

Abstract
Metastasis is a key step in cancer progression, and was traditionally attributed to the accumulation of genetic and epigenetic changes within individual cancer cells. These changes promoted invasiveness, immune evasion and survival at distant sites. However, recent studies reveal that metastasis is not achieved by the cancer cell in isolation, but requires intervention from the immune system. The myeloid cell population in particular is now implicated in many aspects of metastasis. Here, we bring together the evidence for the importance of various myeloid cell sub-populations throughout the metastatic process, from initiation of cancer cell invasiveness, to priming the tissue site for colonization.
Keywords: myeloid, MDSC, macrophages, cancer, metastasis, EMT, tumor
Introduction
Metastasis is the multi-step process by which cancer cells spread from the primary tumor site to other parts of the body, and is responsible for around 90% of cancer deaths.1 To be successful, the process requires the individual cancer cell to acquire a host of new characteristics and abilities that are needed in order to overcome the considerable barriers to the establishment of tumors within the body. However, it is now apparent that these invading cancer cells are not acting alone. Recent studies have highlighted the requirement for support from certain cells of the immune system which aid in each of the major steps of metastasis. This timely review will bring together the evidence for a role of the myeloid cell compartment in the spread of cancer around the body.
The myeloid compartment of the immune system is comprised of a heterogeneous population of cells, all of which originate from the bone marrow, but mature into sub-populations with diverse and unique properties. Neutrophils, eosinophils, dendritic cells (DC) and mast cells are known to play some part in the different phases of metastasis, but in recent years it is the tumor-associated macrophages and myeloid-derived suppressor cells which have received the most attention as key players in supporting the metastatic process.
Tumor-associated macrophages (TAMs) share some similarities with the M2 macrophage subset, also known as alternatively activated macrophages. Broadly speaking, M2 macrophages exhibit anti-inflammatory features; downregulating expression of their major histocompatibility complex (MHC) molecules and interleukin [IL-12, while expressing high levels of IL-10, scavenger receptor A, and arginase (Arg)]. M2 macrophages are involved in wound-healing and angiogenesis.2 This is in contrast to the other main macrophage phenotype, M1, which is associated with anti-tumor responses and production of high levels of pro-inflammatory cytokines including TNFα, IL-1, IL-6, IL-12 and inducible nitric oxide synthase.2 For more information on macrophage polarization and subsets, see the recent review by Sica and Mantovani.3
Myeloid-derived suppressor cells (MDSCs) also exhibit anti-inflammatory properties, and have come into prominence in recent years as their role in cancer progression has been uncovered.4 A significant amount of evidence now indicates that MDSCs accumulate in most malignant murine and human tumors.5,6 Human MDSCs are defined by their CD11b+CD33+HLA-DR- phenotype.7 The murine MDSC population express the phenotypic markers CD11b and Gr-1 but is in fact a heterogeneous group of cells comprising myeloid progenitors, immature macrophages, immature granulocytes and immature dendritic cells.4 Despite their heterogeneity, MDSC are unified by their activated phenotype [characterized by high expression of Arg1, reactive oxygen species (ROS) and nitrogen species (nitric oxide, NO)] and their ability to suppress T cell functions. In both healthy mice and in humans, immature myeloid cells normally lack this suppressive activity and primarily reside in the bone marrow, but in pathological conditions, of which cancer has been the most studied, MDSCs accumulate in secondary lymphoid organs, in blood and in tumors. This redistribution is associated with the production of various pro-tumoral growth factors, chemokines and cytokines.4 A recent in-depth review of the subsets and functions of MDSCs can be found in Zhi, et al.8
We will now review how the various subsets of myeloid cells contribute to the many steps of metastasis, namely delamination from the primary tumor mass, invasion of the surrounding tissues, intra- and extra-vasation and eventually colonization of the metastatic site (Fig. 1).
