

Abstract
Significance: Oxidation and reduction events are critical to physiological and pathological processes and are highly regulated. Herein, we present evidence for the role of Ras and Rho GTPases in controlling these events and the unique underlying mechanisms. Evidence for redox regulation of Ras GTPases that contain a redox-sensitive cysteine (X) in the conserved NKXD motif is presented, and a growing consensus supports regulation by a thiyl radical-mediated oxidation mechanism. We also discuss the debate within the literature regarding whether 2e− oxidation mechanisms also regulate Ras GTPase activity. Recent Advances: We examine the increasing in vitro and cell-based data supporting oxidant-mediated activation of Rho GTPases that contain a redox-sensitive cysteine at the end of the conserved phosphoryl-binding loop (p-loop) motif (GXXXXG[S/T]C). While this motif is distinct from Ras, these data suggest a similar 1e− oxidation-mediated activation mechanism. Critical Issues: We also review the data showing that the unique p-loop placement of the redox-sensitive cysteine in Rho GTPases supports activation by 2e− cysteine oxidation. Finally, we examine the role that Ras and Rho GTPases play in controlling key oxidant-regulating enzymes in the cell, and we speculate on a feedback mechanism. Future Directions: Given that these GTPases and redox-regulating enzymes are involved in multiple physiological and pathological processes, we discuss future experiments that may clarify the interplay between them. Antioxid. Redox Signal. 18, 250–258.
Introduction
Oxidation and reduction reactions are important in numerous signaling pathways that regulate critical physiological processes, and the balance between oxidation and reduction reactions (redox state) is a critical contributor to many diseases. The reactive intermediates (RIs) involved in these processes include reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive thiols. Moreover, protein-RI reactions are diverse and can generate many products. While a number of review articles have concentrated on the diverse RI sources and elimination mechanisms found in cells, this review will focus on the regulation of select Ras and Rho proteins by reversible thiol oxidation.
Ras and Rho GTPases are involved in many fundamental signaling pathways that affect cell cycle, growth, apoptosis, motility, and morphology (25). Dysregulation of these proteins can lead to numerous disease states, including cancer, cardiovascular disease, and neurological disorders. Ras and Rho GTPases are molecular “switches.” GTP binding alters their conformation, which promotes effector binding and turns downstream signaling “on,” whereas the GDP-bound form reduces effector binding, turning signaling “off.” In general, three distinct classes of proteins regulate the level of activated GTPases in the cell (Fig. 1).
FIG. 1.

GTPase nucleotide cycle. GTPases are active in the GTP-bound form. GTPase activating proteins (GAPs) promote GTP hydrolysis, which results in the inactive, GDP-bound form. Guanine nucleotide exchange factors (GEFs) facilitate nucleotide dissociation, and, as the in-cell GTP:GDP ratio is typically high (≥10:1), GTPases bind GTP by mass action and become activated. GDIs can also regulate Rho GTPases by sequestering them, which prevents nucleotide exchange and membrane insertion. We hypothesize that physiological levels of RIs facilitate exchange, which typically populates GTPases in the active conformation, whereas oxidative stress conditions inactivate the GTPases. RhoA is shown in green. The regions that undergo nucleotide-dependent changes in conformation are highlighted in red (switch I) and magenta (switch II). GEF is indicated in yellow, GAP is colored orange, and GDI is in teal. The following structures (PDB ID) were used to generate this model: RhoA-GTP (1A2B), RhoA-GTP with a GAP (1OW3), RhoA-GDP (1FTN), RhoA-GDP complexed with GDI (1CC0), and RhoA-GDP complexed with a GEF (1LB1). GDP, guanosine diphosphate; GDI, GDP dissociation inhibitor; RIs, reactive intermediates. (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars.)
Herein, we describe redox mechanisms involved in regulating Ras and Rho GTPases. We discuss how RIs alter the biochemical properties of these GTPases and the consequences on cell function. For Ras GTPases, recent publications that demonstrate a direct role for RI regulation of Ras in cancer pathogenesis are described. We also discuss evidence for indirect and direct RI regulation of Rho GTPases, highlight cell-based studies that show a strong role for Rho GTPases in regulating key redox-modulating enzymes, and propose a feedback mechanism for redox status in the cell. We conclude by suggesting areas for future research and experimental approaches to address remaining questions.
Redox Regulation of Ras and Rho GTPases: In Vitro Studies
Select free radical oxidants (1e−) can regulate the activity of Ras superfamily GTPases, which possess key cysteine thiol(s). In these GTPases, NO•/O2 and •NO2 have been shown to induce guanine nucleotide oxidation and dissociation (21). As the intracellular GTP:GDP ratio is greater than or equal to 10:1 (50), redox-mediated nucleotide dissociation likely populates these GTPases in the active GTP-bound state; however, inactivation has also been observed, often correlated with oxidative stress. While there is still much debate on whether Ras GTPases are regulated by 2e− (covalent) oxidation, a subset of Rho GTPases are activated by 1e− and 2e− cysteine oxidation. As RIs modulate the activity of several GTPases (Ras and Rho subclasses) involved in cell growth and motility, elucidating the mechanisms for GTPase regulation by redox control may provide new directions for drug targeting.
RI function, regulation, and production
RIs can be beneficial and harmful in biology. They comprise a number of reactive molecules that can interconvert into other RIs and control physiological processes by interacting with cellular components, including lipids, organelles, proteins, and nucleic acids (43, 54). Further, to maintain redox homeostasis in the cell and counterbalance the deleterious effects from RIs, cells have evolved a system of antioxidant molecules and enzymes. Excess RIs can cause changes in the cell redox potential, resulting in various disease states, such as atherosclerosis, cancer, diabetes, and neurodegenerative disorders. As previous reviews have detailed the chemistry underlying these reactions (4, 10, 53), this review will focus on RI-mediated cysteine oxidation and the effects on Ras and Rho GTPases.
