proteins [27].
The same patterns of duplication/divergence can be seen when analyzing complexes from 3D structures [28]. Moreover, these structural studies suggest that the evolutionary path toward larger complexes can be predicted by the size of the interfaces, with larger interfaces being more likely to be conserved across homologous complexes [28]. The group II chaperonins provide a striking example of the general trend observed for the evolution of a protein complex by duplication and divergence of its subunits (Fig. 7.3). These chaperonins form multi-subunit protein folding assemblies that have very conserved structural features. Archeal complexes, termed thermosomes, form an eight-membered ring from a set of 1 to 3 homologous chaperonins, while eukaryotic complexes (called TriC or CCT) form an identical eight-membered ring from a set of eight chaperonin paralogs. Evolutionary analysis suggests that, in archeal genomes, duplication events have occurred in multiple independent lineages while the eight paralogous eukaryotic chaperonins are the result of ancient duplication events that are not likely to have occurred independently in different lineages [29, 30].
Fig. 7.3. Evolution of co-complex interactions in the group II chaperonins.
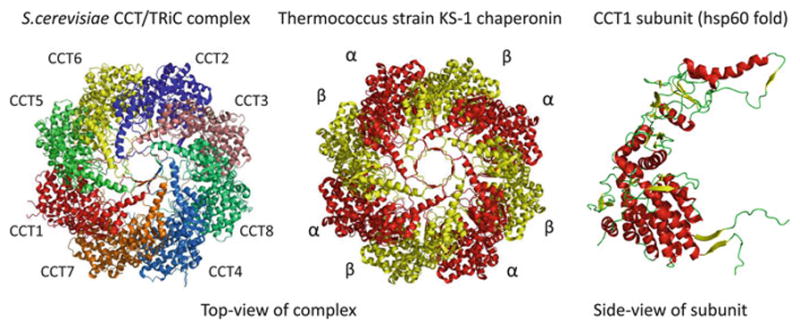
Computational studies have shown that protein complexes usually evolve by duplication and divergence of their subunits. The group II chaperonin complexes provide a good illustration of this general trend. The archeal group II chaperonin complexes (termed thermosomes) usually contain 1–3 homologous chaperonins and it represented here by the thermosome of Thermococcus strain KS-1 (PDB:1Q2V). The eukaryotic complexes (called TriC or CCT) are composed of eight chaperonin paralogs, represented here by the S. cerevisiae CCT complex (PDB:3P9E). All of the subunits are structurally similar, exemplified here by the S. cerevisiae CCT1 subunit structure
Novel complexes might also be the result of partial complex duplications. In support of this, it has been observed that 7–20% of protein complexes in S. cerevisiae are partially homologous. However, the duplication of whole protein complexes is expected to be rare since the simultaneous duplication of all the subunits of a complex is unlikely. In line with this, even for budding yeast that has gone through a whole-genome-duplication event, duplication of whole protein complexes is rarely observed [24–26]. This suggests that this phenomenon is not a common route for the generation of novel complexes.
These different observations result in an apparent contradiction. Global rates of change of all PPIs suggest that these can diverge rapidly after gene duplication but the subset composed of co-complex interactions is highly conserved across species. These opposing results can be reconciled if one notes that the evolutionary rate of change of protein interactions is not uniform [21, 31] with more promiscuous interactions diverging faster than more specific interaction types.
Protein–peptide interactions are an example of promiscuous interactions that are often specified by a few key amino acids within the interacting peptide region. These peptide regions are usually referred to as linear motifs and can determine, for example, sites of posttranslational modifications (PTMs) (i.e., phosphorylation), localization signals (i.e., nuclear import/export), or more generally determine the interaction with specific domain families [32]. Since these interactions are specified by a small number of amino acids within an accessible region, intuitively one can imagine that these can be easily created and destroyed by a few point mutations. In accordance with this intuition, interactions mediated by linear motifs have been shown to be poorly conserved across species [33]. Unfortunately, lower specificity interactions are also harder to detect experimentally with high confidence than more specific interactions like co-complex membership. For this reason, it is not straightforward to distinguish when PPIs with different specificities have different rates of evolution or just different experimental error rates. In order to address this issue, some studies have focused on the functional consequence of linear-motif interactions (e.g., PTMs, protein localization) and their evolution, since these consequences can be measured more accurately than the underlying PPIs [34–36].
Recent advances in mass spectrometry now allow us to accurately determine protein PTMs, such as phosphorylation, acetylation, or ubiquitylation in very high-throughput fashion [37]. The availability of large numbers of high-confidence phosphorylation sites for different species validated some of the previous observations regarding the evolution of linear-motif interactions. In fact, phosphosites [36, 38], phosphoproteins [34], and kinase–substrate interactions [35] are poorly conserved across species, and this divergence has been linked with the subfunctionalization of duplicated genes [39, 40]. Although specific phosphosites and phosphoproteins diverge rapidly, the average number of phosphosites per protein within a functional group (e.g., protein complex or pathway) appears to be well conserved [34]. This function-based conservation is analogous to the conserved timing of transcriptional regulation of protein complexes during the cell cycle, despite the rapid divergence of the transcriptional regulation of specific subunits [41]. In fact, a strong analogy can be made between the properties and evolution of linear-motif mediated PPIs and transcriptional regulation [42]. These two types of interactions both tend to be: transient, mediated by degenerate sequence motifs, poorly conserved and regulatory in nature.
