

Abstract
Ultraviolet (UV) radiation is the major risk factor for developing skin cancer, the most prevalent cancer worldwide. Several studies indicate mammalian target of rapamycin (mTOR) signaling is activated by UVB and may play an important role in skin tumorigenesis. mTOR exists in two functionally and compositionally distinct protein complexes: the rapamycin-sensitive mTOR complex 1 (mTORC1) and the rapamycin-resistant mTOR complex 2 (mTORC2). The purpose of these studies was to investigate the roles of the two mTOR complexes in UVB-mediated proliferation and apoptosis in the skin. We utilized rapamycin, a pharmacological inhibitor of mTORC1, and an inducible mTOR-deficient (K5-CreERT2;mTORfl/fl) mouse model that allows epidermal-specific disruption of mTOR following topical treatment with 4-hydroxytamoxifen (4OHT). Rapamycin blocked UVB-induced phosphorylation of S6K, the downstream target of mTORC1, and significantly reduced UVB-stimulated epidermal proliferation and cell cycle progression, but had no effect on cell death. In contrast, mTOR deletion, which attenuated UVB-induced phosphorylation of both S6K and the mTORC2 target AKTSer473, significantly increased apoptosis both in vivo and in keratinocyte cultures, in addition to reducing hyperproliferation following UVB irradiation. The role of mTORC2 in UVB-induced pro-survival signaling was verified in Rictor-/- MEFs, which lack functional mTORC2 and were more sensitive to UVB-induced apoptosis than controls. These studies show that mTORC1 and mTORC2 play unique but complementary roles in controlling proliferation and apoptosis in the skin. Our findings underscore the importance of both mTOR complexes in mediating UVB-induced signaling in keratinocytes and provide new insight into the pathogenesis of skin cancer.
Keywords: mTOR, rapamycin, rictor, UVB, keratinocyte
INTRODUCTION
Nonmelanoma skin cancer (NMSC) is the most frequent malignancy worldwide, with more than 1 million cases diagnosed each year in the US alone (1). Squamous cell carcinoma (SCC) and basal cell carcinoma are the most common forms of NMSC and account for greater than 40% of newly diagnosed cancers (1). The most important risk factor for NMSC is ultraviolet (UV) radiation exposure, which is the leading cause of skin cell damage, photoaging, and malignant transformation (2). UV radiation from sunlight is composed of UVB (280-315nm) and UVA (315-400nm). UVB wavelengths are the most energetic and account for the majority of the biologically damaging effects from sun exposure. UVB is a complete carcinogen and has been shown to act as a tumor initiator by inducing mutations in critical target genes, and a tumor promoter by activating signal transduction pathways mediated by kinases and transcription factors that induce cell cycle progression and proliferation (3).
The mammalian target of rapamycin (mTOR), an essential serine/theronine kinase conserved in all eukaryotes, is at the center of an increasingly intricate signaling network that responds to nutrients, growth factors, and cellular stress (4). Hyperactivation of mTOR signaling through mutational activation of upstream signals, such as Ras or PI-3-kinase (PI3K), occurs in many cancers and contributes to increased proliferation and reduced sensitivity to apoptotic stimuli (4, 5). mTOR exists in at least two functionally and compositionally distinct protein-signaling complexes: the rapamycin-sensitive mTOR complex 1 (mTORC1) and the rapamycin-resistant mTOR complex 2 (mTORC2). The mTORC1 complex, consisting of mTOR, Raptor and mLST8, is downstream of both the Raf/MEK/ERK and PI3K/Akt pathways, and controls protein synthesis and ribosome biogenesis by direct phosphorylation of eukaryotic initiation factor 4E binding proteins (4EBPs) and p70 S6 kinase 1 (S6K), respectively (6). The mTORC2 complex also contains mTOR and mLST8, but Raptor is replaced by Rictor and mSin1. While the function of mTORC2 is less well-understood, it has been shown to phosphorylate AKT at Ser473, thus promoting cell survival and proliferation (7). Phosphorylation of AKT by mTORC2 can also serve to activate mTORC1, emphasizing the interdependence of the two mTORCs (8).
Several studies indicate that mTOR signaling may play a critical role in NMSC development. Immunohistochemical analysis of human epidermal tumors showed that mTOR itself, as well as its downstream effectors 4EBP1, S6K, and AKTSer473 are phosphorylated at much higher levels in SCC and precancerous actinic keratosis (AK) than normal skin (9). More recently, reverse phase protein microarray analysis of SCC, AK, and normal skin revealed aberrantly activated mTOR pathways in the precancerous and transformed tissues (10). Data compiled from over 30,000 kidney transplant recipients found the use of mTOR inhibitors as maintenance immunosuppressive therapy produced a remarkable reduction in NMSC incidence compared to the calcineurin inhibitor Cyclosporine A (CsA) (11). A dramatic decrease in the incidence of skin malignancies was also observed in transplant patients who were converted to mTOR inhibitors after three months of treatment with CsA (the CONVERT study) (12). These striking results have led to several ongoing prospective randomized trials evaluating the use of mTOR inhibitors in renal transplant patients, and could significantly impact prevention of NMSC in the general population as well.
Photocarcinogensis is characterized by the inhibition of apoptosis and the enhancement of cell proliferation. Although studies have shown that UVB induces phosphorylation of 4EBP1, S6K, and AKT (13-15), the specific roles of mTORC1- and mTORC2-dependent pathways in UVB-induced proliferation and apoptosis have not been defined. In the present study, we examined the effect of UVB radiation on mTOR signaling in cell culture and in vivo using both the specific mTORC1 inhibitor rapamycin and a genetic model of conditional mTOR deletion in the epidermis, which results in inhibition of both mTORC1 and mTORC2. Our findings indicate that UVB stimulates both mTORC1 and mTORC2 activities. While rapamycin treatment effectively blocked the hyperproliferation response that occurs with UVB exposure, keratinocytes were sensitized to UVB-induced apoptosis only when mTORC2 activity was downregulated. Results obtained using rictor-null cells confirmed that intact mTORC2 is necessary to activate pro-survival signaling. These studies thus provide new insights into the molecular mechanisms of UVB-induced damage leading to skin aging and skin cancer.
