

Abstract
C57BL/6 (B6) mice carrying the Sle1b sub-locus (named B6.Sle1b), which harbors the lupus-associated NZM2410/NZW SLAM family genes, produce anti-nuclear antibodies (ANA). However, the role and mechanism(s) involved in the alteration of the germinal center (GC) tolerance checkpoint in the development of ANAs in these mice is not defined. Here we show significantly higher spontaneously formed GCs (Spt-GCs) in B6.Sle1b female mice compared to B6 controls. We also found a significant increase in CD4+CXCR5hiPD-1hi spontaneously activated follicular helper T (Spt-TFH) cells in B6.Sle1b female mice. Compared to B6 controls, B6.Sle1b female mice had increased numbers of proliferating B cells predominantly located in Spt-GCs. The elevated Spt-GCs in B6.Sle1b female mice were strongly associated with increased ANA-specific antibody forming cells (AFCs) and ANA titers. The increased numbers of Spt-GCs and Spt-TFH cells in B6.Sle1b mice were not the result of a generalized defect in B cells expressing Sle1b. Consistent with the elevated spontaneous response in B6.Sle1b mice, the attenuated GC response characteristic of DNA and p-azophenylarsonate (Ars) reactive B cells from Ig VH knock-in mice (termed HKIR) were relieved in adoptively transferred recipients in the presence of Sle1b. Finally, by generating mixed bone marrow chimeras, we showed that the effect of Sle1b on Spt-GC, TFH cell and autoantibody responses in B6.Sle1b mice was B cell autonomous. These data indicate that the NZM2410/NZW-derived Sle1b sub-locus in conjunction with the female sex primarily affects B cells leading to the alteration of the GC tolerance checkpoint and the generation of ANA-specific AFCs.
Keywords: Rodent, B cells, autoimmunity, Systemic Lupus Erythematosus, antibody forming cell response, germinal center response
Introduction
The lupus-prone New Zealand Black/New Zealand White (NZB/NZW)-derived NZM2410 mouse strain develops a disease phenotype that closely resembles human SLE (systemic lupus erythematosus). Three major genomic intervals (Sle1, Sle2, and Sle3) are responsible for systemic autoimmune disease susceptibility in NZM2410 mice (1–3). B6 mice congenic for the Sle1 locus develop high titers of IgG autoantibodies against chromatin (4) and generate T cells specific for histone (5), implicating Sle1 in the loss of tolerance which leads to the development of anti-nuclear antibodies (ANAs). Genetic recombination of the Sle1 locus has further dissected the locus into four sub-loci termed Sle1a, Sle1FcR, Sle1b, and Sle1c (6, 7). B6 mice congenic for each sub-locus display partial autoimmune phenotypes with B6.Sle1b mice exhibiting gender-biased and highly penetrant ANA production (6). Sle1b results in altered functions in both T and B cells (5, 7–9). Resting B cells from B6.Sle1b mice appear to be more readily activated and have an enhanced ability to present antigen compared to B cells from B6 mice (10). T cells from B6.Sle1b exhibit higher Ca+2 flux response after TCR stimulation (7). In addition, a larger percentage of CD4+ T cells from B6.Sle1b are CD69+CD62LloCD44hi (9). Further confirmation of the importance of the Sle1b sub-locus in SLE pathology is evident in B6.Sle1b mice which also have either the Y-linked autoimmune accelerator (yaa) or lymphoproliferation (lpr) gene mutation, as they develop fatal lupus nephritis (11–13).
The Sle1b sub-locus contains the SLAM (signaling lymphocyte activation molecule) family (Slamf) genes derived from the lupus-prone NZW mice (7). The SLAMF cell surface receptors play an important role in regulating cellular and humoral immunity (14–16). Extensive polymorphisms in the Slamf genes have been demonstrated to be responsible for the loss of tolerance to nuclear antigens and for the induction of an autoimmune phenotype in B6.Sle1b mice (7). The Ly108.1 isoform of Ly108/Slamf6 expressed in B6.Sle1b mice is thought to be one of the strongest mediators involved in the loss of early B cell tolerance while Ly108.2 expression in B6 is believed to play role in the maintenance of tolerance (8). Other studies have implicated the protective role of CD48/Slamf2 as ablation of CD48 renders B6.Sle1b mice susceptible to the development of lupus-like autoimmune disease (17). While several candidate genes in the SLAM family in B6.Sle1b mice may contribute to the loss of tolerance resulting in autoimmune pathology, epistatic interactions between these genes most likely mediate the severity of SLE in these mice.
B cell tolerance to self-Ags (i.e., nuclear-Ags) is maintained through multiple tolerance checkpoints operative centrally in the bone marrow or peripherally in the secondary lymphoid organs (i.e., germinal center (GC) checkpoint). B cells undergo proliferation and somatic hypermutation in GCs, which results in B cells with high and low foreign Ag reactivity and potential autoreactivity. According to the current models of B cell selection in GCs, only high-affinity B cells receive survival signals and are then positively selected for further development into class-switched, high-affinity memory B cells and long-lived antibody forming cells (AFC) (18–20). B cells with low Ag-affinity and/or autoreactivity die via apoptosis (negative selection) (21–23). Altered regulation of positive and negative selection in the GCs in the presence of lupus-associated genes (i.e., lupus alleles of SLAM family genes) may allow autoreactive B cells to escape the GC checkpoint which may lead to the development of autoantibody-producing memory B cells and long-lived AFCs. Strains of mice that develop SLE-like disease spontaneously form GCs in the spleen by 1–2 months of age (24). Autoantibodies detected in lupus patients and lupus-prone mice bind their self-Ag with high affinity, are somatically mutated and class-switched (25–31), thus suggesting a role for the GC pathway in autoantibody production. However, the role and mechanism(s) involved in the alteration of the GC checkpoint in autoantibody production in B6.Sle1b mice is unclear.
Follicular helper T cells (TFH) are a subset of CD4+ T cells specialized to aid GC B cell development through B-T co-stimulatory molecule interactions, which include CD40 ligand, ICOS, PD-1 and SLAM. While a break in peripheral B cell tolerance at the GC checkpoint may allow autoreactive B cells to escape negative selection and enter circulation, TFH cells have also been shown to play a key role in contributing to the development of autoimmunity (32–35). Sanroque mice, which have a mutation in the Roquin (Rc3h1) gene, spontaneously develop GCs in the spleen and lymph nodes and have significantly increased numbers of activated memory T cells (33, 36). These mice develop an autoimmune profile resembling that of human SLE, coincident with increased TFH cell numbers per GC and development of anti-DNA-specific antibodies (33). Mice having the yaa mutation on BXSB background (BXSB.yaa) also develop severe autoimmune disease and have increased TFH cell numbers (34, 37). These data indicate the critical role of TFH cells in contributing to autoimmune pathology in several SLE mouse models. However, the role of TFH cells in the formation of Spt-GCs and ANA production in B6.Sle1b mice is not clear.
In the present study, we performed a detailed analysis of the impact of the Sle1b sub-locus on the loss of tolerance to nuclear antigens with an emphasis on the role in the GC pathway. We studied the formation of spontaneous GCs (Spt-GCs) in B6 and B6.Sle1b mice without any immunization, housing them in a pathogen-free barrier facility up to 9 months. B6.Sle1b female mice exhibited significantly increased percentages of splenic Spt-GCs and spontaneously activated CD4+ helper T cells including follicular helper T (named Spt-TFH) cells compared to age and sex-matched B6 controls. These elevated percentages of Spt-GCs and Spt-TFH cells in B6.Sle1b female mice were strongly correlated with increased numbers of ds-DNA, histone, and nucleosome-specific AFCs of both IgM and IgG isotypes. These mice also exhibited high titers of serum ANA-specific IgG2 Abs. These data suggest that the peripheral tolerance checkpoint that controls the formation of Spt-GCs and Spt-TFH cell numbers is altered by the presence of Sle1b. This effect is significant in female, but not in male mice, indicating a role for gender on Sle1b-mediated alteration of the peripheral tolerance checkpoint which allows for the development of autoantibody producing long-lived AFCs.
To further study the nature of self-Ags and to determine B cell autonomous effect of Sle1b on the alteration of the GC checkpoint, we have used an Ig VH chain knock-in mouse line termed HKIR (38, 39) that develops B cells reactive to the hapten p-azophenyl arsonate (Ars) and also have high avidity for DNA and chromatin-based self-antigens. Because of their dual-reactivity with Ars, HKIR self-reactive B cells can be mobilized into the GC reaction (40, 41) where they can participate but due to their autoreactivity these cells are negatively regulated as characterized by the reduced anti-Ars GC response of DNA-Ars dual-reactive HKIR B cells compared to control B cells that are only reactive to Ars (40–42). Thus, this model allowed us to study the influence of Sle1b on the regulation of HKIR DNA-reactive B cells at the GC checkpoint. Using this system we also showed that reduced anti-Ars GC response characteristic of DNA-Ars dual-reactive HKIR B cells was reversed in the presence of Sle1b, consistent with the data generated through spontaneous model showing alteration of the GC checkpoint by Sle1b. Our bone marrow chimeric experiments further revealed that the alteration of the GC checkpoint by Sle1b was B cell intrinsic and the effect of Sle1b on T cells appeared to be mediated by B cell defect caused by this sub-locus.
Materials and Methods
Mice
C57BL/6 (B6), B6.µMT and B6.TCRβδ−/− mice were purchased from The Jackson Laboratory and then bred in-house. B6 mice congenic for the Sle1b sub-locus (named B6.Sle1b mice) (6, 11) and the Ig VH knock-in mouse line HKIR were described previously (38, 43). The HKIR mice were crossed to B6.Sle1b to generate HKIR+/−.Sle1b+/− (named HKIR.Sle1b) mice. All mice were maintained in a pathogen-free barrier facility and were given only autoclaved food and water. Two to three and six to nine month old B6 and B6.Sle1b mice were used in experiments for studying spontaneous B and T cell activation in these mice. The mice designated for use in the sheep red blood cell (SRBC) response experiments were 5 to 6 weeks old when used. All experimental procedures performed on these animals were conducted according to the guidelines of our Institutional Animal Care and Use Committee.