Together, these observations paint a scenario of a very uneven divergence of PPIs. Protein complexes diverge slowly and mostly through duplication and divergence of their subunits with no “re-wiring” of existing proteins. Lower specificity interactions, such as protein–peptide interactions, display higher evolutionary turnover and can result in the creation of novel interactions in the absence of gene-duplication events. In analogy to the existence of pseudo-genes, it is also expected that a significant fraction of measured interactions serve no biological function but are instead remnants of this evolutionary turnover of interactions. Given this uneven rate of change we would also expect that lower-specificity interactions should also have a higher proportion of nonfunctional interactions when compared to co-complex membership [43]. In line with this expectation, it has been recently suggested that a significant fraction of protein phosphorylation might serve no function [38]. As we describe below, this uneven conservation of PPIs has also observable consequences at the level of epistatic interactions and can be used to better interpret the functional consequences of small molecule perturbations.
3 Comparative Epistatic Interaction Networks
The sequencing of the budding yeast genome facilitated the creation of a genome-wide deletion library, a set of yeast strains each lacking a single gene. This resulted in the finding that yeast is surprisingly robust to deletion of individual genes with only ~18% of genes being essential for growth in rich media [44]. Because there is extensive duplication within the S. cerevisiae genome, one plausible explanation for the apparent dispensability of the remaining genes was that there existed functional redundancy between duplicates. Indeed, initial analysis suggested that single copy genes were significantly more likely to be essential than genes with an identifiable paralog [45]. Another explanation was the existence of redundant pathways—disjoint sets of genes performing similar functions, such that either pathway can be perturbed with little functional consequence, but a simultaneous perturbation of both pathways resulted in disruption of a particular, possibly essential, function.
Fortunately the availability of the yeast deletion library also facilitated the development of new high-throughput technologies to study such functional relationships and to what extent they explain robustness to single gene deletion—e.g., Synthetic Genetic Arrays (SGA) [46]. An epistatic (or genetic) interaction occurs when one gene modifies the effect of another, and can be detected by comparing the effects on a phenotype of interest of perturbing single genes with that of perturbing two or more genes simultaneously (see review [47]). SGA technology is used to identify such relationships in high throughput by mating pairs of deletion mutants and assessing the growth of the resulting colonies. The first screens were qualitative in nature, and sought to identify negative (also termed antagonistic or aggravating) epistatic interactions, where crossing two viable single mutants resulted in an inviable or severely sick mutant. Later, the E-MAP approach [48,49] was developed to measure these interactions in a quantitative fashion, detecting both negative (sicker than expected) and positive (also termed synergistic or alleviating) epistatic interactions (healthier than expected).
Initial analysis revealed that although paralogs were indeed more likely than random gene pairs to share a negative epistatic interaction, this accounted for only a small fraction (~2%) of the observed interactions [50]. As more genetic interaction data have become available, we have gained greater insight into the consequences and nature of gene duplication. For instance, Ihmels and colleagues [51] showed that only ~25% of duplicate gene pairs interact negatively, suggesting that duplication can explain only a small fraction of the dispensability of yeast genes. Furthermore, the authors noted that even in cases where such a relationship existed, the buffering was only partial, with duplicate pairs typically displaying additional nonoverlapping genetic interactions and deletion phenotypes. Additionally, VanderSluis and colleagues showed that these non-buffered epistatic interactions exhibited significant asymmetry, with one member of a duplicate pair has significantly more interactions than the other [52].
If duplicate genes explain only a small fraction of the observed epistatic interactions, what accounts for the rest? Initial results indicated that genes which interact were significantly more likely to be involved in the same biological process (as indicated by GO terms) or protein complex. The availability of large-scale protein–protein interaction data offered additional insight into the nature of both essential genes and epistatic interactions. By mapping the essentiality data from the yeast gene deletion project onto co-complex protein interaction data, Hart et al. noticed an interesting trend—genes in the same protein complex tended to have the same dispensability. Complexes are disproportionately mostly essential, or mostly viable, suggesting that dispensability is a feature of functional modules (complexes) rather than individual genes [53].