METHODS
Cell Culture and drug treatment
HaCaT keratinocytes were obtained from The German Cancer Research Center) were maintained in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Atlanta Biologicals) and Penicillin/Streptomycin (100μg/mL, Invitrogen), and cultured for less than 20 passages. Control (RictorEx3cond/w) and rictor-null (RictorEx3del/Ex3del) mouse embryo fibroblasts (MEFs), abbreviated Rictor+/+ and Rictor-/-, were a generous gift from Dr. Mark Magnuson, Vanderbilt University (16) and maintained in DMEM supplemented with 10% FBS and Penicilin/Streptomycin. No further characterization of cell lines was performed. Primary mouse keratinocytes were isolated from 1-3d old pups as described previously (17). Briefly, fully thickness skin was floated overnight at 4°C in Ca+2-free 0.25% Trypsin without EDTA (Cellgro) to separate the epidermis from the dermis. Epidermal sheets were minced and the cell suspension was strained (100-μm nylon filter, Fisher) and plated in keratinocyte growth media [calcium-free MEM Eagle with Earle's BSS, GIn and non-essential amino acids (Lonza), 8% chelexed FBS (Gibco), and Penicillin/Streptomycin]. Cells were maintained in 7% CO2 at 37°C, and medium was changed every other day. Rapamycin (Developmental Therapeutics Program, National Cancer Institute) or vehicle (DMSO) was added to medium 1 h prior to UVB treatment. Primary keratinocyte cultures from transgenic animals (K5-CreERT2;mTORfl/fl and mTORfl/fl) were supplement with 5nM of 4-hydroxy-tamoxifen (4OHT, Sigma) for 4 days to induce recombination.
Animals and drug treatment
All experiments involving mice were carried out in compliance with the Guide for the Care and Use of Laboratory Animals and protocols were approved by the Animal Care and Use Committee of the Pennsylvania State University College of Medicine.
For mouse experiments, the dorsal surface of FVB/N mice (6-8 weeks) was shaved with electrical clippers and mice were allowed to rest for 24-48 h prior to all experiments. For rapamycin in vivo studies, mice were treated topically with 100 nmol rapamycin (in 100μL DMSO:Acetone [1:9], D:A) or vehicle 1 h prior to UVB exposure. We utilized an inducible Cre-LoxP mouse model to ablate mTOR in the epidermis. Floxed mTOR mice (mTORfl/fl) contain LoxP sites flanking exons 49 and 50 of the mTOR gene (18); recombination results in a frameshift mutation and loss of the essential kinase domain. K5-CreERT2 mice (19) express a tamoxifen-activated Cre recombinase fused to a modified estrogen receptor in the basal layer of the epidermis under the control of the keratin 5 promoter. K5-CreERT2 mice were bred with mTORfl/fl mice to ultimately generate mice hemizygous for the K5-Cre-ERT2 transgene and homozygous for the mTOR floxed allele (K5-CreERT2;mTORfl/fl). K5-CreERT2;mTORfl/fl and mTORfl/fl controls were treated topically with 1 mg 4OHT (in 100μL D:A) or vehicle daily for 5 consecutive days. All animals were backcrossed for at least 9 generations onto the FVB/N background. Primer sequences used for PCR are as follows: mTOR 1 (wild-type, LoxP, ΔLoxP): GTC CAC CAA CTC GGG CCT CAT T, mTOR 2 (wild-type, LoxP): GCA TGG CGA GGA CAT GTC A, and mTOR 3 (ΔLoxP): CCA CGC ATG GCC CAC TGT CTT T.
UVB treatment
Cells were cultured 48-72 h until 70% confluence on 60-mm plates under normal culture conditions. For cell cycle experiments (low-dose UVB), medium was replaced with 0.1% fetal bovine serum (FBS) for 24 h prior to UVB treatment. Cells were washed twice with PBS, then in a minimal volume of PBS exposed to UVB (FS20 UVB bulbs, National Biological) emitting UV light between 290-320nm. The irradiation intensity was monitored using a UVB 500C meter (National Biological). After irradiation, PBS was removed and saved medium with drug treatments was added back. For in vivo studies, 3-4 mice were used for each treatment group and housed together. Mice were exposed to UVB irradiation from UVB lamps (FS20 UVB bulbs, National Biological) at a dose of 120mJ/cm2. Bulb intensity was measured at the beginning of each experiment.
Western blotting
For in vitro studies, cells were washed twice with cold PBS and harvested by scraping into RIPA buffer (Santa Cruz, Biotechnology), centrifuged, and supernatants collected. In selected experiments examining apoptosis, detached cells were collected via centrifugation of medium and combined with the attached cells in RIPA buffer. For in vivo studies, dorsal skin was treated with a depilatory agent for 3 min, washed, excised and underlying fat and connective tissue were removed. Whole skin samples were flash-frozen and later processed in RIPA buffer by homogenizing for 30 sec on ice using a Polytron homogenizer, centrifuged at 30,000 x g for 30 min at 4°C, and supernatants collected. Epidermal samples were collected by scraping the surface of excised skin with a razor blade and placed into ice-cold RIPA buffer. Protein concentrations were determined using Biorad assay. Equal amounts of protein were subjected to electrophoresis. Western blotting was preformed as described previously (20). Antibodies used include AKT, p-AKTSer473, S6K, p-S6KThr389, Caspase-3, cleaved Caspase-3, mTOR, actin (all from Cell Signaling, 1:1000) and GAPDH (Proteintech, 1:2000).
Flow Cytometry
For cell cycle analysis, cells were harvested 18 h after UVB and fixed with ice-cold ethanol overnight. Cells were washed twice with PBS and then incubated with propidium iodide (20μg/mL) containing RNase A (Sigma). The DNA content was determined using a FACSCalibur cytometer (Beckman Coulter) and analyzed with Modfit LT software (Verify Software). Apoptosis was assessed with flow cytometry using the PE Annexin V Apoptosis Detection Kit I (BD Biosciences) according to the manufacturer's instructions.
Cell Viability
The cell viability at 24 h after UVB exposure was determined colorimetrically by MTS assay (CellTiter 96 Aqueous Proliferation Assay, Promega) according to the manufacturer's instructions and monitored at 490nm using a model 3550-UV plate reader (Biorad). Each experiment was done in triplicate and the experiments were repeated at least three times.
