

Abstract
The mammalian gastrointestinal (GI) tract provides a complex and competitive environment for the microbiota1. Successful colonization by pathogens depends on scavenging nutrients, sensing chemical signals, competing with the resident bacteria, and precisely regulating expression of virulence genes2. The GI pathogen enterohemorrhagic E.coli (EHEC) relies on inter-kingdom chemical sensing systems to regulate virulence gene expression3–4. Here we show that these systems control the expression of a novel two-component signal transduction system, named FusKR, where FusK is the histidine sensor kinase (HK), and FusR the response regulator (RR). FusK senses fucose and controls expression of virulence and metabolic genes. This fucose-sensing system is required for robust EHEC colonization of the mammalian intestine. Fucose is highly abundant in the intestine5. Bacteroides thetaiotaomicron (B.theta) produces multiple fucosidases that cleave fucose from host glycans, resulting in high fucose availability in the gut lumen6. During growth in mucin, B.theta contributes to EHEC virulence by cleaving fucose from mucin, thereby activating the FusKR signaling cascade, modulating EHEC’s virulence gene expression. Our findings suggest that EHEC uses fucose, a host-derived signal made available by the microbiota, to modulate EHEC pathogenicity and metabolism.
The GI tract is inhabited by trillions of commensal bacteria that play crucial roles in human physiology1. This fundamental relationship between the host and microbiota relies on chemical signaling and nutrient availability2, and invading pathogens compete for these resources through the precise coordination of virulence traits. EHEC colonizes the colon, leading to hemorrhagic colitis7. EHEC colonization depends on the locus of enterocyte effacement (LEE) pathogenicity island (PAI)7. This PAI encodes a regulator for its own expression, ler, and a molecular syringe, a type-3 secretion system (TTSS), which injects effectors into the host cell, leading to the formation of attaching and effacing (AE) lesions on enterocytes. AE lesions are characterized by remodeling of the host-cell cytoskeleton, leading to the formation of a pedestal-like structure beneath the bacteria7. LEE expression is regulated by an inter-kingdom chemical signaling system involving the host hormones epinephrine and/or norepinephrine and the microbial-flora-produced signal autoinducer-3 (AI-3)8. These signals are sensed by two HKs, QseC3 and QseE4, which initiate a signaling cascade that promotes virulence.
HKs, together with RRs comprise a two-component system (TCS), which play a major role in bacterial signal transduction. Upon sensing a signal, the HK autophosphorylates and then transfers its phosphate to the RR. Subsequently, most RRs bind DNA, promoting changes in gene expression9. The cognate RR for QseC is QseB, and for QseE is QseF (Fig. 1a). QseBC and QseEF repress expression of the z0462/z0463 genes (Fig. 1b)10–11. QseB repression of z0462/z0463 expression is direct, while QseF-mediated repression is indirect (Fig. 1c, d), in agreement with QseF being a σ54-dependent transcriptional activator12. QseF activates the expression of a repressor of z0462/z0463.
Figure 1. The TCS FusKR of EHEC.

a, The QseC/QseE signaling-cascade. QseC senses AI-3/epinephrine(Epi)/NE. QseE senses Epi/NE/SO4/PO4. QseC phosphorylates QseB that activates flagella; KpdE that activates the LEE; and QseF. QseE only phosphorylates QseF. QseBC and QseEF repress expression of z0462/z0463. b, qRT-PCR of z0462 in WT, ΔqseB, ΔqseC, ΔqseE and ΔqseF in DMEM. Gene expression is represented as fold differences normalized to WT. Error bars indicate standard deviations of ΔddCt values. (n=18 biological samples per strain; asterisk, P≤0.01; two asterisks, P≤0.001; Student’s t-test). c, EMSA of z0463 with QseB and QseF. d, EMSA of qseE (positive control) with QseF. e, Autophosphorylation of Z0462 in liposomes (top panel), and Commassie gel of Z0462 (lower panel) (loading control). f, Phosphotransfer from Z0462 (in liposomes) to Z0463 (ratio 1 HK: 4 RR) (top panel), Commassie gel of Z0462 and Z0463 (lower panel) (loading control).
The z0462/z0463 genes are within a PAI [O-island 20 (OI-20)]13, which is found in EHEC O157:H7 strains and enteropathogenic E.coli strains exclusively from the 055:H7 serotype (which gave rise to the O157:H7 serotype), but absent in all other E. coli strains whose genomes are currently publically available. This PAI is organized in three transcriptional units (Supplementary Fig. 1). The genes z0462/z0463 encode for a putative TCS: z0462 encodes a HK with 8 transmembrane domains that shares similarity to a glucose-6-phosphate sensor, UhpB (~30%); z0463 encodes a RR with a receiver and a DNA-binding domain (Supplementary Fig. 2). Z0462 in liposomes is a functional HK (Fig. 1e), and it transfers its phosphate to Z0463 (Fig. 1f). Hence Z0462 and Z0463 constitute a cognate TCS.