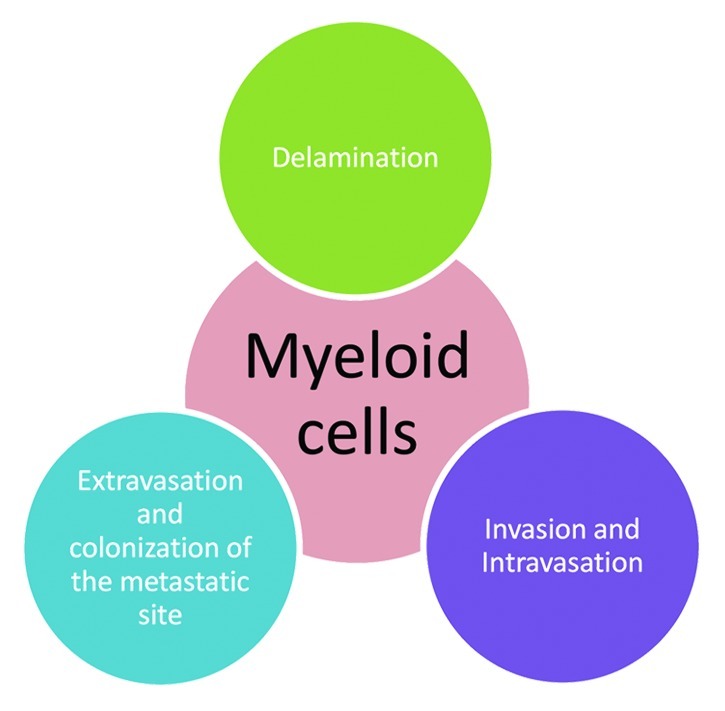
Figure 1. Myeloid cells play important roles in the metastatic process in cancers – from detachment from the primary tumor stroma to colonization of the pre-metastatic niche at distal sites.
Metastasis Step 1—Delamination
Ramón y Cajal, in the 1800s, identified undifferentiated breast carcinomas undergoing physiological changes that allowed them to migrate. “The epithelial islands are not surrounded by a basement membrane… We shall mention the fusiform, pear-like and star-like forms… The cells are not attached to each other…”9 The metastatic process begins with cancer cells acquiring the ability to detach from the primary tumor mass (delamination). This change is often driven by the process of epithelial-mesenchymal transition (EMT),10 and myeloid cells play an important part here. EMT is a coordinated conversion of epithelial cells toward a mesenchymal-like phenotype. Although EMT has mainly been described in embryonic development, it is also involved in cancer progression.10 EMT generally begins with the loss of apical-basal polarity, followed by disassembly of the cell-cell junctions and degradation of the basement membrane.11 Cell surface proteins, such as E-cadherin that mediate epithelial connections to adjacent cells, are replaced with integrins.12 The peripheral actin cytoskeleton is replaced by stress fibers, while cytokeratin intermediate filaments are replaced by vimentin,13 and it is with these changes that the cell acquires the ability to detach from the primary tumor site.
MDSCs can be split into two morphologically distinct populations, monocytic (Mo-) and granulocytic (G-) MDSCs.5 Mo- and G-MDSCs also differ in the expression of selected markers, the mechanisms by which they suppress T-cell responses and their level of differentiation.8 In our studies, we have shown that G-MDSCs induce mesenchymal transition (MT) in a mouse model of spontaneous melanoma.14 Depletion of G-MDSCs resulted in a significant inhibition of tumor cell dissemination and a decrease in metastasis. MT in this model was activated through three pathways: (1) epidermal growth factor (EGF), (2) transforming growth factor (TGF)-β1 and (3) hepatocyte growth factor (HGF). G-MDSCs themselves were the main intra-tumoral source of TGFβ1 and HGF. Like the murine model, EGF, HGF and TGFβ also play important roles in human cancers; HGF, c-Met (the receptor for HGF) and EGF are all expressed in uveal melanoma, where vitreal EGF levels correlate with scleral invasion.15 Both c-Met16 and TGFβ17 activity in melanoma are linked with increased metastatic potential.
TAMs also have the capacity to support EMT. In a mouse mammary tumor model, the tumor cells recruited TAM through the secretion of colony stimulating factor 1 (CSF-1), which in turn stimulated expression of EMT-promoting EGF by TAMs.18 In vitro, macrophage-conditioned medium destabilizes the adherens junctions in a human liver carcinoma cell line by promoting increased tyrosine phosphorylation of β-catenin. This leads to decreased E-cadherin surface expression in the cancer cells. Inhibition of EGF by gefitinib partially blocked the effect of the conditioned medium.19 TGFβ is also involved in macrophage-induced EMT, and is expressed at high levels by M2 macrophages in particular.20 This is not unexpected, as macrophages play an important role in wound-healing, a physiological process that requires cells to undergo EMT.21 In vitro, the macrophage cell line, RAW 264.7, can augment TGFβ-driven EMT by expressing tumor necrosis factor (TNF)-α.22 In human gliomas TAMs at the invasive edge of the tumor express TGF-β that stimulates glioma stem-like cells to express matrix metalloproteinase (MMP)-9, which allows the extravasation of the cancer cells.23 Expression of MMPs by TAMs can also help break-down the extracellular matrix (ECM) and release ECM-bound factors such as TGF-β, thus forming a positive feedback loop. These steps are summarized in Figure 2.