Histologic analysis
Tissue sections were collected from 3-4 mice for each condition at each timepoint (mock animals were not irradiated and collected at 24 h). Dorsal skin was treated with a depilatory agent for 3 min, washed, excised, and fixed overnight in 10% neutral buffered formalin. Skin was embedded in paraffin and 5 μm sections were cut for immunohistochemistry. The effect of rapamycin and mTOR deletion on UVB-mediated epidermal hyperplasia was studied by histopathological examination of hematoxylin and eosin (H&E) stained tissue sections. Epidermal thickness was measured at 5 locations in four different sections for each mouse. Epidermal proliferation was assessed in vivo using 5-bromo-2-deoxyuridine (BrdU) incorporation. Mice received intraperitoneal (i.p.) injections of BrdU (Sigma) at 100 μg/g body wt in 0.9% NaCl 1 h prior to killing. Sections were deparaffinized, rehydrated, and stained with an anti-BrdU antibody as described previously (21). Epidermal proliferation index (PI) was determined by calculating the percentage of basal cells positive for BrdU. A minimum of 1000 basal cells was counted. Apoptosis was assessed using immunohistochemical staining with cleaved caspase-3 antibody. Intrafollicular apoptotic keratinocytes were counted microscopically in at least 5 nonoverlapping low power fields.
Statistical analysis
Data are expressed as the mean of at least three independent experiments analyzed by 2-sided Student's t-test. A p-value of <0.05 was considered significant.
RESULTS
Rapamycin inhibits UVB-induced cell cycle progression and proliferation
In photocarcinogenesis, UVB-stimulated activation of serine/theronine and tyrosine kinase signaling pathways and transcription factors mediate a number of pathologic changes in the skin. The resulting keratinocyte proliferation is responsible for epidermal hyperplasia and tumor promotion (3). We first sought to investigate the role of mTORC1 in UVB-induced cell cycle progression and proliferation using rapamycin. It was previously shown that sub-apoptotic doses of UVB (2.5 to 10 mJ/cm2) activate epidermal growth factor receptor (EGFR) and AKT and induce G1-S cell cycle progression in serum-deprived HaCaT cells (22). To determine whether mTORC1 contributes to UVB-induced cell cycle progression, HaCaT cells were serum-starved to synchronize them in G0 and then subjected to low dose UVB. As shown in Fig. 1A, low dose UVB activates both mTORC1 and mTORC2 signaling pathways as measured by phosphorylation of S6K and AKTSer473 respectively. To determine whether mTORC1 activation is involved in UVB-mediated cell cycle progression, cells were incubated with 50 nM rapamycin to block mTORC1 activation. Rapamycin suppressed UVB-induced S6K phosphorylation, but did not alter phosphorylation of AKTSer473 (Fig. 1B). Low dose UVB stimulated cell cycle progression in vehicle-treated cells, as measured by the percentage of cells in S-phase at 18 h after irradiation (Fig. 1C). The proportion of cells in the S-phase decreased significantly in cells treated with rapamycin (Fig. 1C).
Figure 1. Rapamycin attenuates UVB-induced cell cycle progression in HaCaT cells.
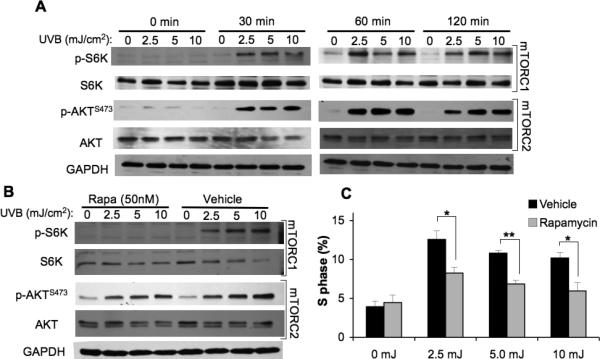
HaCaT cells at 70% confluence in 60-mm dishes were starved in 0.1% FBS DMEM for 24h and then exposed to 2.5, 5, or 10 mJ/cm2 or mock UVB irradiation. A, Immunoblot analysis of mTORC1 and mTORC2 activation markers in HaCaT cells exposed to sublethal doses of UVB (2.5, 5, 10 mJ/cm2). B, Immunoblot analysis of mTOR activation markers at 2h post-irradiation of HaCaT cells pretreated with Rapamycin (Rapa 50 nM) or vehicle (DMSO). C, Cell cycle analysis of S-phase analyzed by flow cytometry 18 h following UVB radiation exposure (mean ± SEM). * p < 0.05, ** p < 0.01; All data are representative from 2-5 independent experiments.
Because cellular proliferation in response to UVB is a key feature of photo-carcinogenesis, we next sought to explore the role of mTORC1 in UVB-mediated responses in vivo. Wild-type FVB/N adult mice were treated topically with rapamycin (100nmol) 1 h prior to irradiation with a single does of UVB (120mJ/cm2) and whole-skin samples were subjected to immunoblot analysis at 6 h post UVB to assess mTOR activation. Concentrations of p-S6K and p-AKTS473 were too low in mock-irradiated samples to discern any noticeable inhibitory effect of rapamycin (Fig. 2A). However, there was a dramatic increase in p-S6K and p-AKTSer473 levels in vehicle-treated skin after UVB irradiation. Rapamycin pretreatment prevented UVB-stimulated phosphorylation of S6K, but had no inhibitory effect on p-AKTSer473. Epidermal hyperplasia (as measured by epidermal thickness) and proliferation (as measured by BrdU incorporation) were examined in skin sections at 24 h and 48 h after UVB exposure. Rapamycin had no effect on epidermal thickness or proliferation in mock-irradiated animals. There was noticeable epidermal thickening in vehicle-treated animals at 24 and 48 h following UVB exposure, but this effect was significantly reduced in animals treated with rapamycin (Fig. 2B,C). Similar blocking effects were seen with rapamycin when epidermal thickening was stimulated by treatment with the chemical tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA, see Supplementary Fig. S1). BrdU labeling of actively proliferating cells at 48 h showed a similar pattern. Vehicle- treated mice showed a significant increase in BrdU-positive cells at 48 h post UVB, and this effect was significantly reduced by rapamycin treatment (Fig. 2D,E). Collectively, these results and those in Fig. 1 suggest that mTORC1 plays an important role in keratinocyte cell cycle progression and epidermal hyperproliferation following UVB irradiation.
Figure 2. Rapamycin inhibits UVB-induced epidermal hyperproliferation in vivo.