Transcriptomic studies (Supplementary Tables 4,5) suggested that Z0462/Z0463 mainly act as repressors of transcription. Z0462/Z0463 represses LEE gene expression (Supplementary Fig. 3). Transcription of all LEE operons is increased inΔz0462 and Δz0463, and complementation restored the expression of ler to WT levels (Fig. 2a–d). Transcription of the LEE genes is activated by Ler14. The RR Z0463 directly represses ler transcription, and subsequently the other LEE operons, and phosphorylation of Z0463 increases its affinity to ler (Fig. 2e, f, Supplementary Figs.6–8). Congruent with the increased LEE transcription, both Δz0462 and Δz0463 secreted more EspB, a LEE-encoded protein (Fig. 2g), and formed more pedestals than WT (Fig. 2h, i). Therefore, Z0462/Z0463 repress AE lesion formation. However, expression of other genes encoding non-LEE-encoded-TTSS effectors, not involved in AE lesion formation, are activated by Z0462/Z0463 (Supplementary Fig. 4).
Figure 2. Z0462/z0463 regulates LEE expression.

a, qRT-PCR of LEE genes in WT and z0462− in DMEM. b, qRT-PCR of ler in WT, z0462− and z0462+ in DMEM. c, qRT-PCR of LEE genes in WT and z0463− in DMEM. d, qRT-PCR of ler in WT, z0463− and z0463+ in DMEM. (n=18 biological samples per strain; two asterisks, P ≤ 0.001; three asterisks, P<0.0001; ns, P>0.05; Student’s t-test). e, Representation of the Ler and Z0463 regulation of the LEE operons. f, EMSA of ler with Z0463 with ler and kan cold probes. g, Western-blot of EspB in supernatants of WT, z0462−, z0462+, z0463− and z0463+ strains. BSA was added as a loading control. h, FAS assay of HeLa cells infected with EHEC WT, z0462−, z0462+, z0463− and z0463+, stained with FITC-phalloidin (actin) and propidium-iodide (bacteria and HeLa DNA).i, Quantification of FAS assay (n=600 cells; three asterisks, P<0.0001; ns, P>0.05; Student’s t-test).
Expression of z0463 is increased by mucus produced by intestinal HT29 cells. EHEC infected undifferentiated HT29 cells were used as negative controls, since they do not produce mucus (Fig. 3a and Supplementary Fig. 9). Z0462 is a predicted hexose-phosphate-sensor, hence, Z0462 may sense sugars in the mucus. Fucose is a major component of mucin glycoproteins, it is abundant in the intestine5, and fucose utilization is important for EHEC intestinal colonization15–16. In E.coli, L-fucose utilization requires the fuc genes, and their activator (FucR)17. Z0462/Z0463 repress the expression of the fuc genes (Fig. 3b), andΔz0462 and Δz0463 grow faster with L-fucose as a sole carbon (C)-source compared to WT (generation times: WT 92.4min, Δz0462 64min and Δz0463 74min) (Fig. 3c). Therefore, Z0462/Z0463 regulates fucose utilization, and this response is specific to fucose, with the mutants and WT growing at similar rates with other C-sources (Supplementary Fig. 10). Z0462 senses fucose, but not glucose nor D-ribose (Fig. 3d, e). The concentration of fucose (100μM) used is physiologically relevant to the mammalian intestine18. Hence we renamed this protein FusK for fucose-sensing-HK, and its cognate RR, FusR for fucose-sensing-RR.
Figure 3. Z0462/Z0463 is a fucose sensing TCS.

a, qRT-PCR of z0463 in WT in the presence of undifferentiated non-mucus-producing HT29 or differentiated HT29 mucus-producing cells. Error bar indicates standard deviations of ΔddCt values. (n=18 biological samples per assay; three asterisks, P<0.0001; Student’s t-test). b, qRT-PCR of fucose-utilization genes in EHEC WT, z0462− and z0463− in DMEM (OD6001.0). (n=18 biological samples per strain; asterisk, P ≤ 0.05; two asterisks, P ≤ 0.01; three asterisks, P ≤ 0.001; Student’s t-test). c, Growth curves of WT, z0462− and z0463− strains in M9-minimal-media with L-fucose as a sole C-source. (n=6 biological samples; significance between generation times calculated through Anova P ≤ 0.01). d, FusK autophosphorylation (in liposomes) in the presence of L-fucose, D-glucose or D-ribose (top panel), and Commassie gel of FusK in liposomes (lower panel) (loading control). e, Quantification of FusK autophosphorylation. Phosphorylation represented at fold-change compared to absence of signal. Error bar indicates the standard deviation of fold-change values. (n=3; three asterisks, P<0.0001; ns, P>0.05; Student’s t-test). f, Schematic representation of the fusRK operon to z0461. g, qRT-PCR of z0461 in WT and ΔfusK. (n=18 biological samples per assay; two asterisks, P<0.001; Student’s t-test). h, Growth curves of WT and Δz0461 in M9-medium with fucose as a sole C-source. (n=6 biological samples; significance between generation times calculated through Anova P ≤ 0.01). i, qRT-PCR of fucA, fucP and fucR in WT and Δz0461. (n=18 biological samples per strain; three asterisks, P<0.001; Student’s t-test).