ROS are produced in vivo by many enzymes and processes; examples include hydrogen peroxide (H2O2), superoxide (O2•−), and hydroxyl radical (•OH). ROS can convert into other ROS forms through enzymes, metal catalysis, and free-radical-mediated reactions; for example, in the presence of transition metals, such as Cu2+ and Fe3+, the Fenton reaction converts H2O2 into •OH, a short-lived and highly reactive free radical (55). NADPH oxidase (Nox) is a major source of O2•− in cells, and O2•− is converted into H2O2 by superoxide dismutase (SOD) (6, 36). Nitric oxide (NO•) is an important RNS in the cell and is produced by inorganic nitrate reduction and nitric oxide synthase (NOS) (38). Nitric oxide modulates multiple cell processes, such as blood pressure regulation, platelet aggregation, and smooth muscle relaxation (14). The effects of NO• in vascular disease and cancer biology have been previously reviewed (32, 52). While the redox potential of nitric oxide is considered too low to directly generate thiyl-radical intermediates from protein thiols, its autooxidation product •NO2 can (4). Peroxynitrite (ONOO−) is another RNS and can be produced from the direct combination of NO• with O2•− (5). Herein, we discuss mechanisms through which cellular NO generation leads to Ras and Rho GTPase activation.
Free radicals modify and activate Ras through a reactive cysteine in the NKCD motif
Ras superfamily GTPases have four conserved nucleotide-binding motifs (29). While the X residue in the NKXD motif is not well-conserved, several Ras subclass members contain a redox-active cysteine at this position (Supplementary Table S1). GTPases with a redox-sensitive NKCD motif can be activated by •NO2 and other RIs; however, NO-mediated regulation is best characterized for Ras. Several cell-based and in vitro studies have shown that •NO2 reacts with Ras through Cys118 to promote nucleotide exchange and Ras activation (10). Our lab has employed NMR and biochemical approaches to show that S-nitrosation of Ras does not affect Ras structure or nucleotide binding (56). We speculated that an intermediate formed during the reaction of Ras with •NO2 modulates Ras activity and investigated several reactions involved in thiol S-nitrosation. Further, we observed that 1e− oxidation (radical-mediated) reactions, which have been postulated to promote thiyl radical formation, enhance nucleotide dissociation from Ras (18). In support of these observations, we have recently confirmed Cys118 radical formation upon exposure of Ras to diethylamine NONOate (NO•-generating agent) and •NO2, using immune-spin trapping and Fourier transform ion cyclotron resonance mass spectrometry (MS) (article in submission). As guanine bases are susceptible to radical-mediated oxidation, we speculated that thiyl radical generation promoted oxidation of the Ras-bound guanine nucleotide. Supporting this premise, we used MS to demonstrate that 5-guanidino-4-nitroimidazole diphosphate (NIm-DP) is formed in the presence of NO•/O2 and •NO2. NIm-DP is a reported breakdown product of 5-nitro-guanine ribose diphosphate, which can be generated upon reaction of guanosine diphosphate with •NO2 (21). Further, mutation of the redox-sensitive cysteine (RasC118S) prevented NO-mediated nucleotide oxidation and dissociation (20). These results suggest that NO•/O2 or •NO2 can produce a Ras thiyl radical (RasC118•) that propagates to the guanine base by electron transfer (Fig. 2A). Guanine base oxidation disrupts critical intermolecular contacts between the nucleotide and Ras nucleotide binding pocket, which enhances nucleotide dissociation. These data suggest that RasC118 thiol nitrosation does not promote nucleotide exchange; rather, exchange is promoted by thiyl radical formation at RasC118 (Fig. 2A, B). Further supporting this model, we demonstrated that nucleotide binding is not altered by either RasC118 mutation or S-nitrosation (20). A proposed thiyl-radical-based mechanism has been described in detail elsewhere (17) but remains inconclusive, as direct evidence to support the proposed radical intermediates has not yet been obtained.
FIG. 2.

Regulation of Ras guanine nucleotide binding by 1e− and 2e− oxidation. (A) Reaction of Ras with •NO2 (a 1e− oxidant) has been shown to generate NIm-DP, a breakdown product of 5-nitro-GDP. In the proposed mechanism, a thiyl radical is generated at Cys118 (asterisk) and transferred to the guanine base bound to Ras (arrow). Phe28 likely maintains the nucleotide orientation for proper radical transfer; however, the exact mechanism of electron transfer is under investigation. The end result of the radial-mediated reaction is oxidation of the guanine base, a loss of interactions between Ras and GDP (depicted by the absence of hydrogen bonds), and dissociation of the bound nucleotide. (B) A 2e− (covalent) oxidation reaction of Ras with CysNO, which results in covalent modification of Cys118 with NO, does not affect the interactions with the bound nucleotide (hydrogen bonds are shown as dotted blue lines). Williams et al. showed using NMR that nitrosation of Cys118, using CysNO, does not perturb the effector and switch regions of Ras (56). This observation was further reinforced by biochemical data that did not show a change in activity after covalent modification. Figure adapted from PDB ID:1CRP using Pymol. The color scheme is white, carbon; blue, nitrogen; salmon, oxygen; orange, phosphate; and yellow, sulfur. All distances are reported in angstroms. 5-nitro-GDP, 5-nitro-guanine ribose diphosphate; NIm-DP, 5-guanidino-4-nitroimidazole diphosphate. (To see this illustration in color the reader is referred to the web version of this article at www.liebertpub.com/ars.)
In contrast to our observations that Cys118 nitrosation does not alter Ras activity, Clavreul et al. suggested that covalent modification of Ras by glutathione enhances nucleotide exchange (8). In these experiments, RasWT, but not RasC118S, was activated by glutathiolation using peroxynitrite, and it was hypothesized that RasC118 glutathiolation by 2e− oxidation (Fig. 2B) perturbs nucleotide binding because glutathione is negatively charged and larger than nitric oxide. Clavreul et al. concluded that glutathiolation significantly altered Ras structure at the nucleotide binding pocket and enhanced nucleotide exchange (8, 9). However, peroxynitrite was used in these experiments, and its breakdown products can generate RIs capable of thiyl radical formation. Peroxynitrite can undergo homolysis to generate •NO2 and •OH. Moreover, in the presence of CO2, peroxynitrite can produce carbonate radicals (39). While Kissner and Koppenol have observed that “radical-free” peroxynitrite homolysis produces nitrite and molecular oxygen (28), Szabo et al. discussed peroxynitrite homolysis products and concluded that it generates radical products (49). Therefore, it is impossible to judge whether the effects of glutathiolation on Ras activity were induced by free radicals or direct Cys118 modification. While Adachi et al. suggested that Ras S-nitrosation may be an intermediate during glutathione modification of Ras, they concluded that covalent (2e−) glutathione modification of Ras leads to Ras activation (1). However, the observation that Ras glutathiolation alters nucleotide exchange contradicts our previous studies, which demonstrated that Cys118 and 1e− oxidants are required for regulation of Ras nucleotide binding and activity. To address this discrepancy, we have recently demonstrated that treatment of Ras with oxidized glutathione (GSSG) results in glutathiolation of Ras at Cys118. However, similar to S-nitrosation, glutathione modification of Ras does not alter Ras nucleotide binding (unpublished observations). Therefore, we propose that Ras activation requires Cys118 thiyl-radical formation for nucleotide dissociation and subsequent activation. One end product of RasC118 thiyl-radical generation is RasSSG. Thus, the presence of RasSSG may reflect Ras activation if it was generated by a reaction with the RasC118 thiyl radical; this is also the most likely mechanism in cells given the slow reaction rate for the Ras thiol and GSSG under physiological conditions (Fig. 2).