SRBC immunization
Five to six week old B6 and B6.Sle1b mice were immunized (i.p) with 200 µl of 10% SRBC (Lampire, Pipersville, PA) in 1XPBS. Mouse spleens were harvested 12 days post immunization for flow cytometric and immunohistological analyses. Serum samples were also collected from these mice prior to sacrifice to measure Ab titers.
Reagents and antibodies for flow cytometry and immunohistology analysis
The following antibodies were utilized for flow cytometric analysis of mouse splenocytes: V500-anti-B220 (RA3-6B2); PeCy7-anti-CD95 (FAS, Jo2); Alexa Fluor 700-anti-CD4 (RM4-5); FITC-Foxp3 (FJK-16s); PE-anti-PD-1 (J43); APC-Cy7- anti-CD25 (PC61); biotin-anti-CXCR5 (2G8); V450-anti-Bcl-6 (K112-91) from BD Pharmingen, San Diego, CA. PerCP-Cy5.5-anti-CD69 (H1.253); PE-Cy5-anti-CD86 (GL1); APC-anti-CD44 (IM7); PE-Cy5-streptavidin (SA) from eBioscience, San Diego, CA. PE-Cy7-anti-CD62L (MEL-14); PE-anti-CD80 (16-10A1) from Biolegend, San Diego, CA. FITC-peanut-agglutinin (PNA) from Sigma-Aldrich, St. Louis, MO.
The follow antibodies were utilized for immunohistochemical analysis of the mouse spleen sections: Biotin-mouse anti-rat IgG (Jackson Immunoresearch Laboratories, West Grove, PA); Alkaline phosphatase (AP)-streptavidin; AP Blue substrate kit III; Vector NovaRED substrate kit (Vector Laboratories, Burlingame, CA); Purified rat anti-mouse Ki67 (ImmunoKontact, Abingdon, UK); FITC-PNA (Sigma-Aldrich, St. Louis, MO). Biotin-MOMA-1 (Abcam, Cambridge, MA); Biotin-anti-IgD (11–26, Southern Biotechnology Associates); PE-Anti-CD4 (GK1.5); FITC-GL7 (RA3-6B2) from BD Pharmingen; PE-Anti-PD-1 (J43), streptavidin Alexa Fluor 633 (Molecular Probes); PE-anti-CD138; Biotin-Anti-BrdU (Bu20a) from Biolegend, San Diego, CA; Purified anti-Foxp3 (FJK-16s; ebioscience, San Diego, CA) and a biotinylated form of the anti-idiotypic mAb E4 (prepared in-house); rabbit anti-mouse Bcl-6 (clone N-3, Santa Cruz Biotechnology, Santa Cruz, CA).
Adoptive transfer
B6 × B6.Sle1b F1 (B6.Sle1b+/−) recipient mice were immunized (i.p.) with 100 µg of Ars-KLH (in alum) 1 week prior to transfer (via retro-orbital i.v.) of MACS-purified 2 × 106 splenic B cells from either HKIR or HKIR mice expressing Sle1b (HKIR.Sle1b) donor mice. These chimeric mice were then injected i.p. with 50 µg of Ars-KLH in PBS immediately after cell transfer. Spleens from the recipient mice were harvested 5 days later and utilized for flow cytometric and immunohistological analysis.
Generation of bone marrow chimeric mice
Ten to twelve week old female B6.µMT (µMT) and B6.TCRβδ-deficient (TCRβδ−/−) mice were lethally irradiated with 10.5 gray of γ-radiation prior to transfer via retro-orbital i.v. injection of 7.5 to 10×106 T cell-depleted mixed bone marrow cells isolated from 8–10 week old female donor mice (i.e., B6, B6.Sle1b, µMT and TCRβδ−/−). µMT recipients received either a 1:1 ratio of B6 and µMT or B6.Sle1b and µMT bone marrow cells. TCRβδ−/− recipients received either a 1:1 ratio of B6 and TCRβδ−/− or B6.Sle1b and TCRβδ−/− bone marrow cells. The recipient chimeric mice were aged for 6 months post transfer of bone marrow cells.
Flow cytometry
Multi-color flow cytometric analysis was done using multiple combinations of the Abs listed above on cell suspensions prepared from spleens of 6–9 month old and SRBC-immunized B6 and B6.Sle1b mice, as well as from recipient B6.Sle1b+/− mice adoptively transferred with either HKIR or HKIR.Sle1b B cells. Biotinylated Abs were detected with streptavidin-conjugated fluorochromes. Stained cells were analyzed using the BD LSRII flow cytometer. Data were analyzed using FlowJo software (Treestar, San Carlos, CA). Intracellular staining for Foxp3 and Bcl-6 was performed through the use of the Foxp3 intracellular staining kit (eBioscience, San Diego, CA) following manufacturer’s directions.
Immunohistology
Spleen cryostat sections (5–6 µm) were prepared as described previously (44). Immunohistology was performed using the Abs listed above and the stained sections were analyzed using a fluorescence microscope (Leica Microsystems) and images were captured as previously described (45).
BrdU cell proliferation experiments
Unimmunized 6–9 month old and SRBC-immunized 5–6 week old B6 and B6.Sle1b mice were injected with BrdU (i.p., 0.6 mg per mouse, BD Bioscience, San Diego, CA) 12 hours and 1–2 hours prior to sacrifice. Immunohistological analysis of spleen sections for BrdU+ cells was performed using BrdU in situ staining kit (BD bioscience, San Diego, CA) following manufacturer’s guidelines.
ELISpot assays
ELISpot assays were performed as described (42). Briefly, splenocyte suspensions from 6–9 month old B6 and B6.Sle1b mice were plated at either 1 × 105 cells/well in IgM and IgG coated or at 1 × 106 cells/well in double-stranded DNA-, histone-, and nucleosome-coated multiscreen 96-well filtration plates (Millipore, Bedford, MA), diluted serially (1:2) and incubated for 6 hr at 37°C. Splenocytes from SRBC-immunized mice were plated at 1×106 cells/well. IgM-producing AFCs were detected using biotinylated anti-mouse IgM (Jackson Immunoresearch, West Grove, PA) and streptavidin (SA)-alkaline phosphatase (Vector Laboratories, Burlingame, CA). IgG-producing AFCs were detected using alkaline-phosphatase conjugated anti-mouse IgG (Molecular Probes, Grand Island, NY). Double-stranded DNA-, histone-, and nucleosome-specific AFCs were detected by biotinylated anti-kappa Ab (Invitrogen, Grand Island, NY) and streptavidin (SA)-alkaline phosphatase (Vector Laboratories, Burlingame, CA). Plates were developed using the Vector Blue Alkaline-phosphatase Substrate Kit III (Vector Laboratories, Burlingame, CA). ELISpots were counted using a computerized imaging video system (Cellular Technology, Cleveland, OH).
ANA titers
Total serum ANA titers from 6–9 month old B6 and B6.Sle1b mice were measured in ELISA plates coated with either double-stranded DNA, histone, or nucleosome and detected with biotinylated anti-kappa Ab (Invitrogen, Grand Island, NY). Similarly, IgG subtype-specific ANA titers were measured by biotinylated-IgG1, biotinylated IgG2b, and AP-IgG2c (Southern Biotech, Birmingham, AL). Biotinylated antibodies were detected by streptavidin (SA)-alkaline phosphatase (Vector Laboratories, Burlingame, CA). The plates were developed by the PNPP (p-Nitrophenyl Phosphate, Disodium Salt) (Thermo Fisher Scientific, Rockford, IL) substrates for alkaline phosphatase. Serum samples were first diluted in PBS and then subsequently two-fold serial dilution was carried out for each sample. The dilution factor for each sample was generated in a logarithmic scale via the software named “Origin” based on the different OD values of 0.8, 0.9 or 1.0 (at 405 nm) set for different isotype-specific ELISA. The OD values of 0.8, 0.9 or 1.0 were determined based on the linear distribution of most of the samples in any given ELISA. In this way, a dilution factor of 400 for a particular sample with an OD value of 1.0 would indicate that this particular serum sample needed to be diluted 400 times to obtain an OD value of 1.0 at 405 nm. In contrast, a serum sample with a dilution factor of 150 needed to be diluted 150 times to obtain the same OD value. Therefore, the higher the dilution factor in an individual mouse the higher the Ab titers for that particular animal.
Statistical analysis
Statistical analysis was done using Student’s t-test. P values of <0.05, <0.01, and <0.001 are depicted as *, **, and *** respectively. Statistical significance for the correlation graphs was performed by using regression (R)/ANOVA (analysis of variance).
Results
Effects of Sle1b and gender on spontaneous activation of B cells and CD4 helper T cells
Previous studies have indicated the influence of Sle1b sub-locus carrying NZM2410/NZW lupus alleles of the SLAM family genes on both B cells and CD4 helper T cells (9, 11, 13). However, the effects of gender on the spontaneous activation of B and T cells in B6.Sle1b mice are not defined. Here we performed a detailed analysis of the influence of Sle1b and gender on B cells and CD4 T cells by analyzing both male and female mice. Splenocytes obtained from 6–9 month old B6 and B6.Sle1b mice were stained with antibodies against B cell activation markers CD69, CD80 and CD86. The percentage of B220+CD69+ (rectangular gates and scatter plot, Fig. 1A) and B220+CD86+ (Fig. 1B) B cells were significantly higher in B6.Sle1b female mice compared to B6.Sle1b males, B6 males and B6 females. In B6 mice, while the percentage of B220+CD69+ cells was similar between the two sexes, females had elevated B220+CD86+ cells compared to males (Fig. 1A and B). We found no difference in the percentage of B220+CD80+ B cells among the four groups of mice (data not shown).
Figure 1. Increased spontaneous activation of B cells and CD4 helper T cells in B6.Sle1b female mice.

Flow cytometric analysis was performed on splenocytes from 6–9 month old, sex-matched B6 and B6.Sle1b mice after staining for activated B cells using mAbs against B220, CD69 and CD86. The percentage of B220+CD69+ (A) and B220+CD86+ (B) activated B cells are shown in rectangular gates (left panels) and in scatter plots (right panels). Activated CD4 T cells were analyzed using antibodies against CD4, CD44, CD62L and CD69. The percentage of CD4+CD44hiCD62Llo short-lived effector and CD4+CD44hiCD62Lhi effector memory helper T cells are shown in rectangular gates (left panel, C) and in scatter plots (middle and right panels, respectively, C). The percentage of CD4+CD69+ activated T cells are shown in rectangular gates (left panel, D) and in scatter plot (D, right panel). Each circle represents an individual mouse and horizontal bars represent the mean values. Statistical analysis was performed as described in Materials and Methods.
