

Abstract
Nitroxyl (HNO) has gained interest as a potential treatment of congestive heart failure through the ability of the HNO donor, Angeli’s salt (AS), to evoke positive inotropic effects in canine cardiac muscle. The release of nitrite during decomposition limits the use of AS requiring other HNO sources. Acyloxy nitroso compounds liberate HNO and small amounts of nitrite upon hydrolysis and the synthesis of the water-soluble 4-nitrosotetrahydro-2H-pyran-4-yl acetate and pivalate allows for pig liver esterase (PLE)-catalysis increasing the rate of decomposition and HNO release. The pivalate derivative does not release HNO, but the addition of PLE catalyzes hydrolysis (t1/2 = 39 min) and HNO formation (65% after 30 min). In the presence of PLE, this compound converts metmyoglobin (MetMb) to iron nitrosyl Mb and oxyMb to metMb indicating these compounds only react with heme proteins as HNO donors. The pivalate in the presence and the absence of PLE inhibits aldehyde dehydrogenase (ALDH) with IC50 values of 3.5 and 3.3 μM, respectively, in a time-dependent manner. Reversibility assays reveal reversible inhibition of ALDH in absence of PLE and partially irreversible inhibition with PLE. Liquid chromatography-mass spectrometry (LC-MS) reveals formation of a disulfide upon incubation of an ALDH peptide without PLE and a mixture of disulfide and sulfinamide in the presence of PLE. A dehydroalanine residue forms upon incubation of this peptide with excess AS. These results identify acyloxy nitroso compounds as unique HNO donors capable of thiol modification through direct electrophilic reaction or HNO release.
Keywords: nitroxyl, nitric oxide, thiol protein, heme proteins, esterase, dehydroalanine
1. INTRODUCTION
Nitroxyl (HNO/NO−), is the one electron reduced relative of nitric oxide (NO), an important biological signaling agent [1-5]. Nitroxyl has distinct biological properties compared to NO and is a potential treatment of myocardial reperfusion injury and congestive heart failure [6-8]. Nitroxyl avidly reacts with heme and thiol containing proteins and these reactions likely mediate its biological actions [8-11]. Nitroxyl must be generated from donors due to its fast dimerization to nitrous oxide (N2O, k = 8 × 106 M−1s−1) and a limited number of donors exist [12-14]. Angeli’s salt (NaN2O3, AS) is the most widely used HNO donor, but at physiological pH AS forms an equivalent of nitrite during decomposition [15], Nitrite reacts with all of the redox forms of hemoglobin and possesses its own biological profile, which must be accounted for in biological evaluations of HNO activity [15-19]. Other limitations of AS include the fast rate of decomposition (k = 10−4 s−1) and difficulty in modifying the structure [14, 15]. The ability of AS to elicit positive inotropic effects in canine cardiac muscle highlights the need for new HNO donors as potential new therapies [7, 20].
Acyloxy nitroso compounds (1-3, Figure 1) act as HNO donors through ester hydrolysis to give an unstable intermediate that decomposes to HNO without generation of nitrite in buffered conditions [21]. Varying the pH and the R group of the ester of these structures varies their stability, HNO donor activity and ability to relax pre-constricted rat aorta [22]. Compounds 1 and 2 only slowly hydrolyze and competitively react with other nucleophiles (thiolates) without HNO formation, but 3 hydrolyzes to HNO under all conditions [22]. While compounds 1-3 provide information concerning the reactivity and HNO release of acyloxy nitroso compounds, their lack of water solubility limits their use in biological systems. Incorporating oxygen into the ring of 1 and 2 gives 4 and 5, and should greatly increase water solubility allowing for esterase mediated hydrolysis to increase the rate of decomposition (Figure 1) [23]1.
Figure 1.

Hydrolytic HNO release from 1-5.
The increased water solubility of 4-5 and their structural simlarlity to C-nitroso compounds, which also react with thiols [24], allows evaluation with myoglobin (Mb), a heme protein, and aldehyde dehydrogenase (ALDH), a thiol containing protein, known HNO targets. in the presence of pig liver esterase (PLE) to enhance HNO release [11, 25, 26]. Our results show that acyloxy nitroso compounds can act as efficient HNO donors in the presence of PLE, which may be useful in further probing HNO biology.
2. EXPERIMENTAL
2.1 General
All chemicals were purchased from Sigma Aldrich Chemical Company and used as received. AS and 15N-AS were prepared as described, and stored dry at −20 °C [27]. Analytical TLC was performed on silica gel plates with C-4 Spectroline 254 indicator. Visualization was accomplished with UV light. Solvents for extraction and purification were technical grade and used as received. Liquid chromatography - mass spectrometry (LC-MS) solvents were HPLC grade. 1H NMR and 13C NMR spectra were recorded using a Bruker Avance 300 MHz NMR spectrometer. Chemical shifts are given in ppm (); multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet). Yeast ALDH (E.C. 1.2.1.5) was purchased from Sigma Aldrich Chemical Company. UV-vis spectrometry was performed on a Cary 100 Bio UV-visible (UV-vis) spectrophotometer (Varian, Walnut Creek, CA). IC50s were obtained by fitting concentration-dependent percent of control data non-linear curve fitting using sigmoidal curve with standard slope algorithm using GraphPad Prism software. ALDH peptide A286-R307 (AIDWIAAGIFYLNSGQNCTANSR) was purchased from Peptide 2.0 (Chantilly, VA) with > 98 % purity. High-resolution mass spectra were obtained using a Thermo Scientific LTQ Orbitrap mass spectrometer equipped with a heated electrospray ionization source operated in positive ion mode.