Similarly, by combining protein–protein interaction data with results from genetic interaction screens, Kelley and Ideker were able to identify “between pathway models” and “within pathway models.” Between pathway models correspond to pairs of physically connected redundant pathways, densely connected by negative epistatic interactions [54]. Within pathway models correspond to sets of genes densely connected by negative genetic interactions, whose products are also physically connected. The authors were able to categorize ~ 40% of the measured epistatic interactions as falling within or between physically connected pathways, and also noted that the between pathway model was significantly more prevalent [54]. Furthermore, Tong et al. noted that although genes which interact genetically more likely encode proteins that physically interact, genes which share similar epistatic interaction profiles were even more likely to do so [50]. That is—genes in the same protein complex tend to have epistatic interactions with the same partners. Again, both of these results indicate that cellular robustness to gene deletion and epistatic interactions can best be understood at the level of functional modules (pathways/complexes) rather than individual gene pairs. It would appear that cellular functions (or systems) are attained by the combined action of multiple molecular entities with partially overlapping roles.
As with most interaction data, S. cerevisiae has by far the greatest coverage of epistatic interactions of any organism. However, methods analogous to SGA have been developed in additional model organisms. Deletion libraries have been exploited in the bacteria E. coli [55, 56] and the fission yeast (S. pombe) to create double mutants whose phenotype can be measured [57, 58]. In metazoans, RNAi technology has been used to create pair-wise knockdowns in both whole organisms (C. elegans) [59] and more recently in Drosophila cell lines [60].
Initial screens revealed similarities in the features of the C. elegans and the S. cerevisiae epistatic interaction networks—duplicate genes are significantly more likely to interact, the degree distribution is apparently scale free and both networks have similar clustering coefficients. However, direct comparison of orthologous interactions revealed a less clear picture. A study of a single gene (MAD1) revealed that ~ 40% of its yeast synthetic lethal interactions caused a noticeable phenotypic enhancement in C. elegans [61]. However, a more comprehensive C. elegans interaction screen suggested that at most 5% of the yeast epistatic interactions were conserved [62]. It is unclear whether this apparent lack of conservation resulted from differences in the methods used (gene deletion vs potentially inefficient RNAi knockdown), the phenotypes measured (colony growth vs whole organism growth), or genuine change of interactions.
Other than budding yeast, the fission yeast S. pombe is the only other eukaryote for which a comprehensive deletion library is available [63]. Although these two species are evolutionarily distant (~400 million years of divergence), ~83% of their single copy orthologs have conserved dispensability. That is, if a gene is essential in one species, it is likely to be essential in the other. Furthermore, changes in dispensability between the two species appear to occur at the level of complexes or pathways. For example, the budding yeast mitochondrial translation machinery is largely nonessential, but it is largely essential in fission yeast [63]. These switches in essentiality likely reflect lifestyle changes between the two yeasts, such as the ability of budding yeast to survive without mtDNA.
This deletion library facilitated a more direct comparison of epistatic interactions across species—the methods developed are similar to those used for S. cerevisiae, and the phenotype measured is also colony growth. Two groups assessed the conservation of interactions across these two species, suggesting that negative genetic interactions were conserved at somewhere between ~ 18% [57] and ~ 30% [58]. By combining the genetics with information from other datasets, Roguev et al. were able to show that different categories of interactions were conserved at different rates. Notably, positive epistatic interactions between genes in the same complex were highly conserved (>50%). Furthermore, pairs of genes with similar epistatic interaction profiles in S. cerevisiae were also likely to have similar epistatic interactions in S. pombe if their products were cocomplexed [57]. Both of these observations, together with the lower overall conservation of genetic interactions, suggest that functional modules (complexes) are conserved between these two species, but the functional interactions between them have changed (Fig. 7.4). These genetic studies argue that protein complexes and pathway membership can be highly conserved, in line with the cross-species protein–protein-interaction studies, but the way in which different modules cooperate inside the cell diverge at a faster rate.
Fig. 7.4. Evolution of epistatic interactions within and between modules.

We compared the genetic interactions within and between modules for the SWR-C, HIR-C, and SET3-C complexes in S. cerevisiae and S. pombe. These illustrate the general trend that genetic interactions within complexes tend to be conserved across species while the genetic interactions between complexes diverge at a higher rate. In this example the positive genetic interactions measured within the SWR-C complex subunits is highly conserved between S. cerevisiae and S. pombe, while the negative genetic interactions between SWR-C and HIR-C and between SET3-C and HIR-C observed in S. pombe are not conserved in S. cerevisiae
4 Comparative Chemical Genetic Interactions
The protein–protein and epistatic interactions obtained to date have taught us much about the function and evolution of cellular interaction networks. We have learned which features of the cellular machinery are more likely to be conserved across species and how combinatorial gene disruption affects cellular function. These findings have the potential to be applied in medically relevant research. We are motivated to rethink our therapeutic strategies [64] since there are a small number of essential genes, but many more combinations that, when perturbed, cause synthetic sickness or lethality. If we can understand how a host cell differs from a pathogen, or how diseased tissue differs from the healthy state, we will be in a better position to devise drug combinations that are more likely to affect the pathogen/disease while leaving the host unharmed. With this in mind, in parallel to the development of high-throughput methods to study physical and epistatic interactions, researchers have also been working on large-scale approaches to measure the effects of small molecule perturbations [65].