Figure 2. MDSC and macrophages induce invasion early in tumor development. Key to this is the secretion of factors that induce EMT. Hepatocyte growth factor (HGF), epidermal growth factor (EGF), transforming growth factor (TGF)-β and migration-stimulating factor (MSF) are all factors secreted by the myeloid cells infiltrating the primary tumors. These factors promote invasiveness of the cancer cells. Matrix metalloproteinases (MMPs) degrade the ECM, releasing pro-invasive factors such as TGF-β. Macrophages might also play a unique role in inducing an invasive phenotype by fusing with cancer cells, transferring their motile phenotype to the latter.
Metastasis Step 2—Invasion and Intravasation
Once they have detached from the primary tumor, cancer cells must navigate on and through the ECM to reach the blood or lymphatic vessels.24 A study by Solinas, et al. identified migration-stimulating factor (MSF) produced by TAMs as an important promoter of tumor cell motility and invasiveness.25 Molecular messengers can also be important in inducing cancer cell invasiveness; Yang, et al. demonstrated that M2 macrophages co-cultured with breast cancer cells exchange miRNA, specifically miR-223, through exosomes. miR-223 that entered the tumor cells had the ability to increase their invasiveness.26
In addition to the roles of MDSCs and TAMs, mast cell numbers are increased in human cancers and this correlates with a poorer prognosis.27 This might be accounted for by the fact that mast cell factors increase the invasiveness of thyroid carcinoma cells in vitro.28 Mast cells also have a similar effect on invasiveness in pancreatic cancer cells.27 It has been postulated that this invasive effect is promoted by tryptase secreted by mast cells and subsequently stimulating MMP-2 production in breast cancer cells, MDA-MB-231, resulting in increased invasion.29
Eosinophils have also been associated with an increased risk of metastasis in a number of cancers,30 however, there are some reports of eosinophils inhibiting tumor growth and reducing the invasive phenotype of cancer cells.31,32 As such, the effect of eosinophils on cancer metastasis is still undetermined.
Other than increasing invasiveness, proteolytic degradation of the ECM is physically important in allowing tumor cells to migrate. Macrophages express a number of proteases such as MMPs, serine proteases and cathepsins.33 These proteinases, especially MMP2, help break down the ECM, permitting metastasis of tumor cells. ECM remodeling also releases matrix-bound factors such as TGFβ which further augment the invasion.34 In a mouse model of epidermal squamous cell carcinoma, TAMs produce MMP-9 which degrades the ECM, releasing pro-angiogenic factors. When tumor-bearing mice were crossed with MMP-9 deficient mice, angiogenesis and invasion were both delayed. This effect was reversed when wild-type bone marrow cells were transferred to the MMP-9 deficient mice, restoring normal malignancy to the tumors.35 Removal of macrophage-derived cathepsin B and S, in vivo, reduces metastasis in mouse models.36 Other MMP-driven forms of ECM remodeling, such as collagen re-structuring, have also been described; however these functions were not directly attributed to macrophage-derived factors.37
Other myeloid cell types further promote the ECM remodeling required for tumor cell migration; in mouse mammary carcinomas, MDSCs accumulate in the primary tumor and promote metastasis through increased production of MMP-14, MMP-13 and MMP-2.38 In a murine model of colorectal cancer, accumulation of immature myeloid cells, distinct from MDSCs, favors invasion through the production of MMP-2 and MMP-9.39 Another mechanism inducing invasiveness relies on urokinase plasminogen activator (uPA) produced by myeloid cells. The uPA receptor is involved in ECM degradation via cell surface plasminogen activation, and has been linked to metastasis and poor prognosis in gastric cancer.40 Mast cells can express proteases directly or induce other cell types to produce proteases. Mouse mast cell protease (mMCP) -4 and mMCP-6 can activate pro-MMP-9 and release other pro-angiogenic factors from the ECM.41
Few studies have shown in vivo that myeloid cell-derived proteases play a direct role in helping tumor cell migration through the ECM. This is confounded by the fact that tumor cells produce MMPs as well. Identifying the source of these proteases in the tumor could help in the therapeutic targeting of the main protease-producing cells. More studies taking advantage of 2-photon intra-vital microscopic techniques, such as those by Wyckoff, et al., might prove instructive in elucidating, specifically, the role of myeloid cell-derived proteases.18