FusKR shares homology to the UhpAB TCS. UhpAB senses glucose-6-phosphate and activates expression of the uhpT gene that encodes a hexose-phosphate-major facilitator-superfamily (MFS) transporter19–21. FusKR represses transcription of the z0461 gene downstream of fusKR, which encodes a predicted MFS (Fig. 3f, g and Supplementary Figs.3,11). Δz0461 has decreased growth with fucose as a sole C-source (generation times: WT is 88.2min and Δz0461 96.6 min) (Fig. 3h), but grows similar to WT with glucose (Supplementary Fig. 12), suggesting that z0461 is involved in optimal fucose import. Transcription of the fuc operons is linked to fucose uptake, fucose yields fuculose-1-phosphate that is the inducer of the FucR activator of the fuc operons17,22–23. Transcription of the fuc genes is decreased in Δz0461 (Fig. 3i). Fucose induces FusKR, which represses z0461, decreasing fucose import and the intracellular levels of the fuculose-1-phosphate inducer of FucR. In further support of this indirect regulation of the fuc genes, FusR does not bind to the fucPIKUR promoter region (Supplementary Fig. 13), in contrast to the direct regulation of the LEE (Fig. 2).
TheΔfusK is irresponsive to fucose, given that expression of ler is repressed by fucose in the WT but not in ΔfusK (Fig. 4a). B.theta produces multiple fucosidases that cleave fucose from host glycans, resulting in high fucose availability in the lumen2. B.theta supplies mucin-derived fucose to EHEC, reducing ler expression, whereas in free fucose there is no change in ler expression whether B.theta is present (Fig. 4b). Of note, expression of ler is decreased when EHEC is grown in mucin compared to fucose (Fig. 4b), consistent with the increased expression of fusR in mucin (Fig. 3a).
Figure 4. FusK in pathogen-microbiota-host associations.
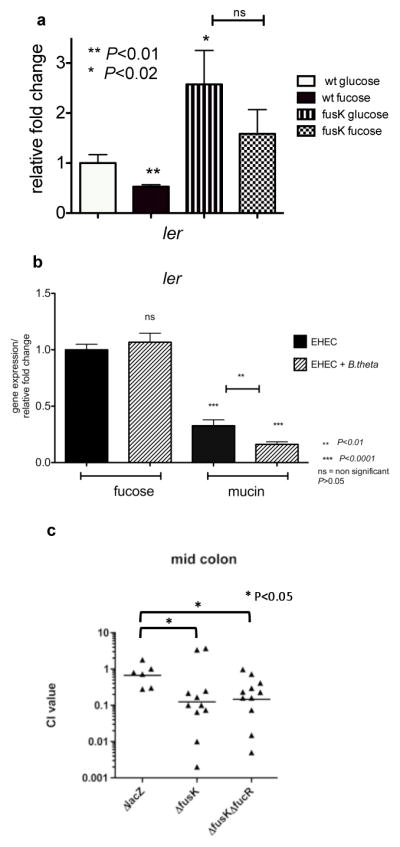
a, qRT-PCR of ler in WT or ΔfusK. RNAs extracted from cultures grown in M9 with either D-glucose or L-fucose as sole C-sources. Error bar indicates standard deviations of ΔddCt values. (n=18 biological samples per assay; asterisk, P<0.02; two asterisks, P<0.01; ns, P>0.05; Student’s t-test). b, qRT-PCR of ler in WT in the absence/presence of B.thetaiotaomicron. RNAs from cultures grown in DMEM containing L-fucose or mucin. Error bar indicates standard deviations of ΔddCt values. (n=18 biological samples per assay; two asterisks, P<0.01; three asterisks, P<0.0001; ns, P>0.05; Student’s t-test). c, Competition assays between WT andΔfusK or ΔfusKfucR. 1:1 mixtures of fusK and WT EHEC or lacZ− and lacZ+ (WT) or fusKfucR and WT were intragastrically inoculated into infant rabbits. CFU in the mid-colon were determined 2-days post-inoculation. Each point represents a competitive index. Bars represent the geometric mean value for each group. (n=2 litters [6–11 animals]ΔlacZ and ΔfusKΔfucR, n=3 litters [11 animals]ΔfusK; asterisk, P<0.05; Mann-Whitney test).