Redox regulation of Rho GTPases by 1e− and 2e− mechanisms through a redox-active cysteine in the phosphoryl-binding loop
Our lab has also demonstrated that select Rho GTPases are redox sensitive; however, Rho GTPases contain a different redox motif than that of Ras (19). Over 50% of Rho GTPases contain a redox-sensitive cysteine at the end of the nucleotide-binding phosphoryl-binding loop (p-loop) motif, GXXXXGK[S/T]C (Supplementary Table S2). Based on the Rac1, Cdc42, and RhoA crystal structures, the solvent-exposed cysteine in the p-loop motif likely has an altered pKa and is accessible to RIs. We have previously shown that the reactive p-loop cysteines in Rho GTPases are sensitive to oxidation, resulting in altered GTPase regulation (19).
Whereas treatment of RhoA with •NO2 can promote nucleotide oxidation and dissociation similar to Rac1 and Cdc42, it can inhibit nucleotide binding in the absence of reducing agents. Distinct from Rac1 and Cdc42, RhoA contains two cysteines in its p-loop (Cys16 and Cys20, Fig. 3; Supplementary Table S1). Exposure of RhoA with •NO2 can promote disulfide bond formation between Cys16 and Cys20, which occludes nucleotide binding (22). However, nucleotide binding can be restored with disulfide reducing agents. Our findings indicate that thiyl radical formation at RhoAC20 upon •NO2 treatment facilitates nucleotide oxidation and release, which increases RhoAC16 accessibility and promotes intramolecular disulfide bond formation with Cys20. RhoA disulfide bond formation occludes nucleotide binding and promotes RhoA inactivation. Based on these observations, we postulate that RhoA is likely activated by thiyl radical-promoting RIs under conditions where the redox potential is capable of promoting disulfide reduction and restoration of RhoA nucleotide binding (2, 44). Redox-mediated inactivation of Ras may occur when the cellular reduction potential is reduced, such as during oxidative stress. It is intriguing to speculate that enzymes, such as thioredoxin and glutaredoxin, which are capable of reducing protein disulfides, may act on RhoA and contribute to activation.
FIG. 3.

The phosphoryl-binding loop of RhoA bound to GDP. The phosphoryl-binding loop of Rho GTPases, including Cys20 and Cys16, makes several critical hydrogen bonds with the bound nucleotide. Oxidation of RhoA by 1e− mediated oxidants results in nucleotide hydrolysis and dissociation similar to Ras GTPases; however, oxidation of RhoAC20 by 2e− oxidants likely results in perturbation of nucleotide binding and an increase in nucleotide exchange. According to data using phenyl-arsine oxide and peroxide (22), a mixed disulfide or disulfide can form between RhoA Cys20 and Cys16 upon oxidation of Cys20. When the disulfide bond is formed, it occludes nucleotide binding and inactivates the GTPase. However, nucleotide binding can be restored upon reduction of the disulfide, and this cycling results in guanine nucleotide exchange (GDP for GTP) and activation of the GTPase. Figure adapted from PDB ID:1FTM using Pymol. The hydrogen bonds are shown as blue dotted lines with the distances in angstroms. Color scheme: carbon, white; sulfur, yellow; nitrogen, blue; oxygen, salmon; phosphate, orange; and magnesium, red. (To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars).
The addition of cisplatin or arsenic trioxide has been shown to inactivate RhoA by promoting mixed disulfide formation between the two p-loop cysteines (22). In contrast to Ras, these results suggest that 2e− oxidation can regulate RhoA activity. In support of this hypothesis, Gerhard et al. showed that phenylarsine oxide generates a mixed disulfide crosslink between RhoA Cys16 and Cys20 using matrix-assisted laser desorption/ionization-MS and demonstrated that the modification inhibits stress fiber formation through the inactivation of RhoA in Caco-2 cells (15).
H2O2 has also been shown to regulate RhoA activity in vitro and in cells (2, 19). Unlike RasC118, RhoAC20 directly interacts with the bound nucleotide (Fig. 4), and 2e− oxidation of this cysteine likely perturbs nucleotide binding. We observed that 2e− oxidation by peroxide at the p-loop cysteines can enhance the rate of nucleotide dissociation by ∼10-fold. In addition, 1e− oxidation has also been shown to induce nucleotide dissociation. Our lab has shown that 1e− oxidants (O2•−, •NO2, and •OH) enhance RhoA nucleotide exchange by nearly 500-fold (19).
FIG. 4.

RhoA signaling pathways. (A) Indirect inhibition of RhoA by Rac1-mediated ROS production. Cell adhesion and resulting Rac1 activation leads to ROS generation through Nox. ROS production inactivates LMW-PTP, resulting in increased phosphorylation of p190 Rho-GAP and, subsequently, its activation. Enhanced GAP activity results in decreased RhoA activity, which promotes cell spreading. (B) Inactivation of RhoA by NO leads to increased p21 expression. p21 expression inhibits smooth muscle cell proliferation. NO is thought to inactivate RhoA directly by S-nitrosation. (C) In addition to indirect regulation by RI, RhoA can be directly activated by ROS or RNS. ROS treatment of cells leads to enhanced nucleotide exchange and RhoA activation. RhoA activation enhances stress fiber formation. LMW-PTP, low-molecular-weight protein tyrosine phosphatase; MLC-p, phosphorylated myosin light-chain; Nox, NADPH oxidase; RNS, reactive nitrogen species; ROCK, Rho-associated protein kinase; ROS, reactive oxygen species; SMC, smooth muscle cells.