2.2 Synthetic Procedures
4-Nitrosotetrahydro-2H-pyran-4-yl Acetate (4)
Ultraviolet-visible (UV-vis) (PBS) λmax = 660 nm, ε= 20.7 M−1 cm−1, 1H NMR (300 MHz, benzene-d6)δ 1.5 (dd, J = 2.5, 14.0 Hz 2H), 1.7 (s, 3H), 1.9 (ddd, J = 5.0, 11.5 Hz, 2H), 3.3 (td, J = 2.7, 11.6 Hz, 2H), 3.6 (ddd, J = 4.6, 11.8 Hz, 2H), 13C NMR (75 MHz, benzene-d6)δ 21.8, 31.3, 64.2, 120.2, 167.6.
4-Nitrosotetrahydro-2H-pyran-4-yl Pivalate (5) [22]
UV-vis (DMF:PBS, 1:29) λmax = 660 nm, ε= 20.7 M−1 cm−1, 1H NMR (300 MHz, benzene-d6)δ 1.2 (s, 9H), 1.5 (dd, J = 2.5, 14.0 Hz, 2H), 1.9 (ddd, J = 5.0, 11.5 Hz, 2H), 3.3 (td, J = 2.4, 11.5 Hz, 2H), 3.6 (ddd, J = 4.8, 11.7 Hz, 2H), 13C NMR (75 MHz, benzene-d6)δ 28.3, 31.1, 40.2, 64.3, 119.6, 175.0.
2.2 UV-Vis Spectroscopy Assay for the decomposition of 4, 5 [21]
A solution of phosphate buffered saline (PBS, 100 mM, pH 7.4) was added to a solution of 4 (5 μmol) in PBS (100 mM, pH 7.4, 100 μL) or 5 (5 μmol) in anhydrous dimethyl formamide (DMF) (50 μL) to give a total volume of 1.5 mL at 37 °C in a sealed cuvette. UV-Vis spectra were taken every 5 min for 6 hr at 660 nm. Similar experiments were performed using a solution of PLE (9 U/μmol) in PBS (100 mM, pH 7.4).
2.3 Gas Chromatographic N2O Analysis [28]
For headspace analysis, a solution of 4 or 5 (0.08 μmol) in anhydrous DMF (8 μL) followed by PBS (10 mM, pH 7.4, 0.8 mL) was injected into a 10 mL round bottom flask sealed with a rubber septum and flushed with inert gas. To some samples PLE (9U/μmol) and GSH (1.60 μmol) were added, the sample was incubated at 37 °C, and at desired timepoints headspace aliquots (100 μL) were injected via a gas-tight syringe onto a 7890A Agilent Technologies gas chromatograph equipped with a micro-electron capture detector and a 30 m × 0.32 m (25 μm) HP-MOLSIV capillary column. The oven was operated at 200 °C for the duration of the run (4.5 minutes). The inlet was held at 250 °C and run in split mode (split ratio 1:1) with a total flow (N2 as carrier gas) of 4 mL/min and a pressure of 37.9 psi. The micro electron capture detector (ECD) was held at 325 °C with a makeup flow (N2) of 5 mL/min. The retention time of nitrous oxide was 3.4 min, and yields were calculated based on a standard curve for nitrous oxide (Matheson Tri-Gas).
2.4 Chemiluminescence NO, NO2− Assay [21]
A solution of 4 or 5 (0.08 μmol) in anhydrous DMF (8 μL) followed by PBS (10 mM, pH 7.4, 0.8 mL) was injected into a 10 mL round bottom flask sealed with a rubber septum and flushed with inert gas. To some samples PLE (9U/μmol) was added and the sample was incubated at 37 °C, and at desired timepoints, aliquots (200 uL headspace, NO or 5 μL reaction mixture, NO2−) were injected into the reaction vessel of a Sievers 280 NOA chemiluminescence detector. This apparatus detects nitrite by reduction of nitrite to NO, followed by chemiluminescence NO detection (the reaction vessel contained 1 % NaI in glacial acetic acid). The yields were calculated based on a standard curve for nitrite.
2.5 UV-vis assay of Mb with 5
2.5.1 UV-vis Assay of Ferrous Nitrosyl Complex from the Reaction of 5 with metmyoglobin (metMb)
Under degassed conditions, a solution of 5 (2.58 μmol) in anhydrous DMF (25.8 μL) was added to a solution of metMb (0.129 μmol) in PBS (100 mM, pH 7.4, 1.47 mL) at 37 °C in a sealed cuvette. In the reactions that contained PLE (9 U/μmol, 85 U/mL in PBS, 100 mM, pH 7.4), it was added directly before UV-vis measurements began. UV-vis spectra were taken every 1.0 min for 4 hrs, and the disappearance of metMb was monitored by the decrease in absorbance at 502 and 630 nm, while the appearance of Mb-NO was monitored by the increase in absorbance at 542 and 583 nm [26].
2.5.2 UV-vis Assay of metmyoglobin (metMb) from the Reaction of 5 with oxymyoglobin (MbO2)
A solution of 5 (84 nmol) in anhydrous DMF (1 μL) was added to a solution of MbO2 (42 nmol) in PBS (100 mM, pH 7.4, 999 μL) at 37 °C in a sealed cuvette. In some reactions, PLE (9 U/μmol, 85 U/mL in PBS, 100 mM, pH 7.4) was added directly before UV-vis measurements began. UV-vis spectra were taken every 2.0 min for 4 hrs, and the disappearance of MbO2 was monitored by the decrease in absorbance at 540 and 580 nm, while the appearance of metMb was monitored by the increase in absorbance at 502 and 630 nm.
2.6 Inhibition Studies
2.6.1 In vitro inhibition of yeast ALDH [24]
Yeast ALDH (0.1 U) was preincubated for 10-120 min with the inhibitor (AS dissolved in 10 mM NaOH, nitrosobenzene (NB) in DMSO, 5 in DMF in the presence or absence of PLE (9 U/μmol, 85 U/mL, 100 mM PBS, pH 7.5)) at 37 °C in the primary reaction mixture (100 mM PBS, pH 7.5) to give a total volume of 0.1 mL. After 10-120 min, 20 μL of the primary mixture was added to a cuvette that contained the secondary mixture (90 mM potassium phosphate buffer, pH 8.0, 1.0 mM EDTA, 0.5 mM NAD+ and 30 % glycerol) to give a final volume of 1.0 mL. The reduction of NAD+ to NADH was activated by the addition of benzaldehyde (0.6 μmol), and the activity of ALDH was followed by the increase in absorbance at 340 nm over 30 minutes Measurements were taken in triplicate and IC50 values were calculated.