Similar flow cytometric analysis of CD4+ T cells using T cell markers, CD4, CD44, CD62L and CD69 revealed significantly higher percentages of both CD4+CD44hiCD62Llo short-lived effector (Fig. 1C, middle panel) and CD4+CD44hiCD62Lhi effector memory (Fig. 1C, right panel) helper T cells in splenocytes from B6.Sle1b males compared to B6 males. CD4+CD44hiCD62Lhi effector memory T cells appeared to be significantly increased in B6.Sle1b females compared to B6 females (Fig. 1C, right panel). Both these effector populations were significantly higher in B6 females compared to males (Fig. 1C). We also observed that the percentage of CD4+CD69+ cells in B6.Sle1b mice (males and females) was significantly increased compared to their B6 control counterparts (rectangular gates and scatter plot, Fig. 1D). These differences were also observed between males and females of each genotype (Fig. 1D).
Significantly higher spontaneously formed GCs (Spt-GCs) in B6.Sle1b mice
GCs are spontaneously formed (named Spt-GC) in lupus-prone mice (24) and human SLE patients (46, 47). Given the increased percentage of activated B cells in 6–9 month old B6.Sle1b mice, we examined whether GCs were spontaneously formed in the presence of Sle1b. Flow cytometric analysis was performed on splenocytes obtained from B6 and B6.Sle1b mice by staining with GC B cell markers B220, PNA and Fas/CD95. The percentage of B220+PNAhiFashi GC B cells in B6.Sle1b males and females was significantly higher compared to their B6 control counterparts (Fig. 2A). Female mice of each genotype also had elevated percentages of B220+PNAhiFashi GC B cells compared to their male counterparts (Fig. 2A). We also performed immunohistological analysis in which spleen sections obtained from 6–9 month old B6 and B6.Sle1b female mice were stained with anti-IgD (blue) and PNA (brown). Consistent with the flow cytometry data, we found increased frequencies of IgDnegPNA+ GCs consisting of predominantly large GCs in B6.Sle1b female mice (lower two panels, Fig. 2C) compared to less frequent and significantly smaller GCs in B6 controls (upper two panels, Fig 2C).
Figure 2. Elevated number of Spt-GCs in B6.Sle1b mice.

(A) Flow cytometric analysis was performed on splenocytes obtained from sex matched, 6–9 month old B6 and B6.Sle1b mice for spontaneous GCs (Spt-GCs). The percentage of B220+ΠNAhiFashi GC B cells are shown in rectangular gates (left panel) and in scatter plots (right panel). (B) In a similar analysis as in (A), splenocytes obtained from 5 to 6 week old SRBC-immunized B6 and B6.Sle1b mice were analyzed 12 days post-immunization to show the percentage of B220+PNAhiFashi GC B cells in these mice. The flow cytometry plots on the left are representative images from each group while the scatter plots on the right showing the range of the GC B cell percentages. The blue dots represent male mice and the red dots represent female mice (right panel, B). Each circle represents an individual mouse and horizontal bars represent the mean values. Statistical analysis was performed as described in Materials and Methods. (C) Spleen sections obtained from 6–9 month old female B6 and B6.Sle1b mice were stained with anti-IgD (blue) and PNA (red). (D) Similar analysis as in (C) was performed on spleen sections obtained from SRBC-immunized 5 to 6 week old female B6 (n=6) and B6.Sle1b (n=6) mice 12 days post immunization. Original magnification of the images was 100×. The data shown in (C and D) represent at least 5 mice of each genotype.
Next, to evaluate whether the increased Spt-GCs in B6.Sle1b mice resulted from a generalized defect in immune responsiveness of B cells expressing Sle1b, we immunized 5–6 wk old B6 and B6.Sle1b mice with the TD-Ag sheep red blood cells (SRBC). The anti-SRBC stimulated GC response was determined 12 days after immunization. We did not find any significant difference in the percentage of B220+PNAhiFashi GC B cells (rectangular gates, Fig. 2B) upon SRBC challenge either between B6.Sle1b and B6 controls, or between males (blue) and females (red) within each genotype (right panel, Fig. 2B). Whereas we observed elevated frequency and size of Spt-GCs in 6–9 month old B6.Sle1b female mice compared to age-matched B6 female controls (Fig. 2C), similar immunohistological analysis of SRBC-immunized B6 and B6.Sle1b spleens revealed no difference in the frequency and size of IgDnegPNA+ GCs between B6.Sle1b and B6 females (Fig. 2D). These data indicate that increased number of Spt-GCs in B6.Sle1b mice was not the result of a generalized defect in the response of B cells expressing Sle1b.
Significantly higher spontaneously activated TFH (Spt-TFH) cells in B6.Sle1b mice
Given the increased percentage of spontaneously activated CD4 T cells in B6.Sle1b mice (Fig. 1C and D) we next sought to evaluate whether B6.Sle1b mice had an elevated percentage of spontaneously activated TFH (named Spt-TFH) cells. Flow cytometric analysis of splenocytes from 6–9 month old B6 and B6.Sle1b mice using Abs against the TFH cell specific markers CD4, CXCR5 and PD-1 exhibited significantly higher percentage of CD4+CXCR5hiPD-1hi Spt-TFH cells in B6.Sle1b mice (males and females) compared to B6 male and B6 female controls (Fig. 3A). In both B6 and B6.Sle1b mice, females also had significantly higher percentage of Spt-TFH cells compared to males (Fig. 3A).
Figure 3. Elevated Spt-TFH cell numbers concomitant with augmented Spt-GCs in B6.Sle1b female mice.

(A) Flow cytometric analysis of splenocytes obtained from sex matched, 6–9 month old B6 and B6.Sle1b mice for the percentages of Spt-TFH cells. (B) The percentage of TFH cells from 5–6 week old SRBC-immunized B6 and B6.Sle1b mice were also analyzed 12 days post-immunization. (A and B) TFH cells are defined as CD4+CXCR5hiPD-1hi and are highlighted by the rectangular gates. The flow cytometry plots on the left are representative images from each group while the scatter plots on the right showing the range of the TFH cell percentages. The blue dots represent male mice and the red dots represent female mice in (B). Each circle represents an individual mouse and horizontal bars represent the mean values. Statistical analysis was performed as described in Materials and Methods. (C) Spleen sections obtained from aged 6–9 month old female B6 and B6.Sle1b mice were stained with anti-IgD (blue), anti-CD4 (red) and anti-PD-1 (green). Representative images from 5 mice of each genotype are shown where original magnification of the images was 200×.
We also examined the TFH cell response in SRBC-immunized 5–6 wk old B6 and B6.Sle1b mice 12 days after immunization. We found no significant difference in the percentage of CD4+CXCR5hiPD-1hi TFH cells between B6.Sle1b mice and B6 controls (Fig. 3B). No significant difference was observed between males (blue) and females (red) of each genotype at this age (Fig. 3B). These data are consistent with the anti-SRBC GC B cell response (Fig. 2B).
To study the anatomical location of CD4+CXCR5hiPD-1hi Spt-TFH cells shown in Fig. 3A, we stained spleen sections from 6–9 month old B6 and B6.Sle1b females with Abs against IgD (blue), CD4 (red) and PD-1 (green). We defined GC by the absence of IgD staining in IgD+ B cell follicles as IgD is down regulated in GC B cells (dashed lines, 1st column, Fig. 3C). Consistent with flow cytometry data we found significantly higher number of CD4+ (lower panel, 2nd column) and PD-1hi (lower panel, 3rd column) TFH cells in B6.Sle1b GCs as evidenced by the overlapped yellow staining (overlay, 4th column) compared to B6 controls (top row, Fig. 3C).
We also evaluated whether increased numbers of Spt-GCs and Spt-TFH cells in B6.Sle1b mice could result from the decrease in CD4+Foxp-3+ T regulatory (Treg) cells. By performing flow cytometric and immunohistological analyses we found no difference between the two strains in the frequency of CD4+Foxp-3+ Tregs within and outside of GCs (Supplementary Fig. 1).
B and T cell abnormalities occur at early age in B6.Sle1b mice
Next, we examined whether the spontaneous responses that we observed in 6–9 month old B6.Sle1b mice also occurred at earlier time points. Splenocytes from 2–3 month old unimmunized female B6.Sle1b mice and B6 controls were analyzed for spontaneous GC, TFH cell and ANA-specific AFC responses. While the overall responses in these mice were lower at these earlier time points compared to later time (i.e., 6–9 month old) we found the difference in the percentages of B220+Fas+PNAhi GC B cells in B6.Sle1b mice compared to sex- and age-matched B6 controls statistically significant at the age of 3 months (Fig. 4A). The percentage of CD4+CXCR5hiPD-1hi TFH cells in B6.Sle1b mice was also increased compared to B6 controls beginning at 2 months of age (Fig. 4B). The difference in the number of autoantibody producing AFCs was not statistically significant at this stage but 25 to 50% B6.Sle1b mice had elevated numbers of ds-DNA, histone and nucleosome-specific AFCs compared to B6 controls at 3 months old (data not shown). These data indicate that the alteration of peripheral tolerance at the GC checkpoint in the presence of Sle1b initiates at the earlier time point (2–3 months), which peaks at 6–9 months old.
Figure 4. Sle1b induces spontaneous GC and TFH cell responses at early age.

Flow cytometric analysis of splenocytes obtained from 2 and 3 months old female B6 and B6.Sle1b mice for the percentages of GC B cells (A) and TFH cells (B). The open circles represent B6 mice while the closed circles represent B6.Sle1b mice. Each circle represents an individual mouse and horizontal bars represent the mean values. Statistical analysis was performed as described in Materials and Methods.