Conclusions
While work from our lab indicates that only 1e− oxidation reactions that generate a thiyl radical at Cys118 in Ras affect Ras activity, discrepancies remain regarding 2e− oxidation. Given the distinct Rho GTPase redox motif, 1e− and 2e− mechanisms can modulate their activity. RhoA is unique because 1e− oxidants can induce a disulfide bond between Cys16 and Cys20, which can inactivate RhoA by occluding nucleotide binding. While the reduction potential under most physiological conditions will likely promote disulfide bond reduction and RhoA activation, under conditions of oxidative stress, radical quenching and/or disulfide reduction may not occur, which could render RhoA inactive (44). Further cell-based studies are needed to clarify the mechanisms of Rho GTPase regulation by redox agents.
Redox Regulation of GTPases: Cell-Based Studies
As we have recently published a detailed review on RNS regulation of Ras activity and biology (10), we will only highlight the most recent and salient data herein. Lander et al. first observed NO-mediated Ras activation in T cells (34), which was transient and concentration dependent. Later, it was shown that treatment with sodium nitroprusside, a nitric-oxide-generating compound, increased Ras downstream signaling through the mitogen-activated kinase pathway (33). An important tool in such studies, RasC118S is a variant that aids in determining the direct and indirect effects of RIs on Ras because it is RI insensitive and does not alter Ras structure and activity (41). A recent study on macrophages demonstrated that Ras S-nitrosation was required for Erk1/2-mediated apoptosis using the NO• donor S-nitrosoglutathione (51). The RasC118S variant reversed the pro-apoptotic effects, indicating that the effects on cell survival were mediated by direct reaction with Ras. In this study, Tsujita et al. suggested that the process of nitrosation, rather than covalent modification, activated Ras (51). Our lab extended these observations and showed that Ras activation depends on 1e− (radical)–mediated oxidation, which induces nucleotide dissociation (18). As most of these earlier studies used exogenous NO• and/or were conducted in cells that overexpressed Ras, it was unclear whether NO• regulated Ras activity under physiological conditions.
However, recent evidence has linked endothelial NOS (eNOS) to Ras nitrosation and activation in Ras-mediated tumorigenesis initiation and maintenance in mice. In the proposed activation pathway, oncogenic Ras activates the phosphoinositide 3-kinases (PI3K) pathway, which results in Akt-mediated (also protein kinase B) eNOS phosphorylation. Through a feedback mechanism, eNOS-generated NO• activates RasWT and stimulates Ras downstream pathways (35). Further, Lim et al. showed that, for H- and N-Ras, introducing the RasC118S mutation circumvented PI3K pathway activation and reduced tumor growth. In a separate cell-based study, eNOS-derived NO• activated N-Ras in T cells engaged with antigen presenting cells; Cys118 was required for activation (24). These data suggest that NO• mediates Ras activation directly through Cys118.
In-cell redox regulation of Rho GTPases
Nimnual et al. first described a pathway where ROS production was postulated to alter Rho GTPase activity by modulating Rho regulatory proteins (Fig. 4) (42). Decreased RhoA activity was observed in HeLa cells overexpressing constitutively active Rac1 (Rac1CA). This effect was abolished when Rac1 was expressed without its “insert” region. The insert region is likely required for Nox activation and ROS production. Rac1CA depression of RhoA activity was inhibited when cells were treated with the ROS scavenger N-acetylcysteine; conversely, RhoA activity was inhibited when cells were exogenously treated with 1 mM H2O2. Moreover, enhanced p190RhoGAP tyrosine phosphorylation was observed with downstream Rac1 activation, and tyrosine phosphorylation was abolished in cells pretreated with an Nox inhibitor (diphenylene iodium) as well as cells expressing Rac1CA without the insert region. The authors further showed that activation of Rac1 inactivates a phosphatase (low-molecular-weight protein tyrosine phosphatase [LMW-PTP]), which may be upstream of p190RhoGAP. However, interpretation of these results may be complicated by the use of peroxide and N-acetylcysteine at high concentrations, which likely altered the cell redox state, redox targets, and signaling pathways. Further, their hypothesis that Rac1 drives ROS production through Nox to affect RhoA relies on the Rac1CA insert-deletion mutant (42). The importance of the insert region in Nox activation has been debated, and at least two reports describe the insert region as dispensable to ROS production (27, 40). However, Nox and Rac isoforms are expressed in different cell types and bind with varying efficiencies, which may account for this discrepancy, and downstream effects from Rac1CA may play a role. An alternative approach to insert-deletion mutants is necessary to interpret the mechanism for ROS regulation of RhoA.
Zuckerbraun et al. observed inactivation of RhoA by NO• in smooth muscle cells after treatment with a pharmacological NO• donor (propylamine propylamine-NONOate) by measuring GTP and effector binding (Fig. 4) (57). Nitric oxide addition to smooth muscle cells decreased stress fiber formation, a phenotype often associated with RhoA inactivation. Further, exogenous NO• enhanced ERK activity, upregulated the cyclin-dependent kinase inhibitor (p21), and decreased smooth muscle cell proliferation. Using RhoACA, the authors showed that NO• inhibits smooth muscle cell proliferation through RhoA activation-dependent p21 upregulation. It was also shown that NO• S-nitrosates RhoA when using NO release and nitrosocysteine immunodetection techniques. In the presence of the NO• donor, GTP association was significantly inhibited, which was reversible with dithiothreitol (DTT). However, DTT reversal does not necessarily imply that S-nitrosation is the RhoA modification post-NO treatment because NO•/O2 can yield a variety of different modifications, many reversible with DTT. An additional missing piece of key data is RhoA S-nitrosation site specificity. Based on work from Gerhard et al. (15) and our lab (22), Cys16 and Cys20 likely form a disulfide, and we hypothesize that Cys20 is the primary site of S-nitrosation.