Following migration through the ECM, cancer cells must reach a blood or lymphatic vessel if they are to be transported to a distant site (intravasation). Therefore, within the primary tumor, angiogenesis, and to a certain extent lymphangiogenesis, are important processes in the dissemination of tumor cells.42,43 Vascular endothelial growth factors (VEGFs) are essential for both angiogenesis (via VEGF-A) and lymphogenesis (via VEGF-C and VEGF-D). MMPs secreted by myeloid cells contribute to this process as well via the ECM degradation that makes available previously-inactive molecules that were bound to the matrix. In dysplasias, MMP-9-expressing CD11b+Gr-1+ cells are involved in the initial angiogenic switching required for pre-malignant lesions to progress; less VEGF to VEGFR binding was found in tumors depleted of these cells.44 The pronociceptive peptide Bv8 (also known as prokineticin 2) is expressed by CD11b+Gr-1+ cells and is known to promote angiogenesis.45 Importantly, this pro-angiogenic function is observed only at the early stages of tumor development, as depletion of Bv8 at later time points had no effect on the tumor vasculature.46
TAMs have been found surrounding blood vessels in necrotic, hypoxic areas of the tumor.47 In human tumors, the density of TAMs correlates with microvessel density,48,49 and the TAMs are known to secrete pro-angiogenic factors including VEGF,47 placental growth factor,50 thymidine phosphorylase49 and platelet-derived endothelial cell growth factor.48 In PyMT mice, which model spontaneous mammary cancer, depletion of macrophages reduced the density of intra-tumoral vessels, while premature infiltration of macrophages in the pre-malignant lesions increased blood vessel density. The authors of this study hypothesized that overexpression of macrophage-derived VEGF may have caused the early blood vessel formation.51
Alongside the TAMs, a specialized subset of highly pro-angiogenic macrophages/monocytes that express the Tie2 (Tyrosine kinase with Ig and EGF homology domain-2) receptor has been identified in both mice and humans. These Tie2-expressing macrophages (TEMs) are found in highly vascularized or stromal regions, but are absent from necrotic tumor regions.52 TEMs are activated by angiopoietin-2, which stimulates the pro-angiogenic activity of TEMs by increasing the expression of thymidine phosphorylase and cathepsin B.53 Removal of TEMs from transplanted human gliomas in a mouse orthotopic model prevented neovascularization and reduced tumor progression.54
In humans, mast cell infiltration is associated with increased microvessel formation in gastric cancer55 and oral squamous cell carcinoma.56 These mast cells are localized around new vessels and correlated with poor prognosis.55 Mast cells produce VEGF,57 angiopoetin-158 and basic fibroblast growth factor59 in the tumor microenvironment, thereby assisting in the angiogenic process. In mouse models, mMCP-4 expressed by mast cells stimulates the angiogenic switch in hyperplastic lesions.41
Tumor-infiltrating DCs, molded by the tumor microenvironment, can be found around capillary-like structures in the tumor. Furthermore, the cells in these structures express both CD31 and CD11c, markers of endothelial cells and dendritic cells respectively. The authors postulated that the tumor-infiltrating DCs acquire an endothelial-like phenotype in the tumor. These DCs were found to be immature and to express VEGF-A, which promoted vascularization of the tumor.60
In terms of lymphangiogenesis, it has been hypothesized that macrophages contribute via at least two mechanisms—by expression of pro-lymphogenic factors, and by acting as progenitors of lymphatic endothelial cells. A subset of TAMs does express VEGF-C and VEGF-D, which are important for lymphangiogenesis and metastasis, at least in transplanted tumor models in mice.61,62 These VEGF-expressing TAMs are located in the peri-tumoral areas and their numbers are correlated with lymphatic vessel density. VEGF-C and -D expression in these macrophages can be induced by TNF-α and VEGF-D.63
In conclusion, we see that myeloid cells play an important role in angiogenesis and, in part, lymphangiogenesis. These processes are invaluable to both tumor growth and the intravasation of tumor cells into the circulation.
Metastasis step 3—Extravasation and Colonization of the Metastatic Site
Metastasis is a very inefficient process. In vivo metastatic assays show that 80% of injected tumor cells survive in the liver microcirculation, but only 2.5% form micrometastases. Of these micrometastases, only 1% goes on to form macrometastases. Overall this adds up to only 0.02% of injected cells achieving tumor formation.64 Although this was illustrated in an artificial transplanted tumor model, the inefficiency of metastasis formation highlights the stringent conditions that are required to support metastatic growth in distal organs. During cancer development, certain sites are induced to become more favorable for distal tumor establishment, and these sites have been termed pre-metastatic niches.65 Myeloid cells play a role in the establishment of this niche.
In cancer patients, tumor-derived factors can alter DC maturation and differentiation. This leads to an accumulation of immature DCs in the primary tumor and the peripheral lymphoid organs. These immature DCs are not able to induce an appropriate anti-tumor response, in addition, immature DCs can tolerize T cells and induce the proliferation of regulatory T cells.66 This results in an overall dampening of the anti-tumor response allowing tumor cells to migrate through the circulation and survive in the distal metastatic site.