2.6.2 In vitro reactivation of inhibited yeast ALDH [25]
The experimental conditions were repeated as in the inhibition assay with each inhibitor at a concentration of 10 times above the calculated IC50. DTT (10mM) was added to the primary mixture after the initial 1-10 min of incubation with the respective inhibitor at 37 °C. After the addition of DTT, the reaction mixture sat for 3 min at room temperature and an aliquot (20 μL) of this reaction mixture was added to the cuvette and the assay was completed as above to measure ALDH activity.
2.7 LC-MS Studies
2.7.1 LC-MS: Reactivity of ALDH peptide A286-R307 with nitrosobenzene (NB), AS, and 5 in the presence and absence of PLE
ALDH peptide A286-R307 (10 μM in 100 mM NH4CO3 buffer) was incubated with NB (10 eq., anhydrous DMF), AS (10, 1000 eq., stock solution in 100 mM NaOH), 15N AS (100 eq., stock solution in 100 mM NaOH), and 5 (10 eq., stock solution in anhydrous DMF) in the presence or absence of PLE (9U/μmol, stock solution in 100 mM NH4CO3 buffer) for 1 hour or 3.5 hours (5 × t1/2 of 5 + PLE) at room temperature (unless noted at 37 °C), and then analyzed by LC-MS on a Thermo Scientific LTQ Orbitrap XL. Separations were achieved using a Thermo Scientific Hypersil GOLD C18 column (150 × 2.1 mm, 1.9 μm). During LC, solvent A (0.1 % formic acid in H2O) and B (0.1 % formic acid in MeOH) were held at a flow rate of 400 μL/min. Conditioning the column involved transitioning from acetonitrile to 95 % A and 5 % B over 15 min, and held for 5 min. LC analysis consisted of injections (10 μL) using a gradient elution of 5 to 95 % B over 10 min, followed by lowering to 5 % B over 10 sec, and held for 1 min. After each analysis two blanks were run. Electrospray ionization (ESI) parameters consisted of a spray voltage of 4 kV, capillary voltage of 40 V, and a capillary temperature of 325 °C. Positive mode high resolution mass spectra were collected with 30,000 Hz resolution from 200-2000m/z. Extracted masses with an error of +/- 5 ppm were found to be 1186.5573 [M(thiol) + 2H]+2, 1202.0613 [M(sulfinamide) + 2H]+2, 1192.0491 [M(dehydroalanine(dhA))+ 2Na]+2, 1240.0791 [M(N-phenyl sulfinamide) + 2H]+2, and 1185.5501 [M(disulfide (RSSR)) + 4H]+4. In addition, the presence of extracted masses for [M(sulfinic acid(SO2−)) + 2H]+2 and [M(sulfonic acid(SO32−)) + 2H]+2 was not observed in these experiments. N-Ethylmalemide (NEM) thiol trapping of the unreacted peptide was performed and analyzed as above.
3. RESULTS and DISCUSSION
3.1 Synthesis
Substitution of the cyclohexane ring of 1-2 with an oxygen-containing tetrahydropyran ring gives new acyloxy nitroso compounds (4-5) that can be prepared using previously reported methodology (Figure 2) [21, 22]. Condensation of tetrahydropyran-4-one with hydroxylamine hydrochloride results in dihydro-2H-pyran-4(3H)-one oxime and lead (IV) tetraacetate oxidation in the absence and presence of 5 equivalents of 2, 2-dimethylpropanoic acid gives 4-nitrosotetrahydro-2H-pyran-4-yl acetate and pivalate (4 and 5, Figure 2). 1H and 13C NMR spectroscopy confirm the structure of the oxime as well as 4 and 5, which exist as bright blue oils. Compounds (4-5) demonstrate significant water solubility (1.7 mM in buffer (compound 4) or a 3 % DMF/buffer mixture (compound 5)) presumably due to the increased polarity and hydrogen bonding interactions of the tetrahydropyran ring oxygen atom with water.
Figure 2.

Synthesis of 4-5.
3.2 Decomposition kinetics
A hydrolytic mechanism provides a reasonable explanation for the observed decomposition rates and HNO formation from 4 and 5 (Figure 3) [21, 22]. Similar to 1-3, HNO release from 4 and 5 requires ester hydrolysis to generate an unstable α-hydroxy nitroso intermediate that collapses to cyclohexanone and HNO (Figure 3) [21]. The improved aqueous solubility of 4 and 5 allows the addition of pig liver esterase (PLE) as a catalyst for hydrolysis (Figure 3). Monitoring the disappearance of the absorption at 660 nm using ultraviolet-visible (UV-Vis) spectroscopy provides kinetic information regarding the decomposition of 4 and 5 in the presence and absence of PLE. Compound 4 decomposes under buffered conditions with a rate constant of k = 3.0 × 10−5 s−1 (t1/2 = 6.4 hr, compared to t1/2 = 14.8 hr for 1) and the addition of PLE increases decomposition with an observed rate constant of k = 5.0 × 10−4 s−1 (t1/2 = 23 min, under these conditions which include 9 U of PLE per μmol of 4). Decomposition of 4 in a completely aqueous environment compared to the aqueous/organic mixture required to dissolve 1 likely enhances hydrolysis. Incubation of 5 in a 3 % mixture of DMF:PBS (100 mM, pH 7.4) at 37 °C shows very slow decomposition over time with a rate constant of k = 6.0 × 10−6 sec−1 (t1/2 = 32 hr, compared to t1/2 = 37.8 hr for 2, Supplemental Data). Under the same reaction conditions, addition of PLE increases the decomposition of 5 50-fold with an observed rate constant of k = 3.0 × 10−4 s−1 (t1/2 = 39 min, under these conditions which include 9 U of PLE per μmol of 5, Figure 3). As expected, the improved water solubility of 5 compared to 2 does not affect hydrolysis of the stable pivalate ester.