We have recently shown that exogenous and endogenous ROS can stimulate RhoA activity (Fig. 4). In fibroblasts, RhoA activation and stress fiber formation were reversible by washing out peroxide and abolished upon N-acetylcysteine treatment. Moreover, we showed that ROS activation of RhoA was abolished when two critical cysteines were mutated (RhoAC16A/C20A). Notably, RhoAC16A/C20A responds normally to guanine nucleotide exchange factor (GEF)–mediated activation and C3 toxin inactivation (2). These results show that ROS can directly activate RhoA through a mechanism involving Cys20 and/or Cys16. These results differ from Nimnual et al., as we observed direct RhoA activation by ROS (Fig. 4). Moreover, we identified the critical cysteines in ROS-mediated RhoA activation (22). Other observations have indicated that RhoA is activated in ROS-exposed cells, although a direct mechanism was not identified. As we have observed RhoA activation by ROS in fibroblasts, we hypothesize that high RI levels or a low cellular reduction potential can inactivate RhoA due to the irreversible formation of an RI-mediated RhoA Cys16/Cys20 disulfide bond. Thus, our results do not implicitly disagree with Zuckerbraun et al. (57). Likely, RI activation and inactivation of GTPases depends on the RI levels and cell redox state. Low RI levels under high-reducing potential conditions may activate RhoA by promoting disulfide reduction and nucleotide exchange, whereas elevated RIs under low reducing potential conditions may inactivate RhoA (44).
Rho GTPases control redox-regulating enzymes
Rho GTPases play a key role in regulating cell redox status; they can modulate cell redox status in response to molecular and mechanical forces as well as an altered redox state. This response is particular to cell type, subcellular location, and initiating event; thus, the downstream effects are diverse and highly regulated. In cell-based and animal experiments, Rac proteins (Rac1 and Rac2 isoforms) and RhoA regulate expression and activity of primary redox-modulating enzymes in the cell: Nox (O2•− generation), NOS (NO• generation), and SOD (O2•− dismutation into H2O2) (Fig. 6). As RIs can modulate the activity of redox-sensitive Rho GTPases in cell-based studies and facilitate guanine nucleotide dissociation in vitro, these GTPases are likely involved in a redox feedback loop.
FIG. 6.
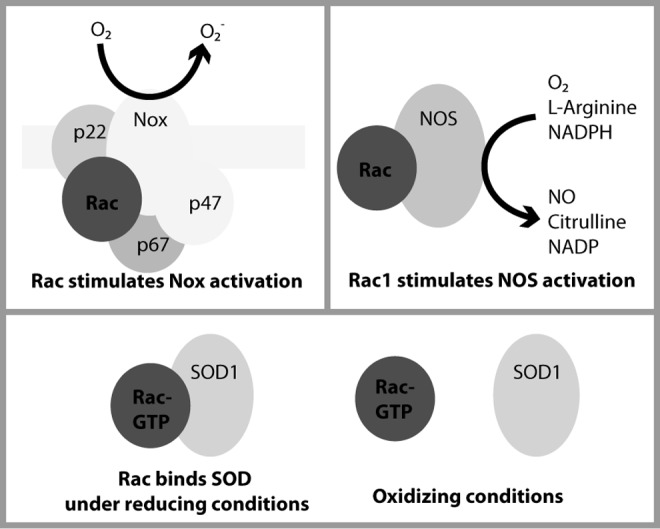
Rac directly interacts with Nox, NOS, and SOD. Rac isoforms have been shown to directly enhance the activities of NOS and Nox isoforms (23, 45, 48). In several Nox isoforms, Rac must interact with the p67 subunit (and possibly flavocytochrome b558) for Nox to convert O2 to O2•− (7, 23). In addition to evidence that Rac indirectly stimulates NOS activity, Rac1 has been shown to increase eNOS and nNOS activity through direct interaction, aiding in the conversion of O2 to NO (45, 48). Finally, recent data show that Rac1 directly interacts with SOD1 in a nucleotide- and redox-dependent manner (16). eNOS, endothelial NOS; nNOS, neuronal NOS; O2•−, superoxide; SOD, superoxide dismutase.
Rac proteins and RhoA regulate NOS expression and activity
The activity and expression of the three NOS isoforms—eNOS, neuronal NOS (nNOS), and inducible NOS (iNOS)—are highly regulated (13). Whereas Rac proteins have been shown to regulate NOS expression and activity, RhoA regulates NOS expression (Figs. 5 and 6). Further, NOS bioavailability is, in part, attributed to its mRNA stability, which Rho GTPases can either enhance (Rac1) or reduce (RhoA) (45, 46). Rac1 promotes eNOS transcription through p21-activated kinase (PAK1), and RhoA reduces mRNA stability through Rho-associated protein kinase (ROCK) (37). Moreover, Rac1 and RhoA work in opposition to regulate NOS activity through the PI3K/Akt redox-sensitive pathway; RhoA inhibits this pathway, and Rac1 activates it (30, 37). Notably, Ras also modulates the PI3K/Akt pathway to regulate eNOS activity (35). As further evidence of RhoA/Rac1 crosstalk in NOS activation, Rac1 regulates cGMP-dependent kinase, which inhibits RhoA activation (37). The Rac1/PAK1 interaction further enhances eNOS activity by stimulating cell uptake of L-Arginine (37). Rac proteins may regulate NOS function in two additional ways, controlling localization and direct protein–protein interaction (31, 48). Rac2 and iNOS interact in a direct, GTP-independent manner, and Rac2 overexpression promotes iNOS localization to cytoskeletal complexes (31). Rac1 has been shown to directly interact with all of the NOS isoforms, and overexpression of Rac1CA enhances eNOS and nNOS activity, likely by direct interaction (31, 45, 48). As NOS isoforms interact with Rac isoforms with varying efficiencies depending upon cell type, stimulating event, and Rac nucleotide status, this interaction may aid in proper compartmentalization for NO generation.
FIG. 5.

GTPase crosstalk in NOS regulation. Shown below are the following: RhoA regulates NOS expression by decreasing mRNA stability through ROCK, and it decreases NOS activity through inactivation of the PI3K/Akt pathway (37, 45, 46). Rac1 enhances NOS mRNA expression through PAK1, and it increases NOS activity by direct interaction (45, 47, 48), activating the PI3K/Akt pathway (30), increasing cell uptake of L-Arginine, and downregulating RhoA activity through cGMP-dependent kinase (37). Ras also plays a role in NOS stimulation through the PI3K/Akt pathway (35). NOS, nitric oxide synthase; PAK1, p21-activated kinase; PI3K, phosphoinositide 3-kinase.