MDSCs accumulate in pre-metastatic mouse lungs through expression of VEGF, TGFβ and TNFα by the primary tumor.67 MDSCs that infiltrate the pre-metastatic niche express the inflammatory molecules S100A8/9 which in turn contribute to priming the site for metastasis. In mice that fail to express S100A9, the number of lung metastases is reduced.68 Interestingly, S100A8/9 expressed by the MDSCs in turn recruits more MDSCs and macrophages to the site, forming a positive feedback loop of pro-tumoral site priming.67
MDSCs recruited to the pre-metastatic lung also condition the lung into an inflammatory and immunosuppressive environment suitable for tumor establishment and growth. MDSCs inhibit IFNγ production in the pre-metastatic lung, while in contrast basic fibroblast growth factor, insulin-like growth factor I and the Th2 cytokines IL-4, IL-5, IL-9 and IL-10 are increased in pre-metastatic lungs.69 All these secreted factors create a microenvironment that suppresses anti-tumoral immune responses.69 MMP-9 secreted by infiltrating MDSCs results in disorganized vessel capillaries with poor pericyte coverage and degraded basement membrane, which leads to a leaky vasculature in pre-metastatic lungs, thereby facilitating extravasation of the cancer cells into the stroma.69
CD11b+VEGFR1+ myeloid cells can also secrete MMP-9 which breaks down the ECM in the pre-metastatic niche, releasing matrix-bound VEGF. Depletion of these cells using anti-VEGFR1 antibodies inhibits the formation of pre-metastatic niches.70 However, there are contradicting reports stating that these myeloid cells are dispensable for the initiation of the pre-metastatic niches, even though they are important for the growth of the established metastases.71 Neutrophils, though intra-vital microscopy, have been seen directly binding to circulating tumor cells in the liver sinusoids.72 This interaction, mediated by Mac-1 and ICAM-1, promotes tumor cell adhesion to the endothelial cells, leading to increased metastasis.72
A subset of CD11b+Gr1−F4/80+CD11cloCX3CR1+CCR2+VEGFR1+ macrophages has been shown to interact with tumor cells in the lung vasculature and help tumor cell extravasation. Depletion of macrophages in this system greatly reduced seeding efficiency in the lung.73 Interestingly, these macrophages do not express Tie2 or CXCR4, thus making them a distinct population from the pro-angiogenic TEMs. Furthermore, these inflammatory macrophages that infiltrated the lungs are distinct from resident lung macrophages, as they do not express CD11c.73
Perspectives
Metastasis is an important event in cancer progression. Until recently, the first step of metastasis (i.e., tumor cell dissemination) was thought to be a late event.74 This time lag was presumably needed to allow selected cancer cells to accumulate the additional mutations required for motility and colonization. However, recent work, including that from our own laboratory, has challenged this paradigm. In fact, cancer cells disseminate even before diagnosis of the primary tumor,75 and so a different, faster mechanism must be driving the development of the motile phenotype. Myeloid cells are prime candidates as inducers of early metastasis in cancer. They are one of the first cells arriving at the primary tumor due to the inflammation present at the cancer site.76 They also play important roles in neo-vasculature formation in this area.51
As seen in this review, myeloid cells are crucial at all stages of the metastatic process (Fig. 3). Targeting myeloid cells in combination with classical therapy could improve current treatments. Rational drug combination strategies may allow lowering of the dosage of toxic chemotherapeutic drugs, while maintaining or increasing efficacy. Furthermore, unlike malignant cells, myeloid cells are not genetically instable, potentially making them better targets than the tumor cells themselves.

Figure 3. Summary of the different factors secreted by myeloid cells that aid in the different stages of metastasis. TAMs, tumor-associated macrophages; MDSC, myeloid-derived suppressor cells; TEMs, Tie2-expressing macrophages; DCs, dendritic cells; MCP, mast cell protease; IL, interleukin; VEGF, vascular endothelial growth factor; TNF, tumor necrosis factor; PIGF, placental growth factor; TYMP, thymidine phosphorylase; PD-ECGF, Platelet-Derived Endothelial Cell Growth Factor; TGF, transforming growth factor; EGF, epidermal growth factor; HGF, hepatocyte growth factor; bFGF, basic fibroblast growth factor; IGF, insulin-like growth factor; IFN, interferon; ANG, angiopoietin; MSF, migration stimulating factor; mIR, microRNA; MMP, matrix metalloproteinase.