Figure 3.

Decomposition of 4-5 in the presence of PLE.
3.3 Gas Chromatographic and Chemiluminescence N2O and NO2− Analysis
Nitrous oxide (N2O), the dimerization and dehydration product of HNO, provides evidence for HNO intermediacy during the decomposition of 4 and 5 (Figure 3) [13]. Figure 4 shows that the decomposition of 4 in buffer generates N2O (17 %, 30 min) as determined by gas chromatography (GC), but the decomposition of 5 does not generate N2O over time. The addition of PLE (9 U/μmol) to 5 results in N2O (65 %) after 30 min, which does not increase over time and suggests other reactions of HNO compete with dimerization under these conditions. Addition of glutathione (GSH) traps HNO and quenches N2O formation, providing further evidence for the presence of HNO (data not shown). Addition of PLE to 4 also increases the amount of N2O formed (Figure 4) and the amount of N2O formation again corresponds to the observed decomposition rates in the presence of PLE. These results strongly suggest that PLE, a serine-based esterase that accepts large hydrophobic substrates catalyzes-hydrolysis and HNO release from these compounds (Figure 3) [29, 30].
Figure 4.

Yields of N2O and NO2− from 4 and 5 as a function of PLE as determined by GC and chemiluminescence analysis.
Figure 4 also shows NO2− formation, as monitored by chemiluminescence, from the decomposition of 4 and 5 in the presence and absence of PLE. In the absence of PLE, decomposition of 4 or 5 only yields small amounts of NO2− (4 - 8 %, Figure 4) and no nitric oxide (Supplemental Data for 5). In the presence of PLE, the amount of NO2− increases over time and 4 yields (17 %, 4 hr, conditions that give 56 % N2O), and under the same conditions 5 produces 10 % NO2− after 4 hr (five-times the half-life of decomposition under these conditions), an amount far less than the one equivalent generated by Angeli’s salt. Nitrite formation appears linked to the presence of the esterase as little nitrite forms in the absence of PLE and the amount of nitrite is proportional to the amount of PLE added (data not shown). Nucleophilic addition of the serine residue of the esterase to the electrophilic nitroso group of 4 or 5 (rather than the ester carbonyl) would yield an addition product that may decompose to alternative products including nitrite [29, 30]. Alternatively, the aerobic oxidation of HNO forms nitrite as the final nitrogen-containing product and presents another path for nitrite formation in these reactions (Figure 3) [31-33]. These results identify 5 as a water-soluble and switchable HNO-donor that does not hydrolyze (or release HNO), and likely behaves as a C-nitroso compound until esterase catalyzed hydrolysis rapidly releases HNO. This information generally segregates the acyloxy nitroso compounds (1-5) into three broad groups regarding kinetic decomposition (and HNO release) at neutral pH with 1 and 4 being slow HNO donors, 2 and 5 (in the absence of PLE) do not release HNO and 3 and 5 (in the presence of PLE) being rapid HNO donors similar to AS [22].
3.4 Reaction of 5 with MetMb and MbO2
The solubility and HNO release properties of 4 and 5 allow evaluation of their interactions with biological targets. Based on the drastic differences in the kinetics of 5 in the absence and presence of PLE, further experiments focused on the reactions of 5 with heme and thiol containing proteins. UV-vis measurements of the incubation with 5 in the presence of PLE with metMb (86 μM) in PBS reveal the disappearance of the peaks at 503 and 632 nm with the appearance of peaks at 542 and 581 nm. These results show the conversion of metMb to the ferrous nitrosyl complex (Figure 5), and support HNO formation from 5 [26]. Similar measurements upon addition of 5 (84 μM) and PLE to a solution of MbO2 (42 μM) show the disappearance of the peaks at 543 and 582 nm with the appearance of peaks at 503 and 633 nm. These results reveal the conversion of oxyMb to metMb and further support HNO formation (Figure 5) [26]. Kinetic analysis of these processes yields rate constants of = 3.0 × 10−4 s−1 (t1/2 = 39 min), which coincide with the rate of PLE-mediated decomposition of 5 supporting HNO formation through PLE catalyzed hydrolysis and decomposition of an -hydroxy nitroso intermediate (Figure 3 and Supplemental Data). Incubation of 5 with metMb or MbO2 in the absence of PLE results in no change in the absorption spectra indicating that the nitroso groups of these acyloxy nitroso compounds do not react with these iron-heme proteins (Supplemental Data). The low level of nitrite, which reacts with both metMb and MbO2, generated from the esterase catalyzed hydrolysis makes 5 a potentially valuable alternative HNO source for studying reactions with iron-heme proteins.
Figure 5.

UV-vis assay of the reaction of 5 with Mb. A) Incubation of metMb (86 μM, 100 mM PBS, pH 7.4, 37 °C) with 5 (1.7 mM) and PLE (9 U/μmol). B) Incubation of MbO2 (42 μM, 100 mM PBS, pH 7.4, 37 °C) with 5 (84 μM) and PLE (9 U/μmol).