Increased Bcl-6 expressing B cells and CD4 helper T cells in B6.Sle1b mice
Bcl-6 is considered to be a master transcriptional regulator for GC B cells (48–51) and TFH cells (52–54). Given the elevated numbers of Spt-GCs and Spt-TFH cells in the presence of Sle1b, we evaluated whether B6.Sle1b mice harbored an increased percentage of Bcl-6 expressing B cells and CD4 helper T cells. Flow cytometric analysis of splenocytes obtained from 6–9 month old B6 and B6.Sle1b mice was performed through surface staining with B220 and anti-CD4, and intracellular staining with anti-Bcl-6. The percentage of B220+Bcl-6+ B cells (Fig. 5A) and CD4+Bcl-6+ T cells (Fig. 5C) in B6.Sle1b females were significantly higher compared to B6.Sle1b males and B6 males and B6 females. No difference was observed between B6 males and B6 females, and B6 males versus B6.Sle1b males.
Figure 5. Increased number of Bcl-6 expressing B and CD4 T cells in B6.Sle1b female mice.

Splenocytes obtained from sex matched, 6–9 month old B6 and B6.Sle1b mice were stained for Abs against either B220 and Bcl-6 (A) or CD4 and Bcl-6 (C) and analyzed by flow cytometry. The left flow cytometry plots are representative images from each group while the scatter plots on the right indicate the range of B220+Bcl-6+ B cell (A) and CD4+Bcl-6+ T cell (C) percentages from each group. Each circle represents an individual mouse and horizontal bars represent the mean values. Statistical analysis was performed as described in Materials and Methods. Spleen sections obtained from 6–9 month old female B6 and B6.Sle1b mice were either stained with GL7 (green) and anti-Bcl-6 (red) (B) or anti-CD4 (red) and anti-Bcl-6 (green) (D). The data shown in B and D were obtained from 4 to 5 female mice of each genotype. Original magnification of the images was 200×.
Furthermore, to determine the follicular-GC (i.e., Spt-GC) vs extrafollicular localization of B220+Bcl-6+ B cells and CD4+Bcl-6+ T cells in the spleen, immunohistological analysis was performed on two consecutive spleen sections obtained from 6–9 month old B6 and B6.Sle1b females. One was stained with the GC B cell marker GL7 and anti-Bcl-6 (Fig. 5B). The other was stained with anti-CD4 and anti-Bcl-6 (Fig. 5D). We found elevated Bcl-6 expressing B cells (Fig. 5B) and T cells (Fig. 5D) in B6.Sle1b mice, predominantly located within GCs as evidenced by the overlapped yellow staining in the overlay images (3rd column, Fig. 5B and D) with a very few such cells in the extrafollicular (EF) location (i.e., T cell zone, marginal zone and red pulp) outside of GCs.
Follicular-germinal center (F-GC) versus extrafollicular (EF) spontaneous B cell proliferation in the presence or absence of Sle1b
Next, we evaluated spontaneous B cell proliferation in the follicular-germinal center (F-GC, also designated as Spt-GC) versus extrafollicular (EF) regions in the spleens of B6 and B6.Sle1b mice. EF areas included T cell zone, marginal zone and red pulp in the spleen. Six to nine month old B6 and B6.Sle1b female mice were injected with BrdU (0.6 mg/mouse) 12 hrs and 1–2 hrs prior to sacrifice to obtain spleens for analysis. Spleen sections obtained from B6 and B6.Sle1b mice were stained with anti-IgD (blue) and anti-BrdU (brown). GCs were characterized by the absence of IgD staining within IgD+ follicles. Low (100×) and high (200×) magnification representative images shown in Fig. 6A revealed significantly higher number of BrdU+ cells in B6.Sle1b mice compared to B6 controls. Interestingly, BrdU+ cells were predominantly located in F-GCs with a very small number of BrdU+ cells in EF regions outside of F-GCs (Fig. 6A).
Figure 6. Significantly higher number of proliferating B cells in B6.Sle1b female mice.
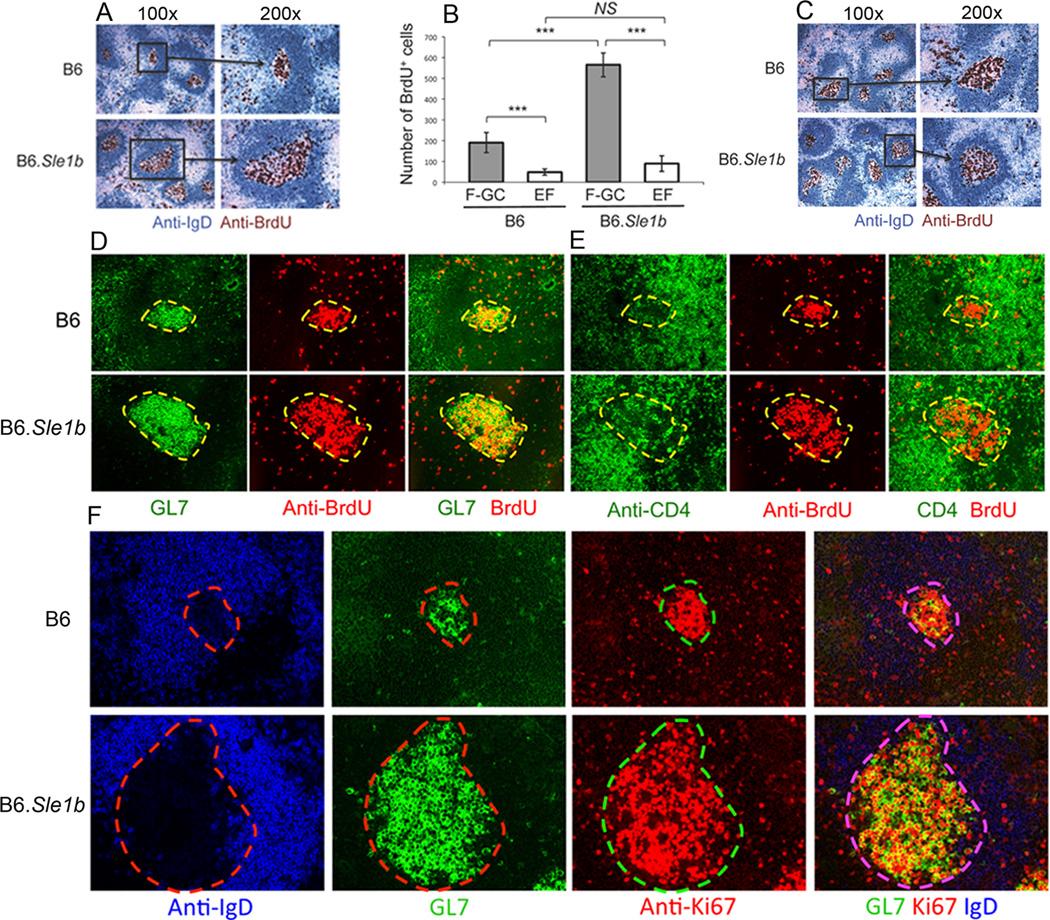
(A) Spleen sections from 6–9 month old B6 and B6.Sle1b mice injected with BrdU 12 hours and 1–2 hours prior to sacrifice were stained for anti-IgD (blue) and anti-BrdU (brown). Original magnification of the images was 100× (left) and 200× (right). (B) Semi-quantitative analysis of the number of BrdU+ cells in follicular-GC (F-GC, gray bars) versus extrafollicular (EF, white bars) regions. BrdU+ cells were counted in two randomly picked areas per spleen at ×100 original magnification. Five B6 and 5 B6.Sle1b female mice were analyzed. (C) Spleen sections obtained from SRBC-immunized 5–6 week old female B6 and B6.Sle1b mice were analyzed for BrdU+ cells inside the GCs as described in A. Parallel spleen sections were obtained from 6–9 month old female B6 and B6.Sle1b mice and stained for either GL7 (green) and anti-BrdU (red) as shown in (D) or anti-CD4 (green) and anti-BrdU (red) as shown in (E). (F) Spleen sections from 6–9 month old female B6 and B6.Sle1b mice were stained for anti-IgD (blue), GL7 (green), and anti-Ki67 (red). Original magnification of the images in C-E was 200×. These data were generated from at least 5 female mice of each genotype.
We performed a semi quantitative analysis in which we counted BrdU+ cells in F-GC and EF regions at ×100 original magnification from 5 B6 and 5 B6.Sle1b female mice. This analysis revealed significantly higher number of BrdU+ cells in F-GCs compared to EF regions in both B6 and B6.Sle1b mice (Fig. 6B). Whereas we did not observe significant difference in BrdU+ cells in EF regions between B6 and B6.Sle1b mice we found B6.Sle1b mice had significantly higher BrdU+ cells in F-GCs/Spt-GCs relative to B6 controls (Fig. 6B). Similar analysis of SRBC-immunized 5–6 week old B6 and B6.Sle1b female mice performed 12 days after immunization showed no difference in the number of BrdU+ cells in F-GCs and EF regions (Fig. 6C and not shown) indicating no generalized defect of B6.Sle1b B cell proliferation against TD-Ag.
Proliferating cells within the GCs are believed to be primarily B cells. However, it is not clear whether the increased number of Spt-TFH cells in Spt-GCs of B6.Sle1b mice (Fig. 3) could result from the proliferation of CD4 T cells located within GCs. To address this issue, we performed immunohistological analysis on two consecutive spleen sections obtained from 6–9 month old B6 and B6.Sle1b females. One was stained with GL7 (green, 1st column) and anti-BrdU (red, 2nd column, Fig. 6D) and the other was stained with anti-CD4 (green, 1st column) and anti-BrdU (red, 2nd column, Fig. 6E). While GL7+ B cells colocalized with BrdU staining as judged by the yellow overlapped staining (3rd column, Fig. 6D) we did not observe such overlapped yellow staining with CD4 and BrdU (3rd column, Fig. 6E). Additionally, when we stained spleen sections from B6.Sle1b and B6 control mice with anti-IgD (blue), GL7 (green) and Ki67 (red), a cell proliferation marker, we found analogous results as in Fig. 6D showing increased numbers of Ki67+ cells in B6.Sle1b relative to B6 control mice (Fig. 6F). Together, these data indicate that BrdU+ and Ki67+ proliferating cells in GCs are B cells and not CD4 T cells. Also, these data together with results shown in Fig. 3C indicate that the increased number of CD4 T cells in 6–9 month old B6.Sle1b GCs is due to spontaneously activated and fully differentiated TFH cells.