Some chemotherapeutic drugs reduce myeloid cell numbers. In humans, sunitinib, a receptor tyrosine kinase inhibitor used in renal cell carcinoma, has been shown to reduce MDSC number and function and alleviate their T-cell suppressive effect. Furthermore, sunitinib reduces MDSC viability and function in vitro.77
Anti-VEGF therapy, in addition to its anti-angiogenic effects, may also help in reducing the numbers of myeloid cells.78 Interestingly, in mouse models, refractoriness to anti-VEGF therapy has been shown to be dependent on MDSCs.79 Furthermore, in a tumor xenograft model in zebrafish, inhibition of VEGF signaling effectively suppresses localized tumor growth but accelerates tumor invasiveness and micrometastasis through the recruitment of neutrophils.80 As seen from these contradictory reports, more studies have to be conducted on the targeting of myeloid cells before we can move on to rationally-designed clinical studies.
The diversity of myeloid cells allows them to play multiple roles in tumor progression. However, they are also crucial in normal physiology and responses to infection. Further studies would have to focus on defining the mechanisms underlying the different functions of myeloid cells with respect to tumor progression. Only then would we be able to safely and effectively incorporate anti-myeloid cell therapy into current treatment regimes.
Acknowledgments
The authors thank Dr. L. Robinson from Insight Editing London for manuscript editing.
Glossary
Abbreviations:
- TAMs
tumor-associated macrophages
- MDSC
myeloid-derived suppressor cells
- TEMs
Tie2-expressing macrophages
- DCs
dendritic cells
- IL
interleukin
- VEGF
vascular endothelial growth factor
- TNF
tumor necrosis factor
- TGF
transforming growth factor
- IFN
interferon
- MMP
matrix metalloproteinase
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/22196
References
- 1.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–64. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 2.Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. 2008;267:204–15. doi: 10.1016/j.canlet.2008.03.028. [DOI] [PubMed] [Google Scholar]
- 3.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–95. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 6.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60:1419–30. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol. 2011;11:802–7. doi: 10.1016/j.intimp.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhi L, Toh B, Abastado JP. Myeloid Derived Suppressor Cells: Subsets, Expansion, and Role in Cancer Progression, Tumor Microenvironment and Myelomonocytic Cells. In: Biswas SK, ed. Tumor Microenvironment and Myelomonocytic Cells. Rijeka, Croatia: INTECH, 2012:63-88. [Google Scholar]
- 9.López-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. 2009;1:303–14. doi: 10.1002/emmm.200900043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 11.Townsend TA, Wrana JL, Davis GE, Barnett JV. Transforming growth factor-beta-stimulated endocardial cell transformation is dependent on Par6c regulation of RhoA. J Biol Chem. 2008;283:13834–41. doi: 10.1074/jbc.M710607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peinado H, Portillo F, Cano A. Transcriptional regulation of cadherins during development and carcinogenesis. Int J Dev Biol. 2004;48:365–75. doi: 10.1387/ijdb.041794hp. [DOI] [PubMed] [Google Scholar]
- 13.Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15:117–34. doi: 10.1007/s10911-010-9178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011;9:e1001162. doi: 10.1371/journal.pbio.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Topcu-Yilmaz P, Kiratli H, Saglam A, Söylemezoglu F, Hascelik G. Correlation of clinicopathological parameters with HGF, c-Met, EGFR, and IGF-1R expression in uveal melanoma. Melanoma Res. 2010;20:126–32. doi: 10.1097/CMR.0b013e328335a916. [DOI] [PubMed] [Google Scholar]
- 16.McGill GG, Haq R, Nishimura EK, Fisher DE. c-Met expression is regulated by Mitf in the melanocyte lineage. J Biol Chem. 2006;281:10365–73. doi: 10.1074/jbc.M513094200. [DOI] [PubMed] [Google Scholar]
- 17.Pinner S, Jordan P, Sharrock K, Bazley L, Collinson L, Marais R, et al. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 2009;69:7969–77. doi: 10.1158/0008-5472.CAN-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–56. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 19.Lin CY, Lin CJ, Chen KH, Wu JC, Huang SH, Wang SM. Macrophage activation increases the invasive properties of hepatoma cells by destabilization of the adherens junction. FEBS Lett. 2006;580:3042–50. doi: 10.1016/j.febslet.2006.04.049. [DOI] [PubMed] [Google Scholar]
- 20.Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer. 2012;12:35. doi: 10.1186/1471-2407-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010;11:97–105. doi: 10.1038/embor.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawata M, Koinuma D, Ogami T, Umezawa K, Iwata C, Watabe T, et al. TGF-β-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW 264.7 macrophage cells. J Biochem. 2012;151:205–16. doi: 10.1093/jb/mvr136. [DOI] [PubMed] [Google Scholar]