3.5 Inhibition Studies
3.5.1 In vitro inhibition of yeast ALDH.
Nitrosobenzene (NB), a C-nitroso compound which cannot hydrolytically release HNO and Angeli’s salt-derived HNO (the simplest nitroso compound) both inhibit the thiol-containing enzyme ALDH (reported IC50 values of 2.5 and 1.8 μM, respectively) [24, 25]. ALDH inhibition arises from a direct reaction of these nitroso compounds with the active site thiol (C302) resulting in inhibition characterized by both an irreversible and reversible (by DTT) component resulting from sulfinamide and disulfide modifications [25]. Previous work defines the reactivity of acyloxy compounds (1-3) with small molecule thiols and while the trifluoroacetate derivative (3) hydrolyzes to form HNO under all conditions (like AS), 1-2 directly react with thiolates (similar to C-nitroso compounds) to give the corresponding disulfide and oxime [22]. Based on this precedent, acyloxy compound 5 should inhibit ALDH both in the presence and absence of PLE through different mechanisms. Nitrosobenzene and AS inhibit ALDH in a concentration dependent manner with IC50 values of 0.2 and 2.1 μM, respectively, and 5 inhibits ALDH (IC50 values of 3.5 and 3.3 μM in the absence and presence of PLE, respectively, Supplemental Data) with similar potency to NB and AS. The inhibition of yeast ALDH by NB, AS, and 5 with or without PLE, (at concentrations of inhibitor one-half [IC50]) increases with increased incubation time suggesting a reactive process resulting in a covalent enzyme modification (Figure 6). For NB and AS, the majority of the inhibition occurs within the first ten minutes of incubation while inhibition with 5 (with and without PLE) progresses over two hours (Figure 6). Experiments with NB and AS at concentrations ten-fold the IC50, show complete inhibition of the enzyme within 10 min (Supplemental Data).
Figure 6.

Effects of incubation time on ALDH inhibition of by various C-nitroso compounds or HNO donors at one-half [IC50]. Control indicates enzyme and substrate incubated at 10 and 120 min.
3.5.2 In vitro reactivation of ALDH activity
Treatment of ALDH inactivated by NB with dithiothreitol (DTT) restores 15% of the enzyme activity revealing a non-reducible modification and reactivation of AS-inactivated ALDH returns 46 % of the enzyme activity suggesting a mixture of reversible and irreversible modifications, as previously reported (Figure 7) [25]. Treatment of ALDH inactivated by 5 alone with DTT restores 63 % of enzyme activity suggesting a reducible disulfide as the predominant modification and suggests that 5 inhibits ALDH via different mechanisms from both NB and AS (Figure 7). Treatment of ALDH inactivated by 5 in the presence of PLE with DTT restores 43 % of the enzyme activity, similar to AS, indicating a mixture of reversible (reducible) and non-reducible modifications, further supporting esterase catalyzed hydrolysis of 5 and HNO formation (Figure 7). Increasing the incubation time of 5 in the presence of PLE to five times the half-life of decomposition does not change DTT reactivation of the enzyme (Supplemental Data). Previous work shows that NO2− from AS does not inhibit ALDH indicating inhibition by 5 with PLE in these experiments occurs via HNO [25]. Overall, these results indicate 5 in the presence of PLE releases HNO and inhibits ALDH in a similar fashion to HNO generated from AS. Nitrosobenzene inhibition arises from the formation of a non-reducible enzyme modification while inhibition with 5 alone likely generates a reducible disulfide revealing that while acyloxy nitroso compounds demonstrate electrophilic behavior similar to C-nitroso compounds, the reactions of acyloxy nitroso and C-nitroso compounds yield different products.
Figure 7.

DTT reactivation of ALDH activity after incubation with C-nitroso compounds or HNO donors (ten-times [IC50]).
3.7 LC-MS Analysis of the reaction of HNO donors with the ALDH peptide A285-R307
High resolution LC-MS experiments provide further molecular insight to the mechanism of ALDH inhibition by nitrosobenzene, AS and 5 in the absence and presence of PLE. Initial experiments using full length intact ALDH followed by tryptic digestion fail to yield clear results regarding the products of these reactions and necessitate the use of ALDH A285-R307 peptide (AIDWIAAGIFYLNSGQNCTANSR), which contains the active site thiol (C302), as a simple model. As this peptide only contains a single cysteine, intramolecular disulfides that play a role in reversible HNO-mediated ALDH inhibition cannot form and results from the reactions of these compounds and this peptide provide structural rather than functional information [25]. Such MS experiments show the expected mass for the unreacted peptide and the addition of N-ethyl maleimide confirms the presence of a thiol (Table 1). Treatment of the ALDH peptide A285-R307 with NB results in the formation of a new peak with a m/z of 1240.0791, which has been assigned the N-phenyl sulfinamide structure (Table 1 and Supplemental Data section, which contains detailed MS data for each incubation). Reaction of the peptide with 5 only forms the disulfide derived from two peptides (m/z = 1185.5496, Table 1). These modifications, formed from electrophilic but non-HNO releasing compounds, explain (within the limitations of this model peptide) the observed inhibition and reactivation results with the full protein as DTT would reduce the disulfide returning activity but not the N-phenyl sulfinamide.
Table 1.
LC-MS Analysis of the Reaction of C-Nitroso Compounds and HNO Donors and AIDH peptide A2B5-R3OT.