Elevated numbers of Spt-GCs and Spt-TFH cells in B6.Sle1b mice strongly correlate with increased number of ANA-specific AFCs
To evaluate whether increased numbers of Spt-GCs and Spt-TFH cells in B6.Sle1b mice correlated with an increased ANA-specific AFC response, we first measured the number of total IgM and IgG-producing AFCs in spleen samples from 6–9 month old B6 and B6.Sle1b mice through ELISpot assay. The number of IgM- and IgG-producing AFCs in B6.Sle1b females was significantly higher compared to B6.Sle1b males and B6 males and B6 females (Fig. 7A). Significant difference was also observed between males and females of each genotype in IgG-producing AFCs. Consistent with the anti-SRBC GC B cell response (Fig. 2B and D) we did not observe any significant difference between B6 and B6.Sle1b mice in SRBC-specific IgM and IgG AFCs (Fig. 7B) 12 days after SRBC immunization. By staining with MOMA-1 (blue), which stains for metalophillic macrophages and defines the border between follicle and marginal zone, anti-CD138 (red), a marker for AFCs and anti-IgG (green) we showed elevated number of CD138+IgG+AFCs located both in the red pulp areas and in the bridging channels of female B6.Sle1b spleens compared to lower number of these cells in B6 controls located primarily in the bridging channels (Fig. 7C).
Figure 7. Increased ANA-specific AFCs in B6.Sle1b female mice.
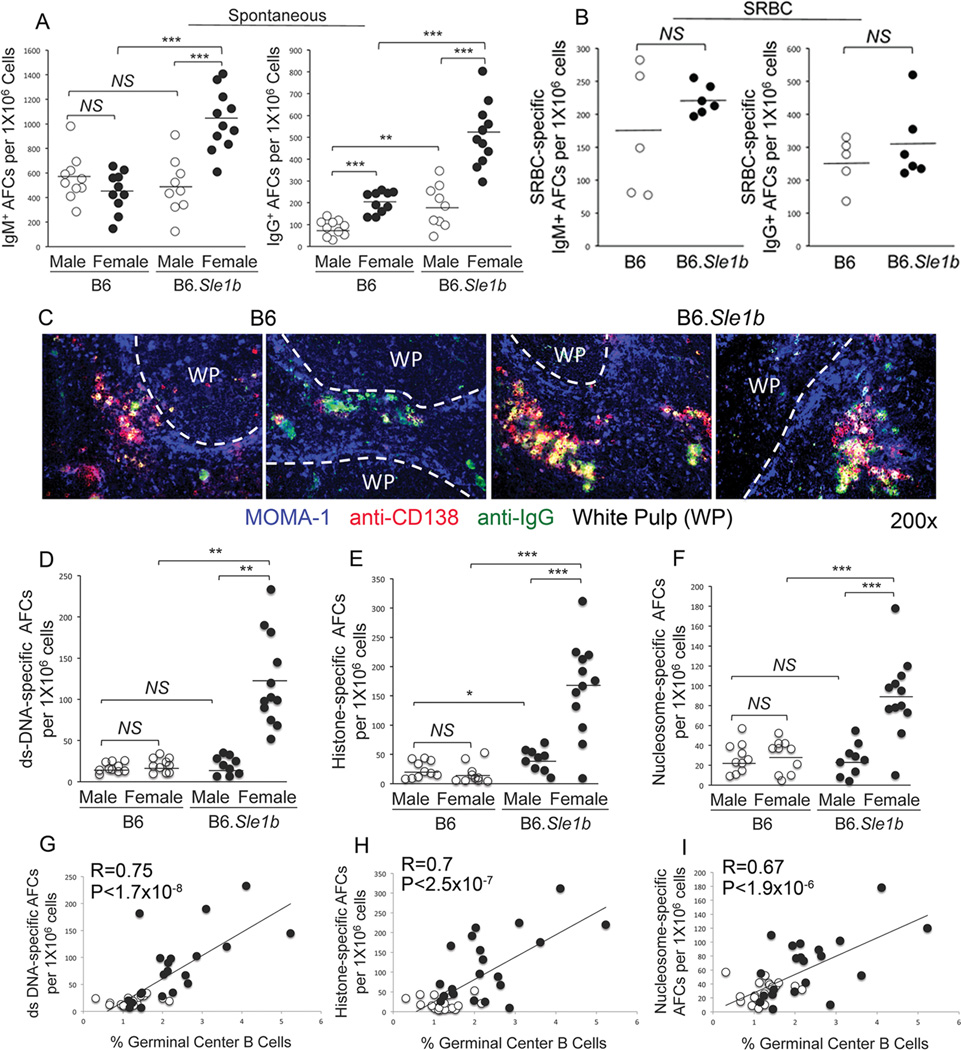
Total number of IgM (left panel) and IgG (right panel) specific AFCs in un-immunized 6–9 month old (A) and SRBC-immunized 5–6 week old (B), sex-matched B6 and B6.Sle1b mice were measured by ELISpot assay. (C) Spleen sections obtained from 6–9 month old female B6 and B6.Sle1b mice were stained for MOMA-1 (blue), anti-CD138 (red), and anti-IgG (green). WP represents white pulp. Original magnification of the images was 200x. Representative data from 5 female mice of each genotype are shown. The number of ds-DNA- (D), histone- (E), and nucleosome- (F) specific AFCs were measured by ELISpot assay. The degree of correlation between GC B cells and ds-DNA- (G), histone- (H), and nucleosome- (I) specific AFCs was analyzed. Each circle represents an individual mouse and B6.Sle1b mice are depicted in black circles while B6 mice are depicted in white circles. Statistical analysis was performed as described in Materials and Methods.
To determine whether increased Spt-GCs and total IgM and IgG AFCs in B6.Sle1b mice led to elevated numbers of ANA-specific AFCs, we measured ds-DNA-, histone- and nucleosome-specific AFCs by ELISpot assay. We found the numbers of AFCs with each of these specificities (Fig. 7D–F) were significantly higher in B6.Sle1b females compared to B6.Sle1b males, B6 males, and B6 females. No difference was observed between B6.Sle1b males and B6 males or B6 males versus B6 females in ds-DNA and nucleosome-specific AFCs. We, however, observed significant difference between B6.Sle1b males and B6 males in histone-specific AFCs (Fig. 7E). Interestingly, we further observed that the increased numbers of ds-DNA-, histone- and nucleosome-specific AFCs strongly correlated with elevated numbers of GC B cells in B6.Sle1b mice (Fig. 7G–I).
Elevated Spt-GCs and ANA-specific AFCs in B6.Sle1b female mice led to increased serum ANA titers
To examine whether the elevated Spt-GC and AFC responses in B6.Sle1b mice correlated with increased serum ANA titers, we first measured ds-DNA-, histone- and nucleosome-specific total (IgM + IgG) Ab titers in sera collected from 6–9 month old B6 and B6.Sle1b mice (Fig. 8A–C). These titers were found to be significantly higher in B6.Sle1b mice compared to B6 controls. Histone-specific Abs were also significantly increased in B6.Sle1b females compared to B6.Sle1b males. No difference was observed between B6 males and B6 females. These data were consistent with results obtained in the ANA detection assay using ANA Hep-2 substrate slides. In this assay, we found significantly higher intensity of nuclear staining with sera from B6.Sle1b females compared to background staining in B6 females (data not shown).
Figure 8. Elevated ANA-specific AFCs in female B6.Sle1b mice correlated with increased ANA-specific Ab titers.

Total double-stranded DNA- (A), histone- (B), and nucleosome- (C) specific serum Ab titers were measured by ELISA in 6–9 month old, sex matched B6 and B6.Sle1b mice. Similarly, ds-DNA-(D), histone- (E) and nuclesosome (F and G)-reactive and IgG subclass-IgG2c/2b-specific ANA titers were measured in mice described in (A–C). Only the ANA-specific IgG subclasses that had elevated levels of Ab titers above baseline are shown. The degree of correlation between the percentage of Spt-GC B cells and IgG2c-2b specific and double-stranded DNA- (H), histone- (I), and nucleosome- (J–K) reactive serum Ab titers in 6–9 month old, sex matched B6 and B6.Sle1b mice are shown. Each circle represents an individual mouse and B6.Sle1b mice are depicted in black circles while B6 mice are depicted in white circles. Statistical analysis was performed as described in Materials and Methods.
Next, we evaluated IgG subclass specific ANA titers in 6–9 month old B6 and Sle1b mice. IgG1 ANA titers remained similar between the four groups of mice (data not shown). In contrast, B6.Sle1b female mice had significantly higher titers of IgG2c Abs specific for ds-DNA, histone and nucleosome compared to B6.Sle1b males, B6 males and B6 females (Fig. 8D–F). We also observed significantly increased IgG2b Abs specific for nucleosome in B6.Sle1b females (Fig. 8G). Finally, the increased IgG2c/2b ANA titers were strongly correlated with the increased number of Spt-GCs in B6.Sle1b mice (Fig. 8H–K).
Reduced GC response characteristic of Ars-DNA dual-reactive HKIR B cells was reversed in the presence of Sle1b
Data described above suggest that peripheral tolerance checkpoints that regulate Spt-GC formation and activation of CD4 helper T cells, including TFH cells, are disrupted in the presence of Sle1b, which may contribute to the development of autoantibody producing AFCs and memory B cells. To determine the role and nature of self-Ags in Spt-GC formation and the alteration of the GC tolerance checkpoint by the Sle1b sub-locus, we used an adoptive transfer system in which we transferred Ars and DNA-dual-reactive HKIR B cells into syngenic recipients (40–42). We previously showed that dual-reactive HKIR B cells can enter GCs upon immunization with Ars-conjugated foreign antigen (i.e., Ars-KLH) but due to their autoreactivity (DNA-reactivity) these cells are negatively regulated and prevented from expanding in GCs presumably by a GC tolerance checkpoint (40–42). Therefore, the HKIR model is ideal to study the effects of Sle1b on GC B cell tolerance pathways of nuclear-Ag-specific B cells.