- 23.Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J Immunol. 2012;189:444–53. doi: 10.4049/jimmunol.1103248. [DOI] [PubMed] [Google Scholar]
- 24.Roussos ET, Condeelis JS, Patsialou A. Chemotaxis in cancer. Nat Rev Cancer. 2011;11:573–87. doi: 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solinas G, Schiarea S, Liguori M, Fabbri M, Pesce S, Zammataro L, et al. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for M2-polarization, influencing tumor cell motility. J Immunol. 2010;185:642–52. doi: 10.4049/jimmunol.1000413. [DOI] [PubMed] [Google Scholar]
- 26.Yang M, Chen J, Su F, Yu B, Su F, Lin L, et al. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol Cancer. 2011;10:117. doi: 10.1186/1476-4598-10-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strouch MJ, Cheon EC, Salabat MR, Krantz SB, Gounaris E, Melstrom LG, et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin Cancer Res. 2010;16:2257–65. doi: 10.1158/1078-0432.CCR-09-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melillo RM, Guarino V, Avilla E, Galdiero MR, Liotti F, Prevete N, et al. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene. 2010;29:6203–15. doi: 10.1038/onc.2010.348. [DOI] [PubMed] [Google Scholar]
- 29.Xiang M, Gu Y, Zhao F, Lu H, Chen S, Yin L. Mast cell tryptase promotes breast cancer migration and invasion. Oncol Rep. 2010;23:615–9. doi: 10.3892/or_00000676. [DOI] [PubMed] [Google Scholar]
- 30.Alrawi SJ, Tan D, Stoler DL, Dayton M, Anderson GR, Mojica P, et al. Tissue eosinophilic infiltration: a useful marker for assessing stromal invasion, survival and locoregional recurrence in head and neck squamous neoplasia. Cancer J. 2005;11:217–25. doi: 10.1097/00130404-200505000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Furbert-Harris PM, Parish-Gause D, Hunter KA, Vaughn TR, Howland C, Okomo-Awich J, et al. Activated eosinophils upregulate the metastasis suppressor molecule E-cadherin on prostate tumor cells. Cell Mol Biol (Noisy-le-grand) 2003;49:1009–16. [PubMed] [Google Scholar]
- 32.Ishibashi S, Ohashi Y, Suzuki T, Miyazaki S, Moriya T, Satomi S, et al. Tumor-associated tissue eosinophilia in human esophageal squamous cell carcinoma. Anticancer Res. 2006;26(2B):1419–24. [PubMed] [Google Scholar]
- 33.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–90. doi: 10.1016/S0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24:241–55. doi: 10.1101/gad.1874010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012;33:119–26. doi: 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H, et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat Genet. 2007;39:467–75. doi: 10.1038/ng1997. [DOI] [PubMed] [Google Scholar]
- 40.Alpízar-Alpízar W, Christensen IJ, Santoni-Rugiu E, Skarstein A, Ovrebo K, Illemann M, et al. Urokinase plasminogen activator receptor on invasive cancer cells: a prognostic factor in distal gastric adenocarcinoma. Int J Cancer. 2012;131:E329–36. doi: 10.1002/ijc.26417. [DOI] [PubMed] [Google Scholar]
- 41.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–97. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dadras SS, Paul T, Bertoncini J, Brown LF, Muzikansky A, Jackson DG, et al. Tumor lymphangiogenesis: a novel prognostic indicator for cutaneous melanoma metastasis and survival. Am J Pathol. 2003;162:1951–60. doi: 10.1016/S0002-9440(10)64328-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park Y, Kitahara T, Takagi R, Kato R. Does surgery for breast cancer induce angiogenesis and thus promote metastasis? Oncology. 2011;81:199–205. doi: 10.1159/000333455. [DOI] [PubMed] [Google Scholar]
- 44.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493–8. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–31. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- 46.Shojaei F, Singh M, Thompson JD, Ferrara N. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc Natl Acad Sci U S A. 2008;105:2640–5. doi: 10.1073/pnas.0712185105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leek RD, Hunt NC, Landers RJ, Lewis CE, Royds JA, Harris AL. Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J Pathol. 2000;190:430–6. doi: 10.1002/(SICI)1096-9896(200003)190:4<430::AID-PATH538>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 48.Matsumura M, Chiba Y, Lu C, Amaya H, Shimomatsuya T, Horiuchi T, et al. Platelet-derived endothelial cell growth factor/thymidine phosphorylase expression correlated with tumor angiogenesis and macrophage infiltration in colorectal cancer. Cancer Lett. 1998;128:55–63. doi: 10.1016/S0304-3835(98)00051-2. [DOI] [PubMed] [Google Scholar]
- 49.Kawahara A, Hattori S, Akiba J, Nakashima K, Taira T, Watari K, et al. Infiltration of thymidine phosphorylase-positive macrophages is closely associated with tumor angiogenesis and survival in intestinal type gastric cancer. Oncol Rep. 2010;24:405–15. doi: 10.3892/or_00000873. [DOI] [PubMed] [Google Scholar]