Rac proteins activate Nox
Rac proteins regulate Nox activity, as they can directly interact with and activate multiple Nox isoforms to produce O2•−, including Nox1, 2, 3, and 4 (23). Rac1 is required for Nox1 activity in vascular smooth muscle cells, and Rac2 activates Nox2 in neutrophils (7, 11). Interestingly, Rap1A, an NKCD-motif-containing Ras subclass GTPase, has been implicated in Nox activation (11). Nox activity can be stimulated by multiple factors, including shear stress, angiotensin II, thrombin, insulin, and vascular endothelial growth factor (3, 6, 26). Various models have been proposed for Rac activation of Nox (Fig. 6), and data support Nox activation by GTP-bound Rac and GDP-bound Rac complexed to GDP dissociation inhibitor (GDI); the authors suggest that GDI association maintains the active Rac conformation even when GDP-bound, as Rac likely binds Nox in this conformation (7, 12). The common steps in the models for Rac1 activation of Nox are as follows: first, upon stimulation, Rac and the cytosolic Nox components translocate simultaneously, but independently, to the cell membrane (7, 11); next, likely through its switch I and insert motifs, Rac directly binds p67 to activate Nox (7). Controversial in these models is whether Rac1 also directly interacts with flavocytochrome b558, whether it activates Nox either through an adaptor function or mediates electron transfer during O2•− production, and whether the Rac insert region is required for Nox activation (7, 11). Depending on cell type, localization, and stimulating event, Rac-dependent Nox activation can promote various signaling pathways with physiological and pathological consequences, such as transcription activation, inflammatory responses, as well as cardiovascular and neurological diseases (46).
Rac1 directly interacts with SOD in a nucleotide- and redox-dependent manner
Recent evidence supports a role for Rac1 in regulating SOD1 (Fig. 6), a key antioxidant enzyme in the cell. In a detailed study, Harraz et al. showed that Rac1 interacts with SOD1 in a nucleotide-dependent manner, indicative of GTPase/effector interactions (Fig. 6) (16). As this study involved cell-based assays, our lab is using in vitro methods to quantitate this interaction and has validated its nucleotide dependence (unpublished observations). Harraz et al. showed that cell redox conditions also affect the Rac1/SOD1 interaction and suggested a feedback mechanism. However, the question remains whether the Rac1/SOD1 interaction enhances SOD1 activity, as in Nox and NOS. Interestingly, these experiments were performed in an amyotrophic lateral sclerosis (ALS) model, and SOD mutations that are common in ALS patients were proposed to uncouple the Rac1/SOD feedback mechanism and enhance cell toxicity. A redox regulatory role for the Rac1/SOD interaction in neural physiology and pathology is consistent with Rac regulation of NOS and Nox in key physiological and pathological pathways.
Conclusions
We have reviewed the cell-based literature demonstrating that RIs regulate Rho GTPase activity and certain Rho GTPases control the activities of the predominant redox-modulating enzymes in cells. As we have recently published a review on RNS regulation of Ras GTPases, we have only noted some of the most recent data. We speculate that Rac is a common control element for regulating cell redox state. Moreover, RhoA has been shown to play a key role in regulating NOS. We postulate that cell redox conditions (oxidative/reducing) regulate the activity of these GTPases; however, the role of these GTPases in controlling RI-regulating enzymes suggests a potential feedback mechanism.
Conclusions and Future Directions
Herein, we discussed the importance of RIs in modulating the activity of select Ras and Rho GTPases and the role of these GTPases in controlling the expression and activity of RI-regulating enzymes. Further, we have previously discussed RNS regulation of Ras in depth (10). As there is a discrepancy in the literature on whether Ras is activated by 1e− (radical) and 2e− (covalent) cysteine oxidation mechanisms, we propose that future experiments address this discrepancy. In addition, our data and a growing body of literature suggest that Rho GTPases are particularly susceptible to redox control given their unique redox-sensitive cysteine-containing p-loop motif. This redox-sensitive cysteine interacts with the bound nucleotide, and 1e− and 2e− redox modification at this cysteine enhances nucleotide exchange, likely by disrupting these interactions. Under physiological conditions, RIs likely activate Ras and Rho GTPases, whereas oxidative stress may promote their inactivation. To aid in discerning whether RI regulation of Rho GTPases is direct or indirect, we have identified redox-insensitive variants that better mimic the wild-type proteins. Moreover, cell-based studies should be performed with endogenous RI sources, redox state characterization, and absent GTPase overexpression. Further, Rho and Ras subclasses also regulate expression and activity of key redox regulating enzymes. RhoA and Ras regulate NOS expression and activity, and Rap1A has been implicated in Nox regulation. Rac controls NOS and Nox, and Rac1 has recently been shown to directly interact with SOD1. Given that Rac1 and SOD1 are ubiquitously expressed proteins involved in numerous cell functions, future experiments should be directed to discerning the mechanism of and effects from this interaction. Further, as the literature is increasingly dedicated to redox-based signaling, perhaps future studies will better define how RIs regulate Ras and Rho GTPase activity and whether these GTPases are regulated by or control additional redox-modulating enzymes.
Supplementary Material
Abbreviations Used
- 5-nitro-GDP
5-nitro-guanine ribose diphosphate
- ALS
amyotrophic lateral sclerosis
- DTT
dithiothreitol
- eNOS
endothelial NOS
- GDI
GDP dissociation inhibitor
- GSSG
oxidized glutathione
- H2O2
hydrogen peroxide
- iNOS
inducible NOS
- LMW-PTP
low-molecular-weight protein tyrosine phosphatase
- MS
mass spectrometry
- NO•
nitric oxide
- nNOS
neuronal NOS
- NOS
nitric oxide synthase
- Nox
NADPH oxidase
- NIm-DP
5-guanidino-4-nitroimidazole diphosphate
- O2•−
superoxide
- •OH
hydroxyl radical
- ONOO−
peroxynitrite
- p-loop
phosphoryl-binding loop
- PAK1
p21-activated kinase
- PI3K
phosphoinositide 3-kinase
- RIs
reactive intermediates
- RNS
reactive nitrogen species
- ROCK
Rho-associated protein kinase
- ROS
reactive oxygen species
- SOD
superoxide dismutase
Acknowledgments
This work has been supported by grants from the National Institutes of Health through grant numbers RCA089614B and GM075431 to SLC; T32 GM008570 to LM and AH; and NRSA F30 HL094063 to AA.