FVB/N mice at 7-weeks of age were treated topically with Rapamycin (Rapa, 100nmol) or vehicle 1h prior to being exposed to 120mJ/cm2 or mock UVB irradation. A, Representative immunoblot analysis of mTORC1 and mTORC2 activation markers in whole-skin extract harvested 6h following UVB radiation exposure from 3 independent experiments. B, Representative H&E images of skin sections (scale bar = 50μm). C, Quantification of epidermal thickness (mean ± SEM) for 3-4 mice/group; * p < 0.05. D, Representative BrdU staining images (scale bar = 50μm). E, Quantification of BrdU Prolliferation Index (PI) (mean ± SEM) for 3-4 mice/group; * p < 0.05,
Inducible mTOR deficiency inhibits UVB-stimulated epidermal proliferation
We next explored the consequences of deletion of mTOR in the basal layer of the skin, targeting both mTORC1 and mTORC2 signaling, to analyze the contribution of both mTOR-signaling complexes in UVB-mediated responses. Because homozygous deletion of mTOR is lethal in utero (23), we used a transgenic system with 4OHT-inducible deletion of mTOR in the epidermis (K5-CreERT2;mTORfl/fl). We verified that the topical treatment with 4OHT, but not vehicle, leads to CreERT2-dependent recombination of the mTOR allele (ΔLoxP) by PCR analysis using DNA from the dorsal epidermis harvested from mice 1 week after the final 4OHT treatment (Fig. 3A). No recombination was observed in the absence of either CreERT2 expression or 4OHT induction (data not shown). Western blot analysis confirmed reduction of mTOR protein levels in 4OHT-treated K5-CreERT2;mTORfl/fl animals (Fig. 3B). Downregulation of both mTORC1 and mTORC2 signaling pathways was verified by examining phosphorylation of S6K and AKTSer473 in whole skin protein extracts. In the absence of UVB stimulation of mTOR pathways, there was no apparent difference between the vehicle-treated and 4OHT-treated animals (Fig. 3C). However, p-S6K and p-AKTser473 levels were dramatically increased at 6 h after UVB (120mJ/cm2) in vehicle-treated mice. This effect was significantly attenuated upon treatment of K5-CreERT2;mTORfl/fl animals with 4OHT (Fig. 3C), confirming that 4OHT treatment in K5-CreERT2;mTORfl/fl mice was sufficient to block UVB-induced activation of both mTORC1 and mTORC2. Histological evaluation of epidermal thickness and proliferation index in mock-irradiated animals revealed no differences between vehicle-treated and 4OHT-treated animals (Fig. 3D-G). UVB irradiation caused a significant increase in epidermal thickness in vehicle-treated K5-CreERT2;mTORfl/fl mice at 24 h and 48 h, while mice treated with 4OHT (Fig. 3D and E) showed much less epidermal thickening in response to UVB. K5-CreERT2;mTORfl/fl mice treated with 4OHT also showed a significant reduction in proliferation index at 48 h after UVB treatment compared to vehicle-treated controls (Fig. 3F and G). All mice used as controls in these experiments responded similarly to each other and to wild-type mice at all timepoints, including mTORfl/fl mice treated with 4OHT, mTORfl/fl mice treated with vehicle, and K5-CreERT2;mTORfl/fl mice treated with vehicle (see Supplementary Fig. S2).
Figure 3. Induction of mTOR deletion in mouse epidermis suppresses UVB-stimulated epidermal proliferation.

K5-CreERT2;mTORfl/fl mice at 7-weeks of age were treated topically with 4OHT (1mg) or vehicle (D:A) daily for 5 days. A, PCR analysis of epidermal DNA harvested 14 d after final 4OHT treatment. ΔLoxP denotes primers specific to the recombined mTOR allele. B, Immunoblot analysis of mTOR in epidermal extracts harvested at 7 d or 14 d after final 4OHT treatment. C, Immunoblot analysis of mTORC1 and mTORC2 activation markers in whole-skin extracts harvested 6h following UVB (120mJ/cm2) radiation exposure. AC data are representative of 2-3 independent experiments. D, Representative H&E images of skin sections (scale bar = 50μm). E, Quantification of epidermal thickness (mean ± SEM) for 3-4 mice/group; * p < 0.05, *** p < 0.005. F, Representative BrdU staining images (scale bar = 50μm). G, Quantification of BrdU Proliferation Index (PI) (mean ± SEM) for 3-4 mice/group; ** p < 0.01.
Rapamycin does not alter UVB-mediated cell death
Because one of the major activities of AKT is to promote cell survival (24), we sought to investigate whether UVB-induced mTOR activation of AKT-dependent pathways plays a role in preventing cell death following high dose UVB. We investigated mTOR-signaling pathways after 50mJ/cm2 UVB exposure by examining the expression of total and phosphorylated S6K and AKT in normal mouse primary keratinocytes by Western blot analysis. Enhanced signaling through both mTORC1 and mTORC2 was indicated by increased levels of p-S6K and p-AKTSer473 at 30 min after 50mJ/cm2 UVB and remained upregulated at 2 h (Fig. 4A). Pretreatment of cells with rapamycin for 1 h completely blocked UVB-induced activation of mTORC1 signaling (p-S6K), but had little if any effect on mTORC2 activity (p-AKTSer473) (Fig. 4B). To determine if inhibition of mTORC1 could sensitize keratinocytes to apoptosis, cells exposed to various rapamycin concentrations were harvested 24 h after UVB exposure and assayed for viability. Treatment with a wide range of rapamycin concentrations did not alter cell viability in mock-irradiated cells (Fig. 4C). Exposure to 50mJ/cm2 UVB resulted in an obvious decrease in cell viability (54% ± 3.6%). However, rapamycin did not enhance UVB-mediated cell death in wild-type primary keratinocytes (Fig. 4C), and this same result was seen in HaCaT cells (Supplementary Fig. S3A). Additionally, increasing the pretreatment time with rapamycin to 24 h did not enhance UVB-mediated cell death (Supplementary Fig. S3B,C). Western blot analysis of cleaved Caspase-3 verified that there was no increase in UVB-mediated apoptosis with rapamycin treatment (Fig. 4D).
Figure 4. Rapamycin does not sensitize keratinocytes to UVB-induced cell death.