In analogy to the quantification of epistatic interactions between gene pairs, a functional link between a gene and drug, or between two drugs, can be quantified as a deviation from the expected effect of the single perturbations on the observed phenotype (usually growth/proliferation). In this way, it is possible to identify genes that, when knocked out, increase the sensitivity or cause resistance to a small molecule, more than expected by a neutral model [65]. Similarly, two bioactive compounds can have a greater than expected (synergistic interaction) or smaller than expected (antagonistic interaction) on an observed phenotype. Unlike the impact of a knockout, the effects of small molecules are usually concentration dependent, so the identification of drug–drug interactions is often determined using dose-matrix experiments which are more laborious are harder to scale up [65].
Chemical-genetic studies provide insight into the function of the genes that are perturbed but can also be used to identify the mode-of-action of bioactive compounds, including medically relevant drugs [66, 67], antifungal [68], or antibiotic compounds [69]. These large-scale chemical-genetic quantitative interactions have been mined to study compound structure–activity relationships [70] and can be integrated with physical [71] and genetic interactions [72] to facilitate functional and mode-of-action studies. These chemical-genetic studies have, so far, focused on single-cell organisms for which knockout collections are available but can be expanded to other species by the use of RNAi [73–75]. One of these RNAi studies identified genes that increased the sensitivity of a human cancer line to paclitaxel, a drug used in the treatment of non-small-cell lung cancer. Among these, the authors identified a subunit of the vacuolar ATPase (v-ATPase) which led them to test and validate a synergistic interaction between paclitaxel and salicylihalamide A, a v-ATPase inhibitor [73]. This example demonstrates how these concepts of epistatic and chemical–genetic interactions can come together to identify potential combinatorial therapeutics with relevance to human disease.
The development of these methods has recently allowed for an exploration of the evolution of chemical–genetic and drug–drug interactions. We have shown that chemical–genetic interactions are poorly conserved across distantly related fungal species (S. cerevisiae and S. pombe), mirroring and validating the results from genetic interaction studies [76]. However, we observed again a pattern of modular conservation with compound–complex functional interactions showing stronger conservation than compound–gene interactions. Interestingly, combining information on compound–complex interactions across species increased the capacity to predict the mode-of-action of the tested compounds [76]. Studies of drug–drug interactions have similarly shown that synergistic drug combinations tend to be poorly conserved across species or even across different cellular states of the same species [77–79]. The availability of large sets of known epistatic and chemical–genetic interactions also allow for development of methods to predict synergistic drug combinations [80, 81]. For example, it has been suggested that genetic interaction information can be mined to predict drug synergies, although a large number of drug–drug interactions might be the result of one of the drugs affecting the bioavailability of the other, instead of mediated by an epistatic interaction of the drug targets [81].
These studies raise some concerns regarding the transfer of knowledge across species by homology. More interestingly however, these also tell us that synergistic drug combinations are an effective way to identify therapies that will specifically target a disease or pathogen but not the host systems. We believe that these lines of research will increase our ability to rationally design therapeutic strategies.
5 Conclusion
We have reviewed here recent progress in the large-scale analysis of protein–protein, genetic, and chemical–genetic interaction networks. These high-throughput methods provide several advantages over small-scale studies. For example, the costs associated with each interaction tend to go down significantly with scale. Also, the scale allows for an unbiased quantification of error rates and statistical approaches to reduce the associated errors that are just not possible with lower-scale experiments [82]. Finally, the standardized nature of the methods make the curation and reutilization of these data easier than the data derived from small-scale experiments that are mostly impossible to access computationally. Indeed, the resulting interaction networks, and the databases that facilitate their easy access, become a resource for the scientific community. Furthermore, the global nature of the experiments means that the resources can be used for purposes which the original authors neither anticipated nor intended. For instance, genome-wide epistatic interaction maps and protein interaction maps have offered insight into the functional consequences of gene duplication, although the data used were not obtained for that explicit purpose.
Although there are significant advantages to these approaches, large-scale, “hypothesis-free” experiments are often dismissed as being little more than fishing expeditions. The exploratory and open-ended nature of these studies has attracted significant criticism. Notably, the Nobel prize winner Sydney Brenner is often quoted, in regard to high-throughput studies, as having said that it was: “a biology I like to call low-input, high-throughput, no-output biology” [83]. We respectfully disagree with this assessment. As we described above, these high-throughput experiments not only tend to reduce costs and error rates but describe the diversity of molecular entities and interactions across species. These studies have resulted in the identification of many novel complexes and pathways and importantly comparing large-scale networks for different species provide us with tremendous insight into the evolutionary process.
Across the different studies described above, we note a consistent trend of conservation of protein modules (i.e., complexes or pathways). Protein–protein and epistatic interactions within complexes are more conserved than across complexes. This conservation is not a general feature of all protein–protein associations as more promiscuous types of interactions, such as those mediated by linear motifs (e.g., PTMs and localization signals), are generally poorly conserved. These differences give us information about how a cell is likely to generate phenotypic diversity. In addition, this modular organization of cellular systems helps us to rationalize the apparent cellular robustness to gene deletion and to environmental perturbations. Together, these results suggest that cellular functions (cell division, cell-fate decisions, response to stress, etc.) can only be understood as the product of the combined action of multiple and interdependent molecular entities that have partially overlapping roles. As such, a cell’s (or system) robustness to gene deletion or environmental stress is not due to any single gene or even pairs of genes (i.e., gene redundancy) but is distributed across many molecules [84].