| Reagent | Retention Time (min) |
m/z | Product(s) | |
|---|---|---|---|---|
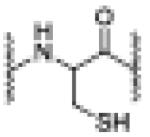
| --- | 6.98 | 1186.5573 [M+2H]+2 |
unreacted peptide |
|
|
| ||||
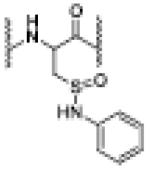
| NB | 7.17 | 1240.0791 [M[N-phenyl sulfinamide + 2H]+2 |
N-phenyl sulfinamide |
|
|
| ||||
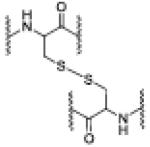
| 5 | 7.54 | 1185.5496 [M(RSSR) + 4H]+4 |
RSSR |
|
|
| ||||
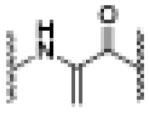
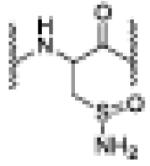
| AS (10eq.) | 6.82 | 1192.0491 [M(dhA)+2Na]+2 |
dhA -major |
|
| 6.94 | 1202.0620 [M(sulfinamide + 2H]+2 |
sulfinamide-minor |
|
|
|
| ||||
| AS(1000 eq.) | 6.89 | 1202.0613 [M(sulfinamide + 2H]+2 |
sulfinamide |
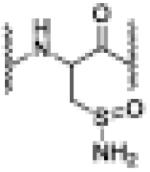
|
|
| ||||
| 15N-AS (1000 eq.) | 6.91 | 1202.5616 [M(15Nsulfinamide + 2H]+2 |
15Nsulfinamide |
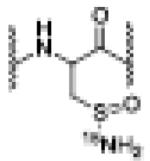
|
|
| ||||
| 5 + PLE | 7.52 | 1185.5514 [M(RSSR) + 4H]+4 |
RSSR |
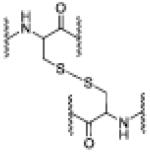
|
| 6.92 | 1202.0618 [M(sulfinamide + 2H]+2 |
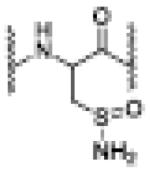
sulfinamide |
|
|
As expected for the reaction of a thiol with excess HNO [31, 34, 35], incubation with AS (1000 eq.) gave a peak with a m/z = 1202.0613, which corresponds to a sulfinamide as the only product (Table 1) and use of 15N-AS yields the 15N-substituted sulfinamide (Table 1) [36]. Interestingly, LC-MS analysis of the reaction of ALDH peptide A285-R307 with AS (10 eq.) does not give the expected disulfide but yields a major product (based on ion count) with a m/z = 1192.0491, which corresponds to the loss of HSONH2, suggesting dehydroalanine (dhA) containing peptide formation (Table 1). A minor product with a m/z = 1202.0620 that corresponds to a sulfinamide also forms, and no evidence of sulfinic or sulfonic acids was detected (Table 1). Addition of L-cysteine to this reaction mixture followed by LC-MS gives an l-cysteine addition product further confirming the structure and revealing dehydroalanine formation occurs before mass spectrometry. MS2 experiments confirm the sequence of these peptides and site of modification (Supplemental Data). No precedent currently exists for dehydroalanine formation from the reaction of HNO with cysteine, although treatment of cysteine containing proteins with the aminating reagent, O-mesitylenesulfonylhydroxylamine, rapidly forms dehydroalanine [37-40]. Whether this modification occurs in full length protein, or why it does not form at higher HNO concentrations remains unknown at this time, but such an HNO-mediated transformation warrants deeper investigation. Sulfinamide and dehydroalanine formation from reaction of AS with the full protein explain the observed irreversible portion of inhibition while disulfide formation (possible in the full protein) explains the reversible part.
Reaction of the peptide with 5 in the presence of PLE at room temperature results in the formation of the major product (m/z = 1185.5514) corresponding to the disulfide derived from two peptides, as well as a minor product with the m/z = 1202.0618, indicating the presence of sulfinamide (Table 1). Based on the above results, the disulfide appears to arise from the direct reaction of the peptide with 5 while the sulfinamide would arise from the reaction with HNO generated through the PLE-mediated hydrolysis of 5. Incubation at 37 °C for longer times (3.5 hr) to assure complete HNO release (and increase the amount of HNO) from 5 yields sulfinamide as the major product (Table 1), as expected if sulfinamide only arises from the reaction with HNO. No evidence of dhA product formation was observed. The formation of these products explains the observed reactivation results with the full length protein and 5 appears to inhibit ALDH in the presence of PLE by two mechanisms: 1) the direct reaction of 5 with ALDH to yield a disulfide and 2) the PLE-catalyzed hydrolysis of 5 to give HNO, which reacts with the protein to give the sulfinamide.
Figure 8 outlines mechanisms for the formation of the observed modifications that include the direct nucleophilic attack of the ALDH peptide thiol/thiolate on an electrophilic nitroso compound. Pathway A shows the addition of this thiolate (more reactive form) to the nitroso group of NB or HNO, generated from AS or the PLE-mediated hydrolysis of 5, to yield an initial N-hydroxysulfenamide that rearranges to the corresponding sulfinamide through dehydration to a sulfenium ion intermediate and rehydration at sulfur [31, 34]. Numerous nucleophiles add to NB and previous work shows the formation of N-substituted sulfinamides upon reaction of thiols with nitrosobenzene [41, 42]. The addition of thiols to HNO yields a similar intermediate that reacts with another thiol to give disulfides or rearranges to the sulfinamide [31, 34], which generally resist DTT reduction resulting in enzyme inactivation [25]. Pathway B shows thiolate addition to the acyloxy nitroso compound (5) to give a product that apparently does not dehydrate but reacts with excess thiol to yield disulfide, which would be reduced by DTT, and the corresponding oxime. This reactivity mirrors previous work that shows disulfide and oxime formation from the reaction of 1 and 2 with various thiolates [22]. The structural difference in the initial addition products of Pathways A and B, with B bearing a suitable carboxylate leaving group that favors decomposition through disulfide formation rather than dehydration, appears to influence this difference in reactivity. Based on these findings, the mixture of disulfide and sulfinamide products observed during the reaction of the ALDH peptide with 5 in the presence of PLE likely form from a mixture of Pathways A and B, with A yielding the sulfinamide via HNO reactions and B yielding disulfide via direct reactions with 5. Indeed, incubation of the ALDH peptide with 5 and PLE for longer times at 37 °C (rather than room temperature) to ensure full HNO release yields sulfinamide, as the major product suggesting an increased contribution from Pathway A (HNO release). Acyloxy nitroso compounds thus act in two separate ways: 1) as electrophiles directly reacting with protein thiol residues to give disulfides (not N-substituted sulfinamides) or 2) sources of electrophilic HNO and both pathways result in thiol modification leading to enzyme inhibition. Acyloxy nitroso compounds demonstrate a distinct direct reactivity with thiols yielding only disulfide products compared to both C-nitroso compounds and HNO allowing these compounds to be considered as “organic nitroxyls” that convert thiols to disulfides. Dehydroalanine formation likely results from the elimination of a sulfur-nitrogen intermediate or the final sulfinamide (Pathway A). Direct amination of cysteine residues yields dehydroalanaine and the loss of HOSNH2 from the sulfinamide finds some precedent in sulfoxide eliminations to yield alkenes [37-40, 43]. Regardless of the mechanism, the formation of dehydroalanine from the reaction of HNO and thiol-containing peptides indicates new HNO chemistry that may be exploited for HNO detection.