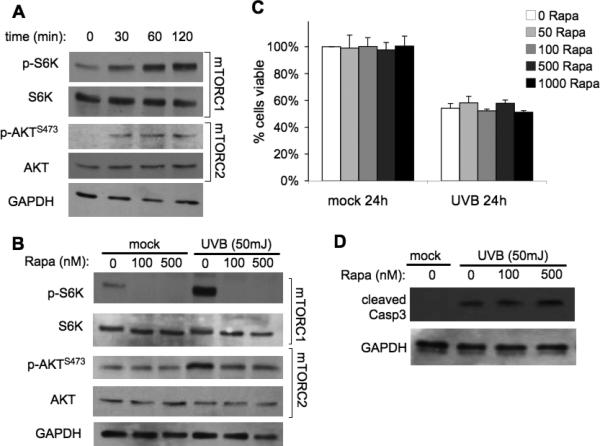
Wild-type primary keratinocytes were harvested from 1-3 day-old pups and plated in low-calcium media. When confluent, cells were treated with Rapamycin (Rapa) for 1h and exposed to 50mJ/cm2 UVB irradiation. A, Immunoblot analysis of mTORC1 and mTORC2 activation markers in primary keratinocytes exposed to UVB. B, Immunoblot analysis of mTOR activation markers at 2h post-irradiation of cells pretreated with various doses of Rapamycin. C, MTS cell viability at 24-h post UVB exposure. D, Immunoblot analysis of cleaved caspase-3, a marker of apoptosis, at 9-h post UVB. All data are representative from 3-5 independent experiments.
Inducible mTOR deficiency sensitizes keratinocytes to UVB-induced apoptosis
To investigate the possible role of mTORC2 in suppressing apoptosis after UVB exposure, we utilized primary keratinocytes isolated from K5-CreERT2;mTORfl/fl mice. PCR analysis verified recombination of the mTOR allele (ΔLoxP) in cells cultured with 4OHT, but not vehicle (Fig. 5A). There was no obvious difference in mTORC1 and mTORC2 activities in 4OHT-treated keratinocytes not exposed to UVB (0 min), as measured by p-S6K and p-AKTSer473 (Fig. 5B). However, when keratinocytes were exposed to UVB (50mJ/cm2) to activate both mTOR complexes, a dramatic reduction in phosphorylation of both S6K and AKTser473 was observed in 4OHT-treated cells compared to vehicle, confirming downregulation of both mTORC1 and mTORC2 signaling (Fig. 5B). Unlike inhibition of mTORC1 with rapamycin (Fig. 4C), deletion of mTOR enhanced UVB-induced cell death (Fig. 5C). Though there was no difference in cell viability in mock irradiated cells, K5-CreERT2;mTORfl/fl keratinocyte cultures treated with 4OHT contained significantly fewer viable cells 24 h after UVB exposure compared to both vehicle-treated K5-CreERT2;mTORfl/fl and 4OHT-treated mTORfl/fl cells. To determine whether the significant decrease in cell viability was due to apoptosis, immunoblot analysis of cleaved caspase-3 was examined (Fig. 5D). Cell extracts harvested at 9 h after UVB showed significantly higher levels of cleaved caspase in mTOR-ablated keratinocytes than in vehicle-treated K5-CreERT2;mTORfl/fl cells.
Figure 5. Induction of mTOR deletion in mouse primary keratinocytes and epidermis sensitizes cells to UVB-induced apoptosis.

K5-CreERT2;mTORfl/fl and mTORfl/fl keratinocytes were harvested from 1-3 day-old pups and cultured in 4OHT (1mM) or vehicle for 3 days prior to UVB (50mJ/cm2) exposure. A, PCR analysis of K5-CreERT2;mTORfl/fl primary keratinocyte DNA harvested 24 h after final 4OHT treatment. ΔLoxP denotes primers specific to the recombined mTOR allele. B, Immunoblot analysis of mTORC1 and mTORC2 activation markers in K5-CreERT2;mTORfl/fl keratinocyte extracts harvested following UVB (50mJ/cm2) exposure. C, MTS cell viability (mean ± SEM) of mTORfl/fl and K5-CreERT2;mTORfl/fl keratinocytes at 24h post UVB; *** p < 0.005. D, Immunoblot analysis (K5-CreERT2;mTORfl/fl primary keratinocytes) of caspase-3 and quantification (mean ± SEM) at 9-h post UVB; * p<0.05. A-D data are representative from 2-4 independent experiments. E, Representative cleaved Caspase-3 staining images of skin sections (scale bar = 200μm) from K5-CreERT2;mTORfl/fl mice treated topically with 4OHT and exposed to UVB (120mJ/cm2). F, Quantification of cleaved capsase-3 (CC3) staining (mean ± SEM) for 3-5 mice/group; ** p < 0.01.
We further investigated the effects of mTOR deficiency on UVB-meditated apoptosis in vivo using our K5-CreERT2;mTORfl/fl mice. UVB irradiation increased the number of epidermal cleaved caspase-3 positive cells 24 h and 48 h after irradiation in K5-CreERT2;mTORfl/fl mice treated with vehicle. The number of apoptotic cells was significantly increased in the epidermis of mTOR-deficient mice (4OHT treated) compared to vehicle controls at 24 h (Fig. 5E and F).
mTORC2 disruption sensitizes cells to UVB-induced apoptosis
Our results demonstrate that K5-CreERT2;mTORfl/fl primary keratinocytes treated with 4OHT to induce mTOR deletion have enhanced sensitivity to UVB-induce apoptosis (Fig 5), but wild-type primary keratinocytes treated with the mTORC1 inhibitor do not (Fig 4). These data suggest that mTORC2, but not mTORC1, influences cell survival signaling following UVB exposure. To further elucidate the specific role of mTORC2 in UVB-induced apoptosis, we utilized rictor-null MEFs (Rictor-/-). UVB exposure (50mJ/cm2) increased phosphorylation of S6K and AKTSer473 in wild-type MEFS (Rictor+/+) (Fig. 6A). UVB induced p-S6K in a similar manner in Rictor-/- cells, but p-AKTSer473 was completely absent, illustrating the loss of mTORC2 signaling (Fig. 6A). Significantly fewer rictor-null cells were viable compared to wild-type cells at 24 h after UVB exposure (Fig. 6B). To determine whether this represents an increased sensitivity to apoptosis in the absence of mTORC2 signaling, cells were analyzed using Annexin V by flow cytometry. There was a significant increase in the percentage of apoptotic Rictor-/- cells following UVB compared to wild-type cells (Fig. 6C). The marked increase in apoptosis was verified by Western blot analysis of caspase-3. The results show that cleaved caspase-3 begins to accumulate in Rictor-/- cells by 6h and continues to increase up to 12 h after UVB exposure (Fig. 6D). In contrast, wild-type MEFs show considerably less caspase-3 cleavage over the same time course. Taken together with the results presented in Figures 4 and 5, these data are consistent with the idea that downregulation of mTORC2 signaling sensitizes cells to UVB-induced apoptosis.