In vitro competitions between ΔfusK and WT, and ΔfusK and Δler (does not express the LEE) in the absence and presence of B.theta, with either fucose or mucin as a sole C-source were performed. The competition index (CI) between ΔfusK and WT was 1 (Supplementary Fig. 14a) both in the absence or presence of B.theta during growth in fucose, suggesting that in the presence of free fucose, B.theta does not impact the competition between ΔfusK and WT, and that the growth advantage of ΔfusK in fucose is counteracted by the WT, which has decreased LEE expression. When these experiments were performed with mucin as a sole C-source the CI between ΔfusK and WT was 0.1 in the absence, and 1 in the presence of B.theta (Supplementary Fig. 14b). In the absence of B.theta there is no free-fucose, hence ΔfusK will not have a growth advantage. Furthermore, ΔfusK over-expresses the LEE, which constitutes an energy burden. Meanwhile, expression of fusKR is activated in mucus (Fig3a), further repressing the LEE in the WT. This scenario, however, reverts to a CI of 1 in the presence of B.theta, which releases fucose from mucin, conferring a growth advantage to ΔfusK, counteracting the WT (Supplementary Fig. 14b). Similar results were obtained in competitions between ΔfusK and Δler, consistent with the role of LEE gene expression being an energy burden in ΔfusK (Supplementary Fig. 14c).
The intricate role of FusK in EHEC’s metabolism and virulence plays a role in intestinal colonization. Competition assays in infant rabbits demonstrated that the WT outcompeted ΔfusK 10-fold (CI of 0.12), which is statistically different (p=0.039) from a control competition assay, where the WT (lacZ+) was competed against a ΔlacZ (CI of 0.7) (Fig. 4c). Hence, a functional FusK is necessary for robust EHEC intestinal colonization. To tease out whether the decreased ability of ΔfusK to colonize the mammalian intestine was due to uncontrolled expression of the LEE and/or fucose utilization, we performed competition experiments between WT and ΔfusKΔfucR, which does not express the fuc genes. The double mutant was outcompeted by the WT with a similar CI to the ΔfusK/WT (Fig. 4c), suggesting that fucose utilization does not play a major role in FusK-mediated intestinal colonization, and the burden of LEE over-expression by ΔfusK is a stronger determinant of its decreased fitness within the intestine.
FusKR repression of LEE expression in the mucus-layer prevents superfluous energy expenditure. Once in close contact to the epithelial surface, the QseCE adrenergic sensing-systems are triggered to activate virulence both directly through the QseCE cascade, and indirectly by repression of fusKR (Supplementary Fig. 15). EHEC competes with commensal E.coli (γ-Proteobacteria), but not B.theta, for the same C-sources (e.g. fucose) within the mammalian intestine15,24–28. Commensal E.coli, however, are not found in close contact with the epithelia, being in the mucus-layer, where it is counterproductive for EHEC to invest resources to utilize fucose, when EHEC can efficiently use other C-sources such as: galactose, hexorunates, and mannose, which are not used by commensal E.coli within the intestine15. Additionally, in contrast to commensal E.coli, EHEC is found closely associated with the intestinal epithelium25. Therefore, EHEC can utilize nutrients exclusively available at the surface of the epithelial cells. Consequently, the decreased expression of the fuc operon through fucose-sensing by FusKR (Fig. 3), may prevent EHEC from expending energy in fucose utilization in the mucus-layer, where it competes with commensal E.coli for this resource, and focus on utilizing other C-sources, not used by this competitor. Thus, the colonization defect of ΔfusK results from its inability to correctly time virulence and metabolic gene expression.
METHODS
Bacterial Strains, Plasmids and Growth Conditions
Strains and plasmids are listed in Supplemental Tables 1 and 2. E. coli strains were grown aerobically at 37°C in DMEM (Gibco) or LB unless otherwise stated. For studies involving fucose utilization, bacterial cultures were grown in M9 minimal media containing 0.4% L-fucose, D-glucose, L-rhamnose, D-galactose or D-xylose (Sigma) as a sole carbon source. For the co-culture experiments between EHEC and B. thetaiotaomicron, these strains were grown anaerobically at 37°C in DMEM (lacking glucose and pyruvate) with or without mucin or free fucose, at a 1:1 ratio. Enumeration of EHEC was performed through serial dilution of these cultures in McConkey agar containing streptomycin (EHEC strain 86-24 is streptomycin resistant, while B. thetaiotaomicron is sensitive to this antibiotic). Enumeration of B. thetaiotaomicron was performed through serial plating in TYG medium supplemented with 10% horse blood in the presence of gentamycin (B. thetaiotaomicron is gentamycin resistant, while EHEC is sensitive to this antibiotic)
Recombinant DNA techniques
Molecular biology techniques were performed as previously described29. Primers used in qRT-PCR and cloning are listed in supplemental Table 2.