Figure 8.

Mechanisms for the formation of the observed modifications of the ALDH peptide thiol when reacted with C-Nitroso Compounds or HNO donors.
4. CONCLUSIONS
In summary, replacement of a methylene group with an oxygen atom (compare compounds 2 and 5) gives new acyloxy nitroso compounds (4 and 5) that demonstrate improved water solubility. The acetate derivative (4) slowly hydrolyzes to HNO but the pivalate (5) does not hydrolyze or release HNO under buffered conditions. Addition of pig liver esterase (PLE) catalyzes hydrolysis of 5 and increases HNO release, making this compound a unique esterase-mediated HNO donor that produces minimal amounts of nitrite. These properties, water-solubility, esterase triggered HNO release and low nitrite formation allow for the evaluation of 5 with both heme and thiol containing proteins. Compound 5 in the presence of PLE converts metMb to iron nitrosyl Mb and oxyMb to metMb indicating that acyloxy nitroso compounds only react with heme proteins as HNO donors. Treatment of ALDH with 5 generates a disulfide resulting in DTT-reversible enzyme inhibition while incubation of ALDH with 5 in the presence of PLE results in inhibition with DTT reversible and irreversible components. The irreversible portion of inhibition appears to arise from HNO mediated sulfinamide formation. These results indicate that acyloxy nitroso compounds interact with thiol containing protein in two distinct ways, direct electrophilic reaction or HNO donation, to yield thiol modifications that influence enzyme activity. These compounds possess reactivity on their own, as well as the ability to release HNO and these properties in terms of solubility, kinetics and by-product release provide valuable alternatives to Angeli’s salt.
Supplementary Material
Highlights.
Water Soluble Acyloxy Nitroso Compounds: HNO Release and Reactions with Heme and Thiol Containing Proteins
synthesis of water-soluble 4-nitrosotetrahydro-2H-pyran-4-yl acetate and pivalate
pig liver esterase-catalyzes hydrolysis and HNO release of these compounds
reaction converts metmyoglobin (Mb) to iron nitrosyl Mb and oxyMb to metMb
reaction inhibits aldehyde dehydrogenase through thiol modification
reaction of HNO with cysteine residue yields dehydro-alanine
ACKNOWLEDGEMENTS
This work was supported by the NIH (HL 062198, SBK) as well as Cardioxyl Pharmaceuticals (SBK). The Bruker NMR spectrometers used in this work were purchased with partial support from the NSF (CHE-9708077) and North Carolina Biotechnology Center (9703-IDG-1007). JFD and SBK kindly thank Dr. Susan Mitroka (Wake Forest University) for helpful discussion.
Footnotes
During this work, the following patent appeared describing the preparation of 4 and 5.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6 REFERENCES
- [1].Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Feelisch M, Fukuto JM, Wink DA. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9196–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Irvine JC, Ritchie RH, Favaloro JL, Andrews KL, Widdop RE, Kemp-Harper BK. Trends Pharmacol. Sci. 2008;29:601–608. doi: 10.1016/j.tips.2008.08.005. [DOI] [PubMed] [Google Scholar]
- [3].Paolocci N, Jackson MI, Lopez BE, Miranda K, Tocchetti CG, Wink DA, Hobbs AJ, Fukuto JM. Pharmacol. Ther. 2007;113:442–458. doi: 10.1016/j.pharmthera.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Irvine JC, Favaloro JL, Kemp-Harper BK. Hypertension. 2003;41:1301–1307. doi: 10.1161/01.HYP.0000072010.54901.DE. [DOI] [PubMed] [Google Scholar]
- [5].Kerwin JF, Lancaster JR, Feldman PL. J. Med. Chem. 1995;38:4343–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- [6].Miller TW, Cherney MM, Lee AJ, Francoleon NE, Farmer PJ, King SB, Hobbs AJ, Miranda KM, Burstyn JN, Fukuto JM. J. Biol. Chem. 2009;284:21788–21796. doi: 10.1074/jbc.M109.014282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM, Espey MG, Fukuto JM, Feelisch M, Wink DA, Kass DA. Proc. Natl. Acad. Sci. U. S. A. 2001;98:10463–10468. doi: 10.1073/pnas.181191198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tocchetti CG, Stanley BA, Murray CI, Sivakumaran V, Donzelli S, Mancardi D, Pagliaro P, Gao WD, van Eyk J, Kass DA, Wink DA, Paolocci N. Antioxid. Redox Signal. 2011;14:1687–1698. doi: 10.1089/ars.2010.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Farmer PJ, Sulc F. J. Inorg. Biochem. 2005;99:166–184. doi: 10.1016/j.jinorgbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- [10].Fukuto JM, Carrington SJ. Antioxid. Redox Signal. 2011;14:1649–1657. doi: 10.1089/ars.2010.3855. [DOI] [PubMed] [Google Scholar]