Figure 6. Loss of Rictor increases sensitivity of MEFs to UVB-induced apoptosis.
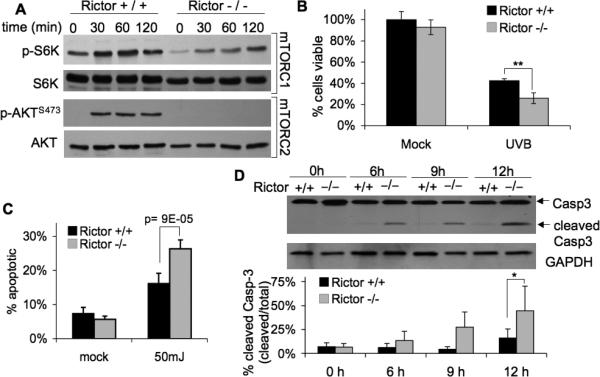
Rictor wild-type (+/+) and knock-out (-/-) MEFs were exposed to UVB (50mJ/cm2) at 70% confluence. A, Immunoblot analysis of mTORC1 and mTORC2 activation markers in cells exposed to UVB. B, MTS cell viability (mean ± SEM) at 24 h post UVB exposure; ** p<0.01. C, Annexin-V flow cytometry (mean ± SEM) at 24-h post UVB exposure. D, Immunoblot analysis of caspase-3 and quantification (mean ± SEM); * p<0.05. All data are representative from 2-4 independent experiments.
DISCUSSION
Better understanding of the signal transduction pathways activated by UVB in keratinocytes is essential for effective prevention of skin cancer. Using rapamycin to inhibit mTORC1, and a Cre/LoxP approach to block both mTORC1 and mTORC2 signaling, the present study demonstrates that the two mTOR complexes play distinct roles in mediating UVB-induced proliferation and pro-survival signaling in the epidermis. Our results fit a model in which mTORC2-dependent pathways maintain the survival of DNA-damaged keratinocytes after exposure to carcinogens, while mTORC1-dependent pathways control their proliferation. These studies validate the potential of both mTOR complexes as prospective chemoprevention targets in non-melanoma skin cancer.
We observed that mTORC1 and mTORC2 downstream pathways were up-regulated in response UVB both in vitro and in vivo. Rapamycin treatment or mTOR ablation inhibited UVB activation of S6K, attenuated UVB-induced cell cycle progression, and blocked the epidermal hyperproliferative response to UVB. Cell proliferation as a result of cell cycle progression is the key process that leads to clonal expansion of initiated cells in tumor promotion, and there is emerging evidence implicating mTORC1 signaling in epithelial tumor promotion. Rapamycin treatment was found previously to inhibit G1-S transition by reducing cyclin D1 mRNA and protein stability (25), while mice with hyperactivation of mTORC1 in the epidermis display epidermal hyperplasia and spontaneous papilloma formation, which is attenuated by treatment with oral RAD001, a rapamycin derivative (26). Other studies have described that mTORC1 inhibition with rapamycin attenuates tumorigenesis in skin, head and neck, and oral squamous carcinoma models (27-31). More recently, Checkley et al. have shown that topical rapamycin is an effective chemoprevention strategy for tumors caused by the classical two-step skin chemical carcinogenesis model by blocking TPA induced proliferation (32). Our findings clearly indicate that mTORC1 activation following UVB positively regulates cell cycle progression and proliferation, confirming and extending previous reports that mTORC1 inhibition blocks tumor promotion.
There was no significant difference in attenuation of UVB-induced hyperproliferation in mice with epidermal mTOR deletion compared to wild-type FVB/N mice receiving topical rapamycin treatment (p=0.11), suggesting UVB-induced cell cycle progression and proliferation are not mediated by mTORC2 signaling. This does not, however, preclude a role for mTORC2 in this process. Although we see no evidence of mTORC2 inhibition in the presence of rapamycin in this model, it is difficult to compare pharmacologic inhibition to genetic ablation. The effects of mTOR deletion may be dampened because it is unlikely that recombination and deletion occurred in all keratinocytes within the epidermis. Additionally, topical rapamycin has been shown to decrease infiltration of dermal inflammatory cells (32), which may contribute to inhibitory effects on keratinocyte proliferation and confound comparisons to our genetic model of mTOR deletion in the skin.
NMSC pathogenesis is characterized by both enhancement of cell proliferation and inhibition of apoptosis. Induction of apoptosis following DNA damage is an essential protective mechanism, ensuring the removal of damaged cells that may harbor oncogenic mutations. However, UVB also activates signaling cascades that promote the survival of these potentially cancerous cells. Previous work in other mouse models has shown that targeting pro-survival pathways induced by UVB increases the sensitivity of DNA-damaged keratinocytes to apoptotic signaling, and is sufficient to inhibit skin carcinogenesis (33). The studies reported here investigate whether activation of mTORC1 and mTORC2 downstream effectors by UVB lead to changes in keratinocyte pro-survival signaling. The effects of mTORC1 inhibition on apoptosis vary greatly depending on the system employed. Enhancement of AKT signaling can occur in the presence of rapamycin, due to relief of an S6K-dependent negative feedback loop targeting PI3-kinase (34, 35). Loss of this feedback inhibition is thought to be responsible for increased mTORC2/AKT activation and decreased sensitivity to apoptotic stimuli in certain malignancies treated with rapamycin (35). On the other hand, there are a number of studies that report enhancement of apoptosis by rapamycin (36-41). It has been shown that prolonged rapamycin treatment reduces mTORC2 complex assembly and AKT activation in approximately 20% of cancer cell lines (42), which could have a direct affect on apoptosis pathways. It is thus possible that the pro-apoptotic effects of rapamycin seen in some previous studies are the result of mTORC2 inhibition rather than a direct affect on mTORC1. This rationale is supported by our results. We see no affect of rapamycin on mTORC2-dependent pathways in our system, and rapamycin treatment does not result in enhanced activation of apoptosis in UVB-treated cells. In contrast, 4OHT-induced mTOR deletion resulted in a significant increase in apoptosis following UVB exposure in both keratinocytes culture and mouse epidermis. Furthermore, rictor-null cells were more sensitive to UVB-induced apoptosis than their wild-type counterparts. These results indicate that mTORC2 activation by UVB plays a critical role in mediating pathways that control keratinocyte survival, and reinforce previous observations of aberrant AKT activation in mouse models of NMSC (43, 44). mTORC2 was also identified as a therapeutic target in prostate cancer induced by loss of the tumor suppressor PTEN, using mice with conditional deletion of either mTOR or rictor (45, 46). The role of mTORC2 in these tumor types may be to maintain high levels of p-AKTSer473, which results in decreased transcription of a number of FOXO1/3-dependent cell cycle arrest and apoptotic genes (47, 48).