Isogenic mutant construction
Construction of isogenic fusK, fusR, z0461, ler and fusKfucR mutants was performed using a lambda-red mediated recombination method as previously described30. Primers used to construct these knockouts are depicted in Supplemental Table 3. Briefly: a mutagenic PCR product was generated using primers containing homologous regions to sequences flaking z0462 (for fusK mutant), z0463 (fusR mutant), z0461, ler, and fucR to amplify a chloramphenicol resistance gene from pKD3. 86-24 cells harboring pKD46 were electroporated using the mutagenic PCR product and selected for chloramphenicol (Cm) resistance. Nonpolar mutants were generated by resolving the Cm resistant clones with resolvase encoded by pCP20. For complementation of the mutants, z0462 and z0463 previously cloned in ZeroBlunt TOPO, digested with BamHI and SalI then cloned into pACYC184, generating the plasmid pARP12 and pARP13, respectively. pARP12 was electroporated into fusK- to generate ARP09 complemented strain; pARP13 was electroporated into fusR- to generate ARP10 complemented strain.
FusR purification
FusR was cloned into ZeroBlunt TOPO, digested using XhoI and HindIII restriction sites then cloned into pBADMycHisA, generating pARP11. pARP11 was subsequently transformed into TOP10 cells, generating the ARP04 strain. ARP04 strain was grown in LB to OD600 0.6 at 37°C, at which point protein expression was induced by addition of a final concentration of 0.2% arabinose and growth overnight at 25°C. FusR was then purified using nickel columns (Qiagen).
Nested deletion analyses
Transcriptional fusions of the ler promoter with promoterless lacZ were described before31. To integrate the transcriptional fusions into the chromosome, E.coli MC4100 was lysogenized with phage λ45 and generating strains FS14 and FS16, respectively.
FusK purification and Reconstitution into Liposomes
FusK was cloned into ZeroBlunt TOPO, digested using XhoI and HindIII restriction sites then cloned into pBADMycHisA. This plasmid was subsequently transformed into TOP10 cells, generating the ARP03 strain. ARP03 strain was grown LB at 37°C until OD600 0.5 then protein expression was induced by addition of a final concentration of 0.2% arabinose and growth for 5 hours at 30°C. Cells were collected, resuspended in 50mL of Lysis buffer (50mM phosphate buffer pH 8.0, 1% Deoxycholic acid, 10mM imidazol, 300mM NaCl, 15% Glycerol, 5mM DTT, 100uL protease inhibitor cocktail), then lysed using emulsiflex. Lysates were incubated 1 hour for solubilization then cleared by centrifugation at 18,000 rpm for 30 minutes. Soluble fraction was collected by ultracentrifugation at 45,000 rpm for 1 hour to obtain membrane fraction, then membranes were resuspended in lysis buffer and incubated with Nickel beads for 1 hour at 4°C with gentle agitation. Membrane suspension and clear lysates were loaded into nickel–NTA columns, washed with Wash Buffer (50mM phosphate buffer pH 8.0, 20mM imidazol, 300mM NaCl, 5mM DTT, 0.1% Deoxycholic acid) and eluted in three steps with elution buffer (250mM Imidazol, 300mM NaCl, 1mM DTT, 0.1% Deoxycholic Acid). Protein was concentrated using centricons with molecular cutoff of 30,000KDa, then its concentration was determined by Bradford. Liposomes were loaded with FusK at ratio 20:1. Liposomes were reconstituted as described previously 32. FusK presence into liposomes was confirmed by western blot using anti-Myc antibody (Invitrogen).
Autophosphorylation and Phosphotransfer Assays
Autophosphorylation assays were performed as described previously3. A concentration of 100μM L-fucose or D-glucose was used. The bands were quantified using IMAGEQUANT version _ software. Quantification of triplets was performed. The Student t-test was used to determine statistical significance. A P-value of less than 0.05 was considered significant.
RNA extraction and Real-Time PCR
RNA from six biological replicates (experiments performed three independent times, total of 18 independent biological replicates) was extracted using RiboPure kit according to manufacturer’s instructions. Primer validation and real time PCR was performed as describe previously 33. Gene expression is represented as fold differences compared to the wild type strain 86-24. The error bars indicate the standard deviations of the ΔΔCT values. The Student t-test was used to determine statistical significance. A P-value of less than 0.05 was considered significant.