We transferred purified B cells (2 × 106) from B6.HKIR and B6.HKIR × B6.Sle1b (named HKIR.Sle1b) female mice into syngenic B6 × B6.Sle1b (B6.Sle1b+/−) female recipients that had been immunized with Ars-KLH one wk prior to transfer. B6.Sle1b+/− mice were used as recipients to avoid allo-rejection and B6.Sle1b+/− mice do not display any autoimmune features, as the influence of Sle1b on lymphoid and accessory cell function is recessive (1). We used the anti-clonotypic mAb E4 to detect dual-reactive HKIR B cells as described (40–42). E4-specific primary AFC and GC responses were determined in spleen samples obtained on day 5 after cell transfer. Flow cytometric analysis of splenocytes revealed a significant increase in the percentage of donor-derived E4+PNA+ GC B cells in HKIR.Sle1b→ B6.Sle1b+/− mice compared with HKIR→ B6.Sle1b+/− controls (Fig. 9A–B). These data were consistent with immunohistology results showing more E4+ (red) and E4+PNA+ (yellow) cells in GCs of HKIR.Sle1b→ B6.Sle1b+/− mice (lower panel, Fig. 9C) compared with HKIR→ B6.Sle1b+/− controls (upper panel, Fig. 9C). The E4-specific short-lived AFC response to Ars was also evaluated by ELISpot assay in the recipient mice. We did not, however, observe any significant difference between HKIR.Sle1b→ B6.Sle1b+/− and HKIR→ B6.Sle1b+/− mice in producing E4-specific short-lived AFCs (data not shown).
Figure 9. Augmented GC response of Ars-DNA dual-reactive HKIR B cells in the presence of Sle1b.

Flow cytometric analysis was performed on splenocytes isolated from B6.Sle1b+/− mice receiving 2×106 HKIR or HKIR.Sle1b B cells and stained with the anti-idiotypic mAb E4 in combination with GC B cell markers B220 and PNA. The percentage of B220hiE4+PNA+ Ars-DNA dual reactive B cells in HKIR→ B6.Sle1b+/− and HKIR.Sle1b → B6.Sle1b+/− GCs is shown in rectangular gates (A) and in scatter plots (B). Each circle represents an individual mouse and horizontal bars represent the mean values. (C) Spleen sections from HKIR→ B6.Sle1b+/− (upper panel) and HKIR.Sle1b → B6.Sle1b+/− (lower panel) recipient mice were stained for E4 (red) and PNA (green). High magnification (200×) representative images from 5 mice of each genotype are displayed.
Sle1b expression in B cells, but not in T cells, resulted in elevated Spt-GCs, TFH cells, ANA-specific AFCs and ANAs in B6.Sle1b mice
Next, we evaluated whether phenotypic changes that were observed in T cells (i.e., activation of T cells and increased percentage of GC TFH cells) in B6.Sle1b mice resulted from the primary effect of Sle1b on T cells or were they influenced by B cells expressing Sle1b. Using µMT and TCRβδ−/− mice which lack B and T cells respectively, we generated mixed bone marrow (BM) chimeric mice where in one group of mice (i.e., B6.Sle1b + B6.µMT marrow → B6.µMT) all reconstituted B cells expressed Sle1b while in the other group (i.e., B6.Sle1b + B6.TCRβδ−/− marrow → B6.TCRβδ−/−) all reconstituted T cells expressed Sle1b in the presence of chimeric accessory compartments. We used B6 + B6.µMT BM marrow → B6.µMT and B6 + B6.TCRβδ−/− marrow → B6.TCRβδ−/− chimeras as controls. Only female mice were used for these experiments. These mice were rested for six months before analyzing for the development of Spt-GCs, TFH cells, autoantibody producing AFCs and ANAs.
Flow cytometry analysis on spleen cells obtained from these mice revealed significantly higher percentages of B220+Fas+PNAhi GC B cells (Fig. 10A) and CD4+CXCR5hiPD-1hi TFH cells (Fig. 9B) in B6.Sle1b + B6.µMT BM → B6.µMT mice compared to B6 + B6.µMT BM → B6.µMT controls. Interestingly, increased frequency of GC B cells and TFH cells was not observed in B6.Sle1b + B6.TCRβδ−/− → B6.TCRβδ −/− chimeras where all T cells expressed Sle1b and no significant difference was observed between B6 + B6.TCRβδ−/− → B6.TCRβδ−/− and B6.Sle1b + B6.TCRβδ−/− → B6.TCRβδ−/− mice (Fig. 10A–B). These results were consistent with immunohistological data obtained from spleen sections showing increased frequencies of IgDnegPNA+ GCs with predominantly large GCs in B6.Sle1b + B6.µMT → B6.µMT mice (upper right, Fig. 10C) compared to other three groups of mice showing less frequent and significantly smaller GCs (Fig. 10C). The number of CD4+ T cells also appeared to be increased in B6.Sle1b + B6.µMT BM → B6.µMT mice compared to other three groups (Fig. 10D).
Figure 10. The Sle1b sub-locus primarily affects on B cells.

Flow cytometric analysis was performed to evaluate the percentages of B220+Fas+PNAhi GC B cells (A) and CD4+CXCR5hiPD-1hi TFH cells (B) in splenocytes obtained from B6 + µMT → µMT, B6.Sle1b + µMT → µMT, B6 + TCRβδ−/− → TCRβδ−/− and B6.Sle1b + TCRβδ−/− → TCRβδ−/− chimeras. Spleen sections from these mice were stained for either IgD (blue) and PNA (red) (C) or B220 (blue) and CD4 (red) (D). Original magnification of the images was 100x. The number of ds-DNA- (E), histone- (F), and nucleosome- (G) specific AFCs were measured by ELISpot assay. Total IgG ds-DNA- (H), histone- (I), and nucleosome- (J) specific ANA titers were measured by ELISA. The open circles represent B6 mice while the closed circles represent B6.Sle1b mice. Each circle represents an individual mouse and horizontal bars represent the mean values. Statistical analysis was performed as described in Materials and Methods.
By performing ELISPot assay, we further observed that B6.Sle1b + B6.µMT → B6.µMT mice had significantly higher ds-DNA-, histone- and nucleosome-specific AFCs compared to B6 + B6.µMT → B6.µMT control mice (Fig. 10E–G). In agreement with the GC B cell and TFH cell data (Fig. 10A–B) we did not find any difference in ANA-specific AFCs between B6 + B6.TCRβδ −/− → B6.TCRβδ −/− and B6.Sle1b + B6.TCRβδ −/− → B6.TCRβδ −/− mice (Fig. 10E–G). Analogous to the AFC data we found significantly higher ds-DNA-, histone- and nucleosome-specific Abs in B6.Sle1b + B6.µMT → B6.µMT mice compared to other three groups of mice (Fig. 10H–J).
These data, together with results described above (Fig. 9) indicate that the alteration of the GC checkpoint by Sle1b is B cell autonomous and the effect of Sle1b on T cells appears to be secondary to the B cell defect.
Discussion
In this study, we determined the influence of the Sle1b genomic interval, which contains the lupus associated NZM2410/NZW SLAM family genes, on the GC tolerance pathway or checkpoint which may lead to the development of elevated ANA titers. We utilized spontaneously developed, B cell adoptive transfer and bone marrow transfer models in order to provide a comprehensive analysis of the effects of Sle1b on the GC checkpoint. In the spontaneously developed GC model, we found significantly increased percentage of GC B cells and GC TFH cells in B6.Sle1b female mice relative to B6 female controls. Strong correlation was observed between the elevated numbers of Spt-GCs in B6.Sle1b mice and the increased numbers of ds-DNA-, histone-, and nucleosome-specific AFCs, as well as high titers of serum ANAs. Using the adoptive transfer model, we found an augmented GC response of Ars-DNA dual-reactive HKIR B cells in the presence of Sle1b. Additionally, by generating mixed bone marrow chimeras, we observed that the effect of Sle1b on the alteration of the GC tolerance checkpoint was B cell autonomous and the effect of Sle1b on T cells including GC TFH cells appeared to be secondary to the B cell defect. Together, these data suggest that genes located in Sle1b contribute to the alteration of the GC checkpoint by primarily affecting B cells leading to enhanced Spt-GCs, Spt-TFH cells and the generation of autoantibody-producing AFCs.
Given the roles of SLAM family co-stimulatory molecules in regulating cellular and humoral immunity (14, 15, 55, 56) and the association of SLAM family genes located in Sle1b with lupus (7–9), these genes are strong candidates for perturbing peripheral tolerance checkpoints (i.e., the GC checkpoint) in B6.Sle1b mice. While the data presented here suggest that the lupus-associated SLAM family genes in Sle1b are presumably responsible for the alteration of peripheral tolerance at the GC checkpoint, previous studies using the HEL immunoglobulin transgenic mouse system revealed that the Ly108.1 isoform of the Ly108 gene in Sle1b alters early or central B cell tolerance (8). Recent studies by Keszei et al. implicated the Ly108.1 gene in altering peripheral tolerance (9). The current studies together with the available literature suggest that the Ly108.1 isoform expressed by B6.Sle1b mice may alter both central and peripheral tolerance checkpoints. The polymorphisms in different members of SLAM family genes may also coordinate in altering B cell tolerance at different checkpoints.
One of the intriguing questions in studies of autoreactive B cell activation in autoimmune mouse models is where the site of a break in tolerance occurs. Whereas defects in the central tolerance checkpoint are proposed in causing autoimmunity, the development of autoantibody producing long-lived IgG+ AFCs and memory B cells in lupus cannot be explained by a defect in the central tolerance checkpoint alone as these cells are usually generated in GCs formed in peripheral lymphoid organs. Therefore, peripheral tolerance checkpoints (i.e., the GC checkpoint) may play a crucial role in the prevention of autoreactive B cells from developing into long-lived AFCs and memory B cells, which can persist for years. Numerous autoimmune mouse models, including (NZB/NZW) F1, BXSB, and sanroque, spontaneously develop GCs in the absence of an infection or immunization (24, 33, 36) and all of these mouse models develop autoantibodies against nuclear-self-antigens. However, the role of a dysregulated GC tolerance pathway in autoantibody production in these mice is unclear. Our current results in B6.Sle1b mice demonstrate that increased GC formation is directly correlated to increased numbers of ds-DNA-, histone-, and nucleosome-specific AFCs, as well as increased ANA titers. Direct evidence for the break in peripheral tolerance at the GC checkpoint is further demonstrated by the increase in HKIR B cells populating GCs in the presence of Sle1b. These results implicate a dysregulated GC checkpoint or pathway in ANA production and autoimmunity in B6.Sle1b mice.