In summary, this is to our knowledge the first study to report that mTORC1- and mTORC2-dependent pathways are both activated by UVB, and play unique roles in controlling proliferation and apoptosis in the skin. These results emphasize the need to further elucidate the roles of mTORC1 and mTORC2 in photocarcinogensis and their links to cell proliferation, apoptosis, and tumor development. Our data provide compelling evidence to support the novel hypothesis that both mTORC1 and mTORC2 act as critical mediators of UVB-activated signal transduction in keratinocytes, and suggest that the combined targeting of both mTOR complexes, or alternatively mTORC1 and AKT, may be an effective chemoprevention strategy against photocarcinogensis.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Patricia Welsh for excellent technical support and Dr. David J. Feith for his helpful suggestions and critical reading of the manuscript.
Financial Support: This work was supported by NIH grants CA133945 (L.M.S.), ES19242 (L.M.S.), DK062880 (C.J.L.), CA37111 (J.D.) and the Pennsylvania Department of Health Tobacco CURE funds (L.M.S.). T.D.C. is the recipient of an MD/PhD predoctoral fellowship (F30 ES19809).
The abbreviations used are
- NMSC
nonmelanoma skin cancer
- mTOR
mammalian target of rapamycin
- 4OHT
4-hydroxytamoxifen
- SCC
squamous cell carcinoma
- AK
actinic keratosis
- CsA
cyclosporine A
- BrdU
5-bromo-2-deoxyuridine
- MEFs
mouse embryo fibroblasts
- TPA
12-O-tetradecanoylphorbol-13-acetate
Footnotes
Conflicts of Interest: No conflicts of interest to disclose.
REFERENCES
- 1.Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signaling. Nat Rev Cancer. 2004;4:23–35. doi: 10.1038/nrc1253. [DOI] [PubMed] [Google Scholar]
- 2.Erb P, Ji J, Kump E, Mielgo A, Wernli M. Apoptosis and Pathogenesis of Melanoma and Nonmelanoma Skin Cancer. Adv Exp Med Biol. 2008;624:283–95. doi: 10.1007/978-0-387-77574-6_22. [DOI] [PubMed] [Google Scholar]
- 3.Ananthaswamy HN, Pierceall WE. Molecular Mechanisms of ultraviolet radiation carcinogenesis. Photochem Photobiol. 1990;52:1119–36. doi: 10.1111/j.1751-1097.1990.tb08452.x. [DOI] [PubMed] [Google Scholar]
- 4.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Albanell J, Dalmases A, Rovira A, Rojo F. mTOR signalling in human cancer. Clin Transl Oncol. 2007;9:484–93. doi: 10.1007/s12094-007-0092-6. [DOI] [PubMed] [Google Scholar]
- 6.Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–71. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- 7.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 8.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 9.Chen SJ, Nakahara T, Takahara M, Kido M, Dugu L, Uchi H, et al. Activation of the mammalian target of rapamycin signalling pathway in epidermal tumours and its correlation with cyclin-dependent kinase 2. Br J Dermatol. 2009;160:442–5. doi: 10.1111/j.1365-2133.2008.08903.x. [DOI] [PubMed] [Google Scholar]
- 10.Einspahr JG, Calvert V, Alberts DS, Curiel-Lewandrowski C, Warneke J, Krouse R, et al. Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev Res (Phila) 5:403–13. doi: 10.1158/1940-6207.CAPR-11-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kauffman HM, Cherikh WS, Cheng Y, Hanto DW, Kahan BD. Maintenance immunosuppression with target-of-rapamycin inhibitors is associated with a reduced incidence of de novo malignancies. Transplantation. 2005;80:883–9. doi: 10.1097/01.tp.0000184006.43152.8d. [DOI] [PubMed] [Google Scholar]
- 12.Alberu J, Pascoe MD, Campistol JM, Schena FP, Rial Mdel C, Polinsky M, et al. Lower malignancy rates in renal allograft recipients converted to sirolimus-based, calcineurin inhibitor-free immunotherapy: 24-month results from the CONVERT trial. Transplantation. 92:303–10. doi: 10.1097/TP.0b013e3182247ae2. [DOI] [PubMed] [Google Scholar]
- 13.Liu G, Zhang Y, Bode AM, Ma WY, Dong Z. Phosphorylation of 4E-BP1 is mediated by the p38/MSK1 pathway in response to UVB irradiation. J Biol Chem. 2002;277:8810–6. doi: 10.1074/jbc.M110477200. [DOI] [PubMed] [Google Scholar]
- 14.Huang C, Li J, Ke Q, Leonard SS, Jiang BH, Zhong XS, et al. Ultraviolet-induced phosphorylation of p70(S6K) at Thr(389) and Thr(421)/Ser(424) involves hydrogen peroxide and mammalian target of rapamycin but not Akt and atypical protein kinase C. Cancer Res. 2002;62:5689–97. [PubMed] [Google Scholar]
- 15.Wan YS, Wang ZQ, Shao Y, Voorhees JJ, Fisher GJ. Ultraviolet irradiation activates PI 3-kinase/AKT survival pathway via EGF receptors in human skin in vivo. Int J Oncol. 2001;18:461–6. doi: 10.3892/ijo.18.3.461. [DOI] [PubMed] [Google Scholar]
- 16.Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell. 2006;11:583–9. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 17.Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc. 2008;3:799–810. doi: 10.1038/nprot.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lang CH, Frost RA, Bronson SK, Lynch CJ, Vary TC. Skeletal muscle protein balance in mTOR heterozygous mice in response to inflammation and leucine. American Journal of Physiology - Endocrinology And Metabolism. 2010;298:E1283–E94. doi: 10.1152/ajpendo.00676.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kataoka K, Kim DJ, Carbajal S, Clifford JL, DiGiovanni J. Stage-specific disruption of Stat3 demonstrates a direct requirement during both the initiation and promotion stages of mouse skin tumorigenesis. Carcinogenesis. 2008;29:1108–14. doi: 10.1093/carcin/bgn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Origanti S, Shantz LM. Ras transformation of RIE-1 cells activates cap-independent translation of ornithine decarboxylase: regulation by the Raf/MEK/ERK and phosphatidylinositol 3-kinase pathways. Cancer Res. 2007;67:4834–42. doi: 10.1158/0008-5472.CAN-06-4627. [DOI] [PubMed] [Google Scholar]
- 21.Feith DJ, Bol DK, Carboni JM, Lynch MJ, Sass-Kuhn S, Shoop PL, et al. Induction of ornithine decarboxylase activity is a necessary step for mitogen-activated protein kinase kinase-induced skin tumorigenesis. Cancer Res. 2005;65:572–8. [PubMed] [Google Scholar]