Microarrays
Microarrays and analysis were performed as previously described 34. The GeneChip E. coli Genome 2.0 array system of the Affymetrix system was used to compare the gene expression in strain 86-24 to that in fusK- and fusR- strains. The output from the scanning of the Affymetrix GeneChip® E. coli 2.0 were obtained using GCOS v 1.4 according to manufacturer’s instructions. Comparisons were performed using the analysis tools within GCOS v 1.4, by selecting the appropriate array, CHP file for comparison, and baseline values. Custom analysis scripts were written in Perl to complete multiple array analyses. Expression data can be accessed using accession number (GSE34991) at the NCBI GEO database.
Fluorescent Actin Staining (FAS) Assay
Fluorescein actin staining (FAS) assays were performed as previously described35. Pedestal enumeration was performed in 600 infected cells. The Student t-test was used to determine statistical significance. A P-value of less than 0.05 was considered significant.
In vitro competition assays
Bacterial strains were grown for 18 hours in LB at 37°C, resuspended in DMEM no glucose and inoculated 1:100 in DMEM (no glucose, no pyruvate) containing fucose or mucin as sole carbon source. B.theta was grown in TYG medium, resuspended in DMEM no glucose and inoculated at 1:9 ratio. In vitro competitions were carried out anaerobically and samples were collected hourly for serial dilution and plating for cfu count. A competition index was determined by the ratio of fusK- to WT EHEC or fusK-to ler-.
Infant rabbit infection studies
Litters of 3-day old infant rabbits were infected as described previously (Ritchie et al 2003). Individual rabbits were oro-gastrically inoculated (approx. 5 × 108 cfu per 90g) with 1:1 mixtures of wild type (lacZ−) EHEC and the fusK- of fusKfucR- mutants. The animals were necrotized 2 days post-inoculation and colonic tissue samples removed and homogenized prior to microbiological analysis. The number of wild type and fusK mutant cells present in the tissue homogenate was determined by serial dilution and plating on media containing Sm and bromo-chloro-indoyl-galactopyranoside (X-gal) as previously described (Ritchie et al 2003). Competition indexes (CI) were calculated as the ratio of fusK to wild type or fusKfucR to wild type in tissue homogenates divided by the ratio of fusK to wild type or fusKfucR to wild type in the input. The CI was compared to the CI value obtained when otherwise isogenic lacZ+ (wild type) and lacZ− strains were given to rabbits. Differences in CIs were compared using the Mann-Whitney test, where a P-value of less than 0.05 was considered significant. All animal experiments were performed were approved by the IACUC offices of UT Southwestern Medical Center and Brigham and Women’s Hospital. (n=2 litters [6–11 animals] ΔlacZ and ΔfusKΔfucR, n=3 litters [11 animals])
β-galactosidase activity assays
The bacterial strains FS14, FS15 and FS16 were transformed with pFusR or empty vector (pBADMycHisA) and grown in aerobically in DMEM containing 0.2% arabinose at 37°C to an OD600 of 0.8. The cultures were diluted 1:100 in Z buffer (60mM Na2HPO4.7H2O, 50mM β-mercaptoethanol) and assayed for β-galactosidase activity by using o-nitrophenyl-β-D-galactopyranoside as substrate as previously described 36.
Electrophoretic Mobility Shift Assay (EMSA)
EMSAs were performed using purified FusR-Myc-His and radiolabelled probes. Primers were end-labelled using [γ32P]ATP and T4 polynucleotide kinase (NEB), and subsequently used on a PCR to generate radiolabelled probes. End-labelled amplicons were run on a 6% polyacrylamide gel, excised, and purified using Qiagen Gel Extraction kit. To test the ability of FusR to directy bind to its target promoters, increasing amounts of FusR (0 to 4.35uM) were incubated with end-labelled probe (10 ng) for 20 minutes at 4°C in binding buffer (500ug/mL BSA, 50ng poly-dIdC, 6-mM HEPES pH 7.5, 5mM EDTA, 3mM DTT, 300mM KCl and 25mM MgCl2). A sucrose solution was used to stop the reaction 29. The reactions were run on a 6% polyacrylamide gel for 6 hours and 30 minutes at 180V. The gels were dried under vacuum and EMSAs were visualized by autoradiography.