These cross-species studies also highlight the difference between the conservation of function versus the conservation of the specific molecular mechanisms that implement those functions. It has been observed that phosphorylation level of protein complexes can be conserved despite the rapid divergence of individual phosphosites. Similarly, compound–complex interactions show higher conservation than the respective underlying compound–gene epistatic interactions. These general trends are consistent with small-scale studies of specific systems. It has been shown that even crucial and highly conserved functions like mating [85] or licensing of the origins of DNA replication [86] can diverge in the molecular mechanisms that give rise to these functions in different species. We envision that these cellular interaction networks in conjunction with structural information and computer simulation studies will increasingly be used to rationalize the consequences of genetic variability observed in natural populations.
Large-scale analysis of interaction networks allow us, for the first time, to bridge the gap between phenotype and genotype and let us study how genetic variability is propagated through molecular structures and interaction networks to have phenotypic consequences. They are our modern world, molecular equivalent of Darwin’s Beagle expedition. Darwin’s voyage and subsequent evolutionary theories transformed biology in a very fundamental way. However, Darwin did not set out with the intention of observing species diversity in order to demonstrate his existing theories. Rather, his observations inspired and facilitated the development of his theories. It is exciting to realize that the interaction networks we described here are just the first few, of many to come. Our voyage has just begun.
Acknowledgments
We thank J. Haber for critically reading the manuscript and funding from the NIH (GM082250, GM084448, GM084279, AI090935, GM081879, AI091575, GM098101). NJK is a Searle Scholar and Keck Young Investigator. PB is supported by the Human Frontiers Science Program. CR is supported by IRCSET.
Contributor Information
Pedro Beltrao, Email: pedro.beltrao@ucsf.edu, Department of Cellular and Molecular Pharmacology, California Institute for Quantitative Biomedical Research, University of California, San Francisco, 1700 4th Street, San Francisco, CA 94158, USA.
Colm Ryan, Email: colm.ryan@ucd.ie, Department of Cellular and Molecular Pharmacology, California Institute for Quantitative Biomedical Research, University of California, San Francisco, 1700 4th Street, San Francisco, CA 94158, USA. School of Computer Science and Informatics, University College Dublin, Dublin, Ireland.
Nevan J. Krogan, Email: krogan@cmp.ucsf.edu, Department of Cellular and Molecular Pharmacology, California Institute for Quantitative Biomedical Research, University of California, San Francisco, 1700 4th Street, San Francisco, CA 94158, USA. J. David Gladstone Institutes, San Francisco, CA 94158, USA
References
- 1.Koonin EV. Darwinian evolution in the light of genomics. Nucleic Acid Res. 2009;37:1011–1034. doi: 10.1093/nar/gkp089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch M. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 3.Dietrich FS, Voegeli S, Brachat S, Lerch A, Gates K, et al. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science (New York, NY) 2004;304:304–307. doi: 10.1126/science.1095781. [DOI] [PubMed] [Google Scholar]
- 4.Kellis M, Birren BW, Lander ES. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004;428:617–624. doi: 10.1038/nature02424. [DOI] [PubMed] [Google Scholar]
- 5.Lynch M, Conery JS. The origins of genome complexity. Science (New York, NY) 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- 6.King N, Westbrook MJ, Young SL, Kuo A, Abedin M, et al. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature. 2008;451:783–788. doi: 10.1038/nature06617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cock JM, Sterck L, Rouzé P, Scornet D, Allen AE, et al. The Ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature. 2010;465:617–621. doi: 10.1038/nature09016. [DOI] [PubMed] [Google Scholar]
- 8.Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- 9.Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478(7370):476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Consortium TIH. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuch BB, Li H, Johnson AD. Evolution of eukaryotic transcription circuits. Science (New York, NY) 2008;319:1797–1799. doi: 10.1126/science.1152398. [DOI] [PubMed] [Google Scholar]
- 12.Tirosh I, Barkai N. Inferring regulatory mechanisms from patterns of evolutionary divergence. Mol Syst Biol. 2011;7:1–10. doi: 10.1038/msb.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 14.Kerppola TK. Complementary methods for studies of protein interactions in living cells. Nat Meth. 2006;3:969–971. doi: 10.1038/nmeth1206-969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gingras A-C, Gstaiger M, Raught B, Aebersold R. Analysis of protein complexes using mass spectrometry. Nat Rev Mol Cell Biol. 2007;8:645–654. doi: 10.1038/nrm2208. [DOI] [PubMed] [Google Scholar]