Activated B cells can also develop into memory B cells and plasma cells independent of the GC outside of the follicle (58–60). The AFCs generated from this extrafollicular pathway tend to be short-lived and the memory B cells remain largely un-switched (57). Several studies have implicated the role of the extrafollicular pathway of B cell activation in autoimmunity. Autoreactive B cells in MRL.Faslpr mice do not involve the GC pathway, but instead they undergo somatic hypermutation in the T cell zone (58, 59). The (NZB/NZW) F1 mice develop short-lived plasmablasts via an extrafollicular pathway that are autoreactive for ds-DNA and contribute to the autoimmunity (60). Our data indicate that the Sle1b sub-locus primarily affects the GC checkpoint leading to increased numbers of ANA-specific and class-switched IgG+ AFCs and ANAs. While a role of extrafollicular ANA-specific B cells cannot be entirely discounted in our autoimmune model, our data suggest that a major contributor to the break in peripheral tolerance leading to the production of ANA in B6.Sle1b mice is through the alteration of the GC tolerance pathway.
TFH cells play an integral role in the formation of the GC as well as for the maturation and development of GC B cells into long-lived AFCs and memory B cells. Several studies have implicated dysregulated TFH cells in the development of autoimmunity (12, 33, 35–37). The autoimmune mouse models, including mice expressing two copies of TLR7 (i.e., BXSB.yaa mice) and sanroque mice have increased numbers of TFH cells coincident with high titers of ANAs (33, 36, 37). B6.Sle1b mice have increased numbers of Spt-GC B cells along with a significantly higher numbers of Spt-TFH cells. Consistent with studies in other autoimmune mice (33, 36, 37), our current data implicate a role for dysregulated TFH cell numbers in developing autoantibody producing IgG+ AFCs and high titers of ANAs in B6.Sleb mice.
Sle1 or Sle1b appears to affect both B and T cells (5, 7–9). In support for a predominant B cell intrinsic effect, Kumar et al. have demonstrated that the presence of the Ly108.1 isoform from the Sle1 locus renders B cells unresponsive to normal regulation at early B cell tolerance checkpoints (8). We previously showed that the presence of Sle1 on HKIR B cells enables them to escape GC tolerance mechanisms and allows increased participation of HKIR B cells expressing Sle1 in the GC response (42). Our current results from B cell adoptive transfer experiments (Fig. 9) showing increased participation of HKIR.Sle1b B cells in the anti-Ars GC response provide evidence for a B cell intrinsic effect on the breach in peripheral tolerance at the GC checkpoint. These results are in agreement with our bone marrow chimeric data (Fig. 10) showing B cell autonomous effect of Sle1b on increased Spt-GCs, TFH cells, and ANA-specific AFCs and Abs. In contrast, studies by Keszei et al. recently suggested the role for peripheral CD4+ T cells from B6.Sle1b mice in autoantibody responses by transferring purified Sle1b CD4+ T cells or CD62L+ naive CD4+ T cells into bm12 mice (9). However, our bone marrow chimeric data revealed no primary effect of Sle1b on T cells (Fig. 10B) rather T cells were indirectly influenced by the expression of Sle1b in B cells, providing evidence for the requirement of B cell intrinsic defect caused by Sle1b in the development of Spt-GCs, TFH cell expansion and autoantibody response.
One central theme that we observed in these studies is that only the female B6.Sle1b mice had the highest percentages of spontaneously formed GCs, TFH numbers, ANA-specific AFCs and serum ANA titers. Even though male B6.Sle1b mice had higher numbers of Spt-GCs and Spt-TFH cells compared to B6 males the numbers of ANA-specific AFCs were not significantly different from B6 controls. Therefore, Sle1b appears to have the highest penetrance in female mice. In our knowledge, this is the first thorough study showing the differences in B cell/T cell activation, GC formation, and ANA-specific AFC numbers between male and female B6.Sle1b mice. Our results are in accordance with previous report by Grimaldi et al. showing that estrogen alters thresholds for B cell apoptosis and activation (61). The importance of sex hormones in the immune system homeostasis is evident through differences between males and females in B6 mice, as female B6 mice tend to have higher spontaneously activated phenotypes. This is consistent with the findings that an overwhelming percentage of people with autoimmune diseases are female. We cannot rule out the potential role of female chromosome as the inactivation of the second X-chromosome in females can sometimes be incomplete leading to dysregulated gene repression (62) that may contribute to enhanced Spt-GC, Spt-TFH and ANA titers observed in female mice in our studies. We are also unable to exclude the potential protective role of male hormones or chromosome in this process. It is of great interest to further dissect the role of the lupus-associated SLAM family genes in conjunction with sex hormones (i.e., estrogen) or chromosomes in breaking peripheral B cell tolerance at the GC checkpoint leading to the development of autoantibody producing long-lived AFC and memory B cells.
Supplementary Material
Acknowledgements
We thank Drs. Cathie Calkins and Chris Snyder for their critical reading of the manuscript and constructive comments. We also thank Dr. Chetna Soni from our laboratory for her indirect contribution. We thank Dr. Tim Manser (Thomas Jefferson University, Philadelphia, PA) for providing the HKIR mice and Dr. Edward Wakeland for providing B6.Sle1b mice.
These studies were supported by grants from the NIH (AI091670) and from the Arthritis National Research Foundation to Z.S.M.R.
Non standard abbreviations
- GC
germinal center
- Spt-GC
spontaneous germinal center
- AFC
antibody forming cell
- MZ
marginal zone
- PNA
peanut lectin (agglutinin)
- ANA
anti-nuclear antibody
- SRBC
sheep red blood cells
- TFH
follicular helper T cells
- Spt-TFH
spontaneously activated follicular helper T cells
References
- 1.Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. 1994;1:219–229. doi: 10.1016/1074-7613(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 2.Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, Wakeland EK. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6670–6675. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morel L, Wakeland EK. Lessons from the NZM2410 model and related strains. Int. Rev. Immunol. 2000;19:423–446. doi: 10.3109/08830180009055506. [DOI] [PubMed] [Google Scholar]
- 4.Mohan C, Alas E, Morel L, Yang P, Wakeland EK. Genetic dissection of SLE pathogenesis. Sle1 on murine chromosome 1 leads to a selective loss of tolerance to H2A/H2B/DNA subnucleosomes. J. Clin. Invest. 1998;101:1362–1372. doi: 10.1172/JCI728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sobel ES, Satoh M, Chen Y, Wakeland EK, Morel L. The major murine systemic lupus erythematosus susceptibility locus Sle1 results in abnormal functions of both B and T cells. J. Immunol. 2002;169:2694–2700. doi: 10.4049/jimmunol.169.5.2694. [DOI] [PubMed] [Google Scholar]
- 6.Morel L, Blenman KR, Croker BP, Wakeland EK. The major murine systemic lupus erythematosus susceptibility locus, Sle1, is a cluster of functionally related genes. Proc. Natl. Acad. Sci. U. S. A. 2001;98:1787–1792. doi: 10.1073/pnas.031336098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wandstrat AE, Nguyen C, Limaye N, Chan AY, Subramanian S, Tian XH, Yim YS, Pertsemlidis A, Garner HR, Jr, Morel L, Wakeland EK. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity. 2004;21:769–780. doi: 10.1016/j.immuni.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Kumar KR, Li L, Yan M, Bhaskarabhatla M, Mobley AB, Nguyen C, Mooney JM, Schatzle JD, Wakeland EK, Mohan C. Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. 2006;312:1665–1669. doi: 10.1126/science.1125893. [DOI] [PubMed] [Google Scholar]
- 9.Keszei M, Detre C, Rietdijk ST, Munoz P, Romero X, Berger SB, Calpe S, Liao G, Castro W, Julien A, Wu YY, Shin DM, Sancho J, Zubiaur M, Morse HC, 3rd, Morel L, Engel P, Wang N, Terhorst C. A novel isoform of the Ly108 gene ameliorates murine lupus. J. Exp. Med. 2011;208:811–822. doi: 10.1084/jem.20101653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jennings P, Chan A, Schwartzberg P, Wakeland EK, Yuan D. Antigen-specific responses and ANA production in B6.Sle1b mice: a role for SAP. J. Autoimmun. 2008;31:345–353. doi: 10.1016/j.jaut.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croker BP, Gilkeson G, Morel L. Genetic interactions between susceptibility loci reveal epistatic pathogenic networks in murine lupus. Genes Immun. 2003;4:575–585. doi: 10.1038/sj.gene.6364028. [DOI] [PubMed] [Google Scholar]
- 12.Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, Schultz RA, Wakeland EK. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. U. S. A. 2006;103:9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie C, Patel R, Wu T, Zhu J, Henry T, Bhaskarabhatla M, Samudrala R, Tus K, Gong Y, Zhou H, Wakeland EK, Zhou XJ, Mohan C. PI3K/AKT/mTOR hypersignaling in autoimmune lymphoproliferative disease engendered by the epistatic interplay of Sle1b and FASlpr. Int. Immunol. 2007;19:509–522. doi: 10.1093/intimm/dxm017. [DOI] [PubMed] [Google Scholar]
- 14.Cannons JL, Tangye SG, Schwartzberg PL. SLAM family receptors and SAP adaptors in immunity. Annu. Rev. Immunol. 2011;29:665–705. doi: 10.1146/annurev-immunol-030409-101302. [DOI] [PubMed] [Google Scholar]
- 15.Detre C, Keszei M, Romero X, Tsokos GC, Terhorst C. SLAM family receptors and the SLAM-associated protein (SAP) modulate T cell functions. Semin. Immunopathol. 2010;32:157–171. doi: 10.1007/s00281-009-0193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma CS, Nichols KE, Tangye SG. Regulation of cellular and humoral immune responses by the SLAM and SAP families of molecules. Annu. Rev. Immunol. 2007;25:337–379. doi: 10.1146/annurev.immunol.25.022106.141651. [DOI] [PubMed] [Google Scholar]