DNAseI Footprinting
DNAseI footprint was performed as previously described 37. Briefly: primer Ler-18FP-R (Table 2) was end-labeled using [γ32P]ATP and T4 polynucleotide kinase (NEB) and used in a PCR with cold primer Ler-299FP-F (Table 2) to generate probe LerFP. The resulting end-labeled probe was used in binding reaction (described above in EMSA) for 20 min at room temperature. At this time, 1:100 dilution of DNAseI (NEB) and the manufacturer-supplied buffer were added to the reaction and digestion proceeded for 7 min at room temperature. The digestion reaction was stopped by addition of 100uL of stop buffer (200mM NaCl, 2mM EDTA and 1% SDS). Protein extraction was performed by phenol-chroloform and DNA was precipitated using 5M NaCl, 100% ethanol and 1uL glycogen. The DNAse reactions were run in a 6% polyacrylamide-urea gel next to a sequencing reaction (Epicentre). Amplicon generated using primers Ler-299FP-F and Ler-18FP-R (radiolabeled) was used as a template for the sequencing reaction. Footprint was visualized by autoradiography.
Supplementary Material
Acknowledgments
We thank M. Kendall for comments. We thank the Microarray-Core Facility. This work was supported by the National Institutes of Health (NIH) Grants AI053067, AI77853 and AI077613, and the Burroughs Wellcome Fund. M.M.C. was supported through NIH Training Grant 5 T32 AI7520-14. Its contents are solely the responsibility of the authors and do not represent the official views of the NIH NIAID.
Footnotes
Author contributions. A.R.P. led the project and contributed to all aspects. M.M.C. J.M.R., D.M., M.K.W. and C.G.M. helped with some experiments. V.S. designed experiments and wrote the paper.
Microarray data are deposited in the Gene Expression Omnibus under accession number GSE34991. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of this article at www.nature.com/nature.
References
- 1.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fischbach MA, Sonnenburg JL. Eating for two: how metabolism establishes interspecies interactions in the gut. Cell Host Microbe. 2011;10:336–347. doi: 10.1016/j.chom.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clarke MB, Hughes DT, Zhu C, Boedeker EC, Sperandio V. The QseC sensor kinase: A bacterial adrenergic receptor. Proc Natl Acad Sci U S A. 2006:10420–10425. doi: 10.1073/pnas.0604343103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reading NC, Rasko DA, Torres AG, Sperandio V. The two-component system QseEF and the membrane protein QseG link adrenergic and stress sensing to bacterial pathogenesis. Proc Natl Acad Sci U S A. 2009;106:5889–5894. doi: 10.1073/pnas.0811409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robbe C, Capon C, Coddeville B, Michalski JC. Structural diversity and specific distribution of O-glycans in normal human mucins along the intestinal tract. Biochem J. 2004;384:307–316. doi: 10.1042/BJ20040605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, et al. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 7.Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 8.Sperandio V, Torres AG, Jarvis B, Nataro JP, Kaper JB. Bacteria-host communication: the language of hormones. Proc Natl Acad Sci USA. 2003;100:8951–8956. doi: 10.1073/pnas.1537100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 10.Hughes DT, Clarke MB, Yamamoto K, Rasko DA, Sperandio V. The QseC adrenergic signaling cascade in Enterohemorrhagic E. coli (EHEC) PLoS Pathog. 2009;5:e1000553. doi: 10.1371/journal.ppat.1000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reading NC, Rasko D, Torres AG, Sperandio V. A transcriptome study of the QseEF two-component system and the QseG membrane protein in enterohaemorrhagic Escherichia coli O157: H7. Microbiology. 2010;156:1167–1175. doi: 10.1099/mic.0.033027-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrios H, Valderrama B, Morett E. Compilation and analysis of sigma(54)-dependent promoter sequences. Nucleic Acids Res. 1999;27:4305–4313. doi: 10.1093/nar/27.22.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perna NT, Plunkett G, 3rd, Burland V, Mau B, Glasner JD, Rose DJ, et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409:529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 14.Mellies JL, Barron AM, Carmona AM. Enteropathogenic and enterohemorrhagic Escherichia coli virulence gene regulation. Infect Immun. 2007;75:4199–4210. doi: 10.1128/IAI.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fabich AJ, Jones SA, Chowdhury FZ, Cernosek A, Anderson A, Smalley D, et al. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect Immun. 2008;76:1143–1152. doi: 10.1128/IAI.01386-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snider TA, Fabich AJ, Conway T, Clinkenbeard KD. E. coli O157:H7 catabolism of intestinal mucin-derived carbohydrates and colonization. Vet Microbiol. 2009;136:150–154. doi: 10.1016/j.vetmic.2008.10.033. [DOI] [PubMed] [Google Scholar]