- 22.Han W, He YY. Requirement for metalloproteinase-dependent ERK and AKT activation in UVB-induced G1-S cell cycle progression of human keratinocytes. Photochem Photobiol. 2009;85:997–1003. doi: 10.1111/j.1751-1097.2008.00531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF, et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol. 2004;24:9508–16. doi: 10.1128/MCB.24.21.9508-9516.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci. 2001;26:657–64. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- 25.Hashemolhosseini S, Nagamine Y, Morley SJ, Desrivieres S, Mercep L, Ferrari S. Rapamycin inhibition of the G1 to S transition is mediated by effects on cyclin D1 mRNA and protein stability. J Biol Chem. 1998;273:14424–9. doi: 10.1074/jbc.273.23.14424. [DOI] [PubMed] [Google Scholar]
- 26.Lu ZH, Shvartsman MB, Lee AY, Shao JM, Murray MM, Kladney RD, et al. Mammalian target of rapamycin activator RHEB is frequently overexpressed in human carcinomas and is critical and sufficient for skin epithelial carcinogenesis. Cancer Res. 70:3287–98. doi: 10.1158/0008-5472.CAN-09-3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amornphimoltham P, Leelahavanichkul K, Molinolo A, Patel V, Gutkind JS. Inhibition of Mammalian target of rapamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clin Cancer Res. 2008;14:8094–101. doi: 10.1158/1078-0432.CCR-08-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wulff BC, Kusewitt DF, VanBuskirk AM, Thomas-Ahner JM, Duncan FJ, Oberyszyn TM. Sirolimus reduces the incidence and progression of UVB-induced skin cancer in SKH mice even with co-administration of cyclosporine A. J Invest Dermatol. 2008;128:2467–73. doi: 10.1038/jid.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Gruijl FR, Koehl GE, Voskamp P, Strik A, Rebel HG, Gaumann A, et al. Early and late effects of the immunosuppressants rapamycin and mycophenolate mofetil on UV carcinogenesis. Int J Cancer. 127:796–804. doi: 10.1002/ijc.25097. [DOI] [PubMed] [Google Scholar]
- 30.Amornphimoltham P, Patel V, Sodhi A, Nikitakis NG, Sauk JJ, Sausville EA, et al. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res. 2005;65:9953–61. doi: 10.1158/0008-5472.CAN-05-0921. [DOI] [PubMed] [Google Scholar]
- 31.Raimondi AR, Molinolo A, Gutkind JS. Rapamycin prevents early onset of tumorigenesis in an oral-specific K-ras and p53 two-hit carcinogenesis model. Cancer Res. 2009;69:4159–66. doi: 10.1158/0008-5472.CAN-08-4645. [DOI] [PubMed] [Google Scholar]
- 32.Checkley LA, Rho O, Moore T, Hursting S, DiGiovanni J. Rapamycin is a potent inhibitor of skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Prev Res (Phila) 4:1011–20. doi: 10.1158/1940-6207.CAPR-10-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim DJ, Kataoka K, Sano S, Connolly K, Kiguchi K, Digiovanni J. Targeted disruption of Bcl-x(L) in mouse keratinocytes inhibits both UVB- and chemically induced skin carcinogenesis. Mol Carcinog. 2009 doi: 10.1002/mc.20527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–8. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 35.O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Avellino R, Romano S, Parasole R, Bisogni R, Lamberti A, Poggi V, et al. Rapamycin stimulates apoptosis of childhood acute lymphoblastic leukemia cells. Blood. 2005;106:1400–6. doi: 10.1182/blood-2005-03-0929. [DOI] [PubMed] [Google Scholar]
- 37.Treeck O, Wackwitz B, Haus U, Ortmann O. Effects of a combined treatment with mTOR inhibitor RAD001 and tamoxifen in vitro on growth and apoptosis of human cancer cells. Gynecol Oncol. 2006;102:292–9. doi: 10.1016/j.ygyno.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 38.Hahn M, Li W, Yu C, Rahmani M, Dent P, Grant S. Rapamycin and UCN-01 synergistically induce apoptosis in human leukemia cells through a process that is regulated by the Raf-1/MEK/ERK, Akt, and JNK signal transduction pathways. Mol Cancer Ther. 2005;4:457–70. doi: 10.1158/1535-7163.MCT-04-0137. [DOI] [PubMed] [Google Scholar]
- 39.Beuvink I, Boulay A, Fumagalli S, Zilbermann F, Ruetz S, O'Reilly T, et al. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell. 2005;120:747–59. doi: 10.1016/j.cell.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 40.Thimmaiah KN, Easton J, Huang S, Veverka KA, Germain GS, Harwood FC, et al. Insulin-like growth factor I-mediated protection from rapamycin-induced apoptosis is independent of Ras-Erk1-Erk2 and phosphatidylinositol 3'-kinase-Akt signaling pathways. Cancer Res. 2003;63:364–74. [PubMed] [Google Scholar]
- 41.Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–7. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 42.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki A, Itami S, Ohishi M, Hamada K, Inoue T, Komazawa N, et al. Keratinocyte-specific Pten deficiency results in epidermal hyperplasia, accelerated hair follicle morphogenesis and tumor formation. Cancer Res. 2003;63:674–81. [PubMed] [Google Scholar]
- 44.Segrelles C, Lu J, Hammann B, Santos M, Moral M, Cascallana JL, et al. Deregulated activity of Akt in epithelial basal cells induces spontaneous tumors and heightened sensitivity to skin carcinogenesis. Cancer Res. 2007;67:10879–88. doi: 10.1158/0008-5472.CAN-07-2564. [DOI] [PubMed] [Google Scholar]
- 45.Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, et al. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15:148–59. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nardella C, Carracedo A, Alimonti A, Hobbs RM, Clohessy JG, Chen Z, et al. Differential requirement of mTOR in postmitotic tissues and tumorigenesis. Sci Signal. 2009;55:1–10. doi: 10.1126/scisignal.2000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–37. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 48.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–25. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.