References
- 1.Adachi T. Pimentel DR. Heibeck T. Hou X. Lee YJ. Jiang B. Ido Y. Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 2.Aghajanian A. Wittchen ES. Campbell SL. Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PloS one. 2009;4:e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amin JK. Xiao L. Pimental DR. Pagano PJ. Singh K. Sawyer DB. Colucci WS. Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J Mol Cell Cardiol. 2001;33:131–139. doi: 10.1006/jmcc.2000.1285. [DOI] [PubMed] [Google Scholar]
- 4.Augusto O. Bonini MG. Amanso AM. Linares E. Santos CC. De Menezes SL. Nitrogen dioxide and carbonate radical anion: two emerging radicals in biology. Free Radic Biol Med. 2002;32:841–859. doi: 10.1016/s0891-5849(02)00786-4. [DOI] [PubMed] [Google Scholar]
- 5.Beckman JS. Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 6.Bedard K. Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 7.Bokoch GM. Diebold BA. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood. 2002;100:2692–2696. doi: 10.1182/blood-2002-04-1149. [DOI] [PubMed] [Google Scholar]
- 8.Clavreul N. Adachi T. Pimental DR. Ido Y. Schoneich C. Cohen RA. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J. 2006;20:518–520. doi: 10.1096/fj.05-4875fje. [DOI] [PubMed] [Google Scholar]
- 9.Clavreul N. Bachschmid MM. Hou X. Shi C. Idrizovic A. Ido Y. Pimentel D. Cohen RA. S-glutathiolation of p21ras by peroxynitrite mediates endothelial insulin resistance caused by oxidized low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2006;26:2454–2461. doi: 10.1161/01.ATV.0000242791.28953.4c. [DOI] [PubMed] [Google Scholar]
- 10.Davis MF. Vigil D. Campbell SL. Regulation of Ras proteins by reactive nitrogen species. Free Radic Biol Med. 2011;51:565–575. doi: 10.1016/j.freeradbiomed.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeLeo FR. Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J Leukoc Biol. 1996;60:677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- 12.Di-Poi N. Faure J. Grizot S. Molnar G. Pick E. Dagher MC. Mechanism of NADPH oxidase activation by the Rac/Rho-GDI complex. Biochemistry. 2001;40:10014–10022. doi: 10.1021/bi010289c. [DOI] [PubMed] [Google Scholar]
- 13.Dudzinski DM. Igarashi J. Greif D. Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 14.Forstermann U. Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2011 doi: 10.1093/eurheartj/ehr304. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerhard R. John H. Aktories K. Just I. Thiol-modifying phenylarsine oxide inhibits guanine nucleotide binding of Rho but not of Rac GTPases. Mol Pharmacol. 2003;63:1349–1355. doi: 10.1124/mol.63.6.1349. [DOI] [PubMed] [Google Scholar]
- 16.Harraz MM. Marden JJ. Zhou W. Zhang Y. Williams A. Sharov VS. Nelson K. Luo M. Paulson H. Schoneich C. Engelhardt JF. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heo J. Redox control of GTPases: from molecular mechanisms to functional significance in health and disease. Antioxid Redox Signal. 2011;14:689–724. doi: 10.1089/ars.2009.2984. [DOI] [PubMed] [Google Scholar]
- 18.Heo J. Campbell SL. Mechanism of p21Ras S-nitrosylation and kinetics of nitric oxide-mediated guanine nucleotide exchange. Biochemistry. 2004;43:2314–2322. doi: 10.1021/bi035275g. [DOI] [PubMed] [Google Scholar]
- 19.Heo J. Campbell SL. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J Biol Chem. 2005;280:31003–31010. doi: 10.1074/jbc.M504768200. [DOI] [PubMed] [Google Scholar]
- 20.Heo J. Campbell SL. Ras regulation by reactive oxygen and nitrogen species. Biochemistry. 2006;45:2200–2210. doi: 10.1021/bi051872m. [DOI] [PubMed] [Google Scholar]
- 21.Heo J. Prutzman KC. Mocanu V. Campbell SL. Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J Mol Biol. 2005;346:1423–1440. doi: 10.1016/j.jmb.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 22.Heo J. Raines KW. Mocanu V. Campbell SL. Redox regulation of RhoA. Biochemistry. 2006;45:14481–14489. doi: 10.1021/bi0610101. [DOI] [PubMed] [Google Scholar]
- 23.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 24.Ibiza S. Perez-Rodriguez A. Ortega A. Martinez-Ruiz A. Barreiro O. Garcia-Dominguez CA. Victor VM. Esplugues JV. Rojas JM. Sanchez-Madrid F. Serrador JM. Endothelial nitric oxide synthase regulates N-Ras activation on the Golgi complex of antigen-stimulated T cells. Proc Natl Acad Sci U S A. 2008;105:10507–10512. doi: 10.1073/pnas.0711062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaffe AB. Hall A. Rho GTPases: biochemistry and biology. Ann Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 26.Jiang F. Zhang Y. Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 27.Karnoub AE. Der CJ. Campbell SL. The insert region of Rac1 is essential for membrane ruffling but not cellular transformation. Mol Cell Biol. 2001;21:2847–2857. doi: 10.1128/MCB.21.8.2847-2857.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kissner R. Koppenol WH. Product distribution of peroxynitrite decay as a function of pH, temperature, and concentration. J Am Chem Soc. 2002;124:234–239. doi: 10.1021/ja010497s. [DOI] [PubMed] [Google Scholar]
- 29.Kjeldgaard M. Nyborg J. Clark BF. The GTP binding motif: variations on a theme. FASEB J. 1996;10:1347–1368. [PubMed] [Google Scholar]
- 30.Kou R. Michel T. Epinephrine regulation of the endothelial nitric-oxide synthase: roles of RAC1 and beta3-adrenergic receptors in endothelial NO signaling. J Biol Chem. 2007;282:32719–32729. doi: 10.1074/jbc.M706815200. [DOI] [PubMed] [Google Scholar]