- 16.Przytycka TM, Singh M, Slonim DK. Toward the dynamic interactome: it’s about time. Brief Bioinform. 2010;11:15–29. doi: 10.1093/bib/bbp057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braun P, Tasan M, Dreze M, Barrios-Rodiles M, Lemmens I, et al. An experimentally derived confidence score for binary protein-protein interactions. Nat Meth. 2009;6:91–97. doi: 10.1038/nmeth.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reguly T, Breitkreutz A, Boucher L, Breitkreutz B-J, Hon GC, et al. Comprehensive curation and analysis of global interaction networks in Saccharomyces cerevisiae. J Biol. 2006;5:11. doi: 10.1186/jbiol36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King MC, Wilson A. Evolution at two levels humans and chimpanze. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 20.Wagner A. How the global structure of protein interaction networks evolves. Proc Biol Sci/The Royal Society. 2003;270:457–466. doi: 10.1098/rspb.2002.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beltrao P, Serrano L. Specificity and evolvability in eukaryotic protein interaction networks. PLoS Comput Biol. 2007;3:e25. doi: 10.1371/journal.pcbi.0030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dreze M, Carvunis a-R, Charloteaux B, Galli M, Pevzner SJ, et al. Evidence for network evolution in an arabidopsis interactome map. Science. 2011;333:601–607. doi: 10.1126/science.1203877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Dam TJP, Snel B. Protein complex evolution does not involve extensive network rewiring. PLoS Comput Biol. 2008;4:e1000132. doi: 10.1371/journal.pcbi.1000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pereira-Leal JB, Teichmann Sa. Novel specificities emerge by stepwise duplication of functional modules. Genome Res. 2005;15:552–559. doi: 10.1101/gr.3102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pereira-Leal JB, Levy ED, Kamp C, Teichmann Sa. Evolution of protein complexes by duplication of homomeric interactions. Genome Biol. 2007;8:R51. doi: 10.1186/gb-2007-8-4-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pereira-Leal JB, Levy ED, Teichmann Sa. The origins and evolution of functional modules: lessons from protein complexes. Philos Trans R Soc Lond B Biol Sci. 2006;361:507–517. doi: 10.1098/rstb.2005.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yosef N, Kupiec M, Ruppin E, Sharan R. A complex-centric view of protein network evolution. Nucleic Acid Res. 2009;37:e88. doi: 10.1093/nar/gkp414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levy ED, Boeri Erba E, Robinson CV, Teichmann Sa. Assembly reflects evolution of protein complexes. Nature. 2008;453:1262–1265. doi: 10.1038/nature06942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Archibald JM, Logsdon JM, Doolittle WF. Origin and evolution of eukaryotic chaperonins: phylogenetic evidence for ancient duplications in CCT genes. Mol Biol Evol. 2000;17:1456–1466. doi: 10.1093/oxfordjournals.molbev.a026246. [DOI] [PubMed] [Google Scholar]
- 30.Archibald JM, Blouin C, Doolittle WF. Gene duplication and the evolution of group II chaperonins: implications for structure and function. J Struct Biol. 2001;135:157–169. doi: 10.1006/jsbi.2001.4353. [DOI] [PubMed] [Google Scholar]
- 31.Shou C, Bhardwaj N, Lam HYK, Yan K-K, Kim PM, et al. Measuring the evolutionary rewiring of biological networks. PLoS Comput Biol. 2011;7:e1001050. doi: 10.1371/journal.pcbi.1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diella F, Haslam N, Chica C, Budd A, Michael S, et al. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci. 2008;13:6580–6603. doi: 10.2741/3175. [DOI] [PubMed] [Google Scholar]
- 33.Neduva V, Russell RB. Linear motifs: evolutionary interaction switches. FEBS Lett. 2005;579:3342–3345. doi: 10.1016/j.febslet.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Beltrao P, Trinidad JC, Fiedler D, Roguev A, Lim Wa, et al. Evolution of phosphoregulation: comparison of phosphorylation patterns across yeast species. PLoS Biol. 2009;7:e1000134. doi: 10.1371/journal.pbio.1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan CSH, Bodenmiller B, Pasculescu A, Jovanovic M, Hengartner MO, et al. Comparative analysis reveals conserved protein phosphorylation networks implicated in multiple diseases. Sci Signal. 2009;2:ra39. doi: 10.1126/scisignal.2000316. [DOI] [PubMed] [Google Scholar]
- 36.Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, et al. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science (New York, NY) 2009;325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choudhary C, Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat Rev Mol Cell Biol. 2010;11:427–439. doi: 10.1038/nrm2900. [DOI] [PubMed] [Google Scholar]
- 38.Landry CR, Levy ED, Michnick SW. Weak functional constraints on phosphoproteomes. Trends Genet. 2009;25:193–197. doi: 10.1016/j.tig.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Amoutzias GD, He Y, Gordon J, Mossialos D, Oliver SG, et al. Posttranslational regulation impacts the fate of duplicated genes. Proc Natal Acad Sci USA. 2010;107:2967–2971. doi: 10.1073/pnas.0911603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freschi L, Courcelles M, Thibault P, Michnick SW, Landry CR. Phosphorylation network rewiring by gene duplication. Mol Syst Biol. 2011;7:504. doi: 10.1038/msb.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen LJ, Jensen TS, de Lichtenberg U, Brunak S, Bork P. Co-evolution of transcriptional and post-translational cell-cycle regulation. Nature. 2006;443:594–597. doi: 10.1038/nature05186. [DOI] [PubMed] [Google Scholar]