- 17.Chen YM, Zhu Y, Lin EC. The organization of the fuc regulon specifying L-fucose dissimilation in Escherichia coli K12 as determined by gene cloning. Mol Gen Genet. 1987;210:331–337. doi: 10.1007/BF00325702. [DOI] [PubMed] [Google Scholar]
- 18.Jaswal VM, Babbar HS, Mahmood A. Changes in sialic acid and fucose contents of enterocytes across the crypt-villus axis in developing rat intestine. Biochem Med Metab Biol. 1988;39:105–110. doi: 10.1016/0885-4505(88)90064-3. [DOI] [PubMed] [Google Scholar]
- 19.Island MD, Wei BY, Kadner RJ. Structure and function of the uhp genes for the sugar phosphate transport system in Escherichia coli and Salmonella typhimurium. J Bacteriol. 1992;174:2754–2762. doi: 10.1128/jb.174.9.2754-2762.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weston LA, Kadner RJ. Identification of uhp polypeptides and evidence for their role in exogenous induction of the sugar phosphate transport system of Escherichia coli K-12. J Bacteriol. 1987;169:3546–3555. doi: 10.1128/jb.169.8.3546-3555.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weston LA, Kadner RJ. Role of uhp genes in expression of the Escherichia coli sugar-phosphate transport system. J Bacteriol. 1988;170:3375–3383. doi: 10.1128/jb.170.8.3375-3383.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Y, Lin EC. An evolvant of Escherichia coli that employs the L-fucose pathway also for growth on L-galactose and D-arabinose. J Mol Evol. 1986;23:259–266. doi: 10.1007/BF02115582. [DOI] [PubMed] [Google Scholar]
- 23.Chen YM, Tobin JF, Zhu Y, Schleif RF, Lin EC. Cross-induction of the L-fucose system by L-rhamnose in Escherichia coli. J Bacteriol. 1987;169:3712–3719. doi: 10.1128/jb.169.8.3712-3719.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, et al. Regulated Virulence Controls the Ability of a Pathogen to Compete with the Gut Microbiota. Science. 2012 doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miranda RL, Conway T, Leatham MP, Chang DE, Norris WE, Allen JH, et al. Glycolytic and gluconeogenic growth of Escherichia coli O157:H7 (EDL933) and E. coli K-12 (MG1655) in the mouse intestine. Infect Immun. 2004;72:1666–1676. doi: 10.1128/IAI.72.3.1666-1676.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang DE, Smalley DJ, Tucker DL, Leatham MP, Norris WE, Stevenson SJ, et al. Carbon nutrition of Escherichia coli in the mouse intestine. Proc Natl Acad Sci U S A. 2004;101:7427–7432. doi: 10.1073/pnas.0307888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Autieri SM, Lins JJ, Leatham MP, Laux DC, Conway T, Cohen PS. L-fucose stimulates utilization of D-ribose by Escherichia coli MG1655 DeltafucAO and E. coli Nissle 1917 DeltafucAO mutants in the mouse intestine and in M9 minimal medium. Infect Immun. 2007;75:5465–5475. doi: 10.1128/IAI.00822-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fox JT, Drouillard JS, Shi X, Nagaraja TG. Effects of mucin and its carbohydrate constituents on Escherichia coli O157 growth in batch culture fermentations with ruminal or fecal microbial inoculum. J Anim Sci. 2009;87:1304–1313. doi: 10.2527/jas.2008-1166. [DOI] [PubMed] [Google Scholar]
- 29.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 30.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharp FC, Sperandio V. QseA directly activates transcription of LEE1 in enterohemorrhagic Escherichia coli. Infect Immun. 2007;75:2432–2440. doi: 10.1128/IAI.02003-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janausch IG, Garcia-Moreno I, Lehnen D, Zeuner Y, Unden G. Phosphorylation and DNA binding of the regulator DcuR of the fumarate-responsive two-component system DcuSR of Escherichia coli. Microbiology. 2004;150:877–883. doi: 10.1099/mic.0.26900-0. [DOI] [PubMed] [Google Scholar]
- 33.Walters M, Sperandio V. Quorum sensing in Escherichia coli and Salmonella. Int J Med Microbiol. 2006;296:125–131. doi: 10.1016/j.ijmm.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 34.Kendall MM, Rasko DA, Sperandio V. Global effects of the cell-to-cell signaling molecules autoinducer-2, autoinducer-3, and epinephrine in a luxS mutant of enterohemorrhagic Escherichia coli. Infect Immun. 2007;75:4875–4884. doi: 10.1128/IAI.00550-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knutton S, Baldwin T, Williams PH, McNeish AS. Actin accumulation at sites of bacterial adhesion to tissue culture cells: basis ofa new diagnostic test for enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun. 1989;57:1290–1298. doi: 10.1128/iai.57.4.1290-1298.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller JH. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- 37.Sperandio VV. How the bacterial flora and the epithelial cell get along. Trends Microbiol. 2000;8:544. doi: 10.1016/s0966-842x(00)01896-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.