- 17.Koh AE, Njoroge SW, Feliu M, Cook A, Selig MK, Latchman YE, Sharpe AH, Colvin RB, Paul E. The SLAM family member CD48 (Slamf2) protects lupus-prone mice from autoimmune nephritis. J. Autoimmun. 2011;37:48–57. doi: 10.1016/j.jaut.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berek C, Milstein C. Mutation drift and repertoire shift in the maturation of the immune response. Immunol. Rev. 1987;96:23–41. doi: 10.1111/j.1600-065x.1987.tb00507.x. [DOI] [PubMed] [Google Scholar]
- 19.Weiss U, Rajewsky K. The repertoire of somatic antibody mutants accumulating in the memory compartment after primary immunization is restricted through affinity maturation and mirrors that expressed in the secondary response. J. Exp. Med. 1990;172:1681–1689. doi: 10.1084/jem.172.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manser T, Wysocki LJ, Margolies MN, Gefter ML. Evolution of antibody variable region structure during the immune response. Immunol. Rev. 1987;96:141–162. doi: 10.1111/j.1600-065x.1987.tb00513.x. [DOI] [PubMed] [Google Scholar]
- 21.Kelsoe G. Life and death in germinal centers (redux) Immunity. 1996;4:107–111. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- 22.Pulendran B, van Driel R, Nossal GJ. Immunological tolerance in germinal centres. Immunol. Today. 1997;18:27–32. doi: 10.1016/s0167-5699(97)80011-4. [DOI] [PubMed] [Google Scholar]
- 23.Pulendran B, Kannourakis G, Nouri S, Smith KG, Nossal GJ. Soluble antigen can cause enhanced apoptosis of germinal-centre B cells. Nature. 1995;375:331–334. doi: 10.1038/375331a0. [DOI] [PubMed] [Google Scholar]
- 24.Luzina IG, Atamas SP, Storrer CE, daSilva LC, Kelsoe G, Papadimitriou JC, Handwerger BS. Spontaneous formation of germinal centers in autoimmune mice. J. Leukoc. Biol. 2001;70:578–584. [PubMed] [Google Scholar]
- 25.Diamond B, Scharff MD. Somatic mutation of the T15 heavy chain gives rise to an antibody with autoantibody specificity. Proc. Natl. Acad. Sci. U. S. A. 1984;81:5841–5844. doi: 10.1073/pnas.81.18.5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diamond B, Katz JB, Paul E, Aranow C, Lustgarten D, Scharff MD. The role of somatic mutation in the pathogenic anti-DNA response. Annu. Rev. Immunol. 1992;10:731–757. doi: 10.1146/annurev.iy.10.040192.003503. [DOI] [PubMed] [Google Scholar]
- 27.Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, Weigert M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J. Exp. Med. 1990;171:265–292. doi: 10.1084/jem.171.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shlomchik MJ, Marshak-Rothstein A, Wolfowicz CB, Rothstein TL, Weigert MG. The role of clonal selection and somatic mutation in autoimmunity. Nature. 1987;328:805–811. doi: 10.1038/328805a0. [DOI] [PubMed] [Google Scholar]
- 29.Olee T, Lu EW, Huang DF, Soto-Gil RW, Deftos M, Kozin F, Carson DA, Chen PP. Genetic analysis of self-associating immunoglobulin G rheumatoid factors from two rheumatoid synovia implicates an antigen-driven response. J. Exp. Med. 1992;175:831–842. doi: 10.1084/jem.175.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim SJ, Zou YR, Goldstein J, Reizis B, Diamond B. Tolerogenic function of Blimp-1 in dendritic cells. J. Exp. Med. 2011;208:2193–2199. doi: 10.1084/jem.20110658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tiller T, Kofer J, Kreschel C, Busse CE, Riebel S, Wickert S, Oden F, Mertes MM, Ehlers M, Wardemann H. Development of self-reactive germinal center B cells and plasma cells in autoimmune Fc gammaRIIB-deficient mice. J. Exp. Med. 2010;207:2767–2778. doi: 10.1084/jem.20100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat. Rev. Immunol. 2005;5:853–865. doi: 10.1038/nri1714. [DOI] [PubMed] [Google Scholar]
- 33.Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, Schwartzberg PL, Cook MC, Walters GD, Vinuesa CG. Follicular helper T cells are required for systemic autoimmunity. J. Exp. Med. 2009;206:561–576. doi: 10.1084/jem.20081886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat. Rev. Rheumatol. 2012;8:337–347. doi: 10.1038/nrrheum.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King C, Tangye SG, Mackay CR. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu. Rev. Immunol. 2008;26:741–766. doi: 10.1146/annurev.immunol.26.021607.090344. [DOI] [PubMed] [Google Scholar]
- 36.Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, Yu D, Domaschenz H, Whittle B, Lambe T, Roberts IS, Copley RR, Bell JI, Cornall RJ, Goodnow CC. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435:452–458. doi: 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 37.Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC, 3rd, Leonard WJ, Roopenian DC. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc. Natl. Acad. Sci. U. S. A. 2009;106:1518–1523. doi: 10.1073/pnas.0807309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heltemes-Harris L, Liu X, Manser T. Progressive surface B cell antigen receptor down-regulation accompanies efficient development of antinuclear antigen B cells to mature, follicular phenotype. J. Immunol. 2004;172:823–833. doi: 10.4049/jimmunol.172.2.823. [DOI] [PubMed] [Google Scholar]
- 39.Liu X, Manser T. Antinuclear antigen B cells that down-regulate surface B cell receptor during development to mature, follicular phenotype do not display features of anergy in vitro. J. Immunol. 2005;174:4505–4515. doi: 10.4049/jimmunol.174.8.4505. [DOI] [PubMed] [Google Scholar]
- 40.Alabyev B, Rahman ZS, Manser T. Quantitatively reduced participation of anti-nuclear antigen B cells that down-regulate B cell receptor during primary development in the germinal center/memory B cell response to foreign antigen. J. Immunol. 2007;178:5623–5634. doi: 10.4049/jimmunol.178.9.5623. [DOI] [PubMed] [Google Scholar]
- 41.Rahman ZS, Alabyev B, Manser T. FcgammaRIIB regulates autoreactive primary antibody-forming cell, but not germinal center B cell, activity. J. Immunol. 2007;178:897–907. doi: 10.4049/jimmunol.178.2.897. [DOI] [PubMed] [Google Scholar]
- 42.Vuyyuru R, Mohan C, Manser T, Rahman ZS. The lupus susceptibility locus Sle1 breaches peripheral B cell tolerance at the antibody-forming cell and germinal center checkpoints. J. Immunol. 2009;183:5716–5727. doi: 10.4049/jimmunol.0804215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Notidis E, Heltemes L, Manser T. Dominant, hierarchical induction of peripheral tolerance during foreign antigen-driven B cell development. Immunity. 2002;17:317–327. doi: 10.1016/s1074-7613(02)00392-8. [DOI] [PubMed] [Google Scholar]
- 44.Vora KA, Tumas-Brundage KM, Lentz VM, Cranston A, Fishel R, Manser T. Severe attenuation of the B cell immune response in Msh2-deficient mice. J. Exp. Med. 1999;189:471–482. doi: 10.1084/jem.189.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rahman ZS, Shao WH, Khan TN, Zhen Y, Cohen PL. Impaired apoptotic cell clearance in the germinal center by Mer-deficient tingible body macrophages leads to enhanced antibody-forming cell and germinal center responses. J. Immunol. 2010;185:5859–5868. doi: 10.4049/jimmunol.1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cappione A, 3rd, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, Sanz I. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J. Clin. Invest. 2005;115:3205–3216. doi: 10.1172/JCI24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, Kirchner T, Kalden JR, Herrmann M. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 48.Basso K, Dalla-Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv. Immunol. 2010;105:193–210. doi: 10.1016/S0065-2776(10)05007-8. [DOI] [PubMed] [Google Scholar]
- 49.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 50.Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri-Shirazi M, Orazi A, Chaganti RS, Rothman P, Stall AM, Pandolfi PP, Dalla-Favera R. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat. Genet. 1997;16:161–170. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- 51.Onizuka T, Moriyama M, Yamochi T, Kuroda T, Kazama A, Kanazawa N, Sato K, Kato T, Ota H, Mori S. BCL-6 gene product, a 92- to 98-kD nuclear phosphoprotein, is highly expressed in germinal center B cells and their neoplastic counterparts. Blood. 1995;86:28–37. [PubMed] [Google Scholar]
- 52.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, Dong C. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, Ellyard JI, Parish IA, Ma CS, Li QJ, Parish CR, Mackay CR, Vinuesa CG. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 54.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Veillette A, Latour S. The SLAM family of immune-cell receptors. Curr. Opin. Immunol. 2003;15:277–285. doi: 10.1016/s0952-7915(03)00041-4. [DOI] [PubMed] [Google Scholar]
- 56.Engel P, Eck MJ, Terhorst C. The SAP and SLAM families in immune responses and X-linked lymphoproliferative disease. Nat. Rev. Immunol. 2003;3:813–821. doi: 10.1038/nri1202. [DOI] [PubMed] [Google Scholar]
- 57.Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J. Exp. Med. 2012;209:597–606. doi: 10.1084/jem.20111696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- 59.Shlomchik MJ, Euler CW, Christensen SC, William J. Activation of rheumatoid factor (RF) B cells and somatic hypermutation outside of germinal centers in autoimmune-prone MRL/lpr mice. Ann. N. Y. Acad. Sci. 2003;987:38–50. doi: 10.1111/j.1749-6632.2003.tb06031.x. [DOI] [PubMed] [Google Scholar]
- 60.Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, Radbruch A, Hiepe F, Manz RA. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J. Exp. Med. 2004;199:1577–1584. doi: 10.1084/jem.20040168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grimaldi CM, Cleary J, Dagtas AS, Moussai D, Diamond B. Estrogen alters thresholds for B cell apoptosis and activation. J. Clin. Invest. 2002;109:1625–1633. doi: 10.1172/JCI14873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brooks WH. X chromosome inactivation and autoimmunity. Clin. Rev. Allergy Immunol. 2010;39:20–29. doi: 10.1007/s12016-009-8167-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.