- 42.Moses AM, Landry CR. Moving from transcriptional to phosphoevolution: generalizing regulatory evolution? Trends Genet. 2010;26:462–467. doi: 10.1016/j.tig.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 43.Michnick SW, Levy ED, Landry CR. How perfect can protein interactomes be? Sci Signal. 2009;2:pe11. doi: 10.1126/scisignal.260pe11. [DOI] [PubMed] [Google Scholar]
- 44.Giaever G, Chu AM, Ni L, Connelly C, Riles L, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 45.Gu Z, Steinmetz LM, Gu X, Scharfe C, Davis RW, et al. Role of duplicate genes in genetic robustness against null mutations. Nature. 2003;421:63–66. doi: 10.1038/nature01198. [DOI] [PubMed] [Google Scholar]
- 46.Tonga H, Evangelista M, Parsonsa B, Xu H, Bader GD, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science (New York, NY) 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 47.Beltrao P, Cagney G, Krogan NJ. Quantitative genetic interactions reveal biological modularity. Cell. 2010;141:739–745. doi: 10.1016/j.cell.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 49.Collins SR, Schuldiner M, Krogan NJ, Weissman JS. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 2006;7:R63. doi: 10.1186/gb-2006-7-7-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tong AHY, Lesage G, Bader GD, Ding H, Xu H, et al. Global mapping of the yeast genetic interaction network. Science (New York, NY) 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 51.Ihmels J, Collins SR, Schuldiner M, Krogan NJ, Weissman JS. Backup without redundancy: genetic interactions reveal the cost of duplicate gene loss. Mol Syst Biol. 2007;3:86. doi: 10.1038/msb4100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.VanderSluis B, Bellay J, Musso G, Costanzo M, Papp B, et al. Genetic interactions reveal the evolutionary trajectories of duplicate genes. Mol Syst Biol. 2010;6:429. doi: 10.1038/msb.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hart GT, Lee I, Marcotte ER. A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. BMC Bioinform. 2007;8:236. doi: 10.1186/1471-2105-8-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kelley R, Ideker T. Systematic interpretation of genetic interactions using protein networks. Nat Biotechnol. 2005;23:561–566. doi: 10.1038/nbt1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Typas A, Nichols RJ, Siegele DA, Shales M, Collins SR, et al. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat Meth. 2008;5:781–787. doi: 10.1038/nmeth.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Butland G, Babu M, Díaz-Mejía JJ, Bohdana F, Phanse S, et al. eSGA: E. coli synthetic genetic array analysis. Nat Meth. 2008;5:789–795. doi: 10.1038/nmeth.1239. [DOI] [PubMed] [Google Scholar]
- 57.Roguev A, Bandyopadhyay S, Zofall M, Zhang K, Fischer T, et al. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science (New York, NY) 2008;322:405–410. doi: 10.1126/science.1162609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dixon SJ, Fedyshyn Y, Koh JLY, Prasad TSK, Chahwan C, et al. Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proc Natal Acad Sci USA. 2008;105:16653–16658. doi: 10.1073/pnas.0806261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet. 2006;38:896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- 60.Horn T, Sandmann T, Fischer B, Axelsson E, Huber W, et al. Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat Meth. 2011;8(4):341–346. doi: 10.1038/nmeth.1581. [DOI] [PubMed] [Google Scholar]
- 61.Tarailo M, Tarailo S, Rose AM. Synthetic lethal interactions identify phenotypic “interologs” of the spindle assembly checkpoint components. Genetics. 2007;177:2525–2530. doi: 10.1534/genetics.107.080408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tischler J, Lehner B, Fraser AG. Evolutionary plasticity of genetic interaction networks. Nature Genet. 2008;40:390–391. doi: 10.1038/ng.114. [DOI] [PubMed] [Google Scholar]
- 63.Kim D-U, Hayles J, Kim D, Wood V, Park H-O, et al. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2010;28:617–623. doi: 10.1038/nbt.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 65.Lehár J, Stockwell BR, Giaever G, Nislow C. Combination chemical genetics. Nat Chem Biol. 2008;4:674–681. doi: 10.1038/nchembio.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parsons AB, Lopez A, Givoni IE, Williams DE, Gray Ca, et al. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 2006;126:611–625. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 67.Ericson E, Gebbia M, Heisler LE, Wildenhain J, Tyers M, et al. Off-target effects of psychoactive drugs revealed by genome-wide assays in yeast. PLoS Genet. 2008;4:e1000151. doi: 10.1371/journal.pgen.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu D, Jiang B, Ketela T, Lemieux S, Veillette K, et al. Genome-wide fitness test and mechanism-of-action studies of inhibitory compounds in Candida albicans. PLoS Pathog. 2007;3:e92. doi: 10.1371/journal.ppat.0030092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, et al. Phenotypic landscape of a bacterial cell. Cell. 2011;144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hillenmeyer ME, Ericson E, Davis RW, Nislow C, Koller D, et al. Systematic analysis of genome-wide fitness data in yeast reveals novel gene function and drug action. Genome Biol. 2010;11:R30. doi: 10.1186/gb-2010-11-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Han S, Kim D. Inference of protein complex activities from chemical-genetic profile and its applications: predicting drug-target pathways. PLoS Comput Biol. 2008;4:e1000162. doi: 10.1371/journal.pcbi.1000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoon S, Smith AM, Wallace IM, Suresh S, Miranda M, et al. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat Chem Biol. 2008;4:498–506. doi: 10.1038/nchembio.100. [DOI] [PubMed] [Google Scholar]