- [11].Reisz J, Bechtold E, King SB. Dalton Trans. 2010;39:5203–5012. doi: 10.1039/c000980f. [DOI] [PubMed] [Google Scholar]
- [12].Shafirovich V, Lymar SV. Proc. Natl. Acad. Sci. U. S. A. 2002;99:7340–7345. doi: 10.1073/pnas.112202099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bonner FT, Hughes MN. Comments Inorg. Chem. 1988;7:215–234. [Google Scholar]
- [14].DuMond JF, King SB. Antioxid. Redox Signal. 2010;14:1637–1648. doi: 10.1089/ars.2010.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bonner FT, Ravid B. Inorg. Chem. 1975;14:558–563. [Google Scholar]
- [16].Miranda KM, Dutton AS, Ridnour LA, Foreman CA, Ford E, Paolocci N, Katori T, Tocchetti CG, Mancardi D, Thomas DD, Espey MG, Houk KN, Fukuto JM, Wink DA. J. Am. Chem. Soc. 2005;127:722–731. doi: 10.1021/ja045480z. [DOI] [PubMed] [Google Scholar]
- [17].Dutton AS, Suhrada CP, Miranda KM, Wink DA, Fukuto JM, Houk KN. Inorg. Chem. 2006;45:2448–2456. doi: 10.1021/ic051505z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hughes MN, Wimbledon PE. J. Chem. Soc., Dalton Trans. 1976:703–707. [Google Scholar]
- [19].Kim-Shapiro DB, Gladwin MT, Patel RP, Hogg N. J. Inorg. Biochem. 2005;99:237–246. doi: 10.1016/j.jinorgbio.2004.10.034. [DOI] [PubMed] [Google Scholar]
- [20].Feelisch M. Proc. Natl. Acad. Sci. U. S. A. 2003;100:4978–4980. doi: 10.1073/pnas.1031571100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sha X, Isbell TS, Patel RP, Day CS, King SB. J. Am. Chem. Soc. 2006;128:9687–9692. doi: 10.1021/ja062365a. [DOI] [PubMed] [Google Scholar]
- [22].Shoman ME, DuMond JF, Isbell TS, Crawford JH, Brandon A, Honovar J, Vitturi DA, White CR, Patel RP, King SB. J. Med. Chem. 2011;54:1059–1070. doi: 10.1021/jm101432z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Frost LM, Courtney SM, Brookfield FA, Kalish VJ. USA. Cardioxyl Pharmaceuticals Inc.; USA: 2009. p. 112. [Google Scholar]
- [24].Nagasawa HT, Yost Y, Elberling JA, Shirota FN, Demaster EG. Biochem. Pharmacol. 1993;45:2129–2134. doi: 10.1016/0006-2952(93)90026-s. [DOI] [PubMed] [Google Scholar]
- [25].DeMaster EG, Redfern B, Nagasawa HT. Biochem. Pharmacol. 1998;55:2007–2015. doi: 10.1016/s0006-2952(98)00080-x. [DOI] [PubMed] [Google Scholar]
- [26].Doyle MP, Mahapatro SN, Broene RD, Guy JK. J. Am. Chem. Soc. 1988;110:593–599. [Google Scholar]
- [27].King SB, Nagasawa HT. Nitric Oxide, Pt C, Methods in Enzymology. 1999;301:211–220. doi: 10.1016/s0076-6879(99)01084-8. [DOI] [PubMed] [Google Scholar]
- [28].Huang ZJ, Velazquez C, Abdellatif K, Chowdhury M, Jain S, Reisz J, DuMond J, King SB, Knaus E. Org. Biomol. Chem. 2010;8:4124–4130. doi: 10.1039/c005066k. [DOI] [PubMed] [Google Scholar]
- [29].Toone EJ, Werth MJ, Jones JB. J. Am. Chem. Soc. 1990;112:4946–4952. [Google Scholar]
- [30].Moorlag H, Kellogg RM, Kloosterman M, Kaptein B, Kamphuis J, Schoemaker HE. J. Org. Chem. 1990;55:5878–5881. [Google Scholar]
- [31].Miranda KM. Coord. Chem. Rev. 2005;249:433–455. [Google Scholar]
- [32].Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Proc. Natl. Acad. Sci. U. S. A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Naik KG, Shah CC, Patel SZ. J. Indian Chem. Soc. 1946;24:284–287. [Google Scholar]
- [34].Fukuto JM, Bianco CL, Chavez TA. Free Radical Biol. Med. 2009;47:1318–1324. doi: 10.1016/j.freeradbiomed.2009.06.014. [DOI] [PubMed] [Google Scholar]
- [35].Flores-Santana W, Salmon DJ, Donzelli S, Switzer CH, Basudhar D, Ridnour L, Cheng R, Glynn SA, Paolocci N, Fukuto JM, Miranda KM, Wink DA. Antioxid. Redox Signal. 2011;14:1659–1674. doi: 10.1089/ars.2010.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shen B, English AM. Biochem. (Mosc) 2005;44:14030–14044. doi: 10.1021/bi0507478. [DOI] [PubMed] [Google Scholar]
- [37].Chalker JM, Bernardes GJL, Lin YA, Davis BG. Chem. Asian J. 2009;4:630–640. doi: 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- [38].Chalker JM, Bernardes GJL, Davis BG. Acc. Chem. Res. 2011;44:730–741. doi: 10.1021/ar200056q. [DOI] [PubMed] [Google Scholar]
- [39].Chalker JM, Gunnoo SB, Boutureira O, Gerstberger SC, Fernandez-Gonzalez M, Bernardes GJL, Griffin L, Hailu H, Schofield CJ, Davis BG. Chem. Sci. 2011;2:1666–1676. [Google Scholar]
- [40].Bernardes GJL, Chalker JM, Errey JC, Davis BG. J. Am. Chem. Soc. 2008;130:5052–5053. doi: 10.1021/ja800800p. [DOI] [PubMed] [Google Scholar]
- [41].Yamamoto H, Kawasaki M. Bull. Chem. Soc. Jpn. 2007;80:595–607. [Google Scholar]
- [42].Callan HE, Jenkins RE, Maggs JL, Lavergne SN, Clarke SE, Naisbitt DJ, Park BK. Chem. Res. Toxicol. 2009;22:937–948. doi: 10.1021/tx900034r. [DOI] [PubMed] [Google Scholar]
- [43].Janssen JWAM, Kwart H. J. Org. Chem. 1977;42:1530–1533. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.