- 50.Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19:31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 51.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 52.De Palma M, Naldini L. Tie2-expressing monocytes (TEMs): novel targets and vehicles of anticancer therapy? Biochim Biophys Acta. 2009;1796:5–10. doi: 10.1016/j.bbcan.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 53.Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010;70:5270–80. doi: 10.1158/0008-5472.CAN-10-0012. [DOI] [PubMed] [Google Scholar]
- 54.De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–26. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 55.Yano H, Kinuta M, Tateishi H, Nakano Y, Matsui S, Monden T, et al. Mast cell infiltration around gastric cancer cells correlates with tumor angiogenesis and metastasis. Gastric Cancer. 1999;2:26–32. doi: 10.1007/s101200050017. [DOI] [PubMed] [Google Scholar]
- 56.Kalra M, Rao N, Nanda K, Rehman F, Girish KL, Tippu S, et al. The role of mast cells on angiogenesis in oral squamous cell carcinoma. Med Oral Patol Oral Cir Bucal. 2012;17:e190–6. doi: 10.4317/medoral.17395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sawatsubashi M, Yamada T, Fukushima N, Mizokami H, Tokunaga O, Shin T. Association of vascular endothelial growth factor and mast cells with angiogenesis in laryngeal squamous cell carcinoma. Virchows Arch. 2000;436:243–8. doi: 10.1007/s004280050037. [DOI] [PubMed] [Google Scholar]
- 58.Nakayama T, Yao L, Tosato G. Mast cell-derived angiopoietin-1 plays a critical role in the growth of plasma cell tumors. J Clin Invest. 2004;114:1317–25. doi: 10.1172/JCI22089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qu Z, Huang X, Ahmadi P, Stenberg P, Liebler JM, Le AC, et al. Synthesis of basic fibroblast growth factor by murine mast cells. Regulation by transforming growth factor beta, tumor necrosis factor alpha, and stem cell factor. Int Arch Allergy Immunol. 1998;115:47–54. doi: 10.1159/000023829. [DOI] [PubMed] [Google Scholar]
- 60.Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10:950–8. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 61.Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–8. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- 62.Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186–91. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- 63.Schoppmann SF, Birner P, Stöckl J, Kalt R, Ullrich R, Caucig C, et al. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol. 2002;161:947–56. doi: 10.1016/S0002-9440(10)64255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865–73. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285–93. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Janikashvili N, Bonnotte B, Katsanis E, Larmonier N. The dendritic cell-regulatory T lymphocyte crosstalk contributes to tumor-induced tolerance. Clin Dev Immunol. 2011;2011:430394. doi: 10.1155/2011/430394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8:1369–75. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 68.Ichikawa M, Williams R, Wang L, Vogl T, Srikrishna G. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol Cancer Res. 2011;9:133–48. doi: 10.1158/1541-7786.MCR-10-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, et al. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010;70:6139–49. doi: 10.1158/0008-5472.CAN-10-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2:289–300. doi: 10.1016/S1535-6108(02)00153-8. [DOI] [PubMed] [Google Scholar]
- 71.Dawson MR, Duda DG, Chae SS, Fukumura D, Jain RK. VEGFR1 activity modulates myeloid cell infiltration in growing lung metastases but is not required for spontaneous metastasis formation. PLoS One. 2009;4:e6525. doi: 10.1371/journal.pone.0006525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spicer JD, McDonald B, Cools-Lartigue JJ, Chow SC, Giannias B, Kubes P, et al. Neutrophils Promote Liver Metastasis via Mac-1-Mediated Interactions with Circulating Tumor Cells. Cancer Res. 2012;72:3919–27. doi: 10.1158/0008-5472.CAN-11-2393. [DOI] [PubMed] [Google Scholar]
- 73.Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One. 2009;4:e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 75.Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest. 2010;120:2030–9. doi: 10.1172/JCI42002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010;10:369–73. doi: 10.2174/156652410791316968. [DOI] [PubMed] [Google Scholar]
- 77.Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–57. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 78.Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92:4150–66. [PubMed] [Google Scholar]
- 79.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–20. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 80.He S, Lamers GE, Beenakker JW, Cui C, Ghotra VP, Danen EH, et al. Neutrophil-mediated experimental metastasis is enhanced by VEGFR inhibition in a zebrafish xenograft model. J Pathol. 2012;227:431–45. doi: 10.1002/path.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]