- 73.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446:815–819. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 74.MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- 75.Castoreno AB, Smurnyy Y, Torres AD, Vokes MS, Jones TR, et al. Small molecules discovered in a pathway screen target the Rho pathway in cytokinesis. Nat Chem Biol. 2010;6:457–463. doi: 10.1038/nchembio.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kapitzky L, Beltrao P, Berens TJ, Gassner N, Zhou C, et al. Cross-species chemogenomic profiling reveals evolutionarily conserved drug mode of action. Mol Syst Biol. 2010;6:1–14. doi: 10.1038/msb.2010.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Borisy Aa, Elliott PJ, Hurst NW, Lee MS, Lehar J, et al. Systematic discovery of multicomponent therapeutics. Proc Natal Acad Sci USA. 2003;100:7977–7982. doi: 10.1073/pnas.1337088100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lehár J, Krueger AS, Avery W, Heilbut AM, Johansen LM, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27:659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spitzer M, Griffiths E, Blakely KM, Wildenhain J, Ejim L, et al. Cross-species discovery of syncretic drug combinations that potentiate the antifungal fluconazole. Mol Syst Biol. 2011;7:499. doi: 10.1038/msb.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jansen G, Lee AY, Epp E, Fredette A, Surprenant J, et al. Chemogenomic profiling predicts antifungal synergies. Mol Syst Biol. 2009;5:338. doi: 10.1038/msb.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cokol M, Chua HN, Tasan M, Mutlu B, Weinstein ZB, et al. Systematic exploration of synergistic drug pairs. Mol Syst Biol. 2011;7:1–9. doi: 10.1038/msb.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choi H, Larsen B, Lin Z-Y, Breitkreutz A, Mellacheruvu D, et al. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Meth. 2011;8:70–73. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brenner S. The Next 100 Years of Biology (lecture) 2006 Available: http://www.mc.vanderbilt.edu/discoveryseries/speaker.html?sid=1.
- 84.Wagner A. Distributed robustness versus redundancy as causes of mutational robustness. BioEssays. 2005;27:176–188. doi: 10.1002/bies.20170. [DOI] [PubMed] [Google Scholar]
- 85.Tsong AE, Tuch BB, Li H, Johnson AD. Evolution of alternative transcriptional circuits with identical logic. Nature. 2006;443:415–420. doi: 10.1038/nature05099. [DOI] [PubMed] [Google Scholar]
- 86.Drury LS, Diffley JFX. Factors affecting the diversity of DNA replication licensing control in eukaryotes. Curr Biol. 2009;19:530–535. doi: 10.1016/j.cub.2009.02.034. [DOI] [PubMed] [Google Scholar]
- 87.Haber JE, Koshland DE., Jr An evaluation of the relatedness of proteins based on comparison of amino acid sequences. J Mol Biol. 1970;50(3):617–39. doi: 10.1016/0022-2836(70)90089-6. [DOI] [PubMed] [Google Scholar]
- 88.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natal Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Uetz P, Giot L, Cagney G, Mansfield Ta, Judson RS, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 90.Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, et al. A protein interaction map of Drosophila melanogaster. Science (New York, NY) 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 91.Li S, Armstrong CM, Bertin N, Ge H, Milstein S, et al. A map of the interactome network of the metazoan C. elegans. Science (New York, NY) 2004;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 93.Rual J-F, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 94.Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 95.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 96.Gavin A-C, Bösche M, Krause R, Grandi P, Marzioch M, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 97.Ewing RM, Chu P, Elisma F, Li H, Taylor P, et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol. 2007;3:89. doi: 10.1038/msb4100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Butland G, Peregrín-Alvarez JM, Li J, Yang W, Yang X, et al. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature. 2005;433(7025):531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 99.Guruharsha KG, Rual J-F, Zhai B, Mintseris J, Vaidya P, et al. A protein complex network of Drosophila melanogaster. Cell. 2011;147:690–703. doi: 10.1016/j.cell.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, et al. Global landscape of HIV–human protein complexes. Nature. 2011:1–6. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]