- 31.Kuncewicz T. Balakrishnan P. Snuggs MB. Kone BC. Specific association of nitric oxide synthase-2 with Rac isoforms in activated murine macrophages. Am J Physiol Renal Physiol. 2001;281:F326–F336. doi: 10.1152/ajprenal.2001.281.2.F326. [DOI] [PubMed] [Google Scholar]
- 32.Lancaster JR., Jr. Xie K. Tumors face NO problems? Cancer Res. 2006;66:6459–6462. doi: 10.1158/0008-5472.CAN-05-2900. [DOI] [PubMed] [Google Scholar]
- 33.Lander HM. Milbank AJ. Tauras JM. Hajjar DP. Hempstead BL. Schwartz GD. Kraemer RT. Mirza UA. Chait BT. Burk SC. Quilliam LA. Redox regulation of cell signalling. Nature. 1996;381:380–381. doi: 10.1038/381380a0. [DOI] [PubMed] [Google Scholar]
- 34.Lander HM. Ogiste JS. Teng KK. Novogrodsky A. p21ras as a common signaling target of reactive free radicals and cellular redox stress. J Biol Chem. 1995;270:21195–21198. doi: 10.1074/jbc.270.36.21195. [DOI] [PubMed] [Google Scholar]
- 35.Lim KH. Ancrile BB. Kashatus DF. Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–649. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liochev SI. Fridovich I. The effects of superoxide dismutase on H2O2 formation. Free Radic Biol Med. 2007;42:1465–1469. doi: 10.1016/j.freeradbiomed.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Loirand G. Pacaud P. The role of Rho protein signaling in hypertension. Nat Rev Cardiol. 2010;7:637–647. doi: 10.1038/nrcardio.2010.136. [DOI] [PubMed] [Google Scholar]
- 38.Lundberg JO. Weitzberg E. Nitrite reduction to nitric oxide in the vasculature. Am J Physiol Heart Circ Physiol. 2008;295:H477–H478. doi: 10.1152/ajpheart.00611.2008. [DOI] [PubMed] [Google Scholar]
- 39.Merenyi G. Lind J. Goldstein S. Czapski G. Peroxynitrous acid homolyzes into *OH and *NO2 radicals. Chem Res Toxicol. 1998;11:712–713. doi: 10.1021/tx980043h. [DOI] [PubMed] [Google Scholar]
- 40.Miyano K. Koga H. Minakami R. Sumimoto H. The insert region of the Rac GTPases is dispensable for activation of superoxide-producing NADPH oxidases. Biochem J. 2009;422:373–382. doi: 10.1042/BJ20082182. [DOI] [PubMed] [Google Scholar]
- 41.Mott HR. Carpenter JW. Campbell SL. Structural and functional analysis of a mutant Ras protein that is insensitive to nitric oxide activation. Biochemistry. 1997;36:3640–3644. doi: 10.1021/bi962790o. [DOI] [PubMed] [Google Scholar]
- 42.Nimnual AS. Taylor LJ. Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol. 2003;5:236–241. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- 43.Poli G. Leonarduzzi G. Biasi F. Chiarpotto E. Oxidative stress and cell signalling. Curr Med Chem. 2004;11:1163–1182. doi: 10.2174/0929867043365323. [DOI] [PubMed] [Google Scholar]
- 44.Raines KW. Bonini MG. Campbell SL. Nitric oxide cell signaling: S-nitrosation of Ras superfamily GTPases. Cardiovasc Res. 2007;75:229–239. doi: 10.1016/j.cardiores.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 45.Rao GK. Bender JR. Rac, PAK, and eNOS ACTion. Circ Res. 2008;103:328–330. doi: 10.1161/CIRCRESAHA.108.182238. [DOI] [PubMed] [Google Scholar]
- 46.Rikitake Y. Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res. 2005;97:1232–1235. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sawada N. Salomone S. Kim HH. Kwiatkowski DJ. Liao JK. Regulation of endothelial nitric oxide synthase and postnatal angiogenesis by Rac1. Circ Res. 2008;103:360–368. doi: 10.1161/CIRCRESAHA.108.178897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Selvakumar B. Hess DT. Goldschmidt-Clermont PJ. Stamler JS. Co-regulation of constitutive nitric oxide synthases and NADPH oxidase by the small GTPase Rac. FEBS Letters. 2008;582:2195–2202. doi: 10.1016/j.febslet.2008.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szabo C. Ischiropoulos H. Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 50.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 51.Tsujita M. Batista WL. Ogata FT. Stern A. Monteiro HP. Arai RJ. The nitric oxide-sensitive p21Ras-ERK pathway mediates S-nitrosoglutathione-induced apoptosis. Biochem Biophys Res Commun. 2008;369:1001–1006. doi: 10.1016/j.bbrc.2008.02.117. [DOI] [PubMed] [Google Scholar]
- 52.Tsutsui M. Shimokawa H. Otsuji Y. Yanagihara N. Pathophysiological relevance of NO signaling in the cardiovascular system: novel insight from mice lacking all NO synthases. Pharmacol Ther. 2010;128:499–508. doi: 10.1016/j.pharmthera.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 53.Ullrich V. Kissner R. Redox signaling: bioinorganic chemistry at its best. J Inorg Biochem. 2006;100:2079–2086. doi: 10.1016/j.jinorgbio.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 54.Valko M. Izakovic M. Mazur M. Rhodes CJ. Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 55.Wardman P. Candeias LP. Fenton chemistry: an introduction. Radiat Res. 1996;145:523–531. [PubMed] [Google Scholar]
- 56.Williams JG. Pappu K. Campbell SL. Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc Natl Acad Sci U S A. 2003;100:6376–6381. doi: 10.1073/pnas.1037299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zuckerbraun BS. Stoyanovsky DA. Sengupta R. Shapiro RA. Ozanich BA. Rao J. Barbato JE. Tzeng E. Nitric oxide-induced inhibition of smooth muscle cell proliferation involves S-nitrosation and inactivation of RhoA. Am J Physiol Cell Physiol. 2007;292:C824–C831. doi: 10.1152/ajpcell